

Abstract
Depression is associated with dysregulated hypothalamic-pituitary-adrenal (HPA) axis function, overactivity of the sympathoadrenal system, and increased levels of inflammation markers. It is not known whether these biological processes are disproportionately elevated in response to acute negative emotional arousal by mental stress (MS). The present study investigates responses of neurohormones and inflammatory markers to MS in 14 clinically depressed (age: 42±10 years; 50% female) and 14 non-depressed control participants (age: 39±6 years; 50% female). Heightened acute MS reactivity was documented in depressed participants (adrenocorticotropic hormone, ρ=0.001; Norepinephrine, ρ=0.042; Epinephrine, ρ=0.039), and a delayed increase in cortisol was observed (ρ=0.002). Inflammation markers increased more strongly in depressed vs. non-depressed participants (IL-6, ρ=0.027; tumor necrosis factor-alpha, ρ=0.050; and recovery C-reactive protein, ρ=0.003). It is concluded that depressed individuals display hyper-reactivity of neuroimmunological markers in response to acute negative emotions. This hyper-reactivity may serve a pathologic role in the elevated morbidity and mortality risk associated with depression.
Major depressive disorder is associated with dysregulated hypothalamic-pituitary-adrenal (HPA) axis function and impaired negative cortisol feedback (Leonard & Myint, 2009). Depression is often associated with the sustained hyperactivity of the stress axis caused by the neurotransmitter corticotropin-releasing factor (CRF) (Leonard & Myint, 2009). CRF initiates the changes in the HPA axis and also stimulates the sympatho-adrenal system (SAM) (Bjorntorp, 1999). CRF results in the release of adrenocorticotropic hormone (ACTH). ACTH then activates the adrenal cortex leading to the synthesis and secretion of glucocortioids (i.e., cortisol). The acute rise in cortisol and other glucocorticoids activates the mineralocortioid receptors in the pituitary and hypothalamus, thereby decreasing the release of CRF (i.e., a negative feedback system). This results in the reduction in ACTH release from the anterior pituitary and thereby decreases the secretion of glucocorticoids. In patients with major depression, desensitization of the glucocortioid receptors frequently occurs. Under these conditions the negative feedback regulation malfunctions and the hypersecretion of cortisol persists (Bhagwagar, Hafizi, & Cowen, 2005; Burke, Davis, Otte, & Mohr, 2005; Halbreich, Asnis, Shindledecker, Zumoff, & Nathan, 1985; Pfohl, Sherman, Schlechte, & Winokur, 1985; Stetler & Miller, 2005). Clinical evidence in support of this is based on the lack of suppression of plasma cortisol following the administration of the synthetic glucocorticoid dexamethasone (Carroll, 1982). In addition to hyperactivity of the HPA axis, depressed individuals also display overactivity of the SAM system (Leonard, 2001). Hypersecretion of NE in unipolar depression has been documented by elevated plasma NE and NE metabolite concentrations, as well as elevated urinary concentrations of NE and its metabolites (Ressler & Nemeroff, 1999). Plasma NE and urinary excretion of NE and its metabolites decrease in response to treatment with tricyclic antidepressants (Ressler & Nemeroff, 1999).
Neurohormonal alterations contribute to immune system dysregulation in depression (Herbert & Cohen, 1993; Kop & Gottdiener, 2005; Pace, et al., 2006). Depression is associated with elevated concentrations of pro-inflammatory cytokines, including interleukin-6 (IL-6) (Dentino, et al., 1999; Lutgendorf, et al., 1999; Maes, et al., 1998; Maes, et al., 1995) and C-reactive protein (CRP) (Kop, et al., 2002; Miller, Rohleder, Stetler, & Kirschbaum, 2005). Successful anti-depressive pharmacologic treatment results in reduced IL-6 levels (Sluzewska, et al., 1995). However, the effect of acute negative emotions on inflammation markers is not well documented in individuals with clinical depression.
Acute perturbation tasks, such as physical exercise and mental arousal, result in increased levels of catecholamines (Gerra, et al., 1998; Martin, Spina, Korte, & Ogawa, 1991; Rogers, et al., 1991), cortisol (Sluiter, Frings-Dresen, Meijman, & van der Beek, 2000; Steinacker, Lormes, Reissnecker, & Liu, 2004), and pro-inflammatory cytokines (LeMay, Vander, & Kluger, 1990; Takaki, Huang, Somogyvari-Vigh, & Arimura, 1994; Zhou, Kusnecov, Shurin, DePaoli, & Rabin, 1993) in non-depressed individuals. Evidence of exaggerated acute stress responses in depression have been documented in experimental and clinical studies, including increased levels of neurohormones, such as cortisol and catecholamines (Grewen, Girdler, Hinderliter, & Light, 2004; Hamer, Tanaka, Okamura, Tsuda, & Steptoe, 2007; Light, Kothandapani, & Allen, 1998). Increased “stress” reactivity may be partially explain the increased morbidity and mortality risks in individuals with depression (Joynt, Whellan, & O'Connor, 2003; Vaccarino, et al., 2007).
The present study examines the hypothesis that individuals with depression will have exaggerated responses in neurohormones (ACTH, cortisol, and catecholamines) and inflammation markers to acute aversive negative emotional arousal induced by mental stress. In addition, because acute stress activates the HPA axis and the sympathetic nervous system, the immune system may also be activated. The response of the immune system to stress involves a complex cascade of events in which catecholamines, glucocorticoids, endorphins and other neuropeptides play important parts (Croiset, Heijnen, Veldhuis, de Wied, & Ballieux, 1987). Exogenous stressors may produce diverse changes in the immune system, which is explained in part by different degrees of stress-induced endocrine and sympathetic activation (Mason, 1971). The relationship between neurohormonal activation and immune system responses is bidirectional, such that changes in the endocrine and sympathetic systems can affect the immune system (e.g., release of pro-inflammatory cytokines) and vice versa (Leonard & Myint, 2009; Song, Dinan, & Leonard, 1994). Therefore, it was also hypothesized that the magnitude of exaggerated responses of neurohormones would be associated with increases in inflammatory parameters in individuals with depression. The current study used stressors that have previously been shown to stimulate both endocrine and sympathetic systems (Kop, et al., 2008).
Depression is also associated with a range of adverse health behaviors, such as cigarette smoking, low physical activity, and poor diet (resulting in increased body weight). These health behaviors are interrelated phenomena (Woo, 2000) and important to consider as covariates in studies that examine neurohormonal and immune system related reactivity of depressed patients. Thus, the current project assessed these health behaviors as potential effect modifying or confounding factors.
Methods
Participants
Participants with a DSM-IV-based diagnosis of depression (n = 14, age 41.7 ± 9.6, 50% female) were recruited via advertisements in local newspapers and evaluated for Major Depressive Disorder using the Structured Clinical Interview for DSM-IV (SCID) (First, Spitzer, Williams, & Gibbon, 1995). Control participants (n = 14, age 39.3 ± 5.6, 50% female) were matched on gender, age (within 5 years) and body mass index (BMI-within 5 kg/m2). The SCID was administered to confirm the absence of psychiatric diagnoses in the control participants.
Exclusion criteria were: age < 18 or >80 years, history of coronary artery disease (myocardial infarction, cardiovascular revascularization procedures, and/or chest pain on exertion), use of anti-hypertensive medications, use of immunomodulatory or anti-inflammatory medications other than aspirin, current or past diagnosis or treatment of bipolar disorder or psychosis, and active suicidal ideation. The project was approved by the Institutional Review Board and all participants gave written informed consent prior to participating in the study.
Procedures
Participants were tested between 12 and 4 pm to control for the effects of time of day on biological and behavioral parameters. First, the SCID was administered to determine depression status, and then questionnaires were completed to evaluate demographic and physical activity levels (age, race/ethnicity, height, weight, current medication use, and current physical activity level) (Kohl, Blair, Paffenbarger, Macera, & Kronenfeld, 1988).
An indwelling catheter was then inserted into an antecubital vein and after a 30-minute resting period to establish baseline, mood measures and a blood sample for neurohormonal analyses were obtained. Then, participants performed a five-minute anger recall task (Ironson, et al., 1992; Kop, et al., 2008). This task required participants to give a speech about a recent incident that elicited frustration or anger. Immediately following the anger recall task, a five-minute mental arithmetic task was performed. In the mental arithmetic task, participants were asked to subtract serial sevens from a four-digit number while being encouraged to work as fast and accurately as possible (Gottdiener, et al., 1994; Kop, et al., 2008). The order of these mental stress tasks was counter-balanced. The consecutive performance of anger recall and mental arithmetic was performed because some immune system parameters require stress exposures of greater than five minutes to induce a detectable response (Kunz-Ebrecht, Mohamed-Ali, Feldman, Kirschbaum, & Steptoe, 2003). The anger recall and mental arithmetic stressor tasks were chosen as they are reliable and valid inducers of stress responses of the HPA, SAM, and immune system parameters (Kop, et al., 2008). The anger recall is a challenge task that involves personally relevant information. The mental arithmetic task has no personally relevant component. Since, the combination of these tasks has been used to induce hemodynamic, neurohormonal and immune system activation (Cohen & Hamrick, 2003), these tasks were chosen. Participants completed five 7-point Likert scale items (angry, frustrated, fatigued, depressed, and irritated) that ranged from “not at all” to “extremely” before and after MS to validate whether the tasks produced a state of acute negative emotion (potential summation score range 5-35). After completion of the mental stress tasks, participants rested for 30-minutes while recovery measurements were obtained.
Blood draws were also obtained immediately post-mental stress, and 30 minutes after the completion of the mental stress task. Blood samples were analyzed for ACTH, cortisol, NE, Epi, IL-6, TNF-α, and CRP. However, because catecholamines return to baseline levels within five minutes following mental stress (Kop, et al., 2008), they were not measured during the recovery period.
Assessments
Depressive Symptoms
The mood disorder module from the Structured Clinical Interview for DSM-IV (SCID) (First, et al., 1995) was used to assess the presence or absence of Major Depressive Disorder. The psychotic screen was used to assess the presence of psychotic symptoms. The Hamilton Rating Scale for Depression (HRSD) was also administered to assess the severity of depressive symptoms (Hamilton, 1960, 1967). The HRSD is a 21-item interview assessing emotional, behavioral and somatic symptoms of depression. Overall inter-rater agreement is high, commonly exceeding 0.84 (Hedlund & Virmani, 1979). Total scores range from 0 to 52, with scores from 7-17 reflecting mild depression and scores above 17 indicating clinical depression (Hamilton, 1960, 1967). Participants also completed the Beck Depression Inventory (BDI-II) to assess severity of depression. This inventory consists of 21 items and has demonstrated excellent reliability (Cronbach's α = 0.92-0.93). BDI-II scores exceeding 10 are considered indicative of the potential presence of depression (Beck, Steer, & Brown, 1996).
Blood Parameters
Blood samples were collected in vacuum tubes (ethylenediaminetetraacetic acid (EDTA) 4.5 mmol/l), mixed gently for 30 seconds, and kept at 4° C. The first 5 mL of each blood draw were discarded to avoid artifacts related to dilution. Separation of plasma was performed by temperature-controlled centrifugation at 3000g for 15 minutes. For measurement of catecholamines, plasma was decanted into storage tubes containing 1.25 μmoles sodium EDTA and 1.25 μmoles sodium metabisulfate to inhibit oxidation (Goldberg, et al., 1996; Kaufmann, et al., 1998). Aliquots of plasma were stored at -80° C until analysis (Cushman, Cornell, Howard, Bovill, & Tracy, 1995; Tracy, et al., 1997).
ACTH
ACTH was assayed in duplicate 200-μl aliquots of EDTA plasma by a two-site immunoradiometric method using materials obtained from the Nichols Institute (San Juan Capistrano, CA). This procedure is highly specific and exhibits limited cross-reactivity with related physiologic peptides. Sensitivity of the assay is 1 pg/mL (0.22 pmole/L) with inter- and intra-assay CV <6% (Raff & Findling, 1989).
Cortisol
Cortisol was assayed in duplicate 10 μl aliquots of EDTA plasma by a solid phase radioimmunoassay (RIA) using materials obtained from DiaSorin Corporation (Stillwater, MN). Sensitivity of the assay is 0.2 μg/dl (2 ng/mL) with inter- and intra-assay CV <4%. Use of this assay has been described previously (Goldberg, et al., 1996; Kaufmann, et al., 1998). Standards (range 1 to 30 ng/mL) consist of serum standards diluted with 200 μl of phosphate-buffered saline. Protein concentrations are equalized in standards and samples by adding cortisol-free serum to the samples. Sensitivity is 0.01 μg/dL (0.1 ng/mL) and inter- and intra-assay CV <6%.
Catecholamines
Epinephrine (Epi) and norepinephrine (NE) were measured in plasma following alumina extraction and on-line cation enrichment (Goldberg, et al., 1996; Kaufmann, et al., 1998). Duplicate sample aliquots (0.25 – 1.0 ml) were taken to mildly basic pH with Tris and extracted with acid-washed, screened alumina. Catecholamines were desorbed from the alumina using 0.1 M perchloric acid, and extracts are analyzed by reverse-phase, ion-pair high performance liquid chromatography in combination with a computer-controlled cation-enrichment pre-column and a 3-electrode electrochemical detector connected to a computerized data-acquisition system. NE intra-assay CVs are 6.6% (<400 pg/ml), 6.5% (400 to 800 pg/ml) and 7.1% (>800 pg/ml), mean inter-assay CV for pooled samples in the range 300 to 550 pg/mL =10.3%, and blanks read 6.0 ± 10.3 (S.D.) pg/mL. For epinephrine, mean intra-assay CV are 27.1% (<40 pg/mL), 13.5% (40 to 80 pg/mL) and 9.6% (>80 pg/mL); mean inter-assay CV for pooled samples in the range 60 to 140 pg/mL is 16.3%, and blanks read 7.0 ± 14.5 (S.D.) pg/mL.
Inflammation markers
Plasma IL-6 and TNF-α were measured by enzyme-linked immunosorbent assays (ELISA) (Quantikine HS Human IL-6 Immunoassay; R&D Systems, Minneapolis, MN). The detection range is 0.156-10.0 pg/mL. Prior intra- and interassay coefficients of variance were 5.6% and 8.6%, respectively and the dynamic range of this high sensitivity assay was 0.256 to 10 pg/mL. All samples from a participant were analyzed in one assay to eliminate interassay variations. The laboratory CVs for these assays are approximately 6.3%.
C-reactive protein (CRP) was measured using the BNII nephelometer from Dade Behring, which is a particle enhanced immunonepholometric assay. Polystyrene particles are coated with monoclonal antibodies to CRP that agglutinate, causing an increase in the intensity of scattered light, in the presence of CRP. The increase in scattered light is proportional to the amount of CRP in the sample. The assay range is 0.175–1100 mg/L. Expected values for CRP in normal, healthy individuals are <3 mg/L. Intra-assay and inter-assay CVs range from 2.3–4.4% and 2.1–5.7%, respectively (Harris, et al., 1999; Macy, Hayes, & Tracy, 1997).
Statistical Analyses
Neurohormonal and inflammatory responsiveness to the mental stress protocol was evaluated using mixed model analyses of variance (ANOVA), where the depressed group was compared to the control group (two-level between subjects factor), and assessments made at the three time points were included as a three-level, within-subject factor (baseline, mental stress, and recovery). Interaction terms between group status and time indicate altered reactivity in depressed versus control participants. To reduce statistical Type-I error all neurohormonal measures and inflammatory measures were examined in multivariate models. Therefore, the four neurohormonal dependent variables (NE, Epi, ACTH, and cortisol) were examined in one separate analysis and the three inflammatory measures (IL-6, TNF-α, and CRP) were examined in a second separate multivariate analysis. A significant group (depression vs. control) × neurohormonal or inflammation marker indicates that the stress response is different in depressed versus control participants. If the overall multivariate analyses revealed statistically significant results, then further analyses were conducted on the individual dependent variables. Significant main and interaction effects were examined by using independent and paired t-tests. Because the biochemistry data were not normally distributed, all were log-transformed and analyses were performed on the transformed data. Geometric means and 95% confidence intervals are presented for the non-normally distributed variables.
To examine whether the magnitude of change in neurohormones was related to the change in inflammatory markers, difference scores for these measures were calculated by subtracting baseline levels from levels during mental stress. Pearson's product-moment correlations were used to examine the relationship between these responses. Two-tailed probabilities were examined at an alpha level of < 0.05.
Results
Participant Characteristics
Characteristics of depressed and non-depressed participants are shown in Table 1. Demographic and other potential confounding variables, including smoking status and physical activity level, were not different in depressed participants versus controls (ρ values >0.20). Matching for age, gender, and body-mass index (BMI) was successfully accomplished (Table 1). Consistent with inclusion criteria, the participants with depression had higher HRSD scores (17.1 ± 6.6 vs. 1.1 ± 1.1) and elevated BDI-II scores (27.1 ± 12.0 vs. 1.6 ± 2.4) compared to the control participants. The mental stress tasks induced the anticipated acute state of aversive negative emotional arousal (depressed participants from 13.7 ± 6.9 to 21.5 ± 6.8, ρ = 0.001, non-depressed: from 6.4 ± 1.6 to 12.2 ± 4.6, ρ = 0.001, ρ-interaction = 0.36).
Table 1.
Participant Characteristics
| Controls N = 14 | Depressed N = 14 | ||
|---|---|---|---|
| Age (years) | 39.3 (5.6) | 41.7 (9.6) | |
| Gender | Female | 7 (50%) | 7 (50%) |
| Race | African American | 6 (43%) | 7 (50%) |
| Caucasian | 6 (43%) | 5 (36%) | |
| Other | 2 (14%) | 2 (14%) | |
| Smoking Status | Current Smoker | 2 (14%) | 4 (29%) |
| Body Mass Index (kg/m2) | 25.2 (4.4) | 26.0 (3.8) | |
| Activity Level (min/week) | 272.4 (181.4) | 257.1 (166.7) | |
| Use of Antidepressant Medication | 0 (0%) | 2 (14%) | |
Neurohormonal Response to Acute Mental Stress
Baseline measures of ACTH, cortisol, NE, and Epi were similar in depressed versus control participants (ρ's > 0.10). Multivariate ANOVA examining the four neurohormonal measures simultaneously, indicated a significant depression × time interaction (F(4,19) = 3.21; ρ = 0.041) consistent with elevated reactivity in depressed individuals, as well as a main effect for time (F(4,19) = 7.37; ρ = 0.001), but no main effect for depression status (F(4,19) = 1.05; ρ = 0.37).
As shown in Figure 1, depressed participants displayed a steeper immediate increase in ACTH following mental stress (F interaction(1,23) = 7.00; ρ = 0.001), and an elevated cortisol response at 30 min post mental stress (F interaction(1,24) = 11.48; ρ = 0.002) compared to controls. Depression was also associated with increased NE reactivity (F interaction(1,24) = 4.48; ρ = 0.042) and Epi reactivity (F interaction(1,24) = 4.18; ρ = 0.039) compared to the control participants (Figure 1).
Figure 1.
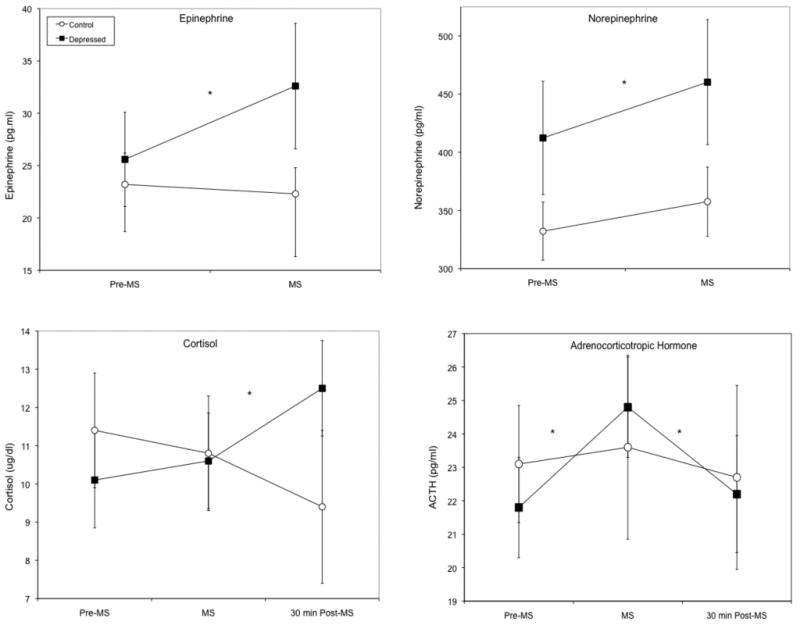
Increased neurohormone concentrations in response to mental stress tasks in depressed versus control participants. Data present geometric means and error bars are 95% confidence intervals. * = ρinteraction < 0.05 depressed versus control participants.
Inflammatory Response to Acute Mental Stress
Baseline measures of inflammatory markers were similar in depressed versus control participants (ρ's > 0.10). Multivariate ANOVA examining the three inflammatory measures simultaneously, indicated a significant depression × time interaction (F(6,14) = 4.07; ρ = 0.014) consistent with elevated reactivity in depressed individuals, as well as a main effect for time (F(6,14) = 3.50; ρ = 0.025), but no main effect for depression status (F(3,17) = 0.94; ρ = 0.45). Figure 2 shows that mental stress induced higher inflammatory responses among depressed individuals as compared to controls: IL-6, Finteraction(1,26) = 4.58, ρ = 0.027 and TNF-α, Finteraction(1,21) = 3.88, ρ = 0.050. Furthermore, a delayed increase in CRP was observed at 30 minutes post-mental stress among depressed participants compared to control participants (Finteraction(1,25) = 11.03, ρ = 0.003 (see Figure 2). Analyses for depressed individuals separately found significant increases in IL-6 (ρ = 0.05), TNF-α (ρ = 0.04), and CRP (ρ = 0.02).
Figure 2.

Increased inflammation in response to mental stress tasks in depressed versus control participants. Data present geometric means and error bars are 95% confidence intervals. * = ρinteraction < 0.05 depressed versus control participants.
Relationship of Neurohormonal and Inflammatory Responses to Acute Mental Stress
Associations between hormonal and inflammatory responses were examined separately for the depressed and control groups, and restricted to the time points when responses were significant (i.e., immediate responses for ACTH, NE, Epi, IL-6, and TNF-α, and 30 minute delayed responses for cortisol and CRP). In depressed participants, statistically significant relationships were observed between increases in cortisol and IL-6 (r = 0.73, ρ = 0.018) and between Epi and TNF-α (r = 0.70, ρ = 0.05) and CRP (r = 0.72, ρ = 0.043). As shown in Table 2, trends were also found for associations between ACTH and CRP (ρ = 0.087), cortisol and CRP (ρ = 0.088), Epi and IL-6 (ρ = 0.081), and NE and TNF-α (ρ = 0.073) (Table 2). In control participants, no statistically significant correlations were noted with the exception of a correlation between ACTH and CRP reactivity (r = 0.74, ρ = 0.004).
Table 2.
Pearson's correlations between the change in neurohormonal and inflammatory responses to an acute mental stress task in individuals with depression.
| ACTH | Cortisol | NE | Epi | ||
|---|---|---|---|---|---|
| Depression | IL-6 | 0.13 | 0.73 * | 0.03 | 0.50 t |
| TNF-α | 0.20 | 0.16 | 0.59 t | 0.70 * | |
| CRP | 0.40 t | 0.44 t | 0.05 | 0.69 * | |
| Control | IL-6 | 0.10 | 0.09 | 0.18 | 0.24 |
| TNF-α | 0.19 | 0.52 t | 0.07 | 0.07 | |
| CRP | 0.74 * | 0.19 | 0.38 | 0.38 | |
ρ < 0.05,
ρ < 0.10.
adrenocorticotropic hormone(ACTH), norepinephrine (NE), epinephrine (Epi), IL-6, and tumor necrosis factor-alpha (TNF-α) immediately post-mental stress, cortisol and C-reactive protein (CRP) and cortisol at 30-min recovery (see text for details).
Discussion
This study demonstrates that depressed individuals have heightened HPA axis and SAM reactivity and increases in markers of inflammation to mental arousal as compared to non-depressed controls. Specifically, the depressed participants showed increased responsiveness in ACTH, NE, and Epi immediately following stress exposure, and in cortisol at 30 minutes post stress. In addition, exaggerated stress-induced increases were observed among depressed individuals immediately in IL-6 and TNF-α, and in CRP concentrations at 30 minutes post stress. A plasma rise in IL-6 is a precursor to a rise in CRP (Pepys & Hirschfield, 2003). Therefore, it follows that an increase in CRP would occur following an increase in IL-6. The biological factors examined in this research may serve a pathologic role in the elevated morbidity and mortality risk associated with depression, particularly adverse cardiovascular disease (Carney, et al., 1988; de Groot, Anderson, Freedland, Clouse, & Lustman, 2001; Frasure-Smith, Lesperance, & Talajic, 1993; Lesperance, Frasure-Smith, Talajic, & Bourassa, 2002; Lustman, et al., 2000; Nicholson, Kuper, & Hemingway, 2006; Wulsin, 2004; Wulsin & Singal, 2003).
This study is consistent with a larger literature documenting increased neurohormonal responsiveness in depressed individuals. Similarities between the acute stress response and depression have been documented in experimental and clinical studies, including increased levels of neurohormones, exaggerated blood pressure, and heart rate, as well as increased arousal and mobilization of energy stores. In addition, critical brain structure involvement may be the same in depression and acute stress, including the locus coeruleus and the central nucleus of the amygdala, which are both innervated by CRF-containing nerve terminals (Pittenger & Duman, 2008). Furthermore, depression is associated with elevations of the normal stress response, which may reflect a failure to respond appropriately to usual counter-regulatory feedback mechanisms, resulting in a sustained version of a usually transient phenomenon, i.e., hyperactivity of the HPA or SAM axis (Strohle & Holsboer, 2003).
Depressed individuals may have hyper-reactivity to challenges for a variety of reasons. One theory on the development of depression posits that depressive symptoms occur when cumulative stressors exceed the individual's distress-related vulnerability threshold (Kovacs & Beck, 1978). Because the vulnerability threshold in depressed individuals may have been reached, exaggerated responses to additional exogenous stressors may develop, as observed in the present study. In addition, depressed individuals typically have more negative expectations regarding their ability to perform tasks. These negative expectations may superimpose additional stress to the mental tasks that control participants without depression do not experience (as the control participants' negative expectations would have been lower).
Depressed individuals may also have dysregulated HPA and SAM systems as a result of a lack of negative feedback regulation (Strohle & Holsboer, 2003). If the negative feedback system is not working properly, then exposure to a stressor could result in an excessive response because the response would not be dampened by negative feedback loops. Although this particular investigation did not assess vulnerability thresholds, negative expectations or the negative feedback system, previous research has demonstrated that these dysfunctional processes are active in individuals with depression.
An alternative explanation of the exaggerated reactivity of neurohormonal and inflammation markers is that individuals with depression may have prolonged involuntary processing of emotional information (including stress-induced negative emotions) (Lyubomirsky, Caldwell, & Nolen-Hoeksema, 1998). Sustained peripheral physiological signals have been found in depression, such as pupil dilation (Siegle, Granholm, Ingram, & Matt, 2001) or central activation in brain areas (such as the amygdala) responsible for encoding emotional features (Siegle, Steinhauer, Thase, Stenger, & Carter, 2002). This prolonged processing of stressors may lead to increases in physiological reactivity to these same stressors when compared to control participants.
The present study has several limitations that merit discussion. First, it did not include a control (non-stress) condition that would allow alternative explanations for stress-related changes to be ruled out, such as passage of time or repeated blood draws. Changes in these variables may be related to diurnal variations rather than acute stress. However, since the control and depressed groups showed different patterns, the diurnal variation may have minimally affected the results. In addition, previous research has shown that HPA axis dysregulation is usually associated specifically with acute depression, while some evidence suggests that individuals with chronic depression do not display abnormal HPA axis function (Watson, et al., 2002). The current study did not assess the duration of depression (acute vs. chronic). Future research is needed to further assess whether the type of depression affects the neuroimmunological responses to mental stress in order to clarify the generalizability of the current results. The sample size of the current investigation is small. Therefore, replication studies with a larger sample size are needed to ensure generalizability. However, statistical significance was found on many of the analyses performed, and therefore the effect sizes appear of sufficient magnitude to be detectable in the relatively small sample size of the present study.
In summary, our findings provide important information regarding the role of depression status on reactivity to mental stressors that induce negative mood states. The health consequences of hyper-reactivity to acute negative emotional arousal in depressed individuals require further investigation in longitudinal studies. Such longitudinal studies would establish whether depressed individuals with hyper-reactivity to stressors develop health problems, including cardiovascular disease, inflammatory diseases, cancer, and premature all-cause mortality. Psychological and pharmacological interventions could be implemented to reduce hyper-reactivity to stressors which may have potential beneficial long-term health consequences in depressed individuals.
Footnotes
The opinions and assertions expressed herein are those of the authors and are not to be construed as reflecting the views of the USUHS or the US Department of Defense.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Beck AT, Steer RA, Brown C. The Beck Depression Inventory Manual. Second. San Antonio, TX: The Psychological Corporation; 1996. [Google Scholar]
- Bhagwagar Z, Hafizi S, Cowen PJ. Increased salivary cortisol after waking in depression. Psychopharmacology (Berl) 2005;182(1):54–57. doi: 10.1007/s00213-005-0062-z. [DOI] [PubMed] [Google Scholar]
- Bjorntorp P. Neuroendocrine perturbations as a cause of insulin resistance. Diabetes Metab Res Rev. 1999;15(6):427–441. doi: 10.1002/(sici)1520-7560(199911/12)15:6<427::aid-dmrr68>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Burke HM, Davis MC, Otte C, Mohr DC. Depression and cortisol responses to psychological stress: a meta-analysis. Psychoneuroendocrinology. 2005;30(9):846–856. doi: 10.1016/j.psyneuen.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Carney RM, Rich MW, Freedland KE, Saini J, teVelde A, Simeone C, et al. Major depressive disorder predicts cardiac events in patients with coronary artery disease. Psychosom Med. 1988;50(6):627–633. doi: 10.1097/00006842-198811000-00009. [DOI] [PubMed] [Google Scholar]
- Carroll BJ. Use of the dexamethasone suppression test in depression. J Clin Psychiatry. 1982;43(11 Pt 2):44–50. [PubMed] [Google Scholar]
- Cohen S, Hamrick N. Stable individual differences in physiological response to stressors: implications for stress-elicited changes in immune related health. Brain Behav Immun. 2003;17(6):407–414. doi: 10.1016/s0889-1591(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Croiset G, Heijnen CJ, Veldhuis HD, de Wied D, Ballieux RE. Modulation of the immune response by emotional stress. Life Sci. 1987;40(8):775–782. doi: 10.1016/0024-3205(87)90305-5. [DOI] [PubMed] [Google Scholar]
- Cushman M, Cornell ES, Howard PR, Bovill EG, Tracy RP. Laboratory methods and quality assurance in the Cardiovascular Health Study. Clin Chem. 1995;41(2):264–270. [PubMed] [Google Scholar]
- de Groot M, Anderson R, Freedland KE, Clouse RE, Lustman PJ. Association of depression and diabetes complications: a meta-analysis. Psychosom Med. 2001;63(4):619–630. doi: 10.1097/00006842-200107000-00015. [DOI] [PubMed] [Google Scholar]
- Dentino AN, Pieper CF, Rao MK, Currie MS, Harris T, Blazer DG, et al. Association of interleukin-6 and other biologic variables with depression in older people living in the community. J Am Geriatr Soc. 1999;47(1):6–11. doi: 10.1111/j.1532-5415.1999.tb01894.x. [DOI] [PubMed] [Google Scholar]
- First MB, Spitzer RL, Williams JB, Gibbon M. Structured Clinical Interview for DSM-IV (SCID) Washington, DC: American Psychiatric Association; 1995. [Google Scholar]
- Frasure-Smith N, Lesperance F, Talajic M. Depression following myocardial infarction. Impact on 6-month survival. Jama. 1993;270(15):1819–1825. [PubMed] [Google Scholar]
- Gerra G, Zaimovic A, Franchini D, Palladino M, Giucastro G, Reali N, et al. Neuroendocrine responses of healthy volunteers to ‘techno-music’: relationships with personality traits and emotional state. Int J Psychophysiol. 1998;28(1):99–111. doi: 10.1016/s0167-8760(97)00071-8. [DOI] [PubMed] [Google Scholar]
- Goldberg AD, Becker LC, Bonsall R, Cohen JD, Ketterer MW, Kaufman PG, et al. Ischemic, hemodynamic, and neurohormonal responses to mental and exercise stress. Experience from the Psychophysiological Investigations of Myocardial Ischemia Study (PIMI) Circulation. 1996;94(10):2402–2409. doi: 10.1161/01.cir.94.10.2402. [DOI] [PubMed] [Google Scholar]
- Gottdiener JS, Krantz DS, Howell RH, Hecht GM, Klein J, Falconer JJ, et al. Induction of silent myocardial ischemia with mental stress testing: relation to the triggers of ischemia during daily life activities and to ischemic functional severity. J Am Coll Cardiol. 1994;24(7):1645–1651. doi: 10.1016/0735-1097(94)90169-4. [DOI] [PubMed] [Google Scholar]
- Grewen KM, Girdler SS, Hinderliter A, Light KC. Depressive symptoms are related to higher ambulatory blood pressure in people with a family history of hypertension. Psychosom Med. 2004;66(1):9–16. doi: 10.1097/01.psy.0000106881.60228.16. [DOI] [PubMed] [Google Scholar]
- Halbreich U, Asnis GM, Shindledecker R, Zumoff B, Nathan RS. Cortisol secretion in endogenous depression. I. Basal plasma levels. Arch Gen Psychiatry. 1985;42(9):904–908. doi: 10.1001/archpsyc.1985.01790320076010. [DOI] [PubMed] [Google Scholar]
- Hamer M, Tanaka G, Okamura H, Tsuda A, Steptoe A. The effects of depressive symptoms on cardiovascular and catecholamine responses to the induction of depressive mood. Biol Psychol. 2007;74(1):20–25. doi: 10.1016/j.biopsycho.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton M. Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol. 1967;6:278–296. doi: 10.1111/j.2044-8260.1967.tb00530.x. [DOI] [PubMed] [Google Scholar]
- Harris TB, Ferrucci L, Tracy RP, Corti MC, Wacholder S, Ettinger WH, Jr, et al. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am J Med. 1999;106(5):506–512. doi: 10.1016/s0002-9343(99)00066-2. [DOI] [PubMed] [Google Scholar]
- Hedlund JL, Virmani R. The Hamilton rating scale for depression: A comprehensive review. Journal of Operational Psychiatry. 1979;11:48–65. [Google Scholar]
- Herbert TB, Cohen S. Depression and immunity: a meta-analytic review. Psychol Bull. 1993;113(3):472–486. doi: 10.1037/0033-2909.113.3.472. [DOI] [PubMed] [Google Scholar]
- Ironson G, Taylor CB, Boltwood M, Bartzokis T, Dennis C, Chesney M, et al. Effects of anger on left ventricular ejection fraction in coronary artery disease. Am J Cardiol. 1992;70(3):281–285. doi: 10.1016/0002-9149(92)90605-x. [DOI] [PubMed] [Google Scholar]
- Joynt KE, Whellan DJ, O'Connor CM. Depression and cardiovascular disease: mechanisms of interaction. Biol Psychiatry. 2003;54(3):248–261. doi: 10.1016/s0006-3223(03)00568-7. [DOI] [PubMed] [Google Scholar]
- Kaufmann PG, McMahon RP, Becker LC, Bertolet B, Bonsall R, Chaitman B, et al. The Psychophysiological Investigations of Myocardial Ischemia (PIMI) study: objective, methods, and variability of measures. Psychosom Med. 1998;60(1):56–63. doi: 10.1097/00006842-199801000-00014. [DOI] [PubMed] [Google Scholar]
- Kohl HW, Blair SN, Paffenbarger RS, Jr, Macera CA, Kronenfeld JJ. A mail survey of physical activity habits as related to measured physical fitness. Am J Epidemiol. 1988;127(6):1228–1239. doi: 10.1093/oxfordjournals.aje.a114915. [DOI] [PubMed] [Google Scholar]
- Kop WJ, Gottdiener JS. The role of immune system parameters in the relationship between depression and coronary artery disease. Psychosom Med. 2005;67 1:S37–41. doi: 10.1097/01.psy.0000162256.18710.4a. [DOI] [PubMed] [Google Scholar]
- Kop WJ, Gottdiener JS, Tangen CM, Fried LP, McBurnie MA, Walston J, et al. Inflammation and coagulation factors in persons > 65 years of age with symptoms of depression but without evidence of myocardial ischemia. Am J Cardiol. 2002;89(4):419–424. doi: 10.1016/s0002-9149(01)02264-0. [DOI] [PubMed] [Google Scholar]
- Kop WJ, Weissman NJ, Zhu J, Bonsall RW, Doyle M, Stretch MR, et al. Effects of acute mental stress and exercise on inflammatory markers in patients with coronary artery disease and healthy controls. Am J Cardiol. 2008;101(6):767–773. doi: 10.1016/j.amjcard.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Kovacs M, Beck AT. Maladaptive cognitive structures in depression. Am J Psychiatry. 1978;135(5):525–533. doi: 10.1176/ajp.135.5.525. [DOI] [PubMed] [Google Scholar]
- Kunz-Ebrecht SR, Mohamed-Ali V, Feldman PJ, Kirschbaum C, Steptoe A. Cortisol responses to mild psychological stress are inversely associated with proinflammatory cytokines. Brain Behav Immun. 2003;17(5):373–383. doi: 10.1016/s0889-1591(03)00029-1. [DOI] [PubMed] [Google Scholar]
- LeMay LG, Vander AJ, Kluger MJ. The effects of psychological stress on plasma interleukin-6 activity in rats. Physiol Behav. 1990;47(5):957–961. doi: 10.1016/0031-9384(90)90024-x. [DOI] [PubMed] [Google Scholar]
- Leonard BE. Stress, norepinephrine and depression. J Psychiatry Neurosci. 2001;26(Suppl):S11–6. S11–S16. [PMC free article] [PubMed] [Google Scholar]
- Leonard BE, Myint A. The psychoneuroimmunology of depression. Hum Psychopharmacol. 2009;24(3):165–175. doi: 10.1002/hup.1011. [DOI] [PubMed] [Google Scholar]
- Lesperance F, Frasure-Smith N, Talajic M, Bourassa MG. Five-year risk of cardiac mortality in relation to initial severity and one-year changes in depression symptoms after myocardial infarction. Circulation. 2002;105(9):1049–1053. doi: 10.1161/hc0902.104707. [DOI] [PubMed] [Google Scholar]
- Light KC, Kothandapani RV, Allen MT. Enhanced cardiovascular and catecholamine responses in women with depressive symptoms. Int J Psychophysiol. 1998;28(2):157–166. doi: 10.1016/s0167-8760(97)00093-7. [DOI] [PubMed] [Google Scholar]
- Lustman PJ, Anderson RJ, Freedland KE, de GM, Carney RM, Clouse RE. Depression and poor glycemic control: a meta-analytic review of the literature. Diabetes Care. 2000;23(7):934–942. doi: 10.2337/diacare.23.7.934. [DOI] [PubMed] [Google Scholar]
- Lutgendorf SK, Garand L, Buckwalter KC, Reimer TT, Hong SY, Lubaroff DM. Life stress, mood disturbance, and elevated interleukin-6 in healthy older women. J Gerontol A Biol Sci Med Sci. 1999;54(9):M434–M439. doi: 10.1093/gerona/54.9.m434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyubomirsky S, Caldwell ND, Nolen-Hoeksema S. Effects of ruminative and distracting responses to depressed mood on retrieval of autobiographical memories. J Pers Soc Psychol. 1998;75(1):166–177. doi: 10.1037//0022-3514.75.1.166. [DOI] [PubMed] [Google Scholar]
- Macy EM, Hayes TE, Tracy RP. Variability in the measurement of C-reactive protein in healthy subjects: implications for reference intervals and epidemiological applications. Clin Chem. 1997;43(1):52–58. [PubMed] [Google Scholar]
- Maes M, Song C, Lin A, De JR, Van GA, Kenis G, et al. The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and a Th1-like response in stress-induced anxiety. Cytokine. 1998;10(4):313–318. doi: 10.1006/cyto.1997.0290. [DOI] [PubMed] [Google Scholar]
- Maes M, Vandoolaeghe E, Ranjan R, Bosmans E, Bergmans R, Desnyder R. Increased serum interleukin-1-receptor-antagonist concentrations in major depression. J Affect Disord. 1995;36(1-2):29–36. doi: 10.1016/0165-0327(95)00049-6. [DOI] [PubMed] [Google Scholar]
- Martin WH, 3rd, Spina RJ, Korte E, Ogawa T. Effects of chronic and acute exercise on cardiovascular beta-adrenergic responses. J Appl Physiol. 1991;71(4):1523–1528. doi: 10.1152/jappl.1991.71.4.1523. [DOI] [PubMed] [Google Scholar]
- Mason JW. A re-evaluation of the concept of “non-specificity” in stress theory. J Psychiatr Res. 1971;8(3):323–333. doi: 10.1016/0022-3956(71)90028-8. [DOI] [PubMed] [Google Scholar]
- Miller GE, Rohleder N, Stetler C, Kirschbaum C. Clinical depression and regulation of the inflammatory response during acute stress. Psychosom Med. 2005;67(5):679–687. doi: 10.1097/01.psy.0000174172.82428.ce. [DOI] [PubMed] [Google Scholar]
- Nicholson A, Kuper H, Hemingway H. Depression as an aetiologic and prognostic factor in coronary heart disease: a meta-analysis of 6362 events among 146 538 participants in 54 observational studies. Eur Heart J. 2006;27(23):2763–2774. doi: 10.1093/eurheartj/ehl338. [DOI] [PubMed] [Google Scholar]
- Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH, et al. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry. 2006;163(9):1630–1633. doi: 10.1176/ajp.2006.163.9.1630. [DOI] [PubMed] [Google Scholar]
- Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest. 2003;111(12):1805–1812. doi: 10.1172/JCI18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfohl B, Sherman B, Schlechte J, Winokur G. Differences in plasma ACTH and cortisol between depressed patients and normal controls. Biol Psychiatry. 1985;20(10):1055–1072. doi: 10.1016/0006-3223(85)90004-6. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 2008;33(1):88–109. doi: 10.1038/sj.npp.1301574. [DOI] [PubMed] [Google Scholar]
- Raff H, Findling JW. A new immunoradiometric assay for corticotropin evaluated in normal subjects and patients with Cushing's syndrome. Clin Chem. 1989;35(4):596–600. [PubMed] [Google Scholar]
- Ressler KJ, Nemeroff CB. Role of norepinephrine in the pathophysiology and treatment of mood disorders. Biol Psychiatry. 1999;46(9):1219–1233. doi: 10.1016/s0006-3223(99)00127-4. [DOI] [PubMed] [Google Scholar]
- Rogers PJ, Tyce GM, Weinshilboum RM, O'Connor DT, Bailey KR, Bove AA. Catecholamine metabolic pathways and exercise training. Plasma and urine catecholamines, metabolic enzymes, and chromogranin-A. Circulation. 1991;84(6):2346–2356. doi: 10.1161/01.cir.84.6.2346. [DOI] [PubMed] [Google Scholar]
- Siegle GJ, Granholm E, Ingram RE, Matt GE. Pupillary and reaction time measures of sustained processing of negative information in depression. Biol Psychiatry. 2001;49(7):624–636. doi: 10.1016/s0006-3223(00)01024-6. [DOI] [PubMed] [Google Scholar]
- Siegle GJ, Steinhauer SR, Thase ME, Stenger VA, Carter CS. Can't shake that feeling: event-related fMRI assessment of sustained amygdala activity in response to emotional information in depressed individuals. Biol Psychiatry. 2002;51(9):693–707. doi: 10.1016/s0006-3223(02)01314-8. [DOI] [PubMed] [Google Scholar]
- Sluiter JK, Frings-Dresen MH, Meijman TF, van der Beek AJ. Reactivity and recovery from different types of work measured by catecholamines and cortisol: a systematic literature overview. Occup Environ Med. 2000;57(5):298–315. doi: 10.1136/oem.57.5.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluzewska A, Rybakowski JK, Laciak M, Mackiewicz A, Sobieska M, Wiktorowicz K. Interleukin-6 serum levels in depressed patients before and after treatment with fluoxetine. Ann N Y Acad Sci. 1995;762:474–6. 474–476. doi: 10.1111/j.1749-6632.1995.tb32372.x. [DOI] [PubMed] [Google Scholar]
- Song C, Dinan T, Leonard BE. Changes in immunoglobulin, complement and acute phase protein levels in the depressed patients and normal controls. J Affect Disord. 1994;30(4):283–288. doi: 10.1016/0165-0327(94)90135-x. [DOI] [PubMed] [Google Scholar]
- Steinacker JM, Lormes W, Reissnecker S, Liu Y. New aspects of the hormone and cytokine response to training. Eur J Appl Physiol. 2004;91(4):382–391. doi: 10.1007/s00421-003-0960-x. [DOI] [PubMed] [Google Scholar]
- Stetler C, Miller GE. Blunted cortisol response to awakening in mild to moderate depression: regulatory influences of sleep patterns and social contacts. J Abnorm Psychol. 2005;114(4):697–705. doi: 10.1037/0021-843X.114.4.697. [DOI] [PubMed] [Google Scholar]
- Strohle A, Holsboer F. Stress responsive neurohormones in depression and anxiety. Pharmacopsychiatry. 2003;36 3:S207–14. S207–S214. doi: 10.1055/s-2003-45132. [DOI] [PubMed] [Google Scholar]
- Takaki A, Huang QH, Somogyvari-Vigh A, Arimura A. Immobilization stress may increase plasma interleukin-6 via central and peripheral catecholamines. Neuroimmunomodulation. 1994;1(6):335–342. doi: 10.1159/000097185. [DOI] [PubMed] [Google Scholar]
- Tracy RP, Lemaitre RN, Psaty BM, Ives DG, Evans RW, Cushman M, et al. Relationship of C-reactive protein to risk of cardiovascular disease in the elderly. Results from the Cardiovascular Health Study and the Rural Health Promotion Project. Arterioscler Thromb Vasc Biol. 1997;17(6):1121–1127. doi: 10.1161/01.atv.17.6.1121. [DOI] [PubMed] [Google Scholar]
- Vaccarino V, Johnson BD, Sheps DS, Reis SE, Kelsey SF, Bittner V, et al. Depression, inflammation, and incident cardiovascular disease in women with suspected coronary ischemia: the National Heart, Lung, and Blood Institute-sponsored WISE study. J Am Coll Cardiol. 2007;50(21):2044–2050. doi: 10.1016/j.jacc.2007.07.069. [DOI] [PubMed] [Google Scholar]
- Watson S, Gallagher P, Del-Estal D, Hearn A, Ferrier IN, Young AH. Hypothalamic-pituitary-adrenal axis function in patients with chronic depression. Psychol Med. 2002;32(6):1021–1028. doi: 10.1017/s0033291702005998. [DOI] [PubMed] [Google Scholar]
- Woo J. Relationships among diet, physical activity and other lifestyle factors and debilitating diseases in the elderly. Eur J Clin Nutr. 2000;54 3:S143–7. S143–S147. doi: 10.1038/sj.ejcn.1601036. [DOI] [PubMed] [Google Scholar]
- Wulsin LR. Is depression a major risk factor for coronary disease? A systematic review of the epidemiologic evidence. Harv Rev Psychiatry. 2004;12(2):79–93. doi: 10.1080/10673220490447191. [DOI] [PubMed] [Google Scholar]
- Wulsin LR, Singal BM. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom Med. 2003;65(2):201–210. doi: 10.1097/01.psy.0000058371.50240.e3. [DOI] [PubMed] [Google Scholar]
- Zhou D, Kusnecov AW, Shurin MR, DePaoli M, Rabin BS. Exposure to physical and psychological stressors elevates plasma interleukin 6: relationship to the activation of hypothalamic-pituitary-adrenal axis. Endocrinology. 1993;133(6):2523–2530. doi: 10.1210/endo.133.6.8243274. [DOI] [PubMed] [Google Scholar]