

Abstract
Mitochondrial DNA (mtDNA) mutations are a common cause of genetic disease with pathogenic mtDNA mutations being detected in approximately 1 in 250 live births1-3 and at least 1 in 10,000 adults in the UK affected by mtDNA disease4. Treatment options for patients with mtDNA disease are extremely limited and are predominantly supportive in nature. MtDNA is transmitted maternally and it has been proposed that nuclear transfer techniques may be an approach to prevent the transmission of human mtDNA disease5,6. Here we show that transfer of pronuclei between abnormally fertilised human zygotes results in minimal carry-over of donor zygote mtDNA and is compatible with onward development to the blastocyst stage in vitro. By optimising the procedure we found the average level of carry-over following transfer of two pronuclei is <2.0%, with many of the embryos containing no detectable donor mtDNA. We believe that pronuclear transfer between zygotes, as well as the recently described metaphase II spindle transfer, has potential to prevent the transmission of mtDNA disease in humans.
MtDNA mutations are maternally transmitted7. MtDNA is present in all cells in multiple copies and in patients with mtDNA disease either all mtDNA copies are mutated (termed homoplasmy) or there is a mixture of wild-type and mutated mtDNA (termed heteroplasmy)8. Studies of human pedigrees with transmitted mtDNA mutations have shown that clinical disease is only seen in those patients with high loads of mutated mtDNA in affected tissues (usually greater than 60% mutated mtDNA)9,10. There has been very limited success in developing effective treatment for mtDNA disease and genetic counselling combined with prenatal or pre-implantation genetic diagnosis is increasingly being offered to women who carry pathogenic mtDNA mutations11. However, these techniques will only be of value to women who have low levels of mtDNA mutations in oocytes.
Following the granting of a research licence by the Human Fertilisation and Embryology Authority (UK), and informed consent by the donors, we used abnormally fertilised (unipronuclear or tripronuclear) human zygotes (one cell embryos) generated from a human IVF programme to study the feasibility of pronuclear transfer to prevent mtDNA disease transmission from mother to child. Unipronuclear and tripronuclear zygotes are not normally used in fertility treatment. Our studies involved the transfer of one or two pronuclei between abnormally fertilised zygotes (Figure 1, Supplementary Figure 1). Following treatment with cytoskeletal inhibitors (nocodazole and cytochalasin B), pronuclei were removed from a donor zygote within a karyoplast containing a small volume of cytoplasm. Karyoplasts were placed under the zona pellucida of a recipient zygote and were fused using inactivated viral envelope proteins of the Hemagglutinating Virus of Japan (HVJ-E). Reconstituted zygotes were cultured for 6-8 days to monitor development in vitro.
Figure 1. Pronuclear transfer using abnormally fertilised human zygotes.
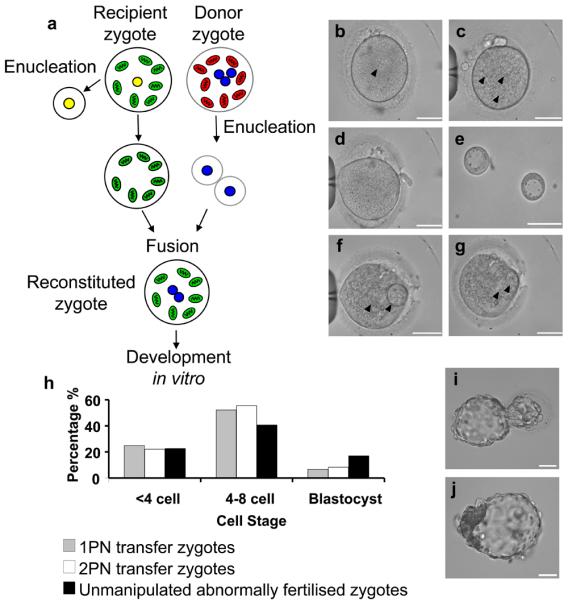
a-g, Transfer of two pronuclei between human zygotes. a, Schematic diagram showing recipient zygote (one pronucleus which is removed) and donor zygote (three pronuclei, two of which are removed and fused with the recipient zygote). b, Recipient zygote containing a single pronucleus (marked with arrow) which is removed by a biopsy pipette to leave an enucleated zygote d. c, Donor zygote with three pronuclei (marked with arrows) and two of these pronuclei removed as karyoplasts e. f, Enucleated recipient zygote with two pronuclear karyoplasts from the donor zygote (arrows) prior to fusion. g, Recipient zygote 20 minutes after transfer already showing fusion of the karyoplast membranes (arrow). h, Development of unmanipulated abnormally fertilised zygotes (n=76; black bars), one pronuclear (n=44; grey bars) and two pronuclear (n=36; white bars) transfer embryos. i, Day 7 hatched blastocyst containing two donor pronuclei. Scale bars are 50μm.
We first confirmed that pronuclear transfer between human zygotes was associated with a change in the nuclear genotype of the embryo by analysing microsatellite markers. In all embryos studied, informative markers confirmed that the reconstituted pronuclear transfer embryo contained donor embryo nuclear genotype (see Supplementary Table 1). We then determined if pronuclear transfer was compatible with onward development in vitro. This was complicated by the fact that abnormally fertilised zygotes have limited potential for development to the blastocyst stage in vitro (17%) compared with normally fertilised embryos (32%). Nonetheless, following pronuclear transfer, zygotes showed onward development with 10 out of 44 (22.7%) of one pronuclear transfer zygotes and 8 out of 36 (22.2%) of two pronuclear transfer zygotes developing to >8 cell stage. We found no difference in embryo development at any stage whether we transferred one or two pronuclei. Following two pronuclear transfer, 8.3% of abnormally fertilised embryos developed to the blastocyst stage (Figure 1h and i). This is approximately 50% of the blastocyst rate for unmanipulated abnormally fertilised embryos; as there is no reliable morphological indicator to distinguish between the male and female pronucleus in the human zygote, it is likely that the decline in blastocyst formation is partly due to absence of either a maternal or paternal genome.
Having established that pronuclear transfer is compatible with onward development of human embryos, we next determined the carry-over of donor mtDNA genotype in the reconstituted pronuclear transfer embryos (Figure 2). We sequenced the non-coding mtDNA control region from both the pronuclear donor and pronuclear recipient embryos (Figure 2b) and identified polymorphic mtDNA variants which were unique to donor or recipient embryo, thereby allowing the determination of mtDNA carry-over in the pronuclear transfer embryo. Hot last cycle-PCR RFLP assays were developed specifically for these mtDNA variants (Figure 2c) and used to analyse mtDNA extracted from whole embryos. We found that there was variation in the amount of mtDNA genotype from the donor zygote which is transferred to the two pronuclear transfer embryo (8.1% ±7.6; mean ± SD n=8) (Figure 2d).
Figure 2. MtDNA analysis of pronuclear transfer embryos.

a, Schematic diagram showing the potential transfer of donor zygote mtDNA to the recipient zygote. b, Sequence electropherograms of mtDNA non-coding control region in donor and recipient zygotes with the sequence variant used for last hot cycle PCR-RFLP assay highlighted. c, Scheme of RFLP designed using the sequence variant. d, Last hot cycle-PCR RFLP analysis of donor mtDNA carry-over detected in two pronuclear transfer embryos with products separated by 12% nondenaturing polyacrylamide gel electrophoresis. U: undigested, C1 and C2: controls (C1: donor embryo for E3, recipient embryo for E1 and E2; C2: donor embryo for E1 and E2, recipient embryo for E3). e, MtDNA copy number in human mature oocytes.
There are many factors which could affect the carry-over of mtDNA following pronuclear transfer. We therefore studied the mtDNA copy number present in human oocytes. Similar to the results in mice and previous studies of human oocytes at various stages of development12 13 14, we found marked variation in the mtDNA copy number (Figure 2e) and this may contribute to variation in level of mtDNA carry-over. Previous studies have investigated heteroplasmy levels in blastomeres obtained from donated heteroplasmic embryos and have reported variation of 0-19% between individual blastomeres from the same embryo11,15. We therefore determined whether the proportion of donor mtDNA genotype also varied between blastomeres in the reconstituted embryos following transfer of two pronuclei (Figure 3a, b). In 1/8 embryos there was no detectable donor mtDNA in any blastomere. In the other seven embryos which contained donor zygote mtDNA, there was variation in level of donor mtDNA genotype between blastomeres (Figure 3b). Although this variation is similar to previous reports on heteroplasmic human embryos11,15, we wished to minimise the carry-over of donor zygote mtDNA and therefore explored techniques to reduce the amount of cytoplasm contained within the pronuclear karyoplast. We focused on careful manipulation of the pronuclear karyoplast and we were able to remove the pronuclei with a minimal amount of cytoplasm (Figure 3c). Using hot last cycle-PCR RFLP assays we demonstrated that the mtDNA carry-over was significantly lower (P<0.005), with 4/9 embryos containing undetectable levels of mtDNA carry-over (Figure 3d, e). The average mtDNA carry-over in all remaining embryos was <2% (mean 1.68 ± 1.81% mean ± SD n=9). These embryos also revealed much less variation in mtDNA carry-over between individual blastomeres (Fig 3e). These levels of mtDNA are equivalent to those seen in unaffected individuals in epidemiological studies1.
Figure 3. MtDNA analysis of individual blastomeres disaggregated from pronuclear transfer embryos.

a, Last hot cycle PCR RFLP of individual blastomeres from a pronuclear transfer embryo showing variable levels of mtDNA donor genotype in individual blastomeres. The arrow indicates the band representing carry-over mtDNA. b, Analysis of levels of donor mtDNA carry-over in individual blastomeres from 8 embryos prior to modifications to minimise levels of donor mtDNA in pronuclear karyoplasts. In some embryos not all blastomeres could be collected. Figures represent the percentage mtDNA carry-over in individual blastomeres following pronuclear transfer. nd: non-detectable. c, Image of pronuclear karyoplasts after additional manipulation showing minimal amount of donor cytoplasm when compared with Figure 1e. Scale bar 25μm. d, Last hot cycle PCR RFLP of individual blastomeres from a pronuclear transfer embryo showing no detectable levels of mtDNA donor genotype in individual blastomeres. The arrow indicates the band representing carry-over mtDNA. e, Analysis of levels of donor mtDNA carry-over in individual blastomeres from 9 embryos following improvements to pronuclear karyoplast removal. In some embryos, not all blastomeres could be collected. Figures represent the percentage of mtDNA carry-over in individual blastomeres following pronuclear transfer. nd: non-detectable.
Very recently a related technique, metaphase II spindle transfer between unfertilised metaphase II oocytes, has been reported using non-human primate oocytes16. This resulted in the birth of live offspring in which the authors were unable to detect donor mtDNA using a less sensitive assay than we have used (lower limit of detection was 3% compared with <0.5%). Whilst our optimised techniques of pronuclear extraction resulted in <3% carry-over, we were nonetheless interested to determine whether the technique of metaphase II spindle transfer might offer the possibility of further reducing the level of mtDNA carry-over. We therefore measured the mtDNA copy number in karyoplasts containing the metaphase II spindle from freshly harvested human oocytes donated to research. We found no significant difference in the mtDNA copy number between metaphase II spindle karyoplasts (13222 ± 5733 mean ± SEM, n=21) compared with double pronuclear karyoplasts (18316 ±4336 mean ± SEM n=12). The wide variation within both groups of karyoplasts is likely due to the vastly different copy numbers observed in human oocytes (Figure 2f). We conclude from this that both approaches would be effective in greatly reducing the risk of mtDNA disease.
Our studies show that in human zygotes, pronuclear transfer has the potential to “treat” human mtDNA disease at a genetic level. The recent development of metaphase II spindle transfer has confirmed in non-human primates that this closely related method also holds great promise. The comparative value of both techniques has not been established in the same animal model or human oocytes, but both have potential advantages. The metaphase II spindle is smaller and technically easier to remove. However, it is not surrounded by a membrane and without the use of a DNA stain, it would be difficult to eliminate the possibility that some chromosomes may not be aligned on the metaphase plate or associated with the spindle as has been previously reported in human oocytes from older women 17 and in response to exposure to ambient conditions18. Studies in mice have shown that pronuclear transfer limits mtDNA transfer to subsequent generations19. In addition, the pronuclei are easier to visualise than the metaphase II spindle but they are also larger and their manipulation may induce more cellular trauma. Our studies in human zygotes have been particularly challenging since working with abnormally fertilised zygotes is technically more difficult than using normally fertilised (two pronuclear) zygotes and is less likely to yield normal embryos due to abnormal chromosomal constitution20. Despite these problems we observed development of the manipulated embryos at approximately 50% of the abnormal embryos which have not been manipulated and shown either no detectable or very low levels of mtDNA carry-over.
In view of the lack of available treatment for these patients and their families21, preventing the transmission of mtDNA disease is a priority. Whilst mtDNA mutations are common, pronuclear or metaphase II spindle transfer is unlikely to be of value for asymptomatic individuals or those with mild mtDNA disease in the family. However, in some families, mtDNA disease can affect multiple family members with catastrophic consequences22 – for these families pronuclear transfer may be an option that mothers who carry mtDNA mutations would consider. MtDNA mutations which are maternally inherited are either homoplasmic or heteroplasmic and high loads of mutated mtDNA are necessary before there is clinical disease (usually >60% of total mtDNA)8. We have shown that we can generate human embryos with donor mtDNA carry-over at levels well below the disease threshold and indeed unlikely to be detected except with very sensitive genetic techniques. With inherited mtDNA mutations there is little evidence of increasing levels of mutated mtDNA in tissues with time, in fact the opposite occurs with loss of mutation in some tissues23, and thus the very low levels of mtDNA carry-over detected in some embryos will not cause mtDNA disease.
We believe the data presented in this paper on human zygotes and their development show that pronuclear transfer has the potential to prevent the transmission of mtDNA disease in humans. Manipulation of human oocytes and zygotes has the potential to cause chromosomal or epigenetic abnormalities24 in the developing embryo which require further study to ensure the safety of different techniques. We believe our study in human zygotes and embryos represents a major advance towards preventing transmission of disease in patients with mtDNA mutations.
METHODS SUMMARY
Human embryos and manipulations
Abnormally fertilised human zygotes and metaphase II oocytes were obtained from patients undergoing fertility treatment at the Newcastle Fertility Centre at Life following informed consent. The projects were licensed by the Human Fertilisation and Embryology Authority (HFEA) and approved by the Newcastle and North Tyneside Local Ethics Committee. Pronuclear transfer was performed using abnormally fertilised human zygotes generated following in vitro fertilisation (IVF) or intracytoplasmic sperm injection (ICSI). Abnormal zygotes were identified on day 1 of development by the presence of one pronucleus (unipronucleate) or three pronuclei (tripronucleate) 18-19 hours after insemination. Karyoplasts containing pronuclei and surrounding cytoplasm were removed from the donor zygote using a biopsy pipette and transferred to a recipient zygote. Following fusion, the reconstituted zygotes were either cultured for 6-8 days to monitor development to the blastocyst stage or were cultured before being disaggregated for analysis of mtDNA in individual blastomeres.
Analysis of mtDNA
To determine the carry-over of donor zygote mtDNA we sequenced the non-coding control region and determined differences between donor and recipient mtDNA sequences. We devised last hot cycle-PCR RFLP assays to differentiate between donor and recipient mtDNA. Following cell lysis, extracted DNA was amplified, digested and separated on polyacrylamide gels. The relative amount of each genotype was determined by quantification of individual bands. MtDNA copy number was determined in oocytes, early embryos and karyoplasts by real-time PCR using a probe to the MT-ND1 gene of the mitochondrial genome.
METHODS
Human oocytes and embryos
Abnormally fertilised human zygotes and metaphase II oocytes were obtained from patients undergoing fertility treatment at the Newcastle Fertility Centre at Life following informed consent. The projects were licensed by the Human Fertilisation and Embryology Authority (HFEA) and approved by the Newcastle and North Tyneside Local Ethics Committee. Pronuclear transfer was performed using abnormally fertilised human zygotes generated following in vitro fertilisation (IVF) or intracytoplasmic sperm injection (ICSI). Abnormal zygotes were identified on day 1 of development by the presence of one pronucleus (unipronucleate) or three pronuclei (tripronucleate) 18-19 hours after insemination. For metaphase II spindle removal, freshly harvested metaphase II oocytes from consenting women were denuded of cumulus cells using hyaluronidase (1X HYASE, Vitrolife). Mature oocytes (metaphase II) oocytes used for the study of mtDNA copy number included in vitro matured oocytes, and oocytes that failed to undergo fertilisation following ICSI
Pronuclear and metaphase II spindle karyoplast removal
Zygotes were transferred to G1v5 Plus medium (Vitrolife) containing cytochalasin B (5μg/ml) and nocodazole (10μg/ml) at 37°C with 7% CO2 immediately before manipulation or for 30 minutes prior to manipulation for improved karyoplast removal. Zygotes were incubated in G1v5 Plus medium (Vitrolife) containing cytochalasin B (5μg/ml) and nocodazole (10μg/ml) at 37°C with 7% CO2 during the procedure. Manipulations were performed using an inverted microscope (Nikon Eclipse TE2000-U) equipped with a micromanipulation system (Integra Ti, Research Instruments, UK). Zygotes were immobilised with a holding pipette and a small ablation made in the zona pellucida using a microsurgical laser (Saturn Active, Research Instruments). A customised biopsy pipette with an inner diameter (ID) of 25μm (Rochford Medical, UK) was inserted under the zona pellucida. The pronucleus and surrounding cytoplasm were then aspirated into the biopsy pipette as a membrane-bound karyoplast. For transfer of a single pronucleus, we removed a pronuclear karyoplast from either a unipronucleate or tripronucleate donor zygote and transferred this to a recipient zygote containing only one pronucleus. The recipient zygote was either a unipronucleate zygote, which required no manipulation prior to transfer, or a tripronucleate zygote from which two pronuclei had been removed. Thus, the reconstituted zygotes contained two pronuclei. In experiments involving transfer of two pronuclei, we removed pronuclei either as two individual pronuclear karyoplasts or a single karyoplast containing both pronuclei. These karyoplasts were then transferred to an enucleated recipient zygote such that the reconstructed zygote contained two pronuclei. For metaphase II spindle removal, oocytes were incubated in G1 medium (Vitrolife) containing 2.5μg/ml cytochalasin B for 10 minutes prior to manipulation and throughout the procedure as above. The spindle was visualized using polarized light birefringence (Oosight Meta Imaging System, Cambridge Research and Instrumentation, CRi). Oocytes were immobilised with a holding pipette and the zona pellucida thinned using a microsurgical laser (Saturn Active, Research Instruments). A biopsy pipette with an ID of 18-20μm was inserted through the zona pellucida and the spindle and surrounding cytoplasm removed from the oocyte as a membrane-bound karyoplast.
Pronuclear karyoplast fusion
Pronuclear karyoplasts were transferred within a biopsy pipette to a 1μl drop of HVJ-E (GenomONE™-CFEX HVJ Envelope Cell Fusion Kit, Cosmo Bio Co) and a small volume of the suspension approximately equal to the volume of the karyoplast aspirated into the pipette. The pipette was then moved to a drop containing a recipient zygote. The pipette was inserted into the zygote through a small ablation in the zona pellucida and the HVJ-E and pronuclear karyoplast aspirated into the perivitelline space ensuring good contact between the karyoplast and plasma membrane. Fusion of the pronuclear karyoplast with the recipient zygote was confirmed visually and usually occurred within 10 minutes up to 1 hour after transfer. Manipulated zygotes were transferred to G1v5 Plus medium (Vitrolife) and cultured at 37°C with 7% CO2. Embryos were transferred to G2v5 Plus medium (Vitrolife) on day 3 of development and cultured at 37°C with 7% CO2 up to day 7. Embryos for mitochondrial DNA analysis were then transferred to sterile 0.5ml microfuge tubes and stored at −80°C until DNA extraction.
Manipulations to obtain individual blastomeres
Pronuclear transfer embryos were disaggregated into individual blastomeres by micromanipulation or removal of the zona pellucida using Acid tyrode's solution. For micromanipulation, the embryo was placed in G-PGD medium (Vitrolife) and immobilised with a holding pipette. A hole was made in the zona pellucida using the microsurgical laser and individual blastomeres removed with a biopsy pipette. For removal of the zona pellucida, the embryo was placed briefly in Acid tyrode's solution until the zona pellucida had dissolved and was then transferred to G-PGD medium. Individual blastomeres were disaggregated by continual pipetting and transferred to sterile 0.5ml microfuge tubes for analysis.
Embryo and blastomeres lysis
Individual embryos or blastomeres were lysed for 2 hours in a lysis buffer (50mM Tris-HCl, pH 8.5, 1mM EDTA, 0.5% Tween-20 and 200μg/ml proteinase K) at 55°C. The enzyme was then inactivated by incubation at 95°C for 10 minutes.
MtDNA sequencing
The non-coding control region of the mitochondrial genome was amplified using two rounds of PCR amplification as described previously25 with the following modification: secondary PCR reactions were performed with 4 sets of overlapping M13-tailed primers (primer nucleotide positions: D1F: 15758-15777 and D1R: 019-001, D2F: 16223-16244 and D2R: 129-110, D3F: 16548-16569 and D3R: 389-370, D4F: 323-343 and D4R: 771-752) with an annealing temperature of 58°C. PCR products were purified using ExoSapIT (Amersham Biosciences) then sequenced on an ABI3170 Genetic Analyser (Applied Biosystems) with BigDye Terminator cycle sequencing chemistries (v3.1, Applied Biosystems). Sequences were directly compared to the revised Cambridge Reference Sequence for human mtDNA26 (GenBank Accession number: AC_000021.2) using SeqScape software (v2.1.1, Applied Biosystems).
Amount of donor zygote mtDNA carry-over in pronuclear transfer embryos
Level of donor zygote mtDNA carry-over was determined by last hot cycle PCR-restriction fragment length polymorphism (RFLP) analysis. Separate assays were developed for each discriminatory mtDNA sequence variant identified and were performed as described previously27,28, with modifications as listed in Supplementary Table 2. Fragments containing the sequence variants of interest were amplified by PCR using primers listed in the table and a last hot cycle performed with 5μCi -dCTP (3,000 Ci/mmol). Equal amounts (1,000 counts) of precipitated labelled products were digested overnight with 10 U of assay-specific restriction enzyme (New England Biolabs, Hitchin, UK). Restriction fragments were separated by 12% non-denaturing polyacrylamide gel electrophoresis, dried onto a support, exposed to a Phosphorimager screen (Molecular Dynamics, Eugene, Oregon) and analysed with ImageQuant software (Molecular Dynamics). Carry-over donor zygote mtDNA was calculated as the percentage of total mtDNA in the recipient embryo.
Quantitative Real-Time PCR
Quantitative real-time PCR was performed using a previously designed TaqMan probe for the MT-ND1 gene (MT-ND1L3506-3529) and PCR primers (forward primer, L3485-3504, reverse primer, H3532-3553)29,30 The reaction mixture consisted of 1μl single cell lysate, 9.5μl nanopure water, 12.5μl TaqMan Universal MasterMix (2.5μl 10 × buffer A, 5μl 10mM MgCl2, 0.5μl each dNTP (10mM), 0.25μl 1U/μl AmpErase uracil-N-glycosylase (UNG), 0.13μl 5U/μl AmpliTaq Gold DNA polymerase, 2.62μl nanopure water; Applied Biosystems, UK), 300nM forward and reverse primer and 100nM fluorgenic probe. Each reaction was completed in triplicate and performed using the ABI PRISM 7000 Sequence Detection System (Applied Biosystems, UK). Amplification conditions were: 50°C for 2mins, 95°C for 10mins, followed by 40 cycles of 95°C for 15sec and 60°C for 1min. A template encompassing the MT-ND1 region was amplified by PCR (forward primer, L3017-3036, reverse primer, H4057-4037) and the gel purified PCR product (QIAEX II Gel Extraction kit, Qiagen) used as a standard control. The concentration of the control template was determined using a spectrophotometer and this value used to calculate the copy number. Serial dilutions of the MT-ND1 template were amplified in triplicate in the same experiment as the samples and a standard curve generated by plotting the logarithm of the copy number against the mean threshold cycle (Ct). The standard curve was then used to calculate the mtDNA copy number for each sample.
Genotyping nuclear DNA
Ovarian follicular cells and sperm were used for donor and recipient nuclear genotype analysis. DNA extraction from follicular cells was performed using the QIAamp® DNA Mini kit according to the manufacturer's instructions (Qiagen). Sperm DNA was extracted in 200μl 5% washed Chelex beads (Sigma), 2μl proteinase K and 7μl 10mM DTT. Following a 4-hour incubation at 56°C, the enzyme was inactivated by incubation at 95°C for 10 minutes. The Chelex beads were removed from the DNA samples by centrifugation at 13,000 rpm for 3 minutes. Whole genome amplification from 4 individual embryos cultured to the 2-8-cell stage, was performed using the REPLI-g® Mini kit (Qiagen, Crawley, UK). Briefly, embryos lacking the zona pellucida were lysed in 2.5μl lysis buffer (200mM NaOH, 50mM DTT) for 10 minutes at 65°C. Lysis was terminated with 2.5μl 200mM Tricine. Genome amplification was achieved in a 50μl reaction for 16 hours at 30°C according to the manufacturer's instructions. The reaction was terminated by incubation at 65°C for 3 minutes. Follicular cell and sperm genomic DNA, and whole genome amplified embryo DNA were analysed for 16 polymorphic microsatellite markers using the PowerPlex® 16 System (Promega). PCR reactions were carried out in a volume of 12.5μl containing 1ng of DNA, 1X Gold Star buffer, 1X PowerPlex® 16 primer pair mix and 2 units of AmpliTaq Gold DNA polymerase (Applied Biosystems). Amplification was performed in a GeneAmp PCR system 9700 thermal cycler (Applied Biosystems) as per manufacturer's instructions. One microlitre of PCR product was diluted in 9μl of Hi-Di™ Formamide (Applied Biosystems) and 1μl of ILS600 size standard (Promega), denatured at 95°C for 3 minutes and immediately cooled on ice for 3 minutes. The same treatment was carried out with 1μl of PowerPlex® 16 System Allelic Ladder (Promega). Amplified fragments were detected using a 3130xl Genetic Analyser (Applied Biosystems) and data analysed using GeneMapper v4.0 software (Applied Biosystems).
Supplementary Material
Acknowledgements
We thank Maria Nesbitt, Linda Burgess and Sam Byerley for help with embryo donation and collection, and Valerie Wilson and Dr Steve Abbs (GSTS Pathology, London) for help with the nuclear genotyping. We thank the patients and staff at Newcastle Fertility Centre and James Lawford-Davies, Kristina Stern, Sir John Burn and Lord Walton of Detchant for helping us to obtain an HFEA research licence and guidance with the legislation. This work was funded by Muscular Dystrophy Campaign, The Wellcome Trust (074454/Z/04/Z), Medical Research Council (G0601157, G0601943), One North East, Marriott Research Fund, UK NIHR Biomedical Research Centre for Ageing and Age-related disease and the Newcastle University Centre for Brain Ageing and Vitality supported by BBSRC, EPSRC, ESRC and MRC (G0700718). PFC is a Wellcome Trust Senior Fellow in Clinical Science
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;83:254–60. doi: 10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vandebona H, et al. Prevalence of mitochondrial 1555A-->G mutation in adults of European descent. N Engl J Med. 2009;360:642–4. doi: 10.1056/NEJMc0806397. [DOI] [PubMed] [Google Scholar]
- 3.Bitner-Glindzicz M, et al. Prevalence of mitochondrial 1555A-->G mutation in European children. N Engl J Med. 2009;360:640–2. doi: 10.1056/NEJMc0806396. [DOI] [PubMed] [Google Scholar]
- 4.Schaefer AM, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–9. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- 5.Sato A, et al. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proc Natl Acad Sci U S A. 2005;102:16765–70. doi: 10.1073/pnas.0506197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown DT, et al. Transmission of mitochondrial DNA disorders: possibilities for the future. Lancet. 2006;368:87–9. doi: 10.1016/S0140-6736(06)68972-1. [DOI] [PubMed] [Google Scholar]
- 7.Wallace DC. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu Rev Biochem. 2007;76:781–821. doi: 10.1146/annurev.biochem.76.081205.150955. [DOI] [PubMed] [Google Scholar]
- 8.DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- 9.Man PY, et al. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am J Hum Genet. 2003;72:333–9. doi: 10.1086/346066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steffann J, et al. Analysis of mtDNA variant segregation during early human embryonic development: a tool for successful NARP preimplantation diagnosis. J Med Genet. 2006;43:244–7. doi: 10.1136/jmg.2005.032326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piko L, Taylor KD. Amounts of mitochondrial DNA and abundance of some mitochondrial gene transcripts in early mouse embryos. Dev Biol. 1987;123:364–74. doi: 10.1016/0012-1606(87)90395-2. [DOI] [PubMed] [Google Scholar]
- 13.Barritt JA, Kokot M, Cohen J, Steuerwald N, Brenner CA. Quantification of human ooplasmic mitochondria. Reprod Biomed Online. 2002;4:243–7. doi: 10.1016/s1472-6483(10)61813-5. [DOI] [PubMed] [Google Scholar]
- 14.Lin DP, et al. Comparison of mitochondrial DNA contents in human embryos with good or poor morphology at the 8-cell stage. Fertil Steril. 2004;81:73–9. doi: 10.1016/j.fertnstert.2003.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Tajima H, et al. The development of novel quantification assay for mitochondrial DNA heteroplasmy aimed at preimplantation genetic diagnosis of Leigh encephalopathy. J Assist Reprod Genet. 2007;24:227–32. doi: 10.1007/s10815-007-9114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tachibana M, et al. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461:367–72. doi: 10.1038/nature08368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Battaglia DE, Goodwin P, Klein NA, Soules MR. Fertilization and early embryology: Influence of maternal age on meiotic spindle assembly oocytes from naturally cycling women. Hum. Reprod. 1996;11:2217–2222. doi: 10.1093/oxfordjournals.humrep.a019080. [DOI] [PubMed] [Google Scholar]
- 18.Almeida PA, Bolton VN. The effect of temperature fluctuations on the cytoskeletal organisation and chromosomal constitution of the human oocyte. Zygote. 1995;3:357–65. doi: 10.1017/s0967199400002793. [DOI] [PubMed] [Google Scholar]
- 19.Meirelles FV, Smith LC. Mitochondrial genotype segregation in a mouse heteroplasmic lineage produced by embryonic karyoplast transplantation. Genetics. 1997;145:445–51. doi: 10.1093/genetics/145.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feenan K, Herbert M. Can ‘abnormally’ fertilized zygotes give rise to viable embryos? Hum Fertil (Camb) 2006;9:157–69. doi: 10.1080/14647270600636269. [DOI] [PubMed] [Google Scholar]
- 21.Chinnery P, Majamaa K, Turnbull D, Thorburn D. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2006:CD004426. doi: 10.1002/14651858.CD004426.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McFarland R, et al. Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat Genet. 2002;30:145–6. doi: 10.1038/ng819. [DOI] [PubMed] [Google Scholar]
- 23.Rahman S, Poulton J, Marchington D, Suomalainen A. Decrease of 3243 A-->G mtDNA mutation from blood in MELAS syndrome: a longitudinal study. Am J Hum Genet. 2001;68:238–40. doi: 10.1086/316930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reik W, et al. Adult phenotype in the mouse can be affected by epigenetic events in the early embryo. Development. 1993;119:933–42. doi: 10.1242/dev.119.3.933. [DOI] [PubMed] [Google Scholar]
- 25.Taylor RW, Taylor GA, Durham SE, Turnbull DM. The determination of complete human mitochondrial DNA sequences in single cells: implications for the study of somatic mitochondrial DNA point mutations. Nucleic Acids Res. 2001;29:E74–4. doi: 10.1093/nar/29.15.e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrews RM, et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 27.Taylor RW, et al. A homoplasmic mitochondrial transfer ribonucleic acid mutation as a cause of maternally inherited hypertrophic cardiomyopathy. J Am Coll Cardiol. 2003;41:1786–96. doi: 10.1016/s0735-1097(03)00300-0. [DOI] [PubMed] [Google Scholar]
- 28.McFarland R, et al. Familial myopathy: New insights into the T14709C mitochondrial tRNA mutation. Ann Neurol. 2004;55:478–84. doi: 10.1002/ana.20004. [DOI] [PubMed] [Google Scholar]
- 29.He L, et al. Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acids Res. 2002;30:e68. doi: 10.1093/nar/gnf067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krishnan KJ, Bender A, Taylor RW, Turnbull DM. A multiplex real-time PCR method to detect and quantify mitochondrial DNA deletions in individual cells. Anal Biochem. 2007;370:127–9. doi: 10.1016/j.ab.2007.06.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.