

Abstract
Epigenetics studies inheritable changes of genes and gene expression that do not concern DNA nucleotide variation. Such modifications include DNA methylation, several forms of histone modification, and microRNAs. From recent studies, we know not only that genetic changes account for heritable phenotypic variation, but that epigenetic changes also play an important role in the variation of predisposition to disease and to drug response. In this review, we discuss recent evidence of epigenetic changes that play an important role in the development of cardiac hypertrophy and heart failure and may dictate response to therapy.
Keywords: DNA methylation, Epigenetics, Heart failure, Histone modification, MicroRNA, Pharmacoepigenetics
Introduction
There is wide variability in an individual’s disease predisposition and response to treatment, which is partially ascribed to heritable factors [1]. However, based on recent whole-genome association studies, we have to conclude that variation in nucleotides does not account for all the heritable phenotypic variation [2–4]. In addition to variation in nucleotides, epigenetic variations may play a critical role in controlling gene expression and therefore offer another mechanism to explain interindividual variation [5].
Epigenetics studies inheritable changes of genes and gene expression that do not concern the original DNA nucleotide variations, such as mutations or polymorphisms. Three different types of epigenetic variations are known to alter gene expression control: methylation of genomic DNA, modification of histone proteins, and regulatory noncoding RNAs, such as microRNAs (Fig. 1). All three mechanisms concern extrinsic factors that can modulate the transcription of genes, even though the protein-encoding regions of the genes themselves are unchanged [6]. These epigenetic control mechanisms may differ among tissues and individuals, but they also may change in time during aging or as a result of environmental interactions or diseases. Because the epigenome plays a critical role in programming the expression of the genome, differences in gene expression among individuals that affect the response to drugs may be modified by epigenomic variations on top of nucleotide mutations or polymorphisms. Therefore, epigenetic changes currently are being considered in clinical medicine complementary to nucleotide variations at the drug response level [7]. This rapidly emerging novel discipline, called pharmacoepigenetics, involves the study of epigenetic factors in the interpersonal variation to drugs but also the discovery of novel pharmacologic targets [8]. It is expected that pharmacoepigenetics (jointly with pharmacogenetics) will play a crucial role in future pharmacology and clinical medicine [7].
Fig. 1.
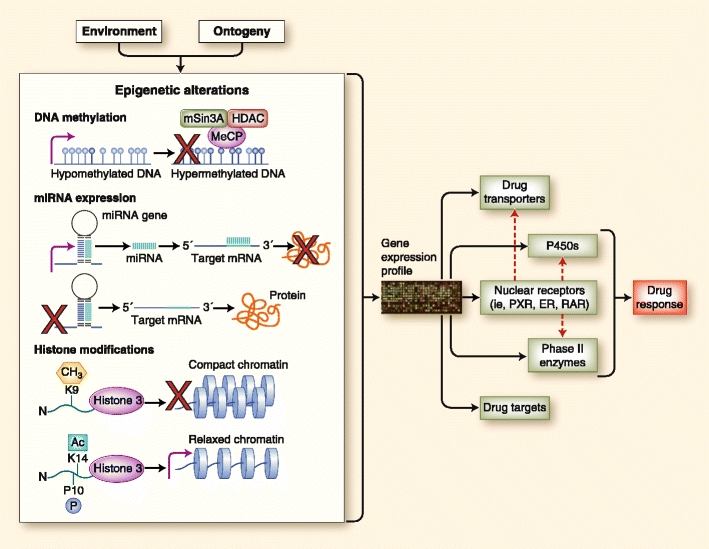
Epigenetic modifications at different genetic levels and their effect on drug response. Environmental factors generate a spectrum of phenotypes in the population through the participation of epigenetic mechanisms such as covalent modification of DNA (ie, methylation) and histones and expression of regulatory noncoding RNA molecules such as microRNAs (miRNAs). Epigenetic factors affect the expression of drug transporters, drug-metabolizing enzymes, nuclear receptors that regulate the expression of various genes, and even drug targets. The dynamic state of epigenetics, as opposed to a rather established nucleotide sequence, provides the basis for an individual’s response to a constantly changing environment. Methylation of DNA provides an impediment to transcription factors and the transcription machinery by attracting proteins that affect chromatin configuration. Posttranslational modifications of the N-terminal of the histone proteins (and their various combinations) affect the compaction of the chromatin; that is, methylation of lysine at position 9 of histone 3 or H3K9me is a signature of heterochromatin or compact DNA, whereas acetylation of lysine at position 14 of histone 3 or H3K14ac—and sometimes in combination with phosphorylation of proline at position 10 of histone 3 or H3P10p—creates a more open chromatin configuration (euchromatin) that allows transcription of genes. miRNA molecules arise from miRNA genes, which, when transcribed, can promote posttranscriptional regulation by complementarity with the 3′-untranslated region of target mRNAs and their subsequent degradation. ER—estrogen receptor; HDAC—histone deacetylase inhibitor; PXR—pregnane X receptor; RAR—retinoic acid receptor. (Adapted from Gomez and Ingelman-Sundberg [35]; with permission from Macmillan Publishers Ltd: Clinical Pharmacology and Therapeutics [85:426–430], © 2009.)
So far, most advances in pharmacoepigenetics have been derived from the oncology field—for example, studies characterizing the interindividual differences of cytochrome P-450 (reviewed by Ingelman-Sundberg et al. [9]). Fortunately, the knowledge of the role of epigenetic modifications is being translated to other complex and heterogeneous conditions, and the applicability is increasing rapidly. Here, we review the most recent work on the epigenetic modifications that have an effect on heart failure (HF) and cardiovascular disease (CVD) therapy.
Epigenetic Modifications and Heart Failure
Histone Modifications
The large eukaryotic genome is compacted tightly as a result of its association with highly conserved histone proteins. In the nucleosomes, genomic DNA is folded and compacted around core histone proteins (two copies of each of the core histones H2A, H2B, H3, and H4), forming the basic repeat units of chromatin. The interaction of genomic DNA with these chromosomal proteins has a major influence on the accessibility of transcriptional factors to their target DNA sequences and thereby regulates transcriptional activity (Fig. 1) [10•]. Through this mechanism, nucleosomes carry epigenetically inherited information in the form of covalent modifications of their core histones. Such modifications include acetylation, methylation, phosphorylation, ubiquitination, and sumoylation of histone proteins [10•]. Core histones have an amino-terminal tail that sticks out from the chromatin fiber and is thought to interact with DNA or other histone or proteins. Lysine and arginine residues within this tail are the main targets for histone modification. Most research was aimed at understanding the role of lysine acetylation and methylation. It turns out that lysine acetylation is associated mainly with chromatin accessibility and transcription, whereas the effect of lysine methylation varies depending on which residue is modified [11].
Interestingly, as reviewed by Mano [10•], the regulation of histone acetylation has been linked to cardiac hypertrophy. The acetylation of histone tails by histone acetyltransferases is required for the induction of hypertrophic changes in cardiac muscle cells by phenylephrine. Consistent with this are the results of studies focused on class II histone deacetylases (HDACs) 5 and 9, which exert antihypertrophic effects by inhibiting the activity of myocyte enhancer factor 2 (MEF2) and further blocking the expression of pro-hypertrophic genes [12]. Contrary to these findings, class I HDACs have rather pro-hypertrophic effects by regulating the expression of phosphatidylinositol (3, 4, 5)-triphosphate phosphatase, which modulates hypertrophy [13]. This means that HDACs control muscle cell size on multiple levels.
DNA Methylation
In eukaryotes, DNA methylation occurs by the addition of a methyl group to the carbon 5′ position of the nucleotide cytosine ring. In mammals, DNA methylation occurs mainly in the sequence 5′-CG-3′, which also is referred to as a CpG dinucleotide; approximately 70% of all CpGs in humans are methylated [14]. On the other hand, unmethylated CpGs are found in the 5′ regulatory regions of many genes as clusters called “CpG islands.” This frequency of CpG dinucleotides in CpG islands is higher than that found in other DNA regions. Notably, differential methylation of CpG islands is part of the epigenetic variation found in humans [15].
DNA cytosine methylation alters the accessibility for transcription factor complexes at a local level and, as with histone modifications, affects chromatin structure at regional and genome-wide levels. Thus, a well-characterized functional effect of DNA methylation is control of gene expression [16]. In this respect, hypermethylation of CpG sites may silence a gene, whereas hypomethylation allows gene transcription. One might say that methylation is a stable and heritable modification, but at the same time, it may be affected by the environment. For example, the mouse agouti locus, which affects coat color, is affected by the methylation status of an upstream transposon. Genetically identical parents in whom agouti genes are in different epigenetic states tend to produce offspring with different coat colors [17].
Experimental evidence for a role in transcriptional regulation for HF-specific genes by DNA methylation came from a recent study by Kao et al. [18]. They showed that the proinflammatory gene TNF-α reduces expression of the sarcoplasmic reticulum Ca2+-ATPase (SERCA2A) by enhancing methylation status of the SERCA2A promoter region. Movassagh et al. [19•] recently showed there are methylation status differences between cardiomyopathy and controls in human cardiac tissue. Furthermore, they identified three loci (PECAM1, PECAM1, AMOTL2) whose differential methylation is correlated with altered gene expression in different cardiac samples.
MicroRNAs
MicroRNAs (miRNAs or miR) are short (∼20 bp) double-strand RNA molecules that originate from nuclear and cytoplasmic larger precursors and act as important regulators of gene expression at a posttranscriptional level. miRNAs fine-tune the expression of 30% to 50% of the protein-coding genes by binding to partly complementary base pairs in 3′ untranslated regions of mRNAs, thereby interfering with translation; targeted mRNAs are either degraded or temporarily silenced [20]. miRNA regulation of protein expression is very complex because several miRNAs can target the same gene and several genes may be targeted by the same miRNA [20]. The expression of miRNAs is tissue and disease specific. In recent years, a molecular miRNA signature has been identified in many pathologic conditions, ranging from various types of cancer to many aspects of CVD [21•].
Growing evidence indicates that miRNAs are involved in basic cell functions [22]. The link between miRNAs and several aspects of HF are now very clear, and the number of publications in this field has increased rapidly in the past few years (Table 1). Research on CVDs has focused mainly on the two miRNA families specifically expressed in heart tissue (miRNA-1/miRNA-133 and miRNA-208). Several studies investigated miRNA expression in the healthy heart, during pressure overload, and in different etiologies of human, mouse, and rat HF, as reviewed by Divakaran and Mann [23]. van Rooij et al. [24] determined that the cardiac-specific miRNA-208 regulates cardiomyocyte hypertrophy, fibrosis, and expression of β-myosin heavy chain (β-MHC) in response to stress and hypothyroidism. This miRNA is encoded by an intron of the α-MHC gene. Thus, the gene encoding α-MHC, in addition to encoding a major cardiac contractile protein, regulates cardiac growth and gene expression in response to stress and hormonal signaling through miRNA-208, via targets of miRNA-208 such as β-MHC. Furthermore, targeted deletion of the muscle-specific miRNA, miRNA-1-2, revealed numerous functions in the heart, including regulation of cardiac morphogenesis, electrical conduction, and cell cycle control [25]. Thum et al. [26] showed that the miRNA signature of failing myocardium closely resembles the miRNA expression pattern of fetal heart, and this underlies the reonset of the fetal gene program in failing hearts. Another interesting study by Thum et al. [27] showed that miRNA-21 regulates the ERK-MAP kinase pathway specifically in cardiac fibroblasts, not in cardiomyocytes, and this affects cardiac structure and function. Increased miRNA-21 levels in fibroblasts activate ERK kinase activity through inhibition-specific genes, and by this mechanism, miRNA-21 regulates interstitial fibrosis and cardiac hypertrophy. These findings revealed an extra level of gene regulation via miRNA-mediated paracrine secretion, in this case via cardiac fibroblasts.
Table 1.
Cardiac microRNAs, their functions, and validated targets
| MiR family | Function | Validated cardiac targets |
|---|---|---|
| All (Dicer) | Regulation of cardiogenesis | Not applicable |
| Cardiomyocyte-specific deletion in mice is embryonically [25] or neonatally [44] lethal (depending on promotor) | ||
| 1 | Embryonic | |
| Modulates cardiogenesis [25, 45–47] | Delta, the Notch ligand, involved in cardiac cell differentiation [45]; Hand2, a cardiac transcription factor involved in cardiomyocyte expansion [25, 46]; and HDAC4, a transcriptional repressor of muscle gene expression [47] | |
| Essential to maintain muscle gene expression [45] | ||
| Deletion in mice is partially embryonically lethal (ventricular–septal defect) [25] | ||
| Regulation of cardiomyocyte cell cycle [25] | ||
| Adult | ||
| Conduction [25]; proarrhythmic [48–51] | Irx5, a cardiac TF that represses the potassium channel Kcnd2 [18]; potassium channels KCNJ2 [48] and KCNE1 [51]; pacemaker channels HCN2 and HCN4 [50]; connexin 43 [48]; and B56α subunit of protein phosphatase 2A [49] | |
| Regulation of cardiomyocyte growth [52, 53] | Pro-hypertrophic genes calmodulin and Mef2a [52]; cell cycle regulators RasGAP and Cdk9 and Rheb [53]; and fibronectin [31] | |
| Proapoptotic [54] | HSP60 and HSP70 [54] | |
| 133 | Embryonic | |
| Modulates cardiogenesis [47, 55] | SRF [47, 55] | |
| Deletion in mice is partially embryonically lethal (ventricular–septal defect) [55] | ||
| Regulation of cardiomyocyte cell cycle [55] | Cyclin D2 [55] | |
| Adult | ||
| Regulation of collagen synthesis [55, 56] | CTGF [56] | |
| Conduction [55]; proarrhythmic [50, 57] | Potassium channels KCNQ [51] and HERG [57] and pacemaker channel HCN2 [50] | |
| Regulation of cardiomyocyte growth [22, 58] | RhoA, a GDP–GTP exchange protein regulating cardiac hypertrophy [22]; Cdc42, a signal transduction kinase implicated in hypertrophy [22]; and Nelf-A/WHSC2, a nuclear factor involved in cardiogenesis [22] | |
| Antiapoptotic [54, 55] | Caspase-9 [54] | |
| 208 | Adult | |
| Regulates cardiomyocyte hypertrophy and fibrosis in response to pressure overload | THRAP1, a thyroid hormone transcription factor [24] | |
GDP—guanosine diphosphate; GTP—guanosine triphosphate; TF—transcription factor.
(From Schroen and Heymans [20]; with permission.)
The profound effects of miRNAs on the heart and the hypertrophic response are subject to further study, and miRNAs are emerging as chief regulators of gene control. Thus far, miRNAs have been shown to affect the myocardium but also the electrical properties of the heart, as well as the modulation of angiogenesis [28].
Methods of Epigenetic Screening
Epigenomic profiles vary from cell type to cell type and may change over time and in response to physiologic, pathologic, and pharmacologic triggers. Therefore, mapping the epigenome, as a follow-up to the human genome sequencing project, is a tremendous task. Although it is possible to determine the epigenetic status of a sequence in the genome, mapping the entire epigenome will require the sequencing of dozens of genomes covering all the different cell types of an organism at different points in life [7].
Bisulfite mapping is the most accurate method for mapping DNA methylation patterns. Genomic DNA is treated with sodium bisulfite, which results in modification and eventual deamination of the unmethylated cytosines to uracil, whereas the methylated cytosines are protected from this conversion. To determine the methylation state of specific genes, gene-specific primers are used to amplify the sequence of interest, which is then subjected to sequencing. Methylated cytosines register as Cs, whereas unmethylated cytosines appear as Ts in the sequence [7].
Several whole-genome approaches to methylation mapping were developed recently that are based on the differential sensitivity of methylated and unmethylated CpGs to restriction enzymes. Restriction length genome scanning uses a double-digestion of DNA with a frequent-cutter methylation-insensitive restriction enzyme and a rare methylation-sensitive enzyme such as Not1, which cleaves its recognition site exclusively when it is unmethylated [7]. A different whole-genome approach that takes advantage of DNA chip technology allows rapid profiling of the status of methylation of thousands of CpG islands at once, using the differential methylation hybridization method [29]. This method is used to identify CpG islands, which are methylated in tumor samples relative to their normal controls.
An alternative to bisulfite conversion is the ChIP-seq (chromatin immunoprecipitation combined with sequencing) technology. Protein-DNA interactions are crosslinked in situ followed by immunoprecipitation and sequencing of genome-wide sites associated with a modification of interest. This approach allows the identification of DNA binding sites of any DNA-associated protein. This technique also provides information on histone modifications (acetylation, methylation, phosphorylation, ubiquitination, and sumoylation). Variations on the ChIP technology have been developed, such as the “DCS” method, which couples ChIP with subtraction polymerase chain reaction (PCR) [30, 31]. This technique aims to avoid the nonspecific signals generated by hybridization of genome-derived fragments to microarrays.
In the same manner, to examine the role of miRNAs in human pathology, most studies have used high-throughput methods to analyze global miRNA expression in clinical samples [32]. High-throughput methods are represented by microRNA microarray and quantitative real-time PCR. Although the discrimination between molecules represents a great challenge, the main advantage of miRNA microarray is represented by the high specificity of the reaction, whereas the main disadvantage is represented by low sensitivity [21•].
Drugs Modifying the Epigenetic Status
Epigenetics modulates phenotypic variation in health and disease, and it seems likely that understanding and manipulating the epigenome hold enormous promise for preventing and treating common human illnesses. Epigenetics also offers an important window to understanding the role of the environment’s interactions with the genome in causing disease, and in modulating those interactions to improve human health [33].
Antagomirs are single-strand RNAs complementary to the miRNA. Chemical modification of antagomirs might represent a very attractive approach to targeting pathologic miRNAs. However, this strategy is challenging because miRNAs all belong to closely related families and the specificity of antagomirs for one miRNA might be difficult to achieve. Furthermore, a single miRNA targets several genes; among them, some might be beneficial for the myocardium. In that regard, chemically modified oligonucleotides that would specifically disrupt the binding between the miRNA and a single mRNA might be good therapeutic candidates. However, the in vivo final effect of the modulation of miRNAs remains unclear, because each of them can target hundreds of genes with different intensity [28]. Finally, the challenges in bringing miRNA antagonists to the clinical arena are similar to the ones we encountered with gene therapy, namely the mode of delivery, vectors, specificity, and toxicity in vivo [34]. At least theoretically, targeting specific miRNAs implicated in ischemic heart disease, cardiac hypertrophy, HF, angiogenesis, and channelopathies might turn out to be an attractive therapeutic tool in the future, aiming to prevent the development of HF [28].
An alternative approach would be to target DNA methylation. Several chemical compounds that affect the genomic DNA methylation landscape already are being used in the clinic. Drugs such as 5-azacytidine and azacytidine inhibit methyltransferases, causing demethylation of DNA sequences. Other drugs inhibit DNA methylation by blocking the maintenance methyltransferase. For more detailed information, see the article by Gomez and Ingelman-Sundberg [35]. The lack of specificity of the current drugs, however, leads to a global effect, which might affect genomic stability. Drugs that can target a specific methylation site have yet to be designed. In addition to work involving drugs that modify DNA methylation, increasing work is being carried out in developing drugs that affect histone modifications.
HDAC inhibitors have been the focus of attention in anticancer drug development because they are seen as presenting a potential strategy to reverse aberrant epigenetic changes associated with cancer [36]. There also is evidence that HDAC inhibitors may restore gene expression programs in cardiomyocyte hypertrophy. Gallo et al. [37] demonstrated in vitro inhibition of cardiac hypertrophy using trichostatin A and sodium butyrate.
Epigenetics and Environment
It is known that environmental factors such as toxins and diet may have an effect on DNA methylation and chromatin modification and that these changes are passed on to the next generation [38, 39]. Estrogenic and antiandrogenic toxins that decrease male fertility alter DNA methylation, and these changes are inherited by subsequent generations [39]. The hypothesis that environmental factors alter heritable epigenetic marks and change patterns of gene expression is an exciting possibility in human disease research. Most common diseases are influenced by both genetic and environmental factors; thus, environmentally induced changes in epigenetic structures may provide a mechanistic link between genes and the environment [40].
Age plays an important role in gene-environment interactions. Common diseases increase with aging, which is related to the accumulation of epigenetic changes through the lifetime of an individual. This idea is supported by a study showing higher levels of variation of total DNA methylation and histone H3K9 acetylation in older monozygotic twins than in younger twins, although that study did not measure epigenetic changes over time in the same individual [41].
Conclusions
The field of epigenetics offers a new playground for researchers to study and modify interindividual variation in clinical effects, variation in drug response and toxicity, and new targets for drug therapy. The increasing knowledge regarding epigenetic mechanisms derived from the Human Epigenome Project is leading to a better understanding of human disease and a new range of molecular targets for epigenetic drugs [42]. Although most advances in pharmacogenomics have been applied to its use in the oncology field, this review presents evidence that in the past years, there has been a significant increase in the number of cardiovascular epigenetic studies, and in miRNAs more specifically [8]. The review by Mishra et al. [43] offers a clear picture of the recent progress made in the microRNomics of CVD and the future of miRNAs as potential therapeutic targets or drug agents.
As epigenetics has an important role in shaping phenotypic variation in health and disease, it seems likely that understanding and manipulating the epigenome hold enormous promise for preventing and treating common human diseases, including CVD and HF.
Acknowledgments
This work was supported by the Netherlands Heart Foundation (grant 2007T046 to Dr. de Boer and grant 2008B065 to Dr. van der Harst) and the Innovational Research Incentives Scheme program of the Netherlands Organization for Scientific Research (NWO VENI, grant 016.106.117 to Dr. de Boer).
Disclosure
No potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Irene Mateo Leach, Phone: +31-50-3615340, FAX: +31-50-3615525, Email: i.v.mateo.leach@thorax.umcg.nl.
Pim van der Harst, Phone: +31-50-3615340, FAX: +31-50-3615525, Email: p.van.der.harst@thorax.umcg.nl.
Rudolf A. de Boer, Phone: +31-50-3615381, FAX: +31-50-3611347, FAX: +31-50-3615525, Email: r.a.de.boer@thorax.umcg.nl
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
- 1.de Boer RA, van der Harst P, van Veldhuisen DJ, van den Berg MP. Pharmacogenetics in heart failure: promises and challenges. Expert Opin Pharmacother. 2009;10:1713–1725. doi: 10.1517/14656560903025171. [DOI] [PubMed] [Google Scholar]
- 2.Wellcome Trust Case Control Consortium Genome-wide association study of 14, 000 cases of seven common diseases and 3, 000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Codd V, Mangino M, van der Harst P, et al. Common variants near TERC are associated with mean telomere length. Nat Genet. 2010;42:197–199. doi: 10.1038/ng.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newton-Cheh C, Johnson T, Gateva V, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennis C. Epigenetics and disease: altered states. Nature. 2003;421:686–688. doi: 10.1038/421686a. [DOI] [PubMed] [Google Scholar]
- 6.Margulies KB, Bednarik DP, Dries DL. Genomics, transcriptional profiling, and heart failure. J Am Coll Cardiol. 2009;53:1752–1759. doi: 10.1016/j.jacc.2008.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szyf M. Toward a discipline of pharmacoepigenomics. Curr Pharmacogenomics. 2004;2:357–377. doi: 10.2174/1570160043377358. [DOI] [Google Scholar]
- 8.Peedicadyl J. Pharmacoepigenetics and pharmacoepigenomics. Pharmacogenomics. 2008;9:1785–1786. doi: 10.2217/14622416.9.12.1785. [DOI] [PubMed] [Google Scholar]
- 9.Ingelman-Sundberg M, Sima SC, Gomez A, Rodriguez-Antona C. Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Therapeut. 2007;116:496–526. doi: 10.1016/j.pharmthera.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Mano H. Epigenetic abnormalities in cardiac hypertrophy and heart failure. Environ Health Prev Med. 2008;13:25–29. doi: 10.1007/s12199-007-0007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernstein BE, Meissner A, Lander ES. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Zhang CL, McKinsey TA, Chang S, et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/S0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trivedi CM, Luo Y, Yin Z, et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med. 2007;13:324–331. doi: 10.1038/nm1552. [DOI] [PubMed] [Google Scholar]
- 14.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 15.Peaston AE, Whitelaw E. Epigenetics and phenotypic variation in mammals. Mamm Genome. 2006;17:365–374. doi: 10.1007/s00335-005-0180-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 17.Ball MP, Li JB, Gao Y, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kao YH, Chen YC, Cheng CC, et al. Tumor necrosis factor-alpha decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit Care Med. 2010;38:217–222. doi: 10.1097/CCM.0b013e3181b4a854. [DOI] [PubMed] [Google Scholar]
- 19.Movassagh M, Choy M-K, Goddard M, et al. Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. PLoS One. 2010;5:e8564. doi: 10.1371/journal.pone.0008564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schroen B, Heymans S. MicroRNAs and beyond: the heart reveals its treasures. Hypertension. 2009;54:1189–1194. doi: 10.1161/HYPERTENSIONAHA.109.133942. [DOI] [PubMed] [Google Scholar]
- 21.Silvestri P, Di Russo C, Rigattieri S, et al. MicroRNAs and ischemic heart disease: towards a better comprehension of pathogenesis, new diagnostic tools and new therapeutic targets. Recent Pat Cardiovasc Drug Discov. 2009;4:109–118. doi: 10.2174/157489009788452977. [DOI] [PubMed] [Google Scholar]
- 22.Carè A, Catalucci D, Felicetti F, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 23.Divakaran V, Mann DL. The emerging role of microRNAs in cardiac remodeling and heart failure. Circ Res. 2008;103:1072–1083. doi: 10.1161/CIRCRESAHA.108.183087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Rooij E, Sutherland LB, Qi X, et al. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 25.Zhao Y, Ransom JF, Li A, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 26.Thum T, Gross C, Fiedler J, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 27.Thum T, Galuppo P, Wolf C, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 28.Zorio E, Medina P, Rueda J, et al. Insights into the role of microRNAs in cardiac diseases: from biological signalling to therapeutic targets. Cardiovasc Hematol Agents Med Chem. 2009;7:82–90. doi: 10.2174/187152509787047676. [DOI] [PubMed] [Google Scholar]
- 29.Huang TH, Perry MR, Laux DE. Methylation profiling of CpG islands in human breast cancer cells. Hum Mol Genet. 1999;8:459–470. doi: 10.1093/hmg/8.3.459. [DOI] [PubMed] [Google Scholar]
- 30.Kaneda R, Toyota M, Yamashita Y, et al. High-throughput screening of genome fragments bound to differentially acetylated histones. Genes Cells. 2004;9:1167–1174. doi: 10.1111/j.1365-2443.2004.00804.x. [DOI] [PubMed] [Google Scholar]
- 31.Kaneda R, Ueno S, Yamashita Y, et al. Genome-wide screening for target regions of histone deacetylases in cardiomyocytes. Circ Res. 2005;97:210–218. doi: 10.1161/01.RES.0000176028.18423.07. [DOI] [PubMed] [Google Scholar]
- 32.Soifer HS, Rossi JJ, Saetrom P. MicroRNAs in disease and potential therapeutic applications. Mol Ther. 2007;15:2070–2079. doi: 10.1038/sj.mt.6300311. [DOI] [PubMed] [Google Scholar]
- 33.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 34.Pucéat M. Pharmacological approaches to regenerative strategies for the treatment of cardiovascular diseases. Curr Opin Pharmacol. 2008;8:189–192. doi: 10.1016/j.coph.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 35.Gomez A, Ingelman-Sundberg M. Pharmacoepigenetics: its role in interindividual differences in drug response. Clin Pharmacol Ther. 2009;85:226–230. doi: 10.1038/clpt.2009.2. [DOI] [PubMed] [Google Scholar]
- 36.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 37.Gallo P, Latronico MV, Gallo P, et al. Inhibition of class I histone deacetylase with an apicidin derivative prevents cardiac hypertrophy and failure. Cardiovasc Res. 2008;80:416–424. doi: 10.1093/cvr/cvn215. [DOI] [PubMed] [Google Scholar]
- 38.Sutherland JE, Costa M. Epigenetics and the environment. Ann NY Acad Sci. 2003;983:151–160. doi: 10.1111/j.1749-6632.2003.tb05970.x. [DOI] [PubMed] [Google Scholar]
- 39.Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466–1469. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turan N, Katari S, Coutifaris C, Sapienza C. Explaining inter-individual variability in phenotype: is epigenetics up to the challenge? Epigenetics. 2010;5:16–19. doi: 10.4161/epi.5.1.10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The American Association for Cancer Research Human Epigenome Task Force and European Union, Network of Excellence, Scientific Advisory Board Moving AHEAD with an international human epigenome project. Nature. 2008;454:711–715. doi: 10.1038/454711a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mishra PK, Tyagi N, Kumar M, Tyagi SC. MicroRNAs as a therapeutic target for cardiovascular diseases. J Cell Mol Med. 2009;13:778–789. doi: 10.1111/j.1582-4934.2009.00744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen JF, Murchison EP, Tang R, et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc Natl Acad Sci U S A. 2008;105:2111–2116. doi: 10.1073/pnas.0710228105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwon C, Han Z, Olson EN, Srivastava D. MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc Natl Acad Sci U S A. 2005;102:18986–18991. doi: 10.1073/pnas.0509535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 47.Chen JF, Mandel EM, Thomson JM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228–233. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang B, Lin H, Xiao J, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 49.Terentyev D, Belevych AE, Terentyeva R, et al. miR-1 overexpression enhances Ca2+ release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56{alpha} and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ Res. 2009;104:514–521. doi: 10.1161/CIRCRESAHA.108.181651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo X, Lin H, Pan Z, et al. Down-regulation of miR-1/miR-133 contributes to re-expression of pacemaker channel genes HCN2 and HCN4 in hypertrophic heart. J Biol Chem. 2008;283:20045–20052. doi: 10.1074/jbc.M801035200. [DOI] [PubMed] [Google Scholar]
- 51.Luo X, Xiao J, Lin H, et al. Transcriptional activation by stimulating protein 1 and post-transcriptional repression by muscle-specific microRNAs of IKs-encoding genes and potential implications in regional heterogeneity of their expressions. J Cell Physiol. 2007;212:358–367. doi: 10.1002/jcp.21030. [DOI] [PubMed] [Google Scholar]
- 52.Ikeda S, He A, Kong SW, et al. microRNA-1 negatively regulates expression of the hypertrophy-associated genes calmodulin and Mef2a. Mol Cell Biol. 2009;29:2193–2204. doi: 10.1128/MCB.01222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sayed D, Hong C, Chen IY, et al. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100:416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 54.Xu C, Lu Y, Pan Z, et al. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J Cell Sci. 2007;120:3045–3052. doi: 10.1242/jcs.010728. [DOI] [PubMed] [Google Scholar]
- 55.Liu N, Bezprozvannaya S, Williams AH, et al. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–3254. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duisters RF, Tijsen AJ, Schroen B, et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104:170–178. doi: 10.1161/CIRCRESAHA.108.182535. [DOI] [PubMed] [Google Scholar]
- 57.Xiao J, Luo X, Lin H, et al. MicroRNA miR-133 represses HERG K+ channel expression contributing to QT prolongation in diabetic hearts. J Biol Chem. 2007;282:12363–12367. doi: 10.1074/jbc.C700015200. [DOI] [PubMed] [Google Scholar]
- 58.Sucharov C, Bristow MR, Port JD. miRNA expression in the failing human heart: functional correlates. J Mol Cell Cardiol. 2008;45:185–192. doi: 10.1016/j.yjmcc.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]