

Abstract
Purpose
Insulin-like growth factor 1 receptor signaling through upregulation of the stimulatory ligand IGF-II has been implicated in the pathogenesis of adrenocortical carcinoma. As there is a paucity of effective therapies, this dose expansion cohort of a phase 1 study was undertaken to determine the safety, tolerability, pharmacokinetics, and effects on endocrine markers of figitumumab in patients with adrenocortical carcinoma.
Methods
Figitumumab was administered on day 1 of each 21-day cycle at the maximal feasible dose (20 mg/kg) to a cohort of patients with metastatic, refractory adrenocortical carcinoma. Serum glucose, insulin, and growth hormone were measured pre-study, at cycle 4 and study end. Pharmacokinetic evaluation was performed during cycles 1 and 4.
Results
Fourteen patients with adrenocortical carcinoma received 50 cycles of figitumumab at the 20 mg/kg. Treatment- related toxicities were generally mild and included hyperglycemia, nausea, fatigue, and anorexia. Single episodes of grade 4 hyperuricemia, proteinuria, and elevated gamma-glutamyltransferase were observed. Pharmacokinetics of figitumumab was comparable to patients with solid tumors other than adrenocortical carcinoma. Treatment with figitumumab increased serum insulin and growth hormone levels. Eight of 14 patients (57%) had stable disease.
Conclusions
The side effect profile and pharmacokinetics of figitumumab were similar in patients with adrenocortical carcinoma in comparison to patients with other solid tumors. While hyperglycemia was the most common adverse event, no clear patterns predicting severity were observed. The majority of patients receiving protocol therapy with single agent figitumumab experienced stability of disease, warranting further evaluation.
Keywords: IGF-1R; Adrenocortical carcinoma; Monoclonal antibody; CP-751,871; Figitumumab
Introduction
The insulin-like growth factor (IGF) system is a complex signaling pathway that is important for human development and growth [1]. Data accumulating over the last three decades have also implicated the IGF system as an important factor in the development, proliferation, survival, and metastatic potential of many human cancers [2-7]. In addition, studies in a variety of pre-clinical models have demonstrated that IGF signaling can provide malignant tumors a mechanism by which they can overcome the anti-tumor effects of various therapies including: cytotoxic chemotherapy, biological or ‘targeted’ therapies, hormonal therapy, and radiation therapy [8-16]. The activation of the IGF system occurs through the interaction between the circulating stimulatory ligands, IGF-1 and IGF-II, and the membrane bound cognate receptor, IGF-1R [17, 18]. IGF-1R activation is necessary for propagation of the mitogenic effects of IGF signaling. In normal tissue, activation of IGF-1R is highly regulated at multiple levels including transcriptional repression of the ligand expression, binding of circulating ligand by binding proteins (i.e., IGFBPs), and a non-signaling ‘sink’ receptor, IGF-IIR. However, in human malignancies these high levels of regulation may be perturbed, leading to over activated IGF-1R signaling [19].
Adrenocortical carcinoma (ACC) is a particularly difficult tumor to treat once resection of a local tumor is no longer an option [20, 21]. In addition to the propensity of this rare malignancy to overproduce functional steroids that define the differentiated state of adrenocortical cells, ACC has the distinction of being a relatively chemotherapy-resistant tumor [22]. While recent reports indicate that the adrenalytic drug mitotane has benefit in reducing the recurrence of ACC after radical surgery, its efficacy in metastatic disease are modest, and, when given at therapeutically effective doses, limited by toxicity [23]. Recent advances in our understanding of ACC have suggested that the IGF system may have a key role in the pathogenesis of ACC. Transcriptional profiling of clinical tumor tissue from ACC patients have shown that the IGF-II gene is the most upregulated transcript in up to 90% of ACC compared to normal adrenal tissue [24-26]. Also, human ACC appears to typically express high levels of the IGF-1R protein, suggesting that the system is activated in the majority of cases [27].
Figitumumab (CP-751,871) is a fully human IgG2 monoclonal antibody that binds and downregulates IGF-1R, as well as blocks its activation by IGF-1 and IGF-II [28]. We have previously reported that figitumumab was very well tolerated and showed a favorable pharmacokinetic profile in patients with solid tumors [29]. In a wide variety of tumor types, figitumumab also demonstrated pharmacodynamic effects consistent with downregulation of IGF-1R and evidence of anti-tumor activity. Armed with our recent pre-clinical data that targeting the IGF-1R in human ACC cells effectively antagonizes downstream activation of AKT and has a marked inhibitory effect on tumor growth in human ACC xenograft models, we aimed to initiate a Phase I trial of IGF-1R inhibition in patients with ACC [30]. Due to the great need for therapies in ACC, the role of IGF-II in ACC, and the tolerability of figitumumab, a dose expansion cohort of patients with refractory ACC was treated with figitumumab at the maximal feasible dose (MFD) of 20 mg/kg. We now report the results of our clinical experience with this agent in patients with ACC.
Patients and methods
Trial design
A phase I, open-label, dose-escalation study of figitumumab administered intravenously in patients with advanced solid tumors in 21-day cycles was conducted. Throughout the study patients received single agent figitumumab by intravenous infusion on day 1, every 21-days. The maximal feasible dose was determined to be 20 mg/kg. The results of the dose-escalation portion of the study have been reported elsewhere [29]. Due to the pre-clinical data supporting the role of IGF signaling in ACC, a cohort of patients with metastatic, refractory adrenocortical carcinoma was accrued at the maximally feasible dose of 20 mg/kg at three institutions (University of Michigan, Mayo Clinic, and Royal Marsden). The goals of this expansion cohort was to determine the safety, tolerability, evaluated the pharmacokinetics, and describe any evidence of clinical benefits of figitumumab in patients with ACC. Patients received single agent figitumumab at a dose of 20 mg/kg, given every 21 days. Figitumumab treatment was continued at this dose as long as it was well tolerated and there was no evidence of disease progression. In addition to the 14 ACC patients, an additional 29 patients with solid tumors other than ACC were treated at the 20 mg/kg dose level. This included 17 patients with sarcoma (5 synovial sarcomas, 4 Ewings sarcomas, 2 desmoplastic small round cell tumors, 2 rhabdomyosarcomas, 2 fibrosarcomas, 1 chondrosarcoma, and 1 peripheral neuroectodermal tumor) and 12 patients other tumor types (3 colorectal carcinomas, 3 non-small cell lung cancers, 2 thymomas, 2 prostate cancers, and one each of bladder and pseudomyxoma peritonei). This report focuses on the results of the 14 adrenocortical carcinoma patients treated in this dose expansion cohort at the maximal feasible dose of 20 mg/kg every 21-days.
Patients
Eligible patients were required to have histological or cytological evidence of metastatic or advanced ACC (for the ACC cohort only) and failed standard effective therapies. Patients were required to be 18 years of age or older with an Eastern Cooperative Oncology Group performance status of 0 or 1. Additional eligibility criteria included: adequate bone marrow, renal and hepatic function (absolute neutrophil count ≥1000/μl, hemoglobin ≥8 g/dl, platelets >75,000/μl, creatinine clearance >30 ml/min, total bilirubin <1.5 × the institution upper limit of normal [ULN], aspartate aminotransferase (AST) and alanine aminotransferase (ALT) <2.5 × ULN); fully recovered from prior anticancer treatments; and use of adequate contraception in patients with reproductive potential. Due to evidence of non-pathological, lymphocyte/granulocyte infiltration of unclear significance in the cardiac valves during pre-clinical toxicology studies with figitumumab, all patients were required to have no evidence of mitral valve regurgitation (≤trivial) as determined by cardiologist evaluation based on precautionary echocardiographic assessments.
Patients were excluded if they were on approved or experimental anti-cancer therapy within 4 weeks of study treatment. Patients were also excluded within 8 weeks of mitomycin C or nitrosoureas, 4 weeks of major surgery, 4 weeks of immunotherapy or other biological therapy, 10 days of palliative radiation therapy or 10 days of hormonal therapy. Additional exclusion criteria were symptomatic or untreated brain metastases, women who were pregnant or breastfeeding, significant active cardiac disease, concomitant high dose corticosteroids (≥100 mg prednisone/day or equivalent), serious active infection, other uncontrolled significant medical illness, psychiatric illness, or social situation that would preclude study participation. Concomitant use of mitotane was allowed.
The study protocol was approved by the institutional review boards of the centers participating in this study. All patients gave written informed consent. The study was conducted in accordance with the Declaration of Helsinki and its amendments.
Patient evaluation
Safety assessments were performed during each treatment cycle. All patients receiving at least one dose of figitumumab were assessed for safety. Treatment-related adverse events were defined as those that were possibly, probably, or definitely related to figitumumab administration and occurred during the period from the time of the first dose until 150 days after the last dose. Dose-limiting toxicity was defined as a treatment-related (either possible, probably, or definite figitumumab attribution) adverse events that occurred in cycle 1 that affected vital organ function or caused a severe infusion reaction. Additionally, due to pre-clinical toxicological, non-pathological findings of lymphocyte infiltration in primate cardiac valves following treatment with high dose figitumumab, echocardiography was performed pre-study, and after every two cycles to ensure identification of any possible cardiac abnormality. The clinical protocol defined dose limiting toxicity as any of the following that occurred during cycle 1 and was treatment-related: (1) National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) hematologic toxicity ≥ grade 4 lasting >7 days or required therapy (growth factor support, transfusion, or hospitalization for complications such as bleeding); (2) Non-hematologic toxicity ≥ grade 3 despite optimal supportive care; (3) Infusion reaction ≥ grade 2 affecting vital organs; (4) Clinically relevant mitral valve regurgitation (>mild). Patients were assessed for subacute and late toxicities up to 150 days following the last dose. Tumor response was assessed every 2 cycles radiographically using Response Evaluation Criteria In Solid Tumors (RECIST) criteria.
Pharmacokinetic analysis
Blood samples were collected in sodium heparin-containing tubes at the following times during cycle 1 and cycle 4: 30 min prior to and 1 h, 24 h, 3 days, 7 days, and 14 days post-end of the figitumumab infusion. Additional samples were collected 30 min prior to and 1 h post-end of the figitumumab infusion in cycles 2 and 3, 30 min prior to figitumumab infusion in cycles 5 and beyond, and at the end of the study.
Plasma concentrations of figitumumab were analyzed by a validated ELISA method, as previously described [28]. The lower limit of quantitation was 120 ng/ml. Plasma concentration–time data of figitumumab were analyzed by non-compartmental methods using WinNonlin V.3.2 (Phar-sight®, Mountain View, CA) [31]. For treatment cycles with sufficient data, area under the plasma concentration–time curve (AUC) from time 0 to the last sampling time point with quantifiable concentration within a cycle (AUClast) and from time 0 to the end of a cycle (AUC0-Day 22) were determined using the linear/log trapezoidal approximation. The accumulation ratio was calculated as the ratio of cycle 4 AUC0-Day22 to cycle 1 AUC0-Day22. The terminal disposition half-life (t1/2) was not estimated as sampling within the 21-day cycles did not sufficiently capture the terminal disposition phase in the majority of the patients at the 20 mg/kg dose level.
Endocrine laboratory studies
When feasible, fasting serum was collected from fasting patients for determinations of glucose, insulin, and human growth hormone (hGH) by the clinical laboratory tests available at the University of Michigan, Mayo Clinic, and Royal Marsden facilities. For the purposes of this study, the “Pre-Study” and “cycle 4” samples were collected within 7 days of receiving figitumumab on cycles 1 and 4, respectively. The “End of Study” samples were collected 21 days (±4 days) after the last dose of figitumumab received. Cycle 4 samples were not collected on patients who did not receive cycle 4 treatment. Additional glucose tests were performed for many patients throughout the study as part of their routine care. When available, these values were reported. Differences in endocrine lab values at pre-study, cycle 4 and end of study were tested by the Wilcoxon Signed Rank Test.
Results
Patients
Fourteen ACC patients, with a median age of 46 (range, 22–77), received a total of 59 cycles of therapy with figitumumab (Table 1). Overall the subjects had a good performance status and received multiple prior therapies for their cancer with evidence of progression at the start of study entry. All but one patient had prior surgery for their cancer, seven (50%) had received chemotherapy and six (43%) were on concomitant mitotane which had been started prior to study entry. The median number of cycles of figitumumab received was 4 (mean–4.2, 95% CI: 3.0, 5.3).
Table 1.
Patient characteristics and treatment summary
| Characteristics number of patients | |
|---|---|
| Age, years | |
| Median | 46 |
| Range | 22–77 |
| Sex | |
| Male | 6 |
| Female | 8 |
| Performance status | |
| 0 | 9 |
| 1 | 5 |
| Prior therapy | |
| Surgery | 13 (4 pts–two surgeries) |
| Radiation therapy | 5 |
| Chemotherapy | 7 |
| Mitotane | 10 (6 on during protocol treatment) |
| Figitumumab (20 mg/kg) | 14 pts |
| Total cycles of therapy | 59 |
| Median, Mean (95% confidence interval) | 4, 4.2 (3.2, 5.2) |
| Range | 1–7 |
Safety
Figure 1 lists the treatment-related toxicities for all evaluable patients for safety assessments (n = 14) and after a total of 59 cycles of figitumumab administered. There were three occurrences of NCI CTCAE grade 4 toxicity; hyperuracemia, proteinuria, and elevated gamma-glutamyltransferase (GGT). All three toxicities were reversed with stopping protocol treatment. The most common adverse events were hyperglycemia, nausea, muscle cramps fatigue, and anorexia. With the exception of muscle cramps, these toxicities are consistent with early investigations with figitumumab.
Fig. 1.
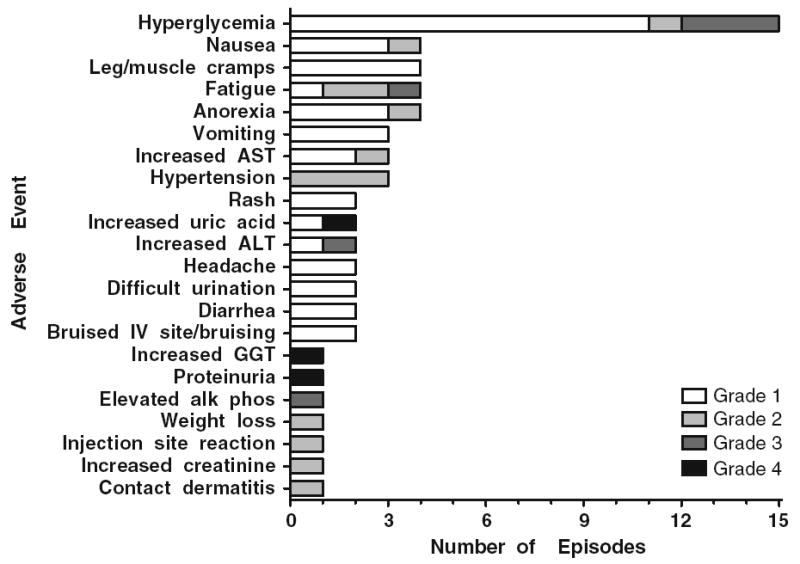
Treatment-related toxicities. Adverse events were graded as per National Cancer Institute Common Toxicity Criteria (version 3.0).
Abbreviations: ALT alanine aminotransferase, AST aspartate aminotransferase, GGT gamma-glutamyltransferase IV intravenous catheter
Pharmacokinetics
Figure 2a shows the mean plasma concentration–time profiles of figitumumab during treatment cycles 1 and 4 in patients with ACC in comparison to patients with other solid tumors. Following intravenous infusion of single agent figitumumab, plasma concentrations decreased multi-exponentially in both ACC and non-ACC patients. During cycle 1, the plasma concentration at 1 h post the end of infusion (C1hr) and the area under the curve through cycle 1 (AUC0-Day22) in patients with ACC were in general comparable to those in patients with other solid tumors (Fig. 2b, c; Table 2). During cycle 4, mean values of C1hr and AUC0-Day22 in ACC patients were 20 and 29%, respectively, lower in patients with ACC (Table 2); meanwhile, there was a relatively larger inter-patient variability in the pharmacokinetic parameters for the non-ACC patient population. The accumulation ratio based on AUC0-Day22 was approximately two for both patient populations (Table 2).
Fig. 2.
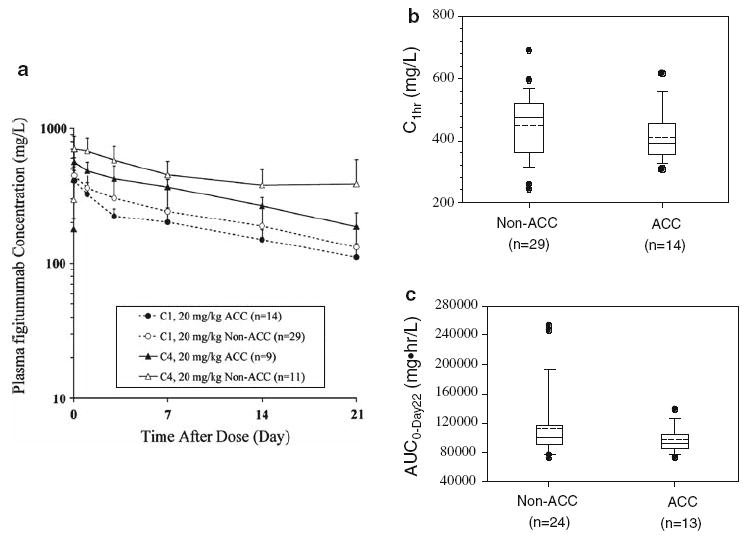
Pharmacokinetic prolife of figitumumab in patients with adrenocortical carcinoma versus other solid tumors. a Mean (± standard deviation) plasma concentration–time profiles of figitumumab in patients with adrenocortical carcinoma (ACC) and non-ACC solid tumors during treatment cycle 1 (C1) and cycle 4 (C4). b, c Comparison of cycle 1 exposure parameters (C1hr and AUC0-Day22) of figitumumab at 20 mg/kg between patients with ACC and patients with other solid tumors. The lower and upper boundaries of the box represent the 25th and 75th percentiles; whiskers above and below the box represent the 90th and 10th percentiles. The solid and dotted lines within the box indicate the median and mean values. Solid dots indicate outlying measurements
Table 2.
Cycles 1 and 4 plasma exposures of figitumumab in patients with ACC, compared to non-ACC solid tumor patients
| Tumor types | Cycle 1 |
Cycle 4 |
Accumulation ratio | ||||
|---|---|---|---|---|---|---|---|
| n | C1hr(mg/l) | AUC0-Day22 (mg hr/l) | n | C1hr(mg/l) | AUC0-Day22 (mg hr/l) | ||
| Non-ACC Solid Tumors | 29 | 447 ± 105 | 112,000 ± 45,5001 | 11 | 701 ± 172 | 226,000 ± 40,0003 | 2.1 ± 0.73 |
| ACC | 14 | 409 ± 78 | 96,500 ± 16,3002 | 9 | 563 ± 44 | 161,000 ± 20,9004 | 1.7 ± 0.34 |
The plasma concentrations at 1 h post figitumumab infusion (C1hr) and area under the curve (AUC0-Day22) was calculated for Cycles 1 and 4. The values represent the mean and standard deviation. The accumulation ratio was calculated as the ratio of cycle 4 AUC0-Day22 to cycle 1 AUC0-Day22
n = 24
n = 13
n = 8
n = 6
Endocrine laboratory values
Based on our previous findings of changes in endocrine laboratory values in clinical investigations with figitumumab, we determined the extent the glucose, insulin, and hGH changes after exposure to figitumumab. Moderate changes in glucose levels were seen in most patients (Fig. 3a). Three patients out of the 14 experienced blood glucose levels over 175. Between them, only one patient with grade 3 hyperglycemia, who had a history of type 2 diabetes, received oral hypoglycemic drugs (glyburide and metformin) which controlled the hyperglycemia. At end of study the range of glucose values in the study subjects was much wider compared with the ranges of pre- and cycle 4 measurements (Fig. 3b). Despite these glucose elevations, all patients, except for one, had resolution of hyperglycemia following their last dose of figitumumab (data not shown). The differences in cycle 4 or end of study glucose compared to the pre-study glucose values were not statistically significant (P = 0.20 and 0.49, respectively). In response to figitumumab, most patients had an increase in insulin secretion (Fig. 3c). This increase stabilized at study end. These differences in insulin from pre-study values were statistically significant (P = 0.03 for both). However, cycle 4 and End of Study insulin levels for three patients with high pre-study insulin levels were not available. Changes in hGH were less apparent and the pre-study values approximated end of study values (Fig. 3c). These differences in values were not statistically significant (P = 0.38 and 0.13, respectively).
Fig. 3.
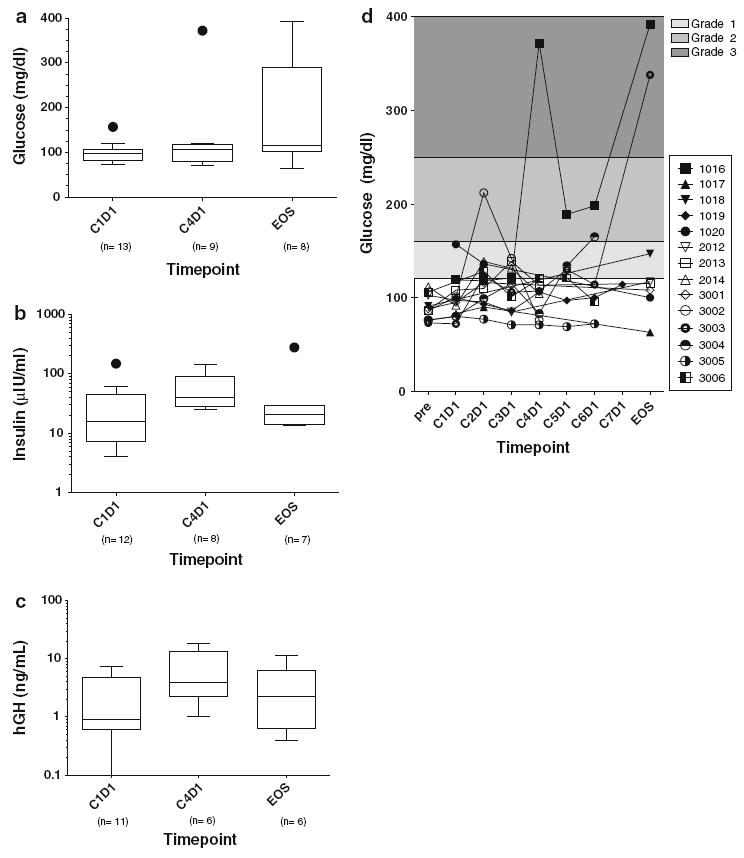
Endocrine laboratory findings. Fasting serum determinations for patients prior to cycles 1 (C1D1) and 4 (C4D1), and at the end of study (EOS) for glucose (a), insulin (b), and human growth hormone (hGH) (c). The lower and upper boundaries of the box represent the 25th and 75th percentiles; whiskers above and below the box represent the 90th and 10th percentiles. The solid lines within the box indicate the median values. Solid dots indicate outlying measurements. d A comprehensive distribution of the glucose values collected for all patients while on study with Common Terminology Criteria for Adverse Events grading ranges indicated. Individual patients are noted by various symbols and 4-digit values. The x-axis denotes the cycle (c) and day number within each cycle (d) from which values are reported. EOS end of study
Anti-tumor activity
All patients presented with metastatic disease. No confirmed responses were seen by RECIST criteria among the 14 ACC patients treated with figitumumab. However, 8 out of 14 patients had stable disease as their best response (Fig. 4a). At 3 months, six patients had stable disease (43%, 95% CI: 21%, 67%). No patients remained on study past seven cycles, due to progressive disease or adverse events. Four patients experience tumor shrinkage, including a patient that demonstrated near resolution of pulmonary metastases (Fig. 4b).
Fig. 4.

a Best response on study. The graph represents the % of tumor increase between consecutive scans taken every 2 cycles or highest percentage of tumor decrease on study when compared to baseline. The total number of cycles of figitumumab by individual patients is noted. † Patients with evaluable disease only on treatment for six cycles. b Chest radiograph for patient # 1020 before and after therapy with figitumumab. The timing of the figures are indicated by the annotated dates, which represent prior to (left) and after (right) four cycles of therapy with figitumumab. The arrows indicate a right upper lobe lung lesion (open single arrow), multiple right lower lobe lesions (three arrows) and a lingular lesion (closed single arrow) that were initially identified by computed tomography scanning and followed during the course of treatment by chest radiograph. After four cycles of therapy, these lesions were notably smaller including a nearly unidentifiable lingular lesion
Discussion
Metastatic adrenocortical carcinoma has poor survival, owing to its relative resistance to standard cytotoxic chemotherapy. The clinical development of therapeutics that may be effective in ACC are also limited by the rarity of the tumor [21]. Due to our pre-clinical data indicating that antagonizing IGF-1R activation effectively blocked downstream AKT activation and tumor growth of ACC xenografts [30], we enrolled 14 patients with ACC in an expansion cohort of the phase I single agent study with anti-IGF-1R antibody antagonist, figitumumab. This portion of the study was carried out at three large referral centers for ACC for timely accrual. The purpose of accruing this cohort was to determine if targeting the IGF-1R in ACC patients was feasible, tolerable and had any evidence of anti-tumor activity that would warrant further investigation either as a single agent or in combination with additional therapies.
Repeated administration of figitumumab was well tolerated by patients with metastatic ACC. Adverse events were generally mild and approximated the phase I experience of patients with other types of solid tumors receiving figitumumab. The exception to this was muscle cramping, which did not occur in non-ACC solid tumor patients, but was seen in multiple myeloma patients receiving figitumumab plus dexamethasone [32]. The mechanism of this adverse event is unclear, but all episodes were CTCAE grade 1 and resolved. Of note, three episodes of grade 4 toxicities, one occurrence of elevated GGT, proteinuria, and hyperuricemia, which were toxicities also seen in other studies with figitumumab [29, 32].
Elevations in glucose were moderate and did not reach statistical significant in this small cohort of patients, although changes in glucose levels should be expected in patients treated with an anti-IGF-1R therapy due to the hypoglycemic effect of IGF-I administration [33]. It has been reported that IGF-I reduces hepatic glucose production and increases peripheral glucose uptake [34]. In this small cohort of patients, increases in insulin were observed, which suggest a compensatory insulin secretion in most of the patients. In addition, this suggests that the insulin receptor (IR) is active in patients treated with figitumumab. Although the IR and IGF-1R are highly homologous, in pre-clinical studies figitumumab did not show any binding to the IR, even at much higher concentrations than the one reached in serum of patients dosed at MFD [28, 29, 35]. Further demonstration of the activity of the IR in patients treated with figitumumab comes from limited data collected in other figitumumab studies where hyperglycemia is controlled by the administration of insulin. Of interest, over-expression of a dominant-negative IR in the skeletal muscle in two different mouse models resulted in insulin resistance. However, these mice did not develop either hyperglycemia or hyperinsulinemia [36, 37]. Taken together the data confirm the pre-clinical studies demonstrating that the IR is not inhibited by figitumumab. It is currently unknown whether the insulin receptor-IGF-1R hybrids that may exist, which according to pre-clinical studies are downregulated by figitumumab, could contribute to hyperinsulinemia and/or hyperglycemia [28, 29].
While no objective responses were seen in this sample of refractory ACC patients, the majority of patients had evidence of anti-tumor activity as evidenced by stability disease and tumor shrinkage that did not meet criteria for a partial response. The median number of cycles received was four, with six patients on study for at least six cycles. One patient received therapy for seven cycles and went off study due to hyperuracemia. Given the relative tolerability of this therapy, further investigation may be warranted in combination with mitotane and/or cytotoxic chemotherapy. In this small cohort of patients, we are unable to conclude with any accuracy whether concomitant mitotane had any effect on the outcomes related to treatment with figitumumab, including frequency of adverse events or likelihood of stable disease. However, it should be noted that all patients who did receive concomitant mitotane with figitumumab, had evidence of progressive disease at the time of trial initiation. Thus, it is possible that the combination of figitumumab with mitotane would have a greater effect in patients naïve to mitotane. Indeed in our pre-clinical studies, significantly enhanced efficacy, as measured by tumoral growth delay, was observed when IGF-1R antagonism was combined with mitotane in a human ACC xenograft model, compared to the single treatment modalities alone [30]. It is hypothesized that by blocking the activation of AKT by IGF-II stimulation of IGF-1R, ACC tumors may be more sensitive to cytotoxic chemotherapy. This hypothesis was recently tested in clinical investigations with figitumumab in combination with chemotherapy in lung cancer [38]. Figitumumab enhanced the anti-tumor activity of cytotoxic chemotherapy in non-small cell lung cancer, which was most profound in the squamous histological subset, which has relatively high expression of IGF-1R [39]. As de novo resistance to chemotherapy limits the activity of chemotherapy in non-small cell lung cancer, it is conceivable that similar improvements in its activity may occur in ACC, which also has evidence of reliance on IGF-1R signaling through IGF-II activation. Further investigations will help us understand if blocking the IGF-1R in patients with ACC with help circumvent prosurvival pathways that will sensitize these highly refractory tumors to chemotherapy and other anti-tumor therapies.
Acknowledgments
Paul Haluska and Johann S. de Bono received research funds from Pfizer Inc.
Contributor Information
Paul Haluska, Division of Medical Oncology, Mayo Clinic College of Medicine, 200 First Street SW, Rochester, MN 55905, USA, haluska.paul@mayo.edu.
Frank Worden, University of Michigan Medical School, BSRB 1502, 109 Zina Pitcher Place, Ann Arbor, MI 48109-2200, USA.
David Olmos, Royal Marsden NHS Foundation Trust, London, UK.
Donghua Yin, Pfizer Global Research and Development, New London, CT, USA.
David Schteingart, University of Michigan Medical School, BSRB 1502, 109 Zina Pitcher Place, Ann Arbor, MI 48109-2200, USA.
Gretchen N. Batzel, Division of Medical Oncology, Mayo Clinic College of Medicine, 200 First Street SW, Rochester, MN 55905, USA
M. Luisa Paccagnella, Pfizer Global Research and Development, New London, CT, USA
Johann S. de Bono, Royal Marsden NHS Foundation Trust, London, UK
Antonio Gualberto, Pfizer Global Research and Development, New London, CT, USA.
Gary D. Hammer, University of Michigan Medical School, BSRB 1502, 109 Zina Pitcher Place, Ann Arbor, MI 48109-2200, USA, ghammer@umich.edu
References
- 1.Le Roith D. Seminars in medicine of the Beth Israel Deaconess Medical Center. Insulin-like growth factors. N Engl J Med. 1997;336(9):633–640. doi: 10.1056/NEJM199702273360907. [DOI] [PubMed] [Google Scholar]
- 2.Cardillo MR, Monti S, Di Silverio F, Gentile V, Sciarra F, Toscano V. Insulin-like growth factor (IGF)-I, IGF-II and IGF type I receptor (IGFR-I) expression in prostatic cancer. Anticancer Res. 2003;23(5A):3825–3835. [PubMed] [Google Scholar]
- 3.Chang YS, Kong G, Sun S, Liu D, El-Naggar AK, Khuri FR, et al. Clinical significance of insulin-like growth factor-binding protein-3 expression in stage I non-small cell lung cancer. Clin Cancer Res. 2002;8(12):3796–3802. [PubMed] [Google Scholar]
- 4.Durai R, Yang W, Gupta S, Seifalian AM, Winslet MC. The role of the insulin-like growth factor system in colorectal cancer: review of current knowledge. Int J Colorectal Dis. 2005;20(3):203–220. doi: 10.1007/s00384-004-0675-4. [DOI] [PubMed] [Google Scholar]
- 5.Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998;351(9113):1393–1396. doi: 10.1016/S0140-6736(97)10384-1. [DOI] [PubMed] [Google Scholar]
- 6.Kalli KR, Falowo OI, Bale LK, Zschunke MA, Roche PC, Conover CA. Functional insulin receptors on human epithelial ovarian carcinoma cells: implications for IGF-II mitogenic signaling. Endocrinology. 2002;143(9):3259–3267. doi: 10.1210/en.2001-211408. [DOI] [PubMed] [Google Scholar]
- 7.Sachdev D, Li SL, Hartell JS, Fujita-Yamaguchi Y, Miller JS, Yee D. A chimeric humanized single-chain antibody against the type I insulin-like growth factor (IGF) receptor renders breast cancer cells refractory to the mitogenic effects of IGF-I. Cancer Res. 2003;63(3):627–635. [PubMed] [Google Scholar]
- 8.Abe S, Funato T, Takahashi S, Yokoyama H, Yamamoto J, Tomiya Y, et al. Increased expression of insulin-like growth factor i is associated with Ara-C resistance in leukemia. Tohoku J Exp Med. 2006;209(3):217–228. doi: 10.1620/tjem.209.217. [DOI] [PubMed] [Google Scholar]
- 9.Allen GW, Saba C, Armstrong EA, Huang SM, Benavente S, Ludwig DL, et al. Insulin-like growth factor-I receptor signaling blockade combined with radiation. Cancer Res. 2007;67(3):1155–1162. doi: 10.1158/0008-5472.CAN-06-2000. [DOI] [PubMed] [Google Scholar]
- 10.Camirand A, Lu Y, Pollak M. Co-targeting HER2/ErbB2 and insulin-like growth factor-1 receptors cause synergistic inhibition of growth in HER2-overexpressing breast cancer cells. Med Sci Monit. 2002;8(12):BR521–BR526. [PubMed] [Google Scholar]
- 11.Desbois-Mouthon C, Cacheux W, Blivet-Van Eggelpoel MJ, Barbu V, Fartoux L, Poupon R, et al. Impact of IGF-1R/EGFR cross-talks on hepatoma cell sensitivity to gefitinib. Int J Cancer. 2006;119(11):2557–2566. doi: 10.1002/ijc.22221. [DOI] [PubMed] [Google Scholar]
- 12.Gee JM, Robertson JF, Gutteridge E, Ellis IO, Pinder SE, Rubini M, et al. Epidermal growth factor receptor/HER2/insulin-like growth factor receptor signalling and oestrogen receptor activity in clinical breast cancer. Endocr Relat Cancer. 2005;12(suppl 1):S99–S111. doi: 10.1677/erc.1.01005. [DOI] [PubMed] [Google Scholar]
- 13.Knowlden JM, Hutcheson IR, Barrow D, Gee JM, Nicholson RI. Insulin-like growth factor-I receptor signaling in tamoxifen-resistant breast cancer: a supporting role to the epidermal growth factor receptor. Endocrinology. 2005;146(11):4609–4618. doi: 10.1210/en.2005-0247. [DOI] [PubMed] [Google Scholar]
- 14.Wan X, Helman LJ. Effect of insulin-like growth factor II on protecting myoblast cells against cisplatin-induced apoptosis through p70 S6 kinase pathway. Neoplasia. 2002;4(5):400–408. doi: 10.1038/sj.neo.7900242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiseman LR, Johnson MD, Wakeling AE, Lykkesfeldt AE, May FE, Westley BR. Type I IGF receptor and acquired tamoxifen resistance in oestrogen-responsive human breast cancer cells. Eur J Cancer. 1993;29A(16):2256–2264. doi: 10.1016/0959-8049(93)90218-5. [DOI] [PubMed] [Google Scholar]
- 16.Yin D, Tamaki N, Parent AD, Zhang JH. Insulin-like growth factor-I decreased etoposide-induced apoptosis in glioma cells by increasing bcl-2 expression and decreasing CPP32 activity. Neurol Res. 2005;27(1):27–35. doi: 10.1179/016164105X18151. [DOI] [PubMed] [Google Scholar]
- 17.Kurmasheva RT, Houghton PJ. IGF-I mediated survival pathways in normal and malignant cells. Biochim Biophys Acta. 2006;1766(1):1–22. doi: 10.1016/j.bbcan.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Samani AA, Yakar S, Leroith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28(1):20–47. doi: 10.1210/er.2006-0001. [DOI] [PubMed] [Google Scholar]
- 19.Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4(7):505–518. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- 20.Icard P, Goudet P, Charpenay C, Andreassian B, Carnaille B, Chapuis Y, et al. Adrenocortical carcinomas: surgical trends and results of a 253-patient series from the French Association of Endocrine Surgeons study group. World J Surg. 2001;25(7):891–897. doi: 10.1007/s00268-001-0047-y. [DOI] [PubMed] [Google Scholar]
- 21.Wajchenberg BL, Albergaria Pereira MA, Medonca BB, Latronico AC, Campos Carneiro P, Alves VA, et al. Adrenocortical carcinoma: clinical and laboratory observations. Cancer. 2000;88(4):711–736. [PubMed] [Google Scholar]
- 22.Kirschner LS. Emerging treatment strategies for adrenocortical carcinoma: a new hope. J Clin Endocrinol Metab. 2006;91(1):14–21. doi: 10.1210/jc.2005-1739. [DOI] [PubMed] [Google Scholar]
- 23.Terzolo M, Angeli A, Fassnacht M, Daffara F, Tauchmanova L, Conton PA, et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med. 2007;356(23):2372–2380. doi: 10.1056/NEJMoa063360. [DOI] [PubMed] [Google Scholar]
- 24.Giordano TJ, Thomas DG, Kuick R, Lizyness M, Misek DE, Smith AL, et al. Distinct transcriptional profiles of adrenocortical tumors uncovered by DNA microarray analysis. Am J Pathol. 2003;162(2):521–531. doi: 10.1016/S0002-9440(10)63846-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Velazquez-Fernandez D, Laurell C, Geli J, Hoog A, Odeberg J, Kjellman M, et al. Expression profiling of adrenocortical neoplasms suggests a molecular signature of malignancy. Surgery. 2005;138(6):1087–1094. doi: 10.1016/j.surg.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 26.West AN, Neale GA, Pounds S, Figueredo BC, Rodriguez Galindo C, Pianovski MA, et al. Gene expression profiling of childhood adrenocortical tumors. Cancer Res. 2007;67(2):600–608. doi: 10.1158/0008-5472.CAN-06-3767. [DOI] [PubMed] [Google Scholar]
- 27.Weber MM, Auernhammer CJ, Kiess W, Engelhardt D. Insulin-like growth factor receptors in normal and tumorous adult human adrenocortical glands. Eur J Endocrinol. 1997;136(3):296–303. doi: 10.1530/eje.0.1360296. [DOI] [PubMed] [Google Scholar]
- 28.Cohen BD, Baker DA, Soderstrom C, Tkalcevic G, Rossi AM, Miller PE, et al. Combination therapy enhances the inhibition of tumor growth with the fully human anti-type 1 insulin-like growth factor receptor monoclonal antibody CP-751, 871. Clin Cancer Res. 2005;11(5):2063–2073. doi: 10.1158/1078-0432.CCR-04-1070. [DOI] [PubMed] [Google Scholar]
- 29.Haluska P, Shaw HM, Batzel GN, Yin D, Molina JR, Molife LR, et al. Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clin Cancer Res. 2007;13(19):5834–5840. doi: 10.1158/1078-0432.CCR-07-1118. [DOI] [PubMed] [Google Scholar]
- 30.Barlaskar FM, Spalding AC, Heaton JH, Kuick R, Kim AC, Thomas DG, et al. Preclinical targeting of the type 1 insulin-like growth factor receptor in adrenocortical carcinoma. J Clin Endocrinol Metab. 2008;94(1):204–212. doi: 10.1210/jc.2008-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibaldi M, Perrier D, editors. Pharmacokinetics. 2. Marcel Dekker; New York (NY): 1982. [Google Scholar]
- 32.Lacy MQ, Alsina M, Fonseca R, Paccagnella ML, Melvin CL, Yin D, et al. Phase I, pharmacokinetic and pharmacodynamic study of the anti-insulin-like growth factor type 1 Receptor monoclonal antibody CP-751,871 in patients with multiple myeloma. J Clin Oncol. 2008;26(19):3196–3203. doi: 10.1200/JCO.2007.15.9319. [DOI] [PubMed] [Google Scholar]
- 33.Laron Z, Klinger B, Erster B, Anin S. Effect of acute administration of insulin-like growth factor I in patients with Laron-type dwarfism. Lancet. 1988;2(8621):1170–1172. doi: 10.1016/s0140-6736(88)90236-x. [DOI] [PubMed] [Google Scholar]
- 34.Di Cola G, Cool MH, Accili D. Hypoglycemic effect of insulin-like growth factor-1 in mice lacking insulin receptors. J Clin Invest. 1997;99(10):2538–2544. doi: 10.1172/JCI119438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gronborg M, Wulff BS, Rasmussen JS, Kjeldsen T, Gammeltoft S. Structure-function relationship of the insulin-like growth factor-I receptor tyrosine kinase. J Biol Chem. 1993;268(31):23435–23440. [PubMed] [Google Scholar]
- 36.Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell. 1998;2(5):559–569. doi: 10.1016/s1097-2765(00)80155-0. [DOI] [PubMed] [Google Scholar]
- 37.Chang PY, Benecke H, Le Marchand-Brustel Y, Lawitts J, Moller DE. Expression of a dominant-negative mutant human insulin receptor in the muscle of transgenic mice. J Biol Chem. 1994;269(23):16034–16040. [PubMed] [Google Scholar]
- 38.Karp DD, Paz-Ares LG, Novello S, Haluska P, Garland L, Cardenal F, et al. Phase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancer. J Clin Oncol. 2009;27(15):2516–2522. doi: 10.1200/JCO.2008.19.9331. [DOI] [PubMed] [Google Scholar]
- 39.Gualberto A, Melvin CL, Dean A, Ang AL, Reynolds JM, Lee AV, et al. Characterization of NSCLC patients responding to anti-IGF-IR therapy. J Clin Oncol. 2008;26:8000. [Google Scholar]