

Abstract
Transforming growth factor β (TGF-β) superfamily ligands have important roles in regulating cellular homeostasis, embryonic development, differentiation, proliferation, immune surveillance, angiogenesis, motility, and apoptosis in a cell type and context specific manner. TGF-β superfamily signaling pathways also have diverse roles in human cancer, functioning to either suppress or promote cancer progression. The TGF-β superfamily co-receptor, the type III TGF-β receptor (TβRIII, also known as betaglycan) mediates TGF-β superfamily ligand dependent as well as ligand independent signaling to both Smad and non-Smad signaling pathways. Loss of TβRIII expression during cancer progression and direct effects of TβRIII on regulating cell migration, invasion, proliferation, and angiogenesis support a role for TβRIII as a suppressor of cancer progression and/or as a metastasis suppressor. Defining the physiological function and mechanism of TβRIII action and alterations in TβRIII function during cancer progression should enable more effective targeting of TβRIII and TβRIII mediated functions for the diagnosis and treatment of human cancer.
Keywords: TβRIII, TGF-β, BMP, cancer, receptor
Introduction
Transforming growth factor β (TGF-β) superfamily ligands mediate and regulate cellular homeostasis, including embryonic development, differentiation, proliferation, immune surveillance, angiogenesis, motility, and apoptosis, in both a cell type and context specific manner [1]. TGF-β superfamily signaling pathways also have diverse roles in human disease, with both increases and decreases in these signaling pathways resulting in human disease [2]. In human cancers, TGF-β superfamily ligands and their pathways function as both tumor suppressors and tumor promoters [2-5]. TGF-β superfamily ligands function as tumor suppressors through the ability to inhibit cell proliferation, maintain tissue architecture, inhibit genomic instability, and induce senescence and apoptosis [6-8]. Alterations in many components of the TGF-β superfamily signaling pathways, such as mutation or deletion of receptors and other signaling components, are frequent events in human cancers and lead to a loss of the tumor suppressor function of these pathways [7, 9]. Conversely, many late stage human tumors dysregulate or over-express TGF-β superfamily ligands, which can have tumor promoting effects on both the stromal compartment (suppressing immune surveillance, enhancing angiogenesis) and on the cancer cells themselves (inducing epithelial-to-mesenchymal transition (EMT), enhancing migration and invasion) to promote tumor invasiveness and metastasis [6-7, 10-11]. Precise mechanisms for this dual tumor suppressor/tumor promoter role for TGF-β superfamily ligands remains a fundamental problem in the TGF-β superfamily signaling field and major obstacle to targeting these pathways for the treatment of human cancers.
Recently, loss or reduced expression of the TGF-β superfamily co-receptor, the type III TGF-β receptor (TβRIII, also known as betaglycan), has been demonstrated in multiple human cancers (Table 1) [12-17]. TβRIII has also been shown to be an important regulator of cell migration, invasion, cell growth, and angiogenesis in both in vitro and in vivo cancer models. Taken together, these results support a role for TβRIII as a mediator of TGF-β superfamily function during cancer progression [18]. Here we review what is currently known about TβRIII, both in the context of TGF-β superfamily signaling and in human cancer biology, supporting TβRIII as a guardian of the epithelial phenotype, as a suppressor of cancer progression, and as a metastasis suppressor.
Table 1.
TβRIII in Human Cancers
| Cancer | TβRIII Expression (Mechanism) | Effects on Signaling | Proliferation | Migration | Invasion | Angiogenesis | In Vivo/Other TβRIII Effects | References |
|---|---|---|---|---|---|---|---|---|
| Breast | Decreased (LOH) | Decreased | No effect | Decreased | Decreased | Decreased | Xenograft: Reduced tumor invasiveness, angiogenesis, metastasis; sTβRIII reduces tumor growth, angiogenesis, metastasis | [12, 67, 68, 90, 91] |
| Lung (NSCLC) | Decreased (LOH) | No effect | No effect | Decreased | Decreased | Decreased | Xenograft: Reduced tumor incidence, growth, & invasiveness | [20] |
| Prostate | Decreased (LOH; epigenetic regulation) | Not examined | No effect | Decreased | Decreased | Decreased | Xenograft: Reduced tumor incidence, & growth; sTβRIII reduced tumor growth, angiogenesis, & MMP induction, increased apoptosis | [14, 95, 96] |
| Pancreatic | Decreased | Decreased | Not examined | Decreased | Decreased | Not examined | Decreased MMP Induction | [16, 104] |
| Ovarian | Decreased (Epigenetic regulation) | Not examined | Not examined | Decreased | Decreased | Not examined | Decreased MMP Induction | [15, 62, 106] |
| Renal | Decreased | Decreased (p38) | Not examined | Not examined | Not examined | Decreased | Xenograft: Reduced tumor growth, increased apoptosis | [13, 82] |
Regulation, Structure, and Function of the Type III TGF-β Receptor
The gene encoding TβRIII, the TGFBR3 gene, is located at chromosome 1p31-32, consists of 16 exons, and has two promoters, a proximal promoter and a distal promoter, which produce two mRNA species [19-20]. In most tissue types, the proximal transcript is the predominant promoter utilized [19-20]. While no mutations in the TGFBR3 gene have been reported in human disease, the chromosomal locus for the TGFBR3 gene, 1p31-32, frequently is lost in human cancer, and loss of heterozygosity of the TGFBR3 gene has been reported in several cancer including breast, prostate, and lung cancer as discussed below [12, 14, 17, 21]. In addition, several studies have identified single nucleotide polymorphisms (SNPs) in the TGFBR3 3’ untranslated region (UTR), open reading frame (ORF), and in the 5’UTR [22-23]. However, these SNPs have not been linked to any physiological effects [22-23].
TβRIII is ubiquitously expressed on nearly all cell types and is the mostly highly expressed of the TGF-β superfamily receptors on those cells, with at least 200,000 receptors/cell as opposed to 5,000-10,000 receptors/cell for most TGF-β superfamily type I and type II receptors [19, 24-26] However, some cell types, including endothelial cells, appear to have little or no TβRIII expression, but instead express the related TGF-β superfamily co-receptor, endoglin [27].
TβRIII expression is regulated at multiple levels. At the transcriptional level, TβRIII expression is negatively regulated by TGF-β1 through inhibition of the proximal promoter in multiple cell types, including breast, ovarian, and small cell lung cancer cell lines, as well as by bone morphogenetic protein-2 (BMP-2) in osteoblast cell lines [20, 28-29]. Expression of TβRIII mRNA has also been reported to decrease with transdifferentiation of hepatic stellate cells (HSC) to myofibroblasts [30] and during induction of EMT in a pancreatic model [16]. In contrast corticosteroids, specifically dexamethasone, aldosterone, and hydrocortisone, transcriptionally up-regulate TβRIII mRNA expression in hepatic stellate cells (HSCs), myofibroblasts, and osteoblasts while both MyoD and retinoic acid increase TβRIII expression in mouse myoblast cells [28-31]. In addition, follicle-stimulating hormone, estrogen, and luteinizing hormone increase TβRIII mRNA expression in ovarian granulosa-luteal cells [32-33]. TβRIII mRNA expression appears to be highly regulated and required during development, demonstrated by the embryonic lethality of the TβRIII null mouse [34-36]. TβRIII expression is increased during C2C12 skeletal muscle differentiation, with an increase in TβRIII expression during the conversion of myoblasts to myotubes [31]. TβRIII expression is also regulated during palatal fusion, with expression temporo-spatially restricted to the medial edge epithelium, suggesting that TβRIII mediates TGF-β3 effects during palatal fusion [37].
Expression of TβRIII is also regulated at the epigenetic level. Treatment of ovarian and prostate cancer cells that express low levels of TβRIII with methyltransferase inhibitors and/or histone deacetylase inhibitors induces TβRIII expression, indicating that either direct or indirect epigenetic silencing plays a role in the loss of TβRIII expression in these cancer types [14-15]. Interestingly, this methyltransferase and/or histone deacetylase inhibitor mediated induction of TβRIII expression is specific to ovarian cancer cells with low levels of TβRIII, with low to no induction observed in normal ovarian epithelial cells or ovarian cancer cells with basal levels of TβRIII [15]. In addition, the distal TGFBR3 promoter has been shown to be highly methylated, although this has not been shown to correlate with TβRIII expression and effects of epigenetic silencing of this promoter region remain to be examined further [20].
Levels of TβRIII expression are also regulated at the protein level. TβRIII interacts with the PDZ-protein GAIP-interacting protein, C terminus (GIPC), which stabilizes TβRIII at the cell surface and enhances TGF-β signaling [38]. Conversely, interaction with β-arrestin2 mediates co-internalization of TβRIII, TβRII, and TβRI and down-regulation of receptor levels, causing a decrease in TGF-β signaling [39-40]. In addition, the induction of EMT in a pancreatic cell line causes a reduction in cell surface TβRIII levels due to increased ectodomain shedding [16]. TβRIII protein levels also decrease in response to exposure to chronic hypoxia in neonatal and adult rat lung tissues, where TGF-β has an important role in mediating lung development, suggesting that loss of TβRIII at the protein level is a mechanism to regulate TGF-β signaling [41]. The regulation of TβRIII receptor levels by receptor trafficking will be discussed in further detail below.
Regulation of TβRIII expression occurs at multiple levels, via regulation of mRNA expression, epigenetic silencing, protein levels, and receptor trafficking. This multi-tiered regulation supports the importance of regulating TβRIII expression levels to maintain its physiological functions.
TβRIII Structure
TβRIII is an 851 amino acid transmembrane proteoglycan, which contains a large 766 amino acid extracellular domain, a hydrophobic transmembrane domain, and a short 42 amino acid cytoplasmic domain, which lacks intrinsic enzymatic activity [19, 42-44] (Figure 1). TβRIII is expressed on the cell surface as a non-covalently linked homodimer [43, 45].
Figure 1.
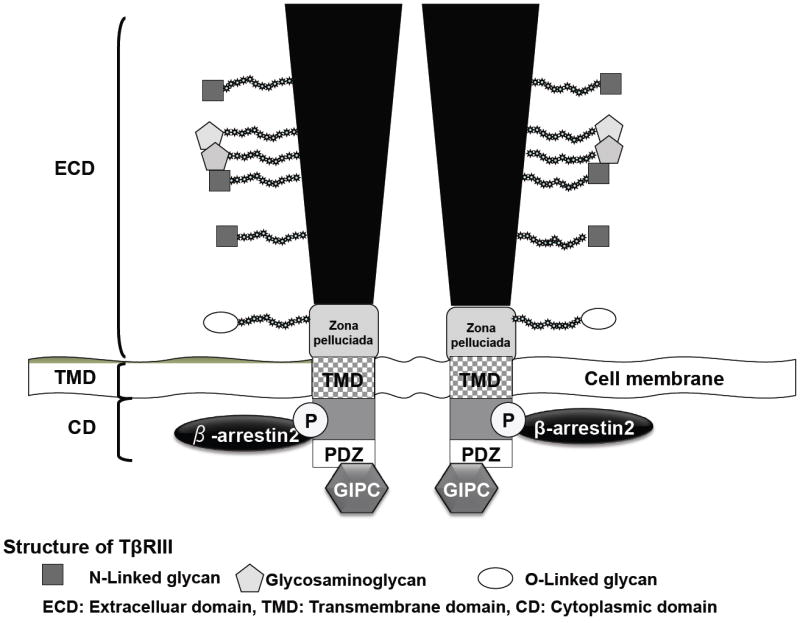
TβRIII Structure. TβRIII consists of a large extracellular domain (ECD), which has multiple sites of glycan modification (proteoglycan and glycosylation), a hydrophobic transmembrane domain, and a short cytoplasmic domain, which interacts with β-arrestin2 via phosphorylation of amino acid site 841, and with GIPC via the PDZ domain.
The extracellular domain of TβRIII contains an N-terminal orphan domain with no known homology, a zona pelucida domain (ZPD), which may be involved in receptor oligomerization, and two independent TGF-β ligand binding domains, a distal binding domain near the N-terminus of the extracellular domain and a proximal binding domain near the C-terminus of the extracellular domain, which are separated by a non-structured linker region [19, 43, 46-50]. TβRIII binds to TGF-β1, TGF-β2, and TGF-β3, BMP-2, BMP-4, and BMP-7, GDF-5, and inhibin A via the ligand binding domains, in addition to bFGF2 via the glycosaminoglycan (GAG) side chain modifications [27, 47, 51-56]. TβRIII contains distinct inhibin and TGF-β binding domains, although these domains overlap, as the inhibin binding site is solely within the ZP domain in the membrane proximal region of TβRIII [57]. The extracellular TβRIII domain contains two sites of heparin and chondroitin sulfate GAG side chain modifications at serine 535 and serine 546 [42, 47]. The TβRIII core protein has a predicted molecular weight of 100 kDa, however fully processed TβRIII migrates at an apparent molecular weight of 180 to 300 kDa due to the glycosaminoglycan post-translational modifications [19, 47]. Mutation of the GAG sites to alanine completely abrogates GAG modification of TβRIII, but does not alter TβRIII stability or the ability to bind ligand [24, 45, 47, 58].
However, post-translational modification of TβRIII does appear to be important for TβRIII function. Oncogenic Ras has been shown to modify TβRIII post-translational modifications, most likely GAG modifications, leading to an increased proliferative response to TGF-β and down-regulation of p21 in colon cancer cells [59]. In addition, TβRIII has also been shown to undergo incomplete post-translational glycosylation modification in goblet cell lines, correlating with their insensitivity to TGF-β1 [60]. Expression of TβRIII in LLC-PK1 renal epithelial cells, which lack endogenous TβRIII, results in alterations in TβRIII GAG chain modifications which prevent the formation of TβRI/TβRII complexes, inhibiting TGF-β signaling, TGF-β induced growth arrest, and altering TGF-β induced collagen deposition [61]. The TβRIII GAG modifications may also contribute to the ability of TβRIII to regulate cell migration, as a TβRIII mutant lacking the GAG chains fails to fully induce alterations in the actin cytoskeleton or inhibit migration in ovarian cells [62].
TβRIII also undergoes ectodomain shedding, with proteolytic cleavage at a site in the extracellular domain proximal to the transmembrane domain releasing the soluble extracellular domain, known as soluble TβRIII (sTβRIII). sTβRIII can be detected in the extracellular matrix and serum [43, 63]. sTβRIII levels have been demonstrated to correlate with the cell surface expression of TβRIII, suggesting constitutive shedding, however little is known about the regulation of sTβRIII production and the specific site of TβRIII cleavage has not been established [43, 63]. Although the mechanism of sTβRIII cleavage is not well understood, TβRIII shedding can be modulated by pervanadate, a tyrosine phosphatase inhibitor, and may be mediated in part by the membrane type matrix metalloproteases (MT-MMP) MT1-MMP and/or MT3-MMP, and plasmin, a serine proteinase which has been shown to cleave the extracellular domain of TβRIII [64-65]. However, in contrast to other shed transmembrane proteins, TβRIII shedding is not enhanced by the protein kinase C activator PMA [66]. The restoration of TβRIII expression in multiple cancer types can inhibit tumor progression in vivo in part through sTβRIII production, which binds to and neutralizes TGF-β, antagonizing the tumor promoting effects of TGF-β signaling in late stage tumors [12, 14-15, 17, 67-68]. The tumor suppressor function of sTβRIII will be discussed further below.
TβRIII has a short cytoplasmic domain that lacks intrinsic enzymatic activity and contains a class I PDZ binding motif [38, 43, 69]. Although the cytoplasmic domain of TβRIII is not essential for mediating ligand presentation, it has been shown to play a role in regulating TGF-β mediated signaling and growth inhibition [38, 47, 69-71]. The cytoplasmic domain of the TβRIII receptor binds to the autophosphorylated cytoplasmic domain of the type II receptor (TβRII), which promotes the formation of an active TβRII-TβRI signaling complex followed by dissociation of TβRIII from this complex [69]. In addition, the TβRIII cytoplasmic domain is phosphorylated by TβRII at threonine (Thr) 841 which facilitates the interaction of TβRIII with the scaffolding protein β-arrestin2 [39, 69]. The phosphorylation of TβRIII at Thr 841 by TβRII is required for the interaction with β-arrestin2, as mutation of the TβRIII 841 threonine to alanine (TβRIII-T841A) completely abrogates the interaction of TβRIII and β-arrestin2 [39]. The interaction of the TβRIII cytoplasmic domain with β-arrestin2 results in the co-internalization of the β-arrestin2/TβRII/TβRIII complex into endocytic vesicles and subsequent down-regulation of TGF-β signaling [39]. Recently, the interaction between TβRIII and β -arrestin2 has been shown to regulate BMP signaling as well as TGF-β signaling [72]. TβRIII complexes with the BMP type I receptor, ALK6, in a β -arrestin2 dependent manner to mediate the internalization of ALK6 and stimulation of specific BMP signaling events downstream of ALK6 [72]. In addition, the ability of TβRIII to negatively regulate NFκ-B signaling in breast cancer is dependent on its interaction with β -arrestin2 [73]. The TβRIII/β-arrestin2 interaction also mediates the ability of TβRIII to regulate cell migration, as TβRIII activates Cdc42, alters the actin cytoskeleton and suppress migration in both normal and cancerous ovarian epithelial cells in a β-arrestin2 dependent manner [62].
The cytoplasmic domain of TβRIII also interacts with GIPC (GAIP-interacting protein C terminus), a PDZ domain-containing protein, via the TβRIII class I PDZ binding domain in the cytoplasmic domain [38]. Interaction with GIPC leads to stabilization of TβRIII cell surface expression and enhancement of TGF-β signaling [38]. The interaction between TβRIII and GIPC is required for the stabilization of TβRIII, as TβRIII-DEL, a mutant which lacks the PDZ binding domain and does not bind to GIPC, is unable to stabilize TβRIII levels or enhance TGF-β signaling [38]. Furthermore, the interaction between TβRIII and GIPC has been shown to play an important role in TβRIII mediated inhibition of TGF-β signaling, cell migration, and invasion during breast cancer progression as discussed further below [71].
Role of TβRIII in TGF-β Superfamily Signaling
TβRIII has a complex and context dependent role in regulating TGF-β superfamily signaling (Figure 2, 3) [1, 74-75]. As the most abundantly expressed TGF-β superfamily receptor, the most well characterized role for TβRIII is as a TGF-β superfamily co-receptor [27, 54]. In this co-receptor role, TβRIII directly binds ligands in the TGF-β superfamily, including TGF-β 1, 2, and 3, inhibin, BMP-2, 4, and 7 and GDF-5. In most cases, the ability of TβRIII to present ligand increases their binding to their respective cognate type I and type II TGF-β superfamily receptors to increase their signaling through the Smad proteins (i.e. Smad-dependent signaling) [27, 47, 51-52, 54, 56]. In the case of inhibin binding, TβRIII is able to inhibit both activin and BMP signaling, by promoting the binding of inhibin to its cognate receptors, the activin type II and BMP type II receptors, as inhibin opposes the action of activin and BMP (Figure 3) [53, 74]. The ability of TβRIII to present ligand is particularly important for some TGF-β superfamily ligands, for example, TGF-β2 and inhibin, as these ligands cannot bind their cognate receptors, TβRII and BMPRII respectively, on their own [47, 53-54, 76]. The importance of TβRIII in regulating TGF-β signaling has been demonstrated through both gain and loss of function studies. For example, endothelial cells can be rendered sensitive to TGF-β2 by expressing TβRIII and pituitary corticotrophe tumor cell and ovarian cells can be rendered sensitive to inhibin by expression of TβRIII [53, 77]. In a reciprocal manner, knockdown of TβRIII expression has been demonstrated to decrease responsiveness to TGF-β, with a reduction in Smad2 phosphorylation and impaired activation of a TGF-β responsive promoter [78]. Cell surface TβRIII may also regulate TGF-β signaling in a context dependent manner, as it has been demonstrated to inhibit TGF-β signaling by preventing the formation of TβRI/TβRII complexes in a GAG modification-dependent manner in LLC-PK1 renal epithelial cells [61]. Mechanisms for the context dependent regulation of TGF-β signaling by TβRIII remain to be fully defined.
Figure 2.

TβRIII mediates both ligand dependent and independent effects. A. TβRIII can mediate cell migration in a β-arrestin2 dependent manner through the activation of Cdc42 in a ligand independent manner. TβRIII activates p38 in both a ligand independent and dependent manner. TβRIII undergoes ectodomain shedding producing soluble TβRIII, which can bind to and sequester ligand, inhibiting TGF-β signaling. B. TβRIII presents ligand to TβRII, which phosphorylates TβRIII, and recruits and phosphorylates TβRI, causing phosphorylation of the R-Smads. Phosphorylation of the R-Smads allows interaction with co-Smad4, mediating nuclear translocation of the Smad complex and regulation of transcriptional activity. Interaction of TβRIII with β-arrestin2 results in the internalization of the TβRIII/TβRII/β-arrestin2 complex and subsequent down-regulation of TGF-β signaling. TβRIII also negatively regulates NF B signaling in a β-arrestin2 dependent manner. Interaction of TβRIII with GIPC stabilizes TβRIII at the cell membrane and enhances TGF-β signaling, as well as mediating effects on migration and invasion.
Figure 3.

TβRIII Regulates BMP Mediated Signaling. A. TβRIII binds to inhibin and promotes the binding of inhibin to the type II receptor. The binding of inhibin to the type II receptor prevents binding of BMP (or activin, not shown) and fails to recruit the type I receptor, antagonizing both BMP (and activin) signaling. B. TβRIII interacts with ALK3 via the extracelluar domain in a β-arrestin2 independent manner and stabilizes ALK3 at the cell surface stimulating ALK3 mediated transcription. C. TβRIII interacts with ALK6 via the extracelluar and cytoplasmic domains in a β-arrestin2 dependent manner. This β-arrestin2- TβRIII-ALK6 complex is internalized and localized to endocytic vesicles, leading to maximal ALK6 mediated BMP signaling.
Just as membrane tethered TβRIII is able to alter TGF-β superfamily ligand binding in a cell autonomous manner, TβRIII is able to regulate TGF-β superfamily ligand binding and signaling in a cell non-autonomous manner by undergoing ectodomain shedding to produce sTβRIII. Indeed as sTβRIII has been detected in serum and milk, in addition to autocrine and paracrine effects, sTβRIII has the potential to have systemic effects on signaling [63, 79]. For ligands in which cell surface TβRIII enhances signaling, sTβRIII has been demonstrated to bind and sequester these ligands to inhibit signaling. Specifically, sTβRIII has been demonstrated to bind TGF-β and BMP and to inhibit TGF-β signaling [47, 80]. However, at low concentrations of TGF-β, sTβRIII has also been shown to enhance TGF-β ligand binding to TβRII, suggesting complex roles for sTβRIII in regulating TGF-β superfamily signaling [46]. The specific role of sTβRIII in regulating BMP and inhibin/activin signaling remains to be defined.
TβRIII has also been shown to have effects on non-Smad signaling pathways. TβRIII negatively regulates NFκ-B signaling in both normal mammary epithelial cells and breast cancer cells in a β -arrestin2 dependent manner [73]. Accordingly, knockdown of TβRIII expression in normal mammary epithelial cells results in an increase in NFκ-B activity, which mediates a reduction in E-cadherin expression, suggesting a role for TβRIII in maintaining an epithelial phenotype through the regulation of E-cadherin expression [73, 81]. Knockdown of TβRIII expression in NMuMG cells, a normal mammary epithelial cell line, also results in the formation of invasive high grade carcinomas an in vivo orthotopic xenograft [81], suggesting that in normal epithelial cells TβRIII plays a role in regulating cell growth, motility, and invasion through the negative regulation of NFκ-B signaling.
TβRIII mediates both ligand dependent and independent p38 pathway signaling, which has been shown to play an important role in regulating growth inhibition and apoptosis [70, 82-84]. Expression of TβRIII in L6 myoblasts causes a ligand independent increase in p38 phosphorylation and enhances TGF-β mediated growth inhibition in a Smad 3/TβRI and p38 pathway dependent manner [70, 83]. In a metastatic renal cancer cell line (UMRC3) TβRIII expression increases phosphorylation of p38 independent of functional TβRII, inducing apoptosis in clear renal cell carcinoma in vitro and in vivo [82]. The effects of TβRIII on p38 phosphorylation are at least partially dependent on the TβRIII cytoplasmic domain [70, 82].
TβRIII has also been shown to mediate effects on cellular migration through β-arrestin2 dependent activation of Cdc42 in a TGF-β signaling independent manner [62]. Knockdown of TβRIII expression in normal ovarian surface epithelial cells alters the actin cytoskeleton, with an increase in stress fibers and lamellapodia, and reduces directional and random cell migration in the absence of ligand stimulation [62]. Reciprocally, increasing the expression of TβRIII inhibits migration in normal epithelial and cancer cells via constitutive activation of Cdc42, independently of TβRII, ALK5, and Smad2 [62]. These effects on migration are mediated in part through the GAG side chains and the cytoplasmic domain, as TβRIII mutants lacking the GAG side chains (TβRIIIΔGAG) or lacking the cytoplasmic domain (TβRIIIΔcyto) do not suppress migration to the extent of full length TβRIII [62].
Regulation of TβRIII Mediated Signaling and Receptor Levels by Receptor Trafficking
TGF-β signaling can be regulated by controlling receptor cell surface expression, receptor endocytosis, and subsequent trafficking events [85]. Many TβRIII-dependent signaling events are regulated through altering these properties for TβRIII, as well as the ability of TβRIII to regulate these events for interacting receptors. Interaction of TβRIII with the PDZ-protein GIPC stabilizes TβRIII at the cell surface to enhance TGF-β signaling [38]. Conversely, internalization of TβRIII, mediated in part by interaction with β -arrestin2, has been shown to result in the co-internalization of TβRII and TβRI and down regulation of TGF-β signaling (Figure 2) [39-40]. TβRIII internalization is both ligand and GAG modification independent and requires the cytoplasmic domain, as deletion of the cytoplasmic domain (TβRIIIΔcyto) results in a slower rate of internalization [40]. Specifically, interaction with β-arrestin2, which is mediated by phosphorylation of threonine 841 by TβRII, increases TβRIII internalization and decreases cell surface levels [39-40]. TβRIII endocytosis can occur by both clathrin-mediated and clathrin-independent (lipid raft) pathways [40]. However, inhibition of the clathrin-mediated endocytosis pathway does not affect down-regulation of TβRIII, while inhibition of the clathrin-independent pathway blocks down-regulation of TβRIII, suggesting that the clathrin-independent endocytosis pathway is the preferred pathway mediating TβRIII trafficking and regulation [40]. In addition, inhibition of the clathrin-independent, but not the clathrin-dependent endocytosis pathway down-regulates Smad dependent and Smad independent (p38) TGF-β mediated signaling [40]. These data demonstrate that TβRIII endocytosis occurs via the clathrin-independent pathway, down-regulating TβRIII receptor levels and subsequent TGF-β signaling in a β-arrestin2 dependent manner [39-40].
Internalization of TβRIII has also been shown to regulate BMP signaling (Figure 3). TβRIII can interact with both type I BMP receptors, ALK3 and ALK6 [72]. TβRIII forms a complex with ALK6 in a β-arrestin2 dependent manner, and this β-arrestin2/TβRIII/ALK6 complex is internalized, where they co-localize in endocytic vesicles [72]. This TβRIII mediated internalization of ALK6 leads to maximal ALK6 mediated BMP signaling, as the ability of ALK6 to induce transcriptional activation is dependent on clathrin-mediated endocytosis of the receptor [72, 86]. Both the extracellular and cytoplasmic domains of TβRIII are required to interact with ALK6 [72]. In contrast to ALK6, TβRIII interacts with ALK3 via the extracellular domain in a β-arrestin2 independent manner and stabilizes ALK3 at the cell surface, stimulating distinct ALK-3 mediated transcription [72]. These studies demonstrate a role for TβRIII in mediating distinct TGF-β and BMP receptor trafficking events resulting in distinct and downstream signaling events. The precise mechanisms by which alteration of receptor trafficking results in distinct signaling events remains to be defined.
Phenotype of TβRIII Null Mice
To examine the role of TβRIII in vivo, several groups have created TβRIII knockout mice. In wild type murine embryos TβRIII mRNA is expressed widely throughout gestation, but is localized to the heart and liver during midgestation [87]. Deletion of TβRIII by the disruption of exon 2 of TβRIII resulted in embryonic lethality for the majority of the null mice, with only 0.3% of TβRIII -/- mice viable [87]. The TβRIII null mice that survived perinatally had a reduction in fertility, however were otherwise pathologically normal. Beginning at day 13.5, TβRIII null embryos began to exhibit enhanced apoptosis in the liver and defects in the development of the myocardial wall of the heart ventricles, with both phenotypes thought to contribute to embryonic lethality [87]. In addition, MEFs (murine embryonic fibroblasts) derived from null TβRIII mice exhibited a reduced sensitivity to TGF-β2, with alterations in growth inhibition and Smad2 nuclear localization. This correlates with previous data in demonstrating that blocking the binding of TGF-β2 to TβRIII or utilizing TβRIII neutralizing antibodies inhibits TGF-β2 induced mesenchymal transformation in chick heart development [34-35]. This suggests that alterations in TβRIII mediated regulation of TGFβ2 signaling may contribute to the liver and cardiac phenotypes observed in the TβRIII null mice.
A second TβRIII knockout mouse supports the observed defects in heart development. Compton et. al. deleted exon 3 of TβRIII, leading to a loss of TβRIII protein expression [36]. Deletion of TβRIII results in embryonic lethality at D14.5 due to defects in coronary vasculogenesis, including reduced size and irregular shape of coronary vessels [36]. TβRIII null embryos also exhibited abnormal epicardium, with increased space between the epicardium and myocardium, abundant subepicardial mesenchyme, and persistent blood islands. The effects of TβRIII on vasculogenesis and angiogenesis appear to be confined to coronary vessels, as the vasculature outside of the coronary circulation appears normal and smooth muscle recruitment is normal in both coronary and extracardiac vessels [36].
These mouse models demonstrate a requirement for TβRIII in murine somatic development, specifically in murine hepatic and cardiac development, which are essential for embryonic viability. In addition, they support essential and non-redundant roles for TβRIII in embryonic development.
TβRIII: A Suppressor of Cancer Progression
Breast Cancer
Breast cancer is the most frequently diagnosed cancer and the second leading cause of cancer death among women [88]. TGF-β signaling plays an important role in mammary gland development through the inhibition of epithelial cell proliferation and the regulation of mammary ductal and alveolar development [89]. The over-expression of TGF-β in mammary tissue potently inhibits mammary gland development, with reduced ductal outgrowth and branching observed in virgin glands of MMTV-TGF-β mice in comparison to wild type mice [89]. In early stage breast tumors TGF-β functions as a tumor suppressor, while in late-stage tumors TGF-β signaling is frequently up-regulated and has a tumor promoting effect, enhancing tumor progression and metastasis [6]. Interestingly, despite the prominent role TGF-β plays in regulating mammary carcinogenesis, mutations or alterations in components of the TGF-β pathway are infrequent in human breast cancers [6]. However, it has been recently demonstrated that the loss of TβRIII expression, due to a loss of heterozygosity (LOH) or through transcriptional down-regulation, is a frequent early event in the development of human breast cancer, beginning during ductal carcinoma in situ (DCIS) [12, 20]. Importantly, the restoration of TβRIII expression in murine mammary cancer cells inhibits tumor invasion, angiogenesis, and metastasis in vivo, in part through sTβRIII production, which binds to and neutralizes TGF-β, antagonizing the tumor promoting effects of TGF-β signaling in late stage mammary tumors [12, 90]. Several studies have demonstrated that treatment with sTβRIII alone inhibits breast cancer tumor growth, angiogenesis, and reduces metastasis in nude mice in vivo xenograft studies of breast cancer cells [67-68, 90-91]. A recent study has also shown a requirement for the TβRIII cytoplasmic domain, specifically interaction with the scaffolding protein GIPC, in mediating the inhibition of TGF-β signaling, cell migration, invasion, and in vivo breast cancer metastasis [71]. These data support that the loss of TβRIII is required for breast cancer progression and metastasis.
In reciprocal studies, knockdown of TβRIII expression in non-tumorigenic NMuMG murine mammary epithelial cells has been demonstrated to down regulate TGF-β signaling resulting in increased growth, motility, and invasion [81]. In contrast, another study demonstrated that knockdown of TβRIII in human and murine breast cancer cells resulted in decreased proliferation, migration, and invasion, and increased apoptosis as well as impairing in vivo tumorigenicity and inhibiting lung metastasis [78]. Mechanistically, loss of TβRIII reduced NF-κB activity and restoration of NF-κB activity restored tumor cell invasiveness and resistance to apoptosis [78]. Although this data is in contrast to previous reports of a role for TβRIII in inhibition of tumor progression, all data supports a role for TβRIII in the regulation of migration and invasion. The effects of TβRIII on tumor cell invasion and migration may be dependent on the activation or inhibition of other oncogenes or tumor suppressors. Taken together, these studies suggest that the loss of TβRIII expression during breast cancer progression alters the regulation of TGF-β signaling and may contribute to the loss of TGF-β mediated inhibition of proliferation and the increase in migration and invasion observed in late stage breast tumors.
Renal cancer
Renal cell carcinoma (RCC) represents approximately 3% of all cancers worldwide [92]. Localized renal cell carcinoma lesions can be surgically cured. However, metastatic disease is incurable and systemic chemotherapy is usually ineffective. Loss of TβRIII expression has also been described during renal cell carcinoma progression. When comparing TβRIII expression between normal renal epithelium, non-metastatic renal cell carcinoma, and primary and disseminated lesions in metastatic renal cell carcinoma, loss of TβRIII was an early event in renal cell carcinoma carcinogenesis, with loss observed in non-metastatic renal cell carcinoma specimens, and subsequent loss of TβRII associated with the acquisition of a metastatic phenotype [13]. In contrast, TβRI levels were not altered across cancer stages [13]. When correlated with TNM score, TβRIII mRNA expression was suppressed in the earliest stage (stage I) and in all tumor stages (I-V) [13]. Restoration of TβRIII expression in clear cell RCC resulted in a marked induction of apoptosis both in vitro and in vivo models [13, 82]. The ability of TβRIII to induce apoptosis was ligand, TβRI, and TβRII independent. The effects of TβRIII were dependent on the cytoplasmic domain, as the expression of the TβRIII cytoplasmic domain, but not the extracellular domain, could mimic the induction of apoptosis by full-length TβRIII in cell culture and inhibited tumor growth in athymic nude mice [82]. This TβRIII associated apoptosis was not dependent on signaling through the canonical TGF-β/Smad pathway but was mediated through the p38 pathway [82]. These studies demonstrate that TβRIII plays an important role in renal cell carcinoma tumorigenesis.
Prostate Cancer
TGF-β signaling plays an important role in the regulation of normal prostate epithelium through the mediation of differentiation, growth arrest, and apoptosis [93]. During prostate cancer, prostate cancer cells become resistant to TGF-β mediated effects, due to in part to a loss of TβRI or TβRII in 30% of tumors [94]. Several studies have shown that the loss of TβRIII is the most common alteration in the TGF-β pathway during prostate cancer progression, with a significant reduction or complete loss of TβRIII in the majority of human prostate cancers [14, 95]. The loss of TβRIII expression has been shown to occur at the mRNA and protein level, and is due to LOH at the genomic locus and either direct or indirect epigenetic regulation of TβRIII expression [14]. Loss of TβRIII correlates with disease stage, including metastatic disease and prostate-specific antigen (PSA) recurrence, suggesting a role for TβRIII in disease progression [14]. In addition, TβRIII expression was shown to decrease further in metastases in comparison to primary prostate tumors.
The knockdown of TβRIII in immortalized prostate epithelial cells causes increased focus formation, although these cells did not form tumors in NOD/SCID mice [95]. In addition, the knockdown of TβRIII led to increased expression of the prostate stem cell marker, CD133, suggesting an expansion of cells with stem cell characteristics [95]. Microarray analysis of changes in gene expression between immortalized prostate epithelial cells expressing TβRIII and cells with knockdown of TβRIII revealed alterations in 101 genes, including down-regulation of genes commonly altered in prostate cancer: bone morphogenic protein 4 (BMP4), latent transforming growth factor beta binding protein 1 (LTBP1), vimentin, coagulation factor C homolog (COCH), SERPINF1, peripheral myelin protein 22 (PMP22), and dihydropyrimidinase-like 3 (DPYSL3) [95]. Interestingly, SERPINF1 is a potent inhibitor of angiogenesis and vimentin expression has been show to decrease in the transition from benign prostate to local prostate cancer in a number of studies [95].
The restoration of TβRIII expression in prostate cancer cells inhibits migration and invasion, both basally and in response to TGF-β treatment, however this has no effect on proliferation [14]. Restoration of TβRIII in an in vivo prostate cancer xenograft model decreased tumor growth, supporting a tumor suppressor role of TβRIII in prostate cancer [14]. In addition, treatment of a prostate cancer xenograft model with sTβRIII significantly inhibited tumor growth, angiogenesis, decreased expression of MMP-9, and enhanced apoptosis [96]. This suggests that loss of TβRIII with disease progression may enable prostate cancer cells to become more motile and invasive. These studies support a role for TβRIII as a suppressor of prostate cancer progression.
Non-small cell lung Cancer (NSCLC)
Lung cancer accounts for the highest number of cancer related deaths in both men and women [88]. The TGF-β signaling pathway plays an important role in lung development and many lung cancers become resistant to the homeostatic effects of TGF-β [97]. This occurs in part through loss of TβRII, Smad2, and Smad4, although these mutations are not common in non-small cell lung cancer (NSCLC), which accounts for greater than 80% of all lung cancers [98-99]. However, TβRIII expression is decreased in NSCLC at both the mRNA level and protein level, due to LOH at the TGFBR3 genomic locus [17]. In addition, decreasing levels of TβRIII expression correlates with increasing tumor grade and disease progression [17]. One study also defined a correlation between decreased TβRIII expression and a decrease in squamous cell carcinoma patients’ 5-year survival [100].
Expression of TβRIII in NSCLC cells, which exhibit normal to low levels of TβRIII, does not affect proliferation or TGF-β dependent Smad phosphorylation [17]. However, TβRIII expression inhibits lung cancer cell motility and invasion in NSCLC cells and knockdown of TβRIII expression increased invasion [17]. The ability of TβRIII to mediate migration and invasion is due, in part, to production of sTβRIII as treatment with conditioned media from cells over-expressing TβRIII was able to inhibit invasion in NSCLC cells. In addition, expression of TβRIII inhibits anchorage-independent growth of NSCLC cells in a soft agar assay [17]. Over-expression of TβRIII in an NSCLS in vivo xenograft assay reduced tumor incidence, tumor growth, and invasiveness of tumors [17]. These studies demonstrate a role for TβRIII in suppression of cancer progression in NSCLC, with TβRIII inhibiting migration, invasion, anchorage independent growth, and in vivo tumorigenicity of lung cancer cells.
Pancreatic Cancer
The expression of multiple components of the TGF-β signaling pathway, including TβRI, TβRII, and Smad4, are altered at both the mRNA and protein levels during pancreatic cancer [101]. TβRIII expression has recently been shown to decrease at the mRNA and protein level in pancreatic adenocarcinomas in comparison to matched normal samples [16]. In addition, the genomic locus for TβRIII on chromosome 1p is deleted in 49% of human pancreatic cancers [102]. The loss of TβRIII increases as tumor grade increases, suggesting that TβRIII plays an important role in mediating the progression of pancreatic cancer [16].
Epithelial to mesenchymal transition (EMT) plays an important role in the metastatic process. During the process of EMT, adherent epithelial cells become motile and invasive, and subsequently lose expression of epithelial E-cadherin cytokeratin and gain expression of vimentin, N-cadherin, and transcription factors Snail and Slug. The TGF-β pathway regulates EMT during development and cancer progression, in multiple systems including pancreatic cancer [11, 103]. TGF-β, BMP2, 4, and 7 have been shown to induce both time and dose dependent EMT in a pancreatic cancer cell line [16, 104]. TβRIII expression decreases during TGF-β and BMP4 induced EMT in PANC-1 cells in a time and dose dependent manner, although expression of TβRI and TβRII is not altered [16]. There is an increase in levels of sTβRIII in pancreatic cancer cells undergoing EMT, suggesting that TβRIII cell surface expression decreases rapidly during EMT due to increased TβRIII shedding [16]. Inhibiting the loss of TβRIII during both TGF-β and BMP4 induced EMT by adenoviral over-expression did not alter ligand induced EMT. However inhibiting loss of TβRIII expression does significantly inhibit the EMT-associated increases in motility and invasiveness in pancreatic cancer cells [16, 104]. Conversely, knockdown of TβRIII expression in pancreatic cancer cells increased motility and invasion in response to TGF-β treatment. A TβRIII mutant lacking the cytoplasmic domain (TβRIII-Δcyto) inhibited basal motility and invasion to the same extent as full length TβRIII, suggesting that the cytoplasmic domain does not play a role in mediating the effects of TβRIII on motility and invasion in pancreatic cells [16]. In addition, maintaining TβRIII expression during EMT inhibits both TGF-β mediated induction of Smad2 phosphorylation and BMP mediated Smad1 phosphorylation. Loss of Smad1 in pancreatic cancer cells inhibits BMP mediated induction of MMPs and cell invasiveness, suggesting that TβRIII may mediate BMP effects on invasiveness through its effects on Smad1 [104]. The ability of TβRIII to inhibit TGF-β mediated motility and invasion occurs at least in part through production of sTβRIII, as conditioned media from cells over-expressing TβRIII inhibits basal and TGF-β mediated increases in invasion [16, 104]. These data demonstrate that while the loss of TβRIII is not required for EMT induction, loss of TβRIII is required for the increase in motility and invasiveness associated with EMT and pancreatic cancer progression.
Ovarian cancer
Ovarian cancer is the most lethal of all gynecological cancers and the fifth leading cause of death among women in United States [88]. Due to the lack of early symptoms or effective screening tests, most ovarian cancer patients are diagnosed with metastatic disease.
TβRIII expression is significantly decreased or lost in human epithelial origin ovarian tumors compared to normal tissue, with loss of TβRIII expression correlating with tumor grade and ovarian cancer progression [15]. Treatment with methyltransferase and histone deacetylase inhibitors induced TβRIII expression in ovarian cancer cell lines, indicating that either direct or indirect epigenetic silencing plays a role in the loss of TβRIII expression in ovarian cancer cells [15]. When TβRIII expression is restored in Ovca429, an ovarian epithelial cell line which lacks TβRIII yet retains high TβRI and TβRII levels, cell invasion and migration were significantly reduced [15]. In addition, re-introduction of TβRIII into Ovca429 cells enhanced the anti-migratory effect of inhibin, which could be due in part to the decrease of MMP2 and MMP9 levels [15]. This anti-migratory effect of TβRIII was independent of TβRI or TβRII and was mediated through the interaction of TβRIII with βarrestin2, resulting in activation of Cdc42, which alters the actin cytoskeleton and reduces directional persistence to inhibit random migration of both cancer and normal ovarian epithelial cells [62].
One possible anti-tumorigenic effect of TβRIII in ovarian cancer is through its enhancement of inhibin binding to ActRII (Activin specific type II receptor serine kinase), which results in enhancement of activin antagonism [53]. Activin and inhibin are structurally related TGF-β superfamily members and are mutually antagonistic regulators of reproductive function. In the ovary, activin and inhibin subunits are expressed mainly in granulose cells but are also detected in normal epithelial cells [105]. Malignant ovarian epithelial tumors have decreased inhibin production relative to activin production in comparison with normal ovarian surface epithelial cells and nonmalignant ovarian surface epithelial cells [105]. Activin binds to ActRII and recruits ALK4, which then activates the Smad pathway, while inhibin binds to ActRII but does not recruit ALK4, thereby inhibiting the pathway. TβRIII binds inhibin with high affinity, enhancing inhibin binding to ActRII and blocking TβRII/I downstream signal transduction.
The effect of TβRIII has also been investigated in ovarian sex cord stromal tumors, specifically granulose cell tumor (GCT), which account for 7% of total ovarian cancer incidences [106]. In this study, 53% (9 of 17) of human GCT samples exhibit significantly lower TβRIII mRNA expression compared to normal premenopausal ovary, suggesting that loss of TβRIII is a feature of clinical disease and may contribute to GCT progression [106]. Restoration of TβRIII expression in COV434 cells derived from metastatic GCT and in KGN cells derived from stage III GCT, increased TGF-β responsiveness and adherence to the extracellular matrix component and decreased migration. However, knockdown of inhibin A abrogated TβRIII mediated inhibition of wound healing and invasion. In addition, when inhibin A and TGF-β were neutralized, this abolished the TβRIII mediated increase in adhesion to substrate [106]. TβRIII did not have affect on COV434 and KGN cell proliferation or apoptosis [106]. These studies support a role for TβRIII in ovarian cancer progression through the inhibition of migration, invasion, and adhesion.
Endometrial carcinoma
In endometrial adenocarcinomas, expression of both TβRIII and inhibin α are reduced at the mRNA and protein level [107]. In normal endometrial tissue, TβRIII is localized to vascular endothelial cells lining vessels in the endometrium and myometrium and also in endometrial epithelial cells. However, TβRIII expression is weak in well-differentiated endometrial carcinoma and lacking in poorly differentiated endometrial carcinoma. [107]. Interestingly, tumor endothelial cells exhibit high TβRIII levels, suggesting that a selective vascular response to TGF-β and inhibin may be possible [107]. This suggests that in addition to the decreased expression of inhibin α, the loss of TβRIII may disrupt the inhibin/activin pathway and TGF-β signaling in endometrial cancer.
Role of TβRIII in Mediating Invasion and Migration
Metastasis is the most common cause of death in cancer patients. The classical view of the metastatic cascade, starting from a primary, epithelial, neoplastic lesion include: EMT and breach of the basement membrane barrier, dissociation of tumor cells from the bulk tumor, invasion of the neighboring tissue, intravasation into pre-existing and newly formed blood and lymph vessels, transport through vessels, extravasation from vessels and establishment of disseminated cells at a secondary anatomic site and outgrowth of micrometastases and macrometastases/secondary tumors [108]. During these processes, cancer cells undergo changes in cell motility parameters, stiffness, and shape as seen during EMT, which allow these cells to enter lymphatic and blood vessels and disseminate. In addition, the cancer cells need to degrade the basement membrane to enter the blood or lymphatic vessels and parenchyma of the target organ where they form secondary tumors. This process can be mediated by secretion or regulation of matrix degrading enzymes including matrix metalloproteinases, proteinases of the plasminogen activator system, and heparanase and by alterations in cell morphology and cytoskeleton remodeling which facilitate the invasion process [109]. Cell-cell adhesion is important for maintaining an epithelial phenotype, and loss of cell-cell adhesion is associated with EMT, increasing migration, and invasion [109]. The TGF-β superfamily, including TGF-β and BMP, regulates EMT during development and cancer progression in multiple systems [11].
TβRIII has been shown to regulate both MMP secretion and cell invasion in multiple cancer systems. Expression of TβRIII in multiple human cancer types that lose TβRIII expression with disease progression has been shown to inhibit cell migration, invasion, and metastasis both in vitro and in vivo [12, 14-17, 106]. In ovarian cancer, TβRIII decreases the level of MMP2 and 9 at both the mRNA and protein level [15]. In pancreatic cancer, reintroduction of TβRIII expression during EMT inhibits BMP mediated Smad1 phosphorylation, which abrogates BMP mediated induction of MMPs and cell invasiveness [104]. Systemic treatment with sTβRIII reduced MMP-9 expression in a prostate cancer xenograft model [96]. Loss of TβRIII expression in human ovarian cancers results in an increase of N-Cadherin and Slug mRNA and also alterations in the cytoskeleton including filopodia and lamelipodia [62]. In pancreatic cancer, loss of TβRIII is associated with TGF-β induced EMT, and this is required for EMT associated increases in migration and invasion possibly via alterations in cell-cell junctions [16].
TβRIII has an emerging role in the regulation of migration in both normal and cancerous cells in both a ligand dependent and independent manner. Re-introduction of TβRIII expression in multiple cancer systems that lose TβRIII expression with disease progression, including breast, ovarian, lung, prostate, and pancreatic cancer, has been shown to inhibit ligand induced migration [12, 14-17, 71]. Recent studies have suggested essential, non-redundant roles for TβRIII in regulating migration signals independently from TβRI/RII in renal cell tumors and in ovarian epithelial and cancer cells [13, 62]. The short TβRIII cytoplasmic domain has been classically considered to lack signaling function. However recent studies suggest that this domain may play an important role in regulation of cell migration. In normal ovarian surface epithelial and ovarian cancer cells, the interaction of the TβRIII cytoplasmic domain and β-arrestin2 regulates the activation of Cdc42 to alter migration [62]. The TβRIII mediated effects on migration are also regulated in part through the GAG side chains, as TβRIII mutants lacking the GAG side chains (TβRIIIΔGAG) or lacking the cytoplasmic domain (TβRIIIΔcyto) do not suppress migration to the extent of full length TβRIII [62]. However, the dependence on the cytoplasmic domain for regulation of cell migration may be cell type dependent as TβRIIIΔcyto was just effective as TβRIII in decreasing migration and invasion in breast and pancreatic cancer cells, in part, via effects of sTβRIII [12, 16].
Soluble TβRIII, generated by ectodomain shedding, has also been demonstrated to regulate cell adhesion and migration. sTβRIII is able to recapitulate the inhibitory effect of membrane bound TβRIII on invasion in breast, pancreatic, and non-small cell lung cancer models [12, 16-17]. sTβRIII may function either by sequestering TGF-β superfamily ligands and suppressing signaling or by binding to extracellular matrix proteins and blocking their ability to interact with TβRIII on the cell surface.
TβRIII has been shown to have an essential role in mediating the migration and invasion of both normal epithelial and epithelial-derived cancer cells. The ability of TβRIII to suppress cancer progression is due in part to inhibition of migration and invasion in cancer cells. Further understanding the role of TβRIII in regulating these mechanisms that contribute to metastasis will increase our ability to target TβRIII and the TGF-β superfamily signaling pathways in human cancer.
Targeting TβRIII in Human Cancers
Cancer progression, including breast, prostate, and glioma progression, is associated with the increased expression of TGF-β and other TGF-β superfamily ligands, which may function through effects on the cancer cells and the stroma to promote cancer progression [6]. Therefore antagonizing TGF-β superfamily ligands is a promising strategy to counteract malignant properties of tumors. TβRIII has been shown to have anti-tumor and anti-metastatic effects through the inhibition of migration, invasion, cell growth, apoptosis, and metastasis in vitro and in vivo. The ability of TβRIII to inhibit tumorigenesis may occur in part through the ability of sTβRIII to sequester TGF-β superfamily ligands and antagonize TGF-β superfamily signaling, suggesting that sTβRIII or agents derived from sTβRIII could be used as therapeutic agents [47, 80].
Several studies have been performed with sTβRIII demonstrating its inhibitory effects on tumor progression. Treatment of mink lung epithelial cells with conditioned medium from MDA-MB-231 cells stably expressing sTβRIII significantly suppressed cell growth in vitro due to the ability of sTβRIII to sequester extracellular TGF-β isoforms and antagonize their function [91]. Treatment of the breast cancer cell line MDA-MB-231 with recombinant sTβRIII or stable expression of sTβRIII in MDA-MB-231 cells antagonized TGF-β signaling, inhibiting anchorage dependent and independent cell growth, and induced apoptosis in vitro and in vivo [91, 110]. MDA-MB-231-sTβRIII cells showed reduced tumor incidence and growth as well as a reduction in lung metastasis compared to MDA-MB-231-Neo cells in vivo [91, 110]. MDA-MB-231-sTβRIII cells also exhibited increased PTEN levels and a concurrent decrease in phosphorylated Akt levels in comparison to the Neo cells [110]. Further, ectopic expression of sTβRIII in human colon (HCT116) and breast carcinoma (MDA-MB-435) in vivo xenograft models inhibited tumor growth and the metastatic potential of tumors, as well as inhibiting angiogenesis in primary tumors [68]. In addition, recombinant sTβRIII was shown to have potential therapeutic utility in breast, colon, and prostate cancer, with systemic treatment of sTβRIII reducing tumor growth, metastasis, and angiogenesis in vivo [91, 96]. Treatment of human endothelial cells with a recombinant sTβRIII significantly inhibited capillary formation, suggesting that the anti-tumorigenic effect of sTβRIII is partially via suppression of TGF-β induced angiogenesis [67-68]. Continuous or bolus administration of sTβRIII inhibited prostate xenograft growth associated with reduced tumor blood volume and microvessel density and an enhanced intra-tumoral apoptosis [96]. Furthermore, sTβRIII treatment inhibited TGF-β induced MMP-9 activity in this system, suggesting inhibition of invasive properties. sTβRIII has also been shown to reduce renal damage in the db/db mouse model of type 2 diabetes [111]. In this model of nephropathy, systemic treatment with sTβRIII prevented up-regulation of TGF-β1, 2, and 3, as well as decreasing fibronectin and collagen IV levels in the kidney, reducing renal damage progression [111]. Treatment of mice with sTβRIII did not cause any deleterious side effects, such as weight loss or behavior changes, suggesting that sTβRIII is well tolerated.
sTβRIII has also been shown to have promise in combination with other agents. For example, treatment of malignant gliomas with sTβRIII alone was not effective, however when combined with soluble TβRII (sTβRII), sTβRIII increased the anti-tumor effect of sTβRII, reducing Smad2 phosphorylation and abrogating TGF-β effects [112]. In addition, combinatorial treatment with sTβRII and sTβRIII in a glioma model blocked the immunosuppressive effect of TGF-β on NK cells, suggesting that sTβRII and sTβRIII may restore an effective anti-glioma immune response by neutralizing TGF-β [112]. This study demonstrated an in vivo survival benefit of treatment with sTβRII and sTβRIII. Although there have been attempts to use other soluble TGF-β receptors as TGF-β inhibitors, their antagonistic potency against different TGF-β isoforms varies and TGF-β isoforms are differentially expressed depending on cell type and context [113-118]. sTβRIII may be more effective than sTβRII, as it can bind and sequester TGF-β2, while sTβRII cannot [80, 119-120]. In addition, TβRIII can bind BMPs and inhibin, suggesting that sTβRIII can bind these ligands as well [52-53].
Recently, a recombinant fusion protein containing the endoglin domain of TβRIII (BGE) and the extracellular domain of TβRII has been constructed for use as a TGF-β ligand antagonist [121]. This fusion protein (BGERII) bound with a higher affinity to TGF-β1 and TGF-β3 than a commercially available sTβRII and with a higher affinity to TGF-β2 than a commercially available sTβRIII [121]. Whereas BGE or TβRII alone showed no antagonistic activity towards TGF-β2, BGERII inhibited the signaling of both TGF-β1 and TGF-β2 in cell-based assays more effectively than equimolar concentrations of either sTβRII or sTβRIII alone, including reducing TGF-β induced phosphorylation of Smad2 and Smad3 and inhibiting transcription from a TGF-β responsive promoter. This study suggests that sTβRIII may be useful for engineering additional reagents which target TGF-β signaling.
It is promising that systemic administration of sTβRIII has been shown to have an anti-tumorigenic effect in breast, prostate, colon, and glioma in vivo cancer models without deleterious side effects. The ability of sTβRIII treatment to reduce tumor growth, inhibit angiogenesis, induce apoptosis, and inhibit metastasis in multiple cancer systems suggests that sTβRIII may be a viable target to inhibit cancer progression.
In addition, TβRIII expression may also have prognostic value as a biomarker, as loss of TβRIII expression increases with clinical stage and correlates with metastatic disease in multiple tumor types, including ovarian, breast, prostate, and NSCLC [12, 14-15, 17, 71]. Levels of TβRIII or sTβRIII expression could be assessed by immunohistochemistry or RT-PCR of tumor tissue or ELISA analysis of blood serum for sTβRIII and could be utilized to determine disease stage or direct treatment.
TβRIII also has functions beyond the production of sTβRIII, including regulation of migration and invasion, therefore restoring the expression of cell surface TβRIII in tumors could have therapeutic benefits. In addition, expression of membrane bound TβRIII has been shown to correlate with production of sTβRIII [43, 63]. As expression of TβRIII is down-regulated via epigenetic silencing in multiple human tumor types, treatment with histone deacetylase inhibitors or DNA methylation inhibitors could be utilized to restore TβRIII expression [14]. Restoration of TβRIII expression inhibits tumor migration, invasion, metastasis, and angiogenesis in multiple in vivo models of tumorigenesis thereby supporting reintroduction of TβRIII expression as a viable therapy [12, 14, 17, 71].
Future Directions
TβRIII mediates and regulates cell migration, invasion, angiogenesis, apoptosis, and metastasis, all mechanisms that contribute to cancer progression. The ability of TβRIII to suppress cancer progression is due in part to the inhibition of migration and invasion of cancer cells. Further understanding the role of TβRIII in regulating these mechanisms that contribute to metastasis will increase our ability to target TβRIII and the TGF-β superfamily signaling pathways in human cancer.
The effects of TβRIII are due at least in part to the production of sTβRIII, as treatment with sTβRIII reduces migration, invasion, and metastasis in in vivo models and is able to recapitulate the inhibitory effect of membrane bound TβRIII on invasion in breast, pancreatic, and non-small cell lung cancer models [12, 16-17]. A number of questions regarding sTβRIII remain to be answered. While TβRIII functions by sequestering TGF-β superfamily ligands and suppressing signaling, the specific roles and the ratio of membrane bound TβRIII and sTβRIII still remain to be fully elucidated. Currently, the specific site of TβRIII cleavage remains unknown. Identification of the cleavage site would allow mutation to inhibit TβRIII cleavage, further enhancing our understanding of the contribution of soluble and membrane bound TβRIII to the inhibition of cancer progression. Finally, the mechanisms regulating ectodomain shedding and production of sTβRIII remain to be discovered. A further understanding of the production of sTβRIII could lead to mechanisms to enhance ectodomain shedding as a therapeutic target.
TβRIII expression is significantly decreased or lost in multiple types of human cancers. Loss of TβRIII expression occurs via epigenetic silencing in multiple human tumor types, suggesting that treatment with histone deacetylase inhibitors or DNA methylation inhibitors could be utilized to restore TβRIII expression [14]. Further understanding the cell type specific regulation of TβRIII mRNA expression may identify possible treatments to enhance or restore TβRIII expression in different human tumors.
Many TβRIII dependent signaling events are regulated through alteration of receptor cell surface expression, receptor endocytosis, and subsequent trafficking events, as well as the ability of TβRIII to regulate these events for interacting receptors [85]. The interaction of TβRIII with GIPC has been shown to stabilize TβRIII at the cell surface resulting in enhanced TGF-β signaling, while interaction with β -arrestin2 leads to co-internalization of TβRIII, TβRII and TβRI and down regulation of TGF-β signaling respectively [39-40]. TβRIII plays a role in mediating distinct TGF-β and BMP receptor trafficking events resulting in distinct and downstream signaling events. The interaction of TβRIII with other scaffolding proteins and subsequent receptor regulation still remains to be discovered. In addition, the precise mechanisms by which alteration of receptor trafficking results in distinct signaling events remains to be defined.
Conclusion
TGF-β signaling plays a dichotomous role in human cancers, functioning as both a tumor suppressor and a tumor promoter. TβRIII plays an important role in mediating ligand dependent TGF-β superfamily signaling as well having as ligand independent functions, including the regulation of migration. The loss of TβRIII expression in early disease stages of multiple human cancer types suggests that TβRIII may inhibit tumor progression [12-17]. Indeed, TβRIII has direct roles in inhibiting cell migration, invasion, angiogenesis, apoptosis, and metastasis in in vivo models suggesting an emerging role for TβRIII as an inhibitor of tumor progression and/or metastasis. Supporting this theory, reintroduction of TβRIII expression or treatment with sTβRIII inhibits metastasis in multiple in vivo tumor models [12, 14, 17, 67, 82, 91, 96]. These studies demonstrate the important and non-redundant roles of TβRIII beyond that of a co-receptor, including regulation of migration, invasion, and metastasis. The anti-tumor and anti-metastatic effects of TβRIII and promising results with sTβRIII treatment suggest that TβRIII may be an important target for treatment of human cancers. Further work remains to more fully understand the role and mechanism of TβRIII in human cancer and to explore its possible role as a therapeutic target.
Abbreviations
- TGF-β
Transforming growth factor β
- TβRIII
Type III TGF-β receptor
- STβRIII
Soluble type III TGF-β receptor
- BMP
Bone morphogenetic protein
- EMT
Epithelial-to-mesenchymal transition
- GIPC
GAIP-interacting protein, C terminus
- GAG
Glycosaminoglycan
- MMP
Matrix metalloproteases
- RCC
Renal cell carcinoma
- NSCLC
Non-small cell lung cancer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Massague J. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 2.Gordon KJ, Blobe GC. Biochim Biophys Acta. 2008;1782(4):197–228. doi: 10.1016/j.bbadis.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Bierie B, Moses HL. Nat Rev Cancer. 2006;6(7):506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 4.Bierie B, Moses HL. Cytokine Growth Factor Rev. 2006;17(1-2):29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Massague J. Cell. 2008;134(2):215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elliott RL, Blobe GC. J Clin Oncol. 2005;23(9):2078–2093. doi: 10.1200/JCO.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 7.Jakowlew SB. Cancer Metastasis Rev. 2006;25(3):435–457. doi: 10.1007/s10555-006-9006-2. [DOI] [PubMed] [Google Scholar]
- 8.Pardali K, Moustakas A. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2007;1775(1):21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Levy L, Hill CS. Cytokine Growth Factor Rev. 2006;17(1-2):41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 10.Rahimi RA, Leof EB. J Cell Biochem. 2007;102(3):593–608. doi: 10.1002/jcb.21501. [DOI] [PubMed] [Google Scholar]
- 11.Xu J, Lamouille S, Derynck R. Cell Res. 2009;19(2):156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong M, How T, Kirkbride KC, Gordon KJ, Lee JD, Hempel N, Kelly P, Moeller BJ, Marks JR, Blobe GC. J Clin Invest. 2007;117(1):206–217. doi: 10.1172/JCI29293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Copland JA, Luxon BA, Ajani L, Maity T, Campagnaro E, Guo H, LeGrand SN, Tamboli P, Wood CG. Oncogene. 2003;22(39):8053–8062. doi: 10.1038/sj.onc.1206835. [DOI] [PubMed] [Google Scholar]
- 14.Turley RS, Finger EC, Hempel N, How T, Fields TA, Blobe GC. Cancer Res. 2007;67(3):1090–1098. doi: 10.1158/0008-5472.CAN-06-3117. [DOI] [PubMed] [Google Scholar]
- 15.Hempel N, How T, Dong M, Murphy SK, Fields TA, Blobe GC. Cancer Res. 2007;67(11):5231–5238. doi: 10.1158/0008-5472.CAN-07-0035. [DOI] [PubMed] [Google Scholar]
- 16.Gordon KJ, Dong M, Chislock EM, Fields TA, Blobe GC. Carcinogenesis. 2008;29(2):252–262. doi: 10.1093/carcin/bgm249. [DOI] [PubMed] [Google Scholar]
- 17.Finger EC, Turley RS, Dong M, How T, Fields TA, Blobe GC. Carcinogenesis. 2008;29(3):528–535. doi: 10.1093/carcin/bgm289. [DOI] [PubMed] [Google Scholar]
- 18.Kirkbride KC, Ray BN, Blobe GC. Trends Biochem Sci. 2005;30(11):611–621. doi: 10.1016/j.tibs.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Wang XF, Lin HY, Ng-Eaton E, Downward J, Lodish HF, Weinberg RA. Cell. 1991;67(4):797–805. doi: 10.1016/0092-8674(91)90074-9. [DOI] [PubMed] [Google Scholar]
- 20.Hempel N, How T, Cooper SJ, Green TR, Dong M, Copland JA, Wood CG, Blobe GC. Carcinogenesis. 2008;29(5):905–912. doi: 10.1093/carcin/bgn049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ragnarsson G, Eiriksdottir G, Johannsdottir JT, Jonasson JG, Egilsson V, Ingvarsson S. Br J Cancer. 1999;79(9-10):1468–1474. doi: 10.1038/sj.bjc.6690234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bayat A, Watson JS, Stanley JK, Ferguson MW, Ollier WE. Eur J Immunogenet. 2002;29(5):445–446. doi: 10.1046/j.1365-2370.2002.00339.x. [DOI] [PubMed] [Google Scholar]
- 23.Zippert R, Baszler A, Holmer SR, Hengstenberg C, Schunkert H. J Hum Genet. 2000;45(4):250–253. doi: 10.1007/s100380070035. [DOI] [PubMed] [Google Scholar]
- 24.Cheifetz S, Like B, Massague J. J Biol Chem. 1986;261(21):9972–9978. [PubMed] [Google Scholar]
- 25.Massague J. J Biol Chem. 1985;260(11):7059–7066. [PubMed] [Google Scholar]
- 26.Massague J, Like B. J Biol Chem. 1985;260(5):2636–2645. [PubMed] [Google Scholar]
- 27.Bernabeu C, Lopez-Novoa JM, Quintanilla M. Biochim Biophys Acta. 2009;1792(10):954–973. doi: 10.1016/j.bbadis.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 28.Ji C, Chen Y, McCarthy TL, Centrella M. J Biol Chem. 1999;274(43):30487–30494. doi: 10.1074/jbc.274.43.30487. [DOI] [PubMed] [Google Scholar]
- 29.Nakayama H, Ichikawa F, Andres JL, Massague J, Noda M. Exp Cell Res. 1994;211(2):301–306. doi: 10.1006/excr.1994.1091. [DOI] [PubMed] [Google Scholar]
- 30.Wickert L, Abiaka M, Bolkenius U, Gressner AM. J Hepatol. 2004;40(1):69–76. doi: 10.1016/j.jhep.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 31.Lopez-Casillas F, Riquelme C, Perez-Kato Y, Ponce-Castaneda MV, Osses N, Esparza-Lopez J, Gonzalez-Nunez G, Cabello-Verrugio C, Mendoza V, Troncoso V, Brandan E. J Biol Chem. 2003;278(1):382–390. doi: 10.1074/jbc.M208520200. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Kuulasmaa T, Kosma VM, Butzow R, Vanttinen T, Hyden-Granskog C, Voutilainen R. J Clin Endocrinol Metab. 2003;88(10):5002–5008. doi: 10.1210/jc.2003-030704. [DOI] [PubMed] [Google Scholar]
- 33.Omori Y, Nakamura K, Yamashita S, Matsuda H, Mizutani T, Miyamoto K, Minegishi T. Endocrinology. 2005;146(8):3379–3386. doi: 10.1210/en.2004-1665. [DOI] [PubMed] [Google Scholar]
- 34.Boyer AS, Runyan RB. Dev Dyn. 2001;221(4):454–459. doi: 10.1002/dvdy.1154. [DOI] [PubMed] [Google Scholar]
- 35.Brown CB, Boyer AS, Runyan RB, Barnett JV. Science. 1999;283(5410):2080–2082. doi: 10.1126/science.283.5410.2080. [DOI] [PubMed] [Google Scholar]
- 36.Compton LA, Potash DA, Brown CB, Barnett JV. Circ Res. 2007;101(8):784–791. doi: 10.1161/CIRCRESAHA.107.152082. [DOI] [PubMed] [Google Scholar]
- 37.Cui XM, Shuler CF. Int J Dev Biol. 2000;44(4):397–402. [PubMed] [Google Scholar]
- 38.Blobe GC, Liu X, Fang SJ, How T, Lodish HF. J Biol Chem. 2001;276(43):39608–39617. doi: 10.1074/jbc.M106831200. [DOI] [PubMed] [Google Scholar]
- 39.Chen W, Kirkbride KC, How T, Nelson CD, Mo J, Frederick JP, Wang XF, Lefkowitz RJ, Blobe GC. Science. 2003;301(5638):1394–1397. doi: 10.1126/science.1083195. [DOI] [PubMed] [Google Scholar]
- 40.Finger EC, Lee NY, You HJ, Blobe GC. J Biol Chem. 2008;283(50):34808–34818. doi: 10.1074/jbc.M804741200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vicencio AG, Eickelberg O, Stankewich MC, Kashgarian M, Haddad GG. J Appl Physiol. 2002;93(3):1123–1130. doi: 10.1152/japplphysiol.00031.2002. [DOI] [PubMed] [Google Scholar]
- 42.Cheifetz S, Andres JL, Massague J. J Biol Chem. 1988;263(32):16984–16991. [PubMed] [Google Scholar]
- 43.Lopez-Casillas F, Cheifetz S, Doody J, Andres JL, Lane WS, Massague J. Cell. 1991;67(4):785–795. doi: 10.1016/0092-8674(91)90073-8. [DOI] [PubMed] [Google Scholar]
- 44.Moren A, Ichijo H, Miyazono K. Biochem Biophys Res Commun. 1992;189(1):356–362. doi: 10.1016/0006-291x(92)91566-9. [DOI] [PubMed] [Google Scholar]
- 45.Pepin MC, Beauchemin M, Plamondon J, O’Connor-McCourt MD. Proc Natl Acad Sci U S A. 1994;91(15):6997–7001. doi: 10.1073/pnas.91.15.6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukushima D, Butzow R, Hildebrand A, Ruoslahti E. J Biol Chem. 1993;268(30):22710–22715. [PubMed] [Google Scholar]
- 47.Lopez-Casillas F, Payne HM, Andres JL, Massague J. J Cell Biol. 1994;124(4):557–568. doi: 10.1083/jcb.124.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pepin MC, Beauchemin M, Collins C, Plamondon J, O’Connor-McCourt MD. FEBS Lett. 1995;377(3):368–372. doi: 10.1016/0014-5793(95)01378-4. [DOI] [PubMed] [Google Scholar]
- 49.Kaname S, Ruoslahti E. Biochem J. 1996;315(Pt 3):815–820. doi: 10.1042/bj3150815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mendoza V, Vilchis-Landeros MM, Mendoza-Hernandez G, Huang T, Villarreal MM, Hinck AP, Lopez-Casillas F, Montiel JL. Biochemistry. 2009 doi: 10.1021/bi901528w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andres JL, DeFalcis D, Noda M, Massague J. J Biol Chem. 1992;267(9):5927–5930. [PubMed] [Google Scholar]
- 52.Kirkbride KC, Townsend TA, Bruinsma MW, Barnett JV, Blobe GC. J Biol Chem. 2008;283(12):7628–7637. doi: 10.1074/jbc.M704883200. [DOI] [PubMed] [Google Scholar]
- 53.Lewis KA, Gray PC, Blount AL, MacConell LA, Wiater E, Bilezikjian LM, Vale W. Nature. 2000;404(6776):411–414. doi: 10.1038/35006129. [DOI] [PubMed] [Google Scholar]
- 54.Lopez-Casillas F, Wrana JL, Massague J. Cell. 1993;73(7):1435–1444. doi: 10.1016/0092-8674(93)90368-z. [DOI] [PubMed] [Google Scholar]
- 55.Wiater E, Vale W. J Biol Chem. 2003;278(10):7934–7941. doi: 10.1074/jbc.M209710200. [DOI] [PubMed] [Google Scholar]
- 56.Esparza-Lopez J, Montiel JL, Vilchis-Landeros MM, Okadome T, Miyazono K, Lopez-Casillas F. J Biol Chem. 2001;276(18):14588–14596. doi: 10.1074/jbc.M008866200. [DOI] [PubMed] [Google Scholar]
- 57.Wiater E, Harrison CA, Lewis KA, Gray PC, Vale WW. J Biol Chem. 2006;281(25):17011–17022. doi: 10.1074/jbc.M601459200. [DOI] [PubMed] [Google Scholar]
- 58.Ponce-Castaneda MV, Esparza-Lopez J, Vilchis-Landeros MM, Mendoza V, Lopez-Casillas F. Biochim Biophys Acta. 1998;1384(2):189–196. doi: 10.1016/s0167-4838(98)00033-8. [DOI] [PubMed] [Google Scholar]
- 59.Yan Z, Deng X, Friedman E. J Biol Chem. 2001;276(2):1555–1563. doi: 10.1074/jbc.M004553200. [DOI] [PubMed] [Google Scholar]
- 60.Deng X, Bellis S, Yan Z, Friedman E. Cell Growth Differ. 1999;10(1):11–18. [PubMed] [Google Scholar]
- 61.Eickelberg O, Centrella M, Reiss M, Kashgarian M, Wells RG. J Biol Chem. 2002;277(1):823–829. doi: 10.1074/jbc.M105110200. [DOI] [PubMed] [Google Scholar]
- 62.Mythreye K, Blobe GC. Proc Natl Acad Sci U S A. 2009;106(20):8221–8226. doi: 10.1073/pnas.0812879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andres JL, Stanley K, Cheifetz S, Massague J. J Cell Biol. 1989;109(6 Pt 1):3137–3145. doi: 10.1083/jcb.109.6.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lamarre J, Vasudevan J, Gonias SL. Biochem J. 1994;302(Pt 1):199–205. doi: 10.1042/bj3020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Velasco-Loyden G, Arribas J, Lopez-Casillas F. J Biol Chem. 2004;279(9):7721–7733. doi: 10.1074/jbc.M306499200. [DOI] [PubMed] [Google Scholar]
- 66.Arribas J, Massague J. J Cell Biol. 1995;128(3):433–441. doi: 10.1083/jcb.128.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bandyopadhyay A, Lopez-Casillas F, Malik SN, Montiel JL, Mendoza V, Yang J, Sun LZ. Cancer Res. 2002;62(16):4690–4695. [PubMed] [Google Scholar]
- 68.Bandyopadhyay A, Zhu Y, Malik SN, Kreisberg J, Brattain MG, Sprague EA, Luo J, Lopez-Casillas F, Sun LZ. Oncogene. 2002;21(22):3541–3551. doi: 10.1038/sj.onc.1205439. [DOI] [PubMed] [Google Scholar]
- 69.Blobe GC, Schiemann WP, Pepin MC, Beauchemin M, Moustakas A, Lodish HF, O’Connor-McCourt MD. J Biol Chem. 2001;276(27):24627–24637. doi: 10.1074/jbc.M100188200. [DOI] [PubMed] [Google Scholar]
- 70.You HJ, Bruinsma MW, How T, Ostrander JH, Blobe GC. Carcinogenesis. 2007;28(12):2491–2500. doi: 10.1093/carcin/bgm195. [DOI] [PubMed] [Google Scholar]
- 71.Lee J, Hempel N, Lee N, Blobe GC. Carncinogenesis. 2009 In Press. [Google Scholar]
- 72.Lee NY, Kirkbride KC, Sheu RD, Blobe GC. Mol Biol Cell. 2009;20(20):4362–4370. doi: 10.1091/mbc.E09-07-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.You HJ, How T, Blobe GC. Carcinogenesis. 2009;30(8):1281–1287. doi: 10.1093/carcin/bgp071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Nature. 1994;370(6488):341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 75.Shi Y, Massague J. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 76.del Re E, Babitt JL, Pirani A, Schneyer AL, Lin HY. J Biol Chem. 2004;279(21):22765–22772. doi: 10.1074/jbc.M401350200. [DOI] [PubMed] [Google Scholar]
- 77.Sankar S, Mahooti-Brooks N, Centrella M, McCarthy TL, Madri JA. J Biol Chem. 1995;270(22):13567–13572. doi: 10.1074/jbc.270.22.13567. [DOI] [PubMed] [Google Scholar]
- 78.Criswell TL, Dumont N, Barnett JV, Arteaga CL. Cancer Res. 2008;68(18):7304–7312. doi: 10.1158/0008-5472.CAN-07-6777. [DOI] [PubMed] [Google Scholar]
- 79.Zhang M, Zola H, Read L, Penttila I. Immunol Cell Biol. 2001;79(3):291–297. doi: 10.1046/j.1440-1711.2001.01013.x. [DOI] [PubMed] [Google Scholar]
- 80.Vilchis-Landeros MM, Montiel JL, Mendoza V, Mendoza-Hernandez G, Lopez-Casillas F. Biochem J. 2001;355(Pt 1):215–222. doi: 10.1042/0264-6021:3550215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Criswell TL, Arteaga CL. J Biol Chem. 2007;282(44):32491–32500. doi: 10.1074/jbc.M704434200. [DOI] [PubMed] [Google Scholar]
- 82.Margulis V, Maity T, Zhang XY, Cooper SJ, Copland JA, Wood CG. Clin Cancer Res. 2008;14(18):5722–5730. doi: 10.1158/1078-0432.CCR-08-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Santander C, Brandan E. Cell Signal. 2006;18(9):1482–1491. doi: 10.1016/j.cellsig.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 84.Yu L, Hebert MC, Zhang YE. EMBO J. 2002;21(14):3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kang JS, Liu C, Derynck R. Trends in Cell Biology. 2009;19(8):385–394. doi: 10.1016/j.tcb.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 86.Hartung A, Bitton-Worms K, Rechtman MM, Wenzel V, Boergermann JH, Hassel S, Henis YI, Knaus P. Mol Cell Biol. 2006;26(20):7791–7805. doi: 10.1128/MCB.00022-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stenvers KL, Tursky ML, Harder KW, Kountouri N, Amatayakul-Chantler S, Grail D, Small C, Weinberg RA, Sizeland AM, Zhu HJ. Mol Cell Biol. 2003;23(12):4371–4385. doi: 10.1128/MCB.23.12.4371-4385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Society AC. Atlanta, GA: 2009. [Google Scholar]
- 89.Pierce DF, Jr, Johnson MD, Matsui Y, Robinson SD, Gold LI, Purchio AF, Daniel CW, Hogan BL, Moses HL. Genes Dev. 1993;7(12A):2308–2317. doi: 10.1101/gad.7.12a.2308. [DOI] [PubMed] [Google Scholar]
- 90.Sun L, Chen C. J Biol Chem. 1997;272(40):25367–25372. doi: 10.1074/jbc.272.40.25367. [DOI] [PubMed] [Google Scholar]
- 91.Bandyopadhyay A, Zhu Y, Cibull ML, Bao L, Chen C, Sun L. Cancer Res. 1999;59(19):5041–5046. [PubMed] [Google Scholar]
- 92.Ljungberg B, Hanbury DC, Kuczyk MA, Merseburger AS, Mulders PF, Patard JJ, Sinescu IC. Eur Urol. 2007;51(6):1502–1510. doi: 10.1016/j.eururo.2007.03.035. [DOI] [PubMed] [Google Scholar]
- 93.Danielpour D. Eur J Cancer. 2005;41(6):846–857. doi: 10.1016/j.ejca.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 94.Guo Y, Jacobs SC, Kyprianou N. Int J Cancer. 1997;71(4):573–579. doi: 10.1002/(sici)1097-0215(19970516)71:4<573::aid-ijc11>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 95.Sharifi N, Hurt EM, Kawasaki BT, Farrar WL. Prostate. 2007;67(3):301–311. doi: 10.1002/pros.20526. [DOI] [PubMed] [Google Scholar]
- 96.Bandyopadhyay A, Wang L, Lopez-Casillas F, Mendoza V, Yeh IT, Sun L. Prostate. 2005;63(1):81–90. doi: 10.1002/pros.20166. [DOI] [PubMed] [Google Scholar]
- 97.Park C, Kim WS, Choi Y, Kim H, Park K. Lung Cancer. 2002;38(2):143–147. doi: 10.1016/s0169-5002(02)00182-4. [DOI] [PubMed] [Google Scholar]
- 98.Anumanthan G, Halder SK, Osada H, Takahashi T, Massion PP, Carbone DP, Datta PK. Br J Cancer. 2005;93(10):1157–1167. doi: 10.1038/sj.bjc.6602831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. CA Cancer J Clin. 2006;56(2):106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 100.Raponi M, Zhang Y, Yu J, Chen G, Lee G, Taylor JM, Macdonald J, Thomas D, Moskaluk C, Wang Y, Beer DG. Cancer Res. 2006;66(15):7466–7472. doi: 10.1158/0008-5472.CAN-06-1191. [DOI] [PubMed] [Google Scholar]
- 101.Rane SG, Lee JH, Lin HM. Cytokine Growth Factor Rev. 2006;17(1-2):107–119. doi: 10.1016/j.cytogfr.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 102.Hilgers W, Tang DJ, Sugar AY, Shekher MC, Hruban RH, Kern SE. Genes Chromosomes Cancer. 1999;24(4):351–355. [PubMed] [Google Scholar]
- 103.Heldin CH, Landstrom M, Moustakas A. Curr Opin Cell Biol. 2009;21(2):166–176. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 104.Gordon KJ, Kirkbride KC, How T, Blobe GC. Carcinogenesis. 2009;30(2):238–248. doi: 10.1093/carcin/bgn274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Steller MD, Shaw TJ, Vanderhyden BC, Ethier JF. Mol Cancer Res. 2005;3(1):50–61. [PubMed] [Google Scholar]
- 106.Bilandzic M, Chu S, Farnworth PG, Harrison C, Nicholls P, Wang Y, Escalona RM, Fuller PJ, Findlay JK, Stenvers KL. Mol Endocrinol. 2009;23(4):539–548. doi: 10.1210/me.2008-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Florio P, Ciarmela P, Reis FM, Toti P, Galleri L, Santopietro R, Tiso E, Tosi P, Petraglia F. Eur J Endocrinol. 2005;152(2):277–284. doi: 10.1530/eje.1.01849. [DOI] [PubMed] [Google Scholar]
- 108.Geiger TR, Peeper DS. Biochim Biophys Acta. 2009;1796(2):293–308. doi: 10.1016/j.bbcan.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 109.Mythreye K, Blobe GC. Cell Signal. 2009;21(11):1548–1558. doi: 10.1016/j.cellsig.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lei X, Bandyopadhyay A, Le T, Sun L. Oncogene. 2002;21(49):7514–7523. doi: 10.1038/sj.onc.1205966. [DOI] [PubMed] [Google Scholar]
- 111.Juarez P, Vilchis-Landeros MM, Ponce-Coria J, Mendoza V, Hernandez-Pando R, Bobadilla NA, Lopez-Casillas F. Am J Physiol Renal Physiol. 2007;292(1):F321–329. doi: 10.1152/ajprenal.00264.2006. [DOI] [PubMed] [Google Scholar]
- 112.Naumann U, Maass P, Gleske AK, Aulwurm S, Weller M, Eisele G. Int J Oncol. 2008;33(4):759–765. [PubMed] [Google Scholar]
- 113.Dallas SL, Zhao S, Cramer SD, Chen Z, Peehl DM, Bonewald LF. J Cell Physiol. 2005;202(2):361–370. doi: 10.1002/jcp.20147. [DOI] [PubMed] [Google Scholar]
- 114.Flanders KC, Wakefield LM. J Mammary Gland Biol Neoplasia. 2009;14(2):131–144. doi: 10.1007/s10911-009-9122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Levine JH, Moses HL, Gold LI, Nanney LB. Am J Pathol. 1993;143(2):368–380. [PMC free article] [PubMed] [Google Scholar]
- 116.Perry KT, Anthony CT, Steiner MS. Prostate. 1997;33(2):133–140. doi: 10.1002/(sici)1097-0045(19971001)33:2<133::aid-pros7>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 117.Rosenthal E, McCrory A, Talbert M, Young G, Murphy-Ullrich J, Gladson C. Mol Carcinog. 2004;40(2):116–121. doi: 10.1002/mc.20024. [DOI] [PubMed] [Google Scholar]
- 118.Saed GM, Collins KL, Diamond MP. Am J Reprod Immunol. 2002;48(6):387–393. doi: 10.1034/j.1600-0897.2002.01090.x. [DOI] [PubMed] [Google Scholar]
- 119.Andres JL, Ronnstrand L, Cheifetz S, Massague J. J Biol Chem. 1991;266(34):23282–23287. [PubMed] [Google Scholar]
- 120.Lin HY, Moustakas A, Knaus P, Wells RG, Henis YI, Lodish HF. J Biol Chem. 1995;270(6):2747–2754. doi: 10.1074/jbc.270.6.2747. [DOI] [PubMed] [Google Scholar]
- 121.Verona EV, Tang Y, Millstead TK, Hinck AP, Agyin JK, Sun LZ. Protein Eng Des Sel. 2008;21(7):463–473. doi: 10.1093/protein/gzn023. [DOI] [PMC free article] [PubMed] [Google Scholar]