

Abstract
Elevated Src kinase activity is linked to the progression of solid tumors, including head and neck squamous cell carcinoma (HNSCC). Src regulates HNSCC proliferation and tumor invasion, with the Src-targeted small molecule inhibitor saracatinib displaying potent anti-invasive effects in preclinical studies. However, the pro-invasive cellular mechanism(s) perturbed by saracatinib are unclear. The anti-proliferative and anti-invasive effects of saracatinib on HNSCC cell lines were therefore investigated in pre-clinical cell and mouse model systems. Saracatinib treatment inhibited growth, cell cycle progression and transwell Matrigel invasion in HNSCC cell lines. Dose-dependent decreases in Src activation and phosphorylation of the invasion-associated substrates focal adhesion kinase, p130 CAS and cortactin were also observed. While saracatinib did not significantly impact HNSCC tumor growth in a mouse orthotopic model of tongue squamous cell carcinoma, impaired perineural invasion and cervical lymph node metastasis was observed. Accordingly, saracatinib treatment displayed a dose-dependent inhibitory effect on invadopodia formation, extracellular matrix degradation and matrix metalloprotease 9 activation. These results suggest that inhibition of Src kinase by saracatinib impairs the pro-invasive activity of HNSCC by inhibiting Src substrate phosphorylation important for invadopodia formation and associated matrix metalloprotease activity.
Keywords: Saracatinib, Src, Head and Neck cancer, Invadopodia, Invasion, MMP
Introduction
Tumor cell invasion and metastasis is a compounding problem in cancer management, with therapeutic intervention of tumor invasion becoming recognized as an increasingly relevant clinical factor (Dolgin, 2009). Increased activation of the protooncogene c-Src (Src) has been established in enhancing tumor progression in human cancer and corresponds with poor clinical outcome (Irby and Yeatman, 2000; Yeatman, 2004). Src is responsible for governing signaling pathways that regulate proliferation, angiogenesis, resistance to apoptosis, adhesion, motility and invasion (Summy and Gallick, 2006). High Src expression and/or activity is observed in metastases, supporting a role for Src in tumor progression by enhancing tumor invasion and metastatic potential (Summy and Gallick, 2003; Yeatman, 2004). Small molecules targeting Src kinase activity suppress proliferation, invasion and metastasis in preclinical settings (Summy and Gallick, 2006), and are currently being evaluated in clinical trials (Kopetz et al., 2007).
Head and neck squamous cell carcinoma (HSNCC) is highly invasive, frequently metastasizing to cervical lymph nodes and corresponds with poor prognosis (Kramer et al., 2005). Src overexpression is common in HNSCC (van Oijen et al., 1998) and is activated following engagement of the epidermal growth factor receptor (EGFR), where it modulates HNSCC growth and invasion through several signaling pathways (Zhang et al., 2004). The small molecule Src kinase inhibitor dasatinib suppresses motility and invasion of HNSCC cells in vitro and in mouse xenografts models, corresponding with decreased Src activation and invasion-associated substrate phosphorylation (Johnson et al., 2005; Sen et al., 2009). Amplification and/or overexpression of Src substrates in HNSCC correlates with poor clinical outcome, potentially serving to magnify Src pathway effects on HNSCC invasion and metastasis (Kelley et al., 2008).
HNSCC invasion and metastatic spread is mediated in part by the action of matrix metalloproteases (MMPs), with MMP1, MMP2, MMP9 and MT1-MMP activity associated with poor outcome (Rosenthal and Matrisian, 2006). MT1-MMP, MMP2 and MMP9 localize to invadopodia, actin-based ventral protrusions in invasive tumor cells that mediate focalized proteolysis of the extracellular matrix (ECM) (Linder, 2007; Weaver, 2006). Invadopodia formation is dependent on Src activity, which enhances MMP2 and MMP9 secretion (Hsia et al., 2003; Mueller et al., 1992) and matrix degradation in HNSCC cells (Clark et al., 2007). The collective localization and action of MMPs at invadopodia allows matrix remodeling to accommodate primary tumor growth and to allow dissemination of encapsulated tumor cells to local and distant sites (Gimona et al., 2008).
Saracatinib (AZD0530) is a recently developed anilinoquinazoline inhibitor designed to disrupt Src kinase activity (Hennequin et al., 2006; Summy and Gallick, 2006). Saracatinib exhibits inhibitory effects on tumor growth in some model systems (Herynk et al., 2006), but several preclinical reports suggest that the primary anticancer effects of saracatinib are impaired tumor cell migration and invasion in HNSCC and other cancer types (Green et al., 2009; Koppikar et al., 2008; Nozawa et al., 2008). The anti-invasive effects of saracatinib are consistent with the effects of Src kinase inhibition in HNSCC by dasatinib, another Src-targeted inhibitor (Johnson et al., 2005). Saracatinib is currently being evaluated in phase I/II clinical trials for efficacy against advanced stage HNSCC and other tumor types (Kopetz et al., 2007).
Although saracatinib and other Src inhibitors are effective anti-invasive compounds, a complete understanding of the how therapeutic Src inhibition perturbs tumor invasion at the cellular level is lacking. We show that saracatinib inhibited Src activation and phosphorylation of the invadopodia regulatory proteins focal adhesion kinase (FAK), p130 Crk-associated substrate (CAS) and cortactin in HNSCC cells. Saracatinib suppressed HNSCC growth and cell cycle progression in a subset of HNSCC cell lines. Administration of saracatinib to nude mice containing orthotopic HNSCC tongue tumors inhibited Src activity, cortactin phosphorylation, perineural invasion and lymph node metastasis. We also demonstrated that saracatinib prevented invadopodia formation and ECM degradation in invasive HNSCC cells, as well as secretion and activation of MMP9. Collectively, these results suggest that saracatinib exhibits anti-tumor effects in HNSCC by inhibiting invasion through the prevention of invadopodia formation. The ability of saracatinib to prevent invadopodia-mediated ECM proteolysis reveals a cellular process perturbed by Src inhibitors that is likely utilized in the progression of HNSCC and other invasive carcinomas containing high Src activity.
Materials and Methods
Cell lines, antibodies and Western blotting
HNSCC cell lines 1483, HN31, UMSCC1, UMSCC19 and MSK 921 were maintained as described (Rothschild et al., 2006). Western blotting of cell lysates was conducted essentially as before (Rothschild et al., 2006). Western blotting of secreted MMP2 and 9 was conducted on conditioned media, with volumes adjusted to compensate for variations in cell numbers using dimethyl sulfoxide (DMSO)-treated cell numbers as controls.
Antibodies for immunoblotting included anti-Src (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-pY418 Src (1:1000; Invitrogen, Carlsbad, CA, USA), anti-p130CAS (1:1000; BD Biosciences, San Jose, CA, USA), anti-pY410 p130CAS (1:1000; Cell Signaling Technology, Danvers, MA, USA), anti-FAK (1:1000; BD Biosciences), anti-pY861 FAK (1:1000; Invitrogen) and anti-cortactin (1 μg/ml; 4F11 (Rothschild et al., 2006)). For detection of human pY421 cortactin, a custom antibody was developed by 21st Century Biochemicals (Marlboro, MA, USA). Briefly, a synthetic cortactin peptide encompassing the sequence around tyrosine 421 (NH2-VpYEDAASFKL-COOH) was synthesized, phosphorylated and injected into rabbits. Immune serum was passed through a column containing agarose beads coupled to the equivalent non-phosphorylated peptide, and then passed over a second agarose column containing a partially overlapping phosphorylated peptide (NH2-LPSSPVpYEDAA-COOH). Bound antibodies were eluted, concentrated and screened for specificity by Western blotting against recombinant cortactin mutant proteins harboring phenylalanine-tyrosine point mutations at tyrosine 421 (Figure 1, Supplemental Material). Anti-ERK1/2 (Cell Signaling) and anti-pT202/pY204 ERK1/2 (Cell Signaling) were used at 1:1000. Anti-MMP2 (1:500; Millipore, Billerica, MA, USA) was used to detect cellular MMP2 levels. Secreted MMP2 was detected with antibody CA-4001 (1:100; Millipore). Cellular and secreted MMP9 was detected with monoclonal antibody 9D4.2 (1:100; Millipore). All Western blots were quantified by densitometry and ImageJ (NIH) analysis, and band intensities determined relative to non-treated controls.
Cell proliferation and cell cycle progression assays
For cell proliferation assays, 4,000 cells were seeded overnight and treated with 0–10 μM saracatinib (AstraZeneca, Cheshire, UK) for 5d. 100 μg of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma-Aldrich, St. Louis, MO, USA) was added to each saracatinib treatment condition for 4h, cells were washed and the reduced dye extracted with a 75% isopropanol/2% HCl/23% H2O mixture. Dye absorbance was read at 490 nm with an automated plate reader.
Cell cycle distribution was determined by fluorescence activated cell sorting (FACS) as previously described (Frederick et al., 2007).
Invasion assays
BioCoat Matrigel invasion chambers (BD Biosciences) were rehydrated with serum-free DMEM media for 2 h. 1 × 105 cells suspended in serum-free media were plated in the chamber insert and incubated for 2 h to allow attachment. The media in the upper and lower chambers was replaced, with serum-free DMEM added to the upper chamber and DMEM containing 5% FBS added to the lower chamber to generate a chemotactic gradient. Increasing doses of saracatinib (0–1 μM) as indicated (Figure 1D) were added to the upper and lower chambers. Cells were allowed to invade for 12–24 h (depending on cell line), fixed with 10% buffered formalin phosphate (Fisher Scientific, Hanover Park, IL, USA) and rinsed with PBS. Non-invasive cells were removed from the interior of the chamber insert with a swab and the remaining cells were stained with 0.4% Crystal Violet solution (Fisher) for 15 min. Invasion was assessed by counting cells in four random 20X microscopic fields.
Figure 1.

Effects of saracatinib on HNSCC proliferation, cell cycle progression, Erk1/2 activation and in vitro invasion. A: IC50 values for cell growth determined by 5 day MTT assays for the indicated HNSCC lines treated with 0–10 μM saracatinib. Mean values are shown for each line from three independent assays. B: Impact of saracatinib on HNSCC cell cycle progression. HNSCC cell lines were treated with the indicated amounts of saracatinib for 24 hours, fixed labeled with propidium iodide to assess DNA content, and analyzed for cell cycle status by fluorescence-activated cell sorting. Results show the average percentage of cells in each cell cycle phase as indicated on the left. Bars, SD of two independent experiments. C: Effects of saracatinib on Erk1/2 activity. HNSCC cells were treated with saracatinib for 24 hours at the indicated doses, lysed and analyzed by Western blotting with phosphorylation-specific (pErk1/2) and total Erk1/2 antibodies. Blots shown are representative of three different experiments, with indicated band intensities shown relative to no treatment (0 μM) for each cell line. D: Saracatinib inhibits in vitro HNSCC invasion. HNSCC cells (1×105) were plated in Matrigel-coated transwells alone or with increasing concentrations of saracatinib. After 2 h, invasion was stimulated with 5% FBS and cells were allowed to invade for 12 h (UMSCC1) or 24 h (HN31 and 1483). Invaded cells were quantified by brightfield microscopy. Bars, SEM of three independent experiments.
Orthotopic xenograft assay of HNSCC invasion
An in vivo mouse model of oral tongue squamous cell carcinoma was established as described (Myers et al., 2002) with minor modification using female athymic Foxn1nu/nu mice 4–5 weeks of age (Harlan Laboratories, Indianapolis, IN, USA). All animal procedures were conducted according to an approved protocol by the West Virginia University Animal Care and Use Committee. Anesthetized mice were injected with 2.5 × 104 UMSCC1 cells suspended in DMEM into the anterior ~1/3 of the tongue. After 10 d, treatment was initiated by daily oral gavage of 25mg/kg saracatinib suspended in a sterile solution of 0.5% methyl cellulose/0.1% polysorbate 80 (Tween 80, Sigma-Aldrich). Control animals were gavaged with the methylcellulose/Tween 80 vehicle. No overt difficulties were encountered when gavaging mice over time as the tumor size increased. Six mice were used for each treatment group. After 30 d of treatment, mice were euthanized by carbon dioxide inhalation and tumor volumes determined as described (Huang et al., 2002). Tongues, sublingual tissue containing the superficial cervical lymph nodes and the tracheoesophageal region, deep cervical and mediastinial lymph nodes, liver and lung were removed from each animal, rinsed, fixed and paraffin embedded for routine histological evaluation.
Immunohistochemistry
Human HNSCC cases were obtained from the West Virginia Tissue Bank and used under the approval of West Virginia University Institutional Review Board. Five-micrometer sections from human HNSCC and mouse tissue blocks were processed and immunolabeled or hematoxylin and eosin (H&E) stained using a Discovery XT automated staining system (Ventana Medical Systems, Tucson, AZ, USA). For immunohistochemistry, primary antibody conditions were: anti-total Src (Cell Signaling) 1:600 in Dako diluent (Dako, Carpinteria, CA, USA) for 1 h, anti-pY416 Src family kinase (SFK) (Cell Signaling) 1:25 in phosphate saline solution (PSS) (Ventana) for 12 h, anti-total cortactin (Novus Biologicals, Littleton, CO, USA) 1:700 in PSS for 1 h, anti-pY421 cortactin 1:50 in Tris buffered saline containing 4% BSA for 1 h, and prediluted anti-cytokeratin 14 (Abcam, Cambridge, MA, USA) for 20 min. Primary antibodies were detected using the Omnimap antibody horseradish peroxidase kit (Ventana) and slides were counterstained with hematoxylin. Images were acquired as described (Rothschild et al., 2006). For quantifying pY416 Src and pY421 cortactin staining intensities, brightfield images from at least 5 randomly selected images were captured on an Olympus ZX70 Provis microscope (Olympus, Center Valley, PA, USA) with a 20x/0.70 UPlanApo objective and an Optronics MicroFire 1600×1200 color CCD camera (Optronics Inc, Goleta, CA, USA) using the StereoInvestigator imaging package (MBF Bioscience, Williston, VT) with the same camera settings and brightfield correction enabled to ensure even illumination across the image. Brown 3,3′-diaminobenzidine (DAB) staining was separated from blue hematoxylin staining using the color deconvolution plug-in function of ImageJ as described (Park et al., 2008). The vector values for the DAB staining were determined from ROIs with brown staining (R=0.425, G=0.600 and B=0.677 for pY421 cortactin; R=0.475, G= 0.653 and B=0.686 for pY416 Src). Brown images were inverted and intensities were measured inside the tumor tissue. The mean DAB intensities were averaged within the group to calculate ratios of phosphorylation-specific staining in treated vs. control tissues.
Immunofluorescence labeling, confocal microscopy and image analysis
UMSCC1 cells were plated on fluorescein isothiocyanate (FITC)-gelatin (Sigma) coated coverslips as described (Artym et al., 2006) for 2 h. Cells were left untreated or treated with saracatinib for 6 h, rinsed and fixed with 4% formaldehyde. Cells were permeabilized with 0.4% Triton-X/PBS for 4 minutes, then blocked in 5% BSA/PBS for 1 h. To identify invadopodia, cells were incubated with rhodamine-conjugated phalloidin (1:1000; Invitrogen), anti-cortactin monoclonal antibody 4F11 (1 μg/ml) and polyclonal pTyr-100 (1:200; BD Biosciences) in 5% BSA/PBS for 1 h. After washing, cells were incubated in 5% BSA/PBS containing AlexaFluor 405 goat anti-rabbit and AlexaFluor 647 goat anti-mouse secondary antibodies (Invitrogen) at 1:1000. Cells were rinsed and mounted in Fluoromount-G (SouthernBiotech, Birmingham, AL, USA).
For quantifying saracatinib effects on invadopodia incidence and matrix degradation, eight-bit 1024×1024 pixel confocal images were acquired with a Zeiss LSM510 confocal microscope using AIM software (Carl Zeiss MicroImaging, Thronwood, NY, USA). Images were scanned with a 63x/1.4 NA Oil Plan-Apochromat objective at 1.3x zoom, yielding a resolution of 10.14 pixels/μm. All images of the FITC-gelatin were taken with the same parameters (pinhole size, laser intensity and gain) so image intensity would be comparable between samples. For invadopodia formation, a minimum of six independent fields comprising ≥ 50 cells were analyzed for cells containing invadopodia compared to total cell number. For matrix degradation, cells were analyzed using ImageJ software. Actin images were adjusted to threshold values to include all cellular regions, and the resulting images were used to calculate total cell areas in μm2. For quantifying matrix degradation, FITC-gelatin images were inverted so that regions with increased degradation would yield higher intensity values, ensuring selection of all areas of matrix degradation. The integrated density was reported as the amount of degradation per total cell area. A minimum of 15 cells were analyzed for each saracatinib concentration.
Gelatin zymography
UMSCC1 and 1483 cells were plated overnight at 5 × 106 in complete media were treated with saracatinib at increasing dosage for 24 h, rinsed and incubated for 24 h in serum-free media containing the equivalent saracatinib dose. Cells were counted, and conditioned media collected and concentrated by ultrafiltration using Amicon Ultra-4 centrifugal filter devices with a 10kDa molecular weight cutoff (Millipore). Zymography was conducted as described (Clark et al., 2007) with minor modification. Conditioned media (35 μl) was diluted in 2X non-reducing SDS-PAGE sample buffer and resolved on 8% SDS-PAGE gels containing 1 mg/ml bovine gelatin (Sigma). Aliquots of serum-free DMEM and DMEM containing 10% FBS were used as negative and positive controls, respectively. MMP activity was renatured by washing gels in 2.5% Triton X-100 for 30 min, followed by washing gels in Developing Buffer (50 mM Tris, 0.2M NaCl, 5mM CaCl2, 0.02% Brij 35) for 30 min at room temperature. Gels were then incubated in renewed Developing buffer for 24h at 37° C to allow MMP activity to proceed. Gels were stained with Coomassie Brilliant Blue R-250 (0.5% w/v) for 30 min, followed by destaining in water. Resulting gels were scanned using a FotoAnalyst Investigator (Fotodyne Inc, Hartland, WI, USA) and areas of gelatinase activity quantified using ImageJ. Results were adjusted relative to control DMSO treatment for gelatinase activity and cell counts for each treatment to compensate for variations in final cell numbers.
Statistical analysis
Differences in mean values between saracatinib treatment groups for invasion, invadopodia and gelatinase assays were evaluated using one-way ANOVA, followed by Student-Newman-Keuls post hoc testing. Differences were considered significant at P<0.05, with all experimentation conducted at least in triplicate.
Results
Saracatinib effects on HNSCC proliferation and invasion
As a first step in our work, we characterized several HNSCC cell lines previously determined to have varying degrees of invasive and metastatic potential (Rothschild et al., 2006; Sano and Myers, 2007; Yang et al., 2004) for their response to saracatinib. To determine the effect of saracatinib treatment on proliferation in these lines, growth inhibition was assessed by 5d 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays for cells treated with increasing doses of saracatinib (Figure 1A). The HN31 and UMSCC1 lines were sensitive to growth inhibition by saracatinib, as indicated by sub-micromolar IC50 values (Figure 1A). In contrast, the 1483 line demonstrated a marked resistance to saracatinib, with an IC50 = 7.60μM. Corresponding effects on cell cycle progression are also observed, with increasing saracatinib concentrations resulting in enhanced G1 checkpoint arrest in HN31 and UMSCC1 lines determined by FACS analysis (Figure 1B). Saracatinib treatment did not alter the percentage of cells undergoing G2-M transition for either line. Arrest of G1 was not evident in 1483 cells treated with saracatinib concentrations up to 1 μM, in agreement with resistance of this line in proliferation analysis (Figure 1A). Erk1/2 activation, a potent driver of mitogenesis, was impaired in HN31 and UMSCC1 cells at concentrations above 0.5 μM a determined by Western blotting (Figure 1C). Similar results were obtained for the invasive HNSCC lines MSK921 and UMSCC19 (data not shown). Erk1/2 activation was elevated in 1483 cells at doses up to 1μM (Figure 1C), in agreement with the high IC50 value for this line (Figure 1A). These data indicate that the HNSCC lines used in this study vary in their proliferative response to saracatinib treatment, and can be segregated into sensitive (HN31 and UMSCC1) and resistant (1483) populations.
Next we evaluated the effect of saracatinib on HNSCC invasion in vitro using modified Boyden chamber transwell assays. In the absence of saracatinib, UMSCC1 and HN31 cells displayed an invasive response to serum in the absence of drug, averaging 4150 cells/aggregate field and 1719 cells/aggregate field, respectively (Figure 1D). 1483 cells were weakly invasive, with an average of 243 cells/aggregate field (Figure 1D). Increased concentrations of saracatinib resulted in dose-dependent inhibition of HNSCC invasion for all tested lines, with reduced invasion compared to control levels of 88% in UMSCC1, 70% in HN31, and 78% in 1483 cells at the highest evaluated dose (1.0 μM) (Figure 1D). These results indicate that saracatinib directly impacts the ability of HNSCC cell lines to invade in an in vitro setting.
Saracatinib inhibits Src activation and substrate phosphorylation in HNSCC cells
To determine the impact of saracatinib on Src activity and phosphorylation of invadopodia-related Src substrates, dose-dependence experiments were performed on HN31, UMSCC1 and 1483 cells (Figure 2). Cell lines were treated with increasing doses of saracatinib for 24 hours, lysed and assessed for phosphorylation by immunoblotting using anti-phosphorylation site-specific antibodies to detect Src activation (pY418) and specific Src phosphorylation sites in downstream substrates (pY410 p130 CAS, pY421 cortactin and pY861 FAK). The phosphorylation of Src at Y418 was inhibited by saracatinib in all tested lines at concentrations between 0.5 and 1.0 μM (Figure 2). Phosphorylation of cortactin at tyrosine 421 and FAK at tyrosine 861 was also reduced within the same range of saracatinib concentrations (FAK phosphorylation in 1483 cells could not be evaluated due to the absence of detectable FAK expression in this line). While phosphorylation of tyrosine 410 in p130 CAS was inhibited within this same dose range in UMSCC1 cells, we observed that p130 CAS phosphorylation was consistently inhibited at lower dose ranges (0.01–0.05 μM) in HN31 and 1483 cells (Figure 2). Immunoblotting with antibodies against total p130 CAS, cortactin and FAK indicate that the expression levels of these proteins are somewhat reduced at high dose levels of saracatinib treatment (0.5–1μM), but not at levels accountable for the resultant decrease of tyrosine phosphorylation at the assayed sites (0.1–0.5μM).
Figure 2.
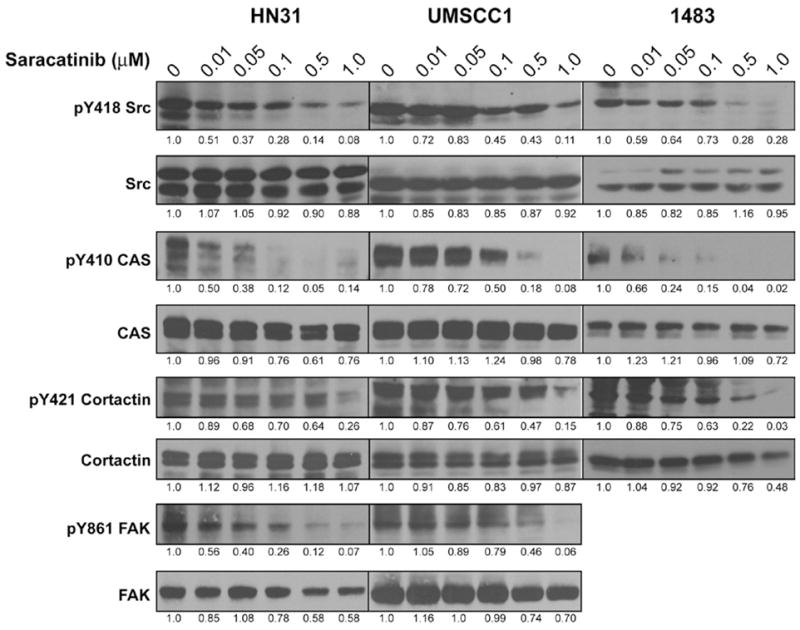
Saracatinib inhibits Src activity and downstream Src substrate phosphorylation in HNSCC cell lines. HN31, UMSCC1 and 1483 cells were treated with DMSO vehicle or saracatinib (0.01–1 μM) for 24 h. Cells were lysed and total protein amounts were analyzed by Western blotting with total or phosphorylation site-specific antibodies for Src and the indicated substrates. Blots shown are representative of at least four independent experiments, with band intensities for each substrate quantified relative to the untreated (0 μM) condition for each cell line.
Saracatinib inhibits Src activation, invasion and cervical lymph node metastasis in an orthotopic mouse model of oral squamous cell carcinoma
A mouse orthotopic model of tongue squamous cell carcinoma (Myers et al., 2002) that phenotypically mimics human HNSCC (Figure 3) was utilized to evaluate the effects of saracatinib on HNSCC progression and invasion in an in vivo setting. Athymic mice with UMSCC1 tongue tumors were randomized and treated with either vehicle or daily with 25mg/kg saracatinib. At the end of treatment (40d), mice from both groups had similar weight (24g) and mean tumor volumes (54.1mm3 ± 2.3mm for controls; 43.7mm3 ± 4.6mm with saracatinib treatment) the modest impact of saracatinib on UMSCC1 tumor growth was not statistically significant. To determine the impact of saracatinib on Src activity, primary tumors were evaluated for Src and cortactin phosphorylation by immunohistochemistry. Active Src (determined by pY416 SFK staining) and pY421 cortactin labeling in UMSCC1 tumors displayed similar patterns compared to human HNSCC (Figure 3A). Saracatinib treatment reduced the ratios of pY416 SFK and pY421 cortactin compared to control-treated mice (Figure 3A).
Figure 3.

Saracatinib inhibits Src activity, perineural invasion and cervical lymph node metastasis in orthotopic UMSCC1 tongue tumors. A: UMSCC1 tongue tumors from representative control-treated or saracatinib-treated mice were sectioned and stained with hematoxylin and eosin (H&E) or by IHC with the indicated antibodies (left). A case of human HNSCC was evaluated in parallel as a positive control. The pY416 SFK and pY421 cortactin ratios from saracatinib treated to control levels are indicated. Bars, 100 μm. B: Locoregional invasion and lymph node metastasis is inhibited by saracatinib. Submental and associated tracheoesophageal tissue from control treated and saracatinib treated mice was immunostained for cytokeratin 14 to detect cells of epithelial origin. Magnified regions containing a single sublingual nerve and superficial cervical lymph node are shown for clarity. Inset shows a magnified cortical region of superficial cervical lymph nodes from control and saracatinib treated mice. Arrowheads denote metastasized UMSCC1 cells. N; sublingual nerve, ED; excretory duct. Bars 100 μm; inset, 50 μm.
Human HNSCC often displays perineural invasion with regional lymph node involvement. Given the invasive nature of UMSCC1 cells (Figure 1D), we evaluated the impact of saracatinib treatment on loco-regional tissue invasion and cervical lymph node metastasis in treated mice. Soft tissues from the submental space through the tracheoesophageal region were evaluated for perineural invasion and cervical lymph node metastasis by immunostaining for the epithelial marker cytokeratin 14. Similar to human tumors, extensive perineural invasion and metastasis to the superficial cervical lymph nodes was evident in 5/6 control treated mice (Figure 3B). Invasion and lymph node metastasis was found in 1/6 saracatinib-treated mice, and the remaining mice displayed a complete absence of cytokeratin-positive cells associated with nerves, sublingual glands, connective tissue or cervical lymph nodes (Figure 3B). These data demonstrate that the in vivo ability of saracatinib to down-regulate Src activity and cortactin phosphorylation correlates with decreased invasion and local lymph node metastasis.
Saracatinib inhibits invadopodia formation and matrix degradation in UMSCC1 cells
UMSCC1 cells plated on fluorescently-labeled gelatin formed centrally localized ventral puncta enriched with cortactin, filamentous (F)-actin and phosphotyrosine (Figure 4A, DMSO), three markers that define invadopodia (Bowden et al., 2006). Spontaneous invadopodia formation was observed in 51% of UMSCC1 cells by confocal microscopy at a given time, corresponding with focalized areas of matrix clearing (FITC-gelatin) (Figure 4B, 0μM dosage point). The gelatin matrix underneath UMSCC1 cells typically displayed degradation encompassing ~53% of the overlying cell area, reflecting the action of invadopodia-associated MMP activity (Figure 4C).
Figure 4.

Saracatinib inhibits invadopodia formation and ECM degradation. A: Representative images of UMSCC1 cells treated with different saracatinib concentrations. UMSCC1 cells plated on FITC-gelatin coated coverslips (pseudocolored white) for 2 h were treated with saracatinib as indicated (left) for 6 h. Cells were labeled to visualize F-actin (red), cortactin (green) and phosphotyrosine (blue). Arrows denote invadopodia and corresponding colocalized areas of focal matrix degradation with invadopodia components. Treatment with 1.0 μM saracatinib resulted in F-actin aggregates lacking cortactin but accumulated at cytoplasmic sites where invadopodia typically occur (arrowheads). Bar, 10 μm. B: UMSCC1 cells treated with increasing concentrations of saracatinib were stained as in A and quantified to determine the percentage of cells that produced functional invadopodia, presented as the mean ± SEM. All treatment groups were significantly different from each another based on a one-way ANOVA (p<0.05) except 0 and 0.01 μM, and 0.5 and 1.0 μM pairs. C: Saracatinib decreases the ability of UMSCC1 cells to degrade ECM. The percentage of gelatin degradation per cell area for the cell population analyzed in B is shown with the mean ± SEM.
UMSCC1 cells plated on FITC-coated gelatin were treated with increasing concentrations of saracatinib, and invadopodia formation and matrix degradation was evaluated by confocal microscopy (Figure 4). The number of cells containing invadopodia was significantly decreased with increasing saracatinib dosage, with < 2% of cells having formed invadopodia at concentrations at or above 0.5 μM (Figure 4B). Effects on matrix degradation were more pronounced, where increased saracatinib dosage resulted in similar incremental decreases in degradation (Figure 4C). UMSCC1 cells treated with 1.0 μM saracatinib did not contain invadopodia and were incapable of degrading matrix (Figure 4A). These cells also lacked focal cortactin localization, had diminished phosphotyrosine levels at focal adhesions and contained disorganized F-actin puncta on the ventral membrane surface where invadopodia typically form (Figure 4A, arrowheads).
Saracatinib inhibits MMP9 secretion from HNSCC cells
In addition to MT1-MMP, secretion and activation of MMP2 and MMP9 at invadopodia has been reported to be partially responsible for the observed effects on matrix degradation (Linder, 2007). Src activity regulates MMP2 and MMP9 secretion in fibroblasts (Hsia et al., 2003). We therefore determined the effect of saracatinib on MMP secretion. Confocal immunofluorescence microscopy indicated that UMSCC1 cells have MMP9-containing vesicles localized to invadopodia at sites that correspond with gelatin degradation (Figure 5A), indicating that MMP9 is concentrated in UMSCC1 invadopodia. To evaluate the impact of saracatinib on MMP2 and MMP9 secretion and activity in HNSCC cells, total cell lysates and conditioned media from saracatinib-treated 1483 and UMSCC1 cells were analyzed for the presence of cellular and secreted MMP2 and MMP9 by Western blotting (Figure 5B). Saracatinib treatment resulted in modest decreases (up to 24%) in cellular MMP2 levels at concentrations to 1 μM, while cellular MMP9 levels demonstrated up to a two-fold increase under the same concentration range. Although secreted MMP2 was not detected in the media of either cell line, both lines secreted detectible amounts of MMP9 (Figure 5B). Treatment of either line with saracatinib inhibited MMP9 secretion, and each line displayed differential drug sensitivity. Detectible MMP9 secretion from 1483 cells was largely absent at the lowest evaluated concentration (0.01 μM) whereas secretion from the more invasive UMSCC1 line was inhibited at concentrations of 0.5 μM and above (Figure 5B). The secreted MMP9 from both lines displayed proteolytic activity when assayed by gelatin zymography (Figure 5C). Saracatinib concentrations up to 0.1μM did not significantly affect MMP9 activity as determined by ANOVA analysis for both lines, although mean values for 1483 cells treated with these lower doses were consistently below control levels (Figure 5C). Higher saracatinib concentrations (0.5 μM and 1.0 μM) reduced MMP9 activity to respective mean values of 39% and 25% for control levels in 1483 cells, and 22% and 12% in UMSCC1 cells (Figure 5B,C). These data indicate that saracatinib treatment of HNSCC cells leads to selective inhibition of MMP9 secretion in 1483 and UMSCC1 cells, preventing efficient enzymatic degradation of extracellular matrix components.
Figure 5.

MMP9 secretion and ECM degradation activity in HNSCC cells is blocked by saracatinib. A: Localization of MMP9-containing vesicles in UMSCC1 invadopodia. Top: UMSCC1 cells plated on FITC-coated gelatin coverslips for 2 h were fixed and labeled with antibodies against cortactin and MMP9. The merged image indicates areas of cortactin and MMP9 co-localization (yellow; white arrows) that correspond with sites of focal gelatin degradation (black arrows). Bar; 10 μM, Asterisk; regions of global matrix degradation due to secreted protease activity. Bottom: Magnified view of indicated Top region. B: Inhibition of MMP9 secretion by saracatinib. Total cell lysates (cell) and aliquots of normalized conditioned media containing secreted MMPs (sec) from 1483 and UMSCC1 cells treated with increasing doses of saracatinib (bottom) were assayed for the presence of MMP2 and MMP9 by immunoblotting. Band intensities relative to control (0 μM) are shown for each treatment condition; secreted MMP2 was not detected and therefore not quantified. C: Gelatin zymography of MMP9 activity. Representative zymograms from conditioned media of 1483 or UMSCC1 cells cultured with the indicated saracatinib concentrations (bottom). DMEM was used as a negative control (M), DMEM containing 10% FBS (FBS) was used as a positive control for zymography. Graphs, densitometric analysis of MMP9 zymography. Percentage of MMP9 gelatin clearing is represented and the mean ± SEM for each cell line from three independent experiments.
Discussion
The present study demonstrates that inhibition of HNSCC invasion in preclinical in vitro and in vivo settings by saracatinib directly corresponds to disruption of HNSCC invadopodia formation and function, identifying invadopodia as a potential downstream target of therapeutic Src kinase inhibition in HNSCC and other invasive human cancers. Invasive HNSCC presents a difficult problem in patient care, given the proximity of most tumors to multiple vital organ sites in the head and neck region. Disregulation of signaling pathways that promote and sustain invasion impinge on adhesion- and cytoskeletal-associated proteins. These proteins function in concert with MMPs to enable tumor cells to degrade and protrude through an encapsulating ECM, allowing movement into neighboring tissues. The ability of saracatinib to ablate invadopodia and the associated invasive behavior of HNSCC cells in mice provides further evidence for a direct link between invadopodia activity and tumor invasion, shedding light on the specific invasion-promoting cellular processes perturbed by Src kinase inhibition.
The HNSCC lines used in this study displayed differential responses to saracatinib in terms of anti-proliferate effects, with some lines (HN31 and UMSCC1) having submicromolar sensitivity, cell cycle inhibition and decreased ERK1/2 activity and others (1483) demonstrating resistance to the drug at concentrations up to 1 μM (Figure 1A–C). While a recent study reported IC50 saracatinib values near 1 μM for five different HNSCC lines (Koppikar et al., 2008), our findings are in line with the wider range of IC50 values reported for different HNSCC lines treated with the non-related Src kinase inhibitor dasatinib (Johnson et al., 2005) as well as in other tumor cell types (Boyer et al., 2002; Johnson et al., 2005; Jones et al., 2002). These reports taken together with our data suggest that the HNSCC lines utilized in this study fall within the typical in vitro proliferative response profile to therapeutic Src inhibition. While the underlying compensatory mechanism for saracatinib resistance in 1483 cells is unknown, future expression profiling of resistant and sensitive lines may provide insight into the molecular nature of saracatinib resistance, as has been recently conducted for the EGFR inhibitor gefitinib in a variety of HNSCC lines (Frederick et al., 2007).
Saracatinib treatment resulted in in vitro anti-invasive activity, impaired Src activation and tyrosine phosphorylation of FAK and p130 CAS in all analyzed HNSCC lines (Figure 1D, Figure 2). This is in accord with other studies on other HNSCC lines utilizing saracatinib or dasatinib as single agents (Johnson et al., 2005; Koppikar et al., 2008; Nozawa et al., 2008). Tyrosine phosphorylation of FAK and p130 CAS have been commonly utilized as downstream indicators for preclinical therapeutic anti-Src efficacy, since Src-mediated phosphorylation of these proteins are critical events in enabling tumor invasiveness (Brabek et al., 2005; Zhao and Guan, 2009). In addition, we show that cortactin tyrosine phosphorylation was also reduced following saracatinib treatment (Figure 2). Cortactin is a Src substrate commonly overexpressed in invasive HNSCC and regulates invadopodia formation (Rodrigo et al., 2000; Rothschild et al., 2006; Weaver, 2008). Cortactin phosphorylation is important for tumor cell motility and matrix degradation at invadopodia (Ayala et al., 2008; Oser et al., 2009; Rothschild et al., 2006). These results suggest that cortactin tyrosine phosphorylation status can serve as an additional downstream monitor of Src activity and invasive potential in HNSCC cells where Src kinase function is impaired.
While saracatinib was able to modestly decrease the size of in vivo UMSCC1 tongue tumors, the reduction in tumor size was not significant compared to untreated controls. UMSCC1 cell growth is inhibited by saracatinib in vitro (Figure 1A), implying that microenvironmental factors such as inflammatory cytokines, growth factors, neoangiogenic and hypoxic aspects responsible for promoting and maintaining HNSCC growth (Pries and Wollenberg, 2006; Timar et al., 2005) may partially circumvent the growth-inhibitory effects of saracatinib in UMSCC1 xenografts. On the other hand, saracatinib displays in vivo anti-invasive properties by potently inhibiting perineural invasion and cervical lymph node metastasis. Suppressed Src activation and cortactin tyrosine phosphorylation in primary tumors (Figure 4A) supports this conclusion, as signaling through these proteins promotes invasion and metastatic spread (Weaver, 2008; Yeatman, 2004). Recent findings in a comparable xenograft system utilizing mice treated with dasatinib demonstrated reduced Src and FAK activity (Sen et al., 2009). EGFR overexpression is common in HNSCC, resulting in enhanced Src activity, cortactin phosphorylation and tumor invasiveness (Koppikar et al., 2008; Rothschild et al., 2006). Clinical EGFR inhibitors also display anti-invasive activity and impair invadopodia formation in preclinical settings (Huang et al., 2002; Yamaguchi et al., 2005; Yang et al., 2004), suggesting in light of our findings that inhibiting EGFR activity in HNSCC impairs Src activation and substrate phosphorylation required for invasion. This is supported by emerging rationale for dual targeting of EGFR and Src in treating advanced HNSCC (Egloff and Grandis, 2008).
Elevated Src activity is necessary and essential for invadopodia formation (Chen et al., 1985; Chen et al., 1984). The ability of saracatinib to ablate invadopodia and associated matrix degradation demonstrates that a clinically utilized Src inhibitor disrupts a vital subcellular structure required for tumor invasion. Src-induced invadopodia formation in carcinoma cells first targets cortactin and F-actin formation at matrix adhesion sites, forming a core preinvadopodia complex. Recruitment of MT1-MMP to preinvadopodia initiates matrix degradation and invadopodia maturation, with further maturation involving dissolution of the cortactin-F-actin complex, focal retention of MT1-MMP and continued proteolytic activity (Artym et al., 2006). While rudimentary invadopodia-like F-actin structures formed in UMSCC1 cells treated with inhibitory concentrations of saracatinib, they do not contain cortactin (Figure 4A), which is essential for invadopodia formation (Artym et al., 2006), indicating that Src kinase activity is required for cortactin localization to invadopodia. Similar results have been shown through the use of kinase-inactive Src constructs (Bowden et al., 2006). Tyrosine phosphorylation of invadopodia proteins is strongly linked with the ability to degrade extracellular matrix (Bowden et al., 2006), with Src phosphorylation of cortactin (Ayala et al., 2008), paxillin (Badowski et al., 2008), and ASAP1 (Bharti et al., 2007) requisite for invadopodia formation and/or proteolytic activity. Src kinase inhibition therefore displays at least a two-fold effect on substrates in invadopodia by impairing proper preinvadopodia targeting and perturbing phosphotyrosine-based signaling dynamics involved in regulating invadopodia maturation and function. Tyrosine phosphorylation in peripheral focal adhesions was is observed in UMSCC1 cells, indicating saracatinib may also perturb focal adhesion formation and/or function given the critical role for Src and related kinases in these structures (Frame, 2004).
MMP activity is essential for HNSCC invadopodia formation and function (Clark et al., 2007). The impairment of MMP9 secretion and activation from HNSCC cells treated with saracatinib indicates that Src kinase activity is required for targeting and secretion of MMP9-containing vesicles at invadopodia. This is in agreement with observed effects of Src kinase inhibition on MMP9 secretion in other tumor types (Cortes-Reynosa et al., 2008; Lee et al., 2005). The lack of apparent MMP2 secretion in the HNSCC lines used in our studies precluded evaluation of this metalloproteinase, and is likely characteristic to these lines since other HNSCC cells secrete MMP2 (Clark et al., 2007). Localization of transmembrane and secreted MMPs to invadopodia involves directed trafficking of vesicles emanating from the trans-Golgi network, where a dynamin-2-N-WASp-Arp2/3-cortactin complex has been implicated in coupling cortical actin regulation with invadopodia membrane dynamics (Buccione et al., 2004). Cortactin has been implicated as a key regulator of MT1-MMP surface expression and MMP2 and MMP9 secretion in HNSCC (Clark and Weaver, 2008; Clark et al., 2007). While it is currently unknown how cortactin regulates the targeting of MMP-containing vesicles, Src-mediated phosphorylation may play a vital role since tyrosine phosphorylation of cortactin increases its binding to vesicle-associated proteins (Ammer and Weed, 2008) and is required for efficient invadopodia-mediated ECM degradation (Ayala et al., 2008; Webb et al., 2007).
Our results indicate that disruption of Src activity by saracatinib impairs HNSCC cell invasion and lymph node metastasis by preventing invadopodia formation and function, identifying a cellular mechanism that may be a universally impacted by Src inhibition in invasive carcinoma cells. In addition to Src and related kinases, saracatinib also inhibits Ablkinase, an activity that has been exploited to evaluate imatinib-resistant chronic myelogenous leukemia (CML) cases expressing the constitutively active BCR-Abl Philadelphia chromosome gene product (Gwanmesia et al., 2009). Along with the tumor-promoting activities in CML, elevated Abl kinase activity has been shown to be important in breast (Srinivasan and Plattner, 2006) and non small cell lung cancer (Lin et al., 2007), indicating Abl kinase activity has a functional role in solid tumor progression (Lin and Arlinghaus, 2008). Abl expression has been evaluated in oral squamous cell carcinoma and correlates with tumor stage (Yanagawa et al., 2000), suggesting that elevated Abl expression may contribute to HNSCC progression. While a mechanism pertaining to Abl function in solid tumor invasion or invadopodia function has not been reported, Abl does bind and phosphorylate cortactin (Boyle et al., 2007), raising the potential for Abl kinase to play a role in cortactin-based invadopodia function in HNSCC and other invasive Abl-expressing solid tumors. Such a role for Abl in regulating tumor invasion would also likely be impaired by saracatinib and other dual Src/Abl inhibitory compounds.
In addition to carcinoma invadopodia, Src-mediated processes in non-cancerous cell types involved in promoting invasion may also be impacted by Src family kinase inhibition. The motility and protease remodeling ability of tumor stromal fibroblasts involved in enabling collective HNSCC invasion could be affected by impairing Src family kinases in vivo (Gaggioli et al., 2007), as well as the proinvasive properties of tumor associated macrophages (Condeelis and Pollard, 2006). The ability of saracatinib and other Src inhibitors to impair functions of different cellular types that propagate tumor invasion provides the opportunity for the future discovery of additional Src-based cellular mechanisms utilized during tumor progression.
Supplementary Material
Acknowledgments
We thank J. Myers and M. Younes (Department of Head and Neck Surgery, University of Texas M.D. Anderson Cancer Center, Houston, TX) for UMSCC1 cells and related help, P. Turner and K. Secrest of the West Virginia University Department of Pathology Tissue Bank for histochemistry services. The contributions of the West Virginia University Microscopic Imaging Facility, Mary Babb Randolph Cancer Center are gratefully acknowledged. This work was supported by a grant from AstraZeneca to DR, BF and SAW and by NIH grants R01 DE014578 and P20 RR16440 to SAW. LCK was supported in part by the West Virginia University Office of Research and Graduate Education.
Abbreviations
- CAS
Crk-Associated Substrate
- c-Src
cellular Src kinase
- ECM
Extracellular Matrix
- FACS
Fluorescence-Activated Cell Sorting
- FAK
Focal Adhesion Kinase
- FITC
Fluorescein Isothiocyanate
- HNSCC
Head and Neck Squamous Cell Carcinoma
- IHC
Immunohistochemistry
- SFK
Src Family Kinase
References
- 1.Ammer AG, Weed SA. Cortactin branches out: roles in regulating protrusive actin dynamics. Cell Motil Cytoskeleton. 2008;65:687–707. doi: 10.1002/cm.20296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Artym VV, Zhang Y, Seillier-Moiseiwitsch F, Yamada KM, Mueller SC. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 2006;66:3034–43. doi: 10.1158/0008-5472.CAN-05-2177. [DOI] [PubMed] [Google Scholar]
- 3.Ayala I, Baldassarre M, Giacchetti G, Caldieri G, Tete S, et al. Multiple regulatory inputs converge on cortactin to control invadopodia biogenesis and extracellular matrix degradation. J Cell Sci. 2008;121:369–78. doi: 10.1242/jcs.008037. [DOI] [PubMed] [Google Scholar]
- 4.Badowski C, Pawlak G, Grichine A, Chabadel A, Oddou C, et al. Paxillin phosphorylation controls invadopodia/podosomes spatiotemporal organization. Mol Biol Cell. 2008;19:633–45. doi: 10.1091/mbc.E06-01-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bharti S, Inoue H, Bharti K, Hirsch DS, Nie Z, et al. Src-dependent phosphorylation of ASAP1 regulates podosomes. Mol Cell Biol. 2007;27:8271–83. doi: 10.1128/MCB.01781-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowden ET, Onikoyi E, Slack R, Myoui A, Yoneda T, et al. Co-localization of cortactin and phosphotyrosine identifies active invadopodia in human breast cancer cells. Exp Cell Res. 2006;312:1240–53. doi: 10.1016/j.yexcr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 7.Boyer B, Bourgeois Y, Poupon MF. Src kinase contributes to the metastatic spread of carcinoma cells. Oncogene. 2002;21:2347–56. doi: 10.1038/sj.onc.1205298. [DOI] [PubMed] [Google Scholar]
- 8.Boyle SN, Michaud GA, Schweitzer B, Predki PF, Koleske AJ. A critical role for cortactin phosphorylation by Abl-family kinases in PDGF-induced dorsal-wave formation. Curr Biol. 2007;17:445–51. doi: 10.1016/j.cub.2007.01.057. [DOI] [PubMed] [Google Scholar]
- 9.Brabek J, Constancio SS, Siesser PF, Shin NY, Pozzi A, et al. Crk-associated substrate tyrosine phosphorylation sites are critical for invasion and metastasis of SRC-transformed cells. Mol Cancer Res. 2005;3:307–15. doi: 10.1158/1541-7786.MCR-05-0015. [DOI] [PubMed] [Google Scholar]
- 10.Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–57. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- 11.Chen WT, Chen JM, Parsons SJ, Parsons JT. Local degradation of fibronectin at sites of expression of the transforming gene product pp60src. Nature. 1985;316:156–8. doi: 10.1038/316156a0. [DOI] [PubMed] [Google Scholar]
- 12.Chen WT, Olden K, Bernard BA, Chu FF. Expression of transformation-associated protease(s) that degrade fibronectin at cell contact sites. J Cell Biol. 1984;98:1546–55. doi: 10.1083/jcb.98.4.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark ES, Weaver AM. A new role for cortactin in invadopodia: Regulation of protease secretion. Eur J Cell Biol. 2008;87:581–90. doi: 10.1016/j.ejcb.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark ES, Whigham AS, Yarbrough WG, Weaver AM. Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 2007;67:4227–35. doi: 10.1158/0008-5472.CAN-06-3928. [DOI] [PubMed] [Google Scholar]
- 15.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 16.Cortes-Reynosa P, Robledo T, Macias-Silva M, Wu SV, Salazar EP. Src kinase regulates metalloproteinase-9 secretion induced by type IV collagen in MCF-7 human breast cancer cells. Matrix Biol. 2008;27:220–31. doi: 10.1016/j.matbio.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 17.Dolgin E. Cancer metastasis scrutinized. Nature. 2009;461:854–5. doi: 10.1038/461854b. [DOI] [PubMed] [Google Scholar]
- 18.Egloff AM, Grandis JR. Targeting epidermal growth factor receptor and SRC pathways in head and neck cancer. Semin Oncol. 2008;35:286–97. doi: 10.1053/j.seminoncol.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frame MC. Newest findings on the oldest oncogene; how activated src does it. J Cell Sci. 2004;117:989–98. doi: 10.1242/jcs.01111. [DOI] [PubMed] [Google Scholar]
- 20.Frederick BA, Helfrich BA, Coldren CD, Zheng D, Chan D, et al. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther. 2007;6:1683–91. doi: 10.1158/1535-7163.MCT-07-0138. [DOI] [PubMed] [Google Scholar]
- 21.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 22.Gimona M, Buccione R, Courtneidge SA, Linder S. Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol. 2008;20:235–41. doi: 10.1016/j.ceb.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Green TP, Fennell M, Whittaker R, Curwen J, Jacobs V, et al. Pre-clinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol. 2009 doi: 10.1016/j.molonc.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gwanmesia PM, Romanski A, Schwarz K, Bacic B, Ruthardt M, et al. The effect of the dual Src/Abl kinase inhibitor AZD0530 on Philadelphia positive leukaemia cell lines. BMC Cancer. 2009;9:53. doi: 10.1186/1471-2407-9-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hennequin LF, Allen J, Breed J, Curwen J, Fennell M, et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. 2006;49:6465–88. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- 26.Herynk MH, Beyer AR, Cui Y, Weiss H, Anderson E, et al. Cooperative action of tamoxifen and c-Src inhibition in preventing the growth of estrogen receptor-positive human breast cancer cells. Mol Cancer Ther. 2006;5:3023–31. doi: 10.1158/1535-7163.MCT-06-0394. [DOI] [PubMed] [Google Scholar]
- 27.Hsia DA, Mitra SK, Hauck CR, Streblow DN, Nelson JA, et al. Differential regulation of cell motility and invasion by FAK. J Cell Biol. 2003;160:753–67. doi: 10.1083/jcb.200212114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang SM, Li J, Harari PM. Molecular inhibition of angiogenesis and metastatic potential in human squamous cell carcinomas after epidermal growth factor receptor blockade. Mol Cancer Ther. 2002;1:507–14. [PubMed] [Google Scholar]
- 29.Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636–42. doi: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- 30.Johnson FM, Saigal B, Talpaz M, Donato NJ. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung cancer cells. Clin Cancer Res. 2005;11:6924–32. doi: 10.1158/1078-0432.CCR-05-0757. [DOI] [PubMed] [Google Scholar]
- 31.Jones RJ, Avizienyte E, Wyke AW, Owens DW, Brunton VG, et al. Elevated c-Src is linked to altered cell-matrix adhesion rather than proliferation in KM12C human colorectal cancer cells. Br J Cancer. 2002;87:1128–35. doi: 10.1038/sj.bjc.6600594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelley LC, Shahab S, Weed SA. Actin cytoskeletal mediators of motility and invasion amplified and overexpressed in head and neck cancer. Clin Exp Metastasis. 2008;25:289–304. doi: 10.1007/s10585-008-9154-6. [DOI] [PubMed] [Google Scholar]
- 33.Kopetz S, Shah AN, Gallick GE. Src continues aging: current and future clinical directions. Clin Cancer Res. 2007;13:7232–6. doi: 10.1158/1078-0432.CCR-07-1902. [DOI] [PubMed] [Google Scholar]
- 34.Koppikar P, Choi SH, Egloff AM, Cai Q, Suzuki S, et al. Combined inhibition of c-Src and epidermal growth factor receptor abrogates growth and invasion of head and neck squamous cell carcinoma. Clin Cancer Res. 2008;14:4284–91. doi: 10.1158/1078-0432.CCR-07-5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kramer RH, Shen X, Zhou H. Tumor cell invasion and survival in head and neck cancer. Cancer Metastasis Rev. 2005;24:35–45. doi: 10.1007/s10555-005-5046-2. [DOI] [PubMed] [Google Scholar]
- 36.Lee JC, Maa MC, Yu HS, Wang JH, Yen CK, et al. Butyrate regulates the expression of c-Src and focal adhesion kinase and inhibits cell invasion of human colon cancer cells. Mol Carcinog. 2005;43:207–14. doi: 10.1002/mc.20117. [DOI] [PubMed] [Google Scholar]
- 37.Lin J, Arlinghaus R. Activated c-Abl tyrosine kinase in malignant solid tumors. Oncogene. 2008;27:4385–91. doi: 10.1038/onc.2008.86. [DOI] [PubMed] [Google Scholar]
- 38.Lin J, Sun T, Ji L, Deng W, Roth J, et al. Oncogenic activation of c-Abl in non-small cell lung cancer cells lacking FUS1 expression: inhibition of c-Abl by the tumor suppressor gene product Fus1. Oncogene. 2007;26:6989–96. doi: 10.1038/sj.onc.1210500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Linder S. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 2007;17:107–17. doi: 10.1016/j.tcb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Mueller SC, Yeh Y, Chen WT. Tyrosine phosphorylation of membrane proteins mediates cellular invasion by transformed cells. J Cell Biol. 1992;119:1309–25. doi: 10.1083/jcb.119.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Myers JN, Holsinger FC, Jasser SA, Bekele BN, Fidler IJ. An orthotopic nude mouse model of oral tongue squamous cell carcinoma. Clin Cancer Res. 2002;8:293–8. [PubMed] [Google Scholar]
- 42.Nozawa H, Howell G, Suzuki S, Zhang Q, Qi Y, et al. Combined inhibition of PLC{gamma}-1 and c-Src abrogates epidermal growth factor receptor-mediated head and neck squamous cell carcinoma invasion. Clin Cancer Res. 2008;14:4336–44. doi: 10.1158/1078-0432.CCR-07-4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oser M, Yamaguchi H, Mader CC, Bravo-Cordero JJ, Arias M, et al. Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J Cell Biol. 2009;186:571–87. doi: 10.1083/jcb.200812176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, et al. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008;68:3323–33. doi: 10.1158/0008-5472.CAN-07-2997. [DOI] [PubMed] [Google Scholar]
- 45.Pries R, Wollenberg B. Cytokines in head and neck cancer. Cytokine Growth Factor Rev. 2006;17:141–6. doi: 10.1016/j.cytogfr.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 46.Rodrigo JP, Garcia LA, Ramos S, Lazo PS, Suarez C. EMS1 gene amplification correlates with poor prognosis in squamous cell carcinomas of the head and neck. Clin Cancer Res. 2000;6:3177–82. [PubMed] [Google Scholar]
- 47.Rosenthal EL, Matrisian LM. Matrix metalloproteases in head and neck cancer. Head Neck. 2006;28:639–48. doi: 10.1002/hed.20365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rothschild BL, Shim AH, Ammer AG, Kelley LC, Irby KB, et al. Cortactin overexpression regulates actin-related protein 2/3 complex activity, motility, and invasion in carcinomas with chromosome 11q13 amplification. Cancer Res. 2006;66:8017–25. doi: 10.1158/0008-5472.CAN-05-4490. [DOI] [PubMed] [Google Scholar]
- 49.Sano D, Myers JN. Metastasis of squamous cell carcinoma of the oral tongue. Cancer Metastasis Rev. 2007;26:645–62. doi: 10.1007/s10555-007-9082-y. [DOI] [PubMed] [Google Scholar]
- 50.Sen B, Saigal B, Parikh N, Gallick G, Johnson FM. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009;69:1958–65. doi: 10.1158/0008-5472.CAN-08-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Srinivasan D, Plattner R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006;66:5648–55. doi: 10.1158/0008-5472.CAN-06-0734. [DOI] [PubMed] [Google Scholar]
- 52.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–58. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 53.Summy JM, Gallick GE. Treatment for advanced tumors: SRC reclaims center stage. Clin Cancer Res. 2006;12:1398–401. doi: 10.1158/1078-0432.CCR-05-2692. [DOI] [PubMed] [Google Scholar]
- 54.Timar J, Csuka O, Remenar E, Repassy G, Kasler M. Progression of head and neck squamous cell cancer. Cancer Metastasis Rev. 2005;24:107–27. doi: 10.1007/s10555-005-5051-5. [DOI] [PubMed] [Google Scholar]
- 55.van Oijen MG, Rijksen G, ten Broek FW, Slootweg PJ. Overexpression of c-Src in areas of hyperproliferation in head and neck cancer, premalignant lesions and benign mucosal disorders. J Oral Pathol Med. 1998;27:147–52. doi: 10.1111/j.1600-0714.1998.tb01931.x. [DOI] [PubMed] [Google Scholar]
- 56.Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis. 2006;23:97–105. doi: 10.1007/s10585-006-9014-1. [DOI] [PubMed] [Google Scholar]
- 57.Weaver AM. Cortactin in tumor invasiveness. Cancer Lett. 2008;265:157–66. doi: 10.1016/j.canlet.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Webb BA, Jia L, Eves R, Mak AS. Dissecting the functional domain requirements of cortactin in invadopodia formation. Eur J Cell Biol. 2007;86:189–206. doi: 10.1016/j.ejcb.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 59.Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, et al. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–52. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yanagawa T, Harada H, Iwasa S, Tabuchi K, Omura K, et al. c-Abl expression in oral squamous cell carcinomas. Oral Oncol. 2000;36:89–94. doi: 10.1016/s1368-8375(99)00067-6. [DOI] [PubMed] [Google Scholar]
- 61.Yang Z, Bagheri-Yarmand R, Wang RA, Adam L, Papadimitrakopoulou VV, et al. The epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 (Iressa) suppresses c-Src and Pak1 pathways and invasiveness of human cancer cells. Clin Cancer Res. 2004;10:658–67. doi: 10.1158/1078-0432.ccr-0382-03. [DOI] [PubMed] [Google Scholar]
- 62.Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470–80. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Q, Thomas SM, Xi S, Smithgall TE, Siegfried JM, et al. SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res. 2004;64:6166–73. doi: 10.1158/0008-5472.CAN-04-0504. [DOI] [PubMed] [Google Scholar]
- 64.Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.