

SUMMARY
Acetylcholine is a ubiquitous cortical neuromodulator implicated in cognition. In order to understand the potential for acetylcholine to play a role in visual attention, we studied nicotinic acetylcholine receptor (nAChR) localization and function in area V1 of the macaque. We found nAChRs presynaptically at thalamic synapses onto excitatory, but not inhibitory, neurons in the primary thalamorecipient layer 4c. Furthermore, consistent with the release enhancement suggested by this localization, we discovered that nicotine increases responsiveness and lowers contrast threshold in layer 4c neurons. We also found that nAChRs are expressed by GABAergic interneurons in V1 but rarely by pyramidal neurons, and that nicotine suppresses visual responses outside layer 4c. All sensory systems incorporate gain control mechanisms, or processes which dynamically alter input/output relationships. We demonstrate that at the site of thalamic input to visual cortex, the effect of this nAChR-mediated gain is an enhancement of the detection of visual stimuli.
INTRODUCTION
As a central nervous system neuromodulator, acetylcholine (ACh) is involved in processes underlying the sleep/wake cycle and arousal (Jasper and Tessier, 1971; Jimenez-Capdeville and Dykes, 1996; Vazquez and Baghdoyan, 2001), reward and addiction (Maskos et al., 2005), attention (Sarter et al., 2005), and learning and memory (Everitt and Robbins, 1997; Hasselmo and McGaughy, 2004; Rezvani and Levin, 2001), and in a number of neuropathologies including Alzheimer’s disease (Gallagher and Colombo, 1995). The rewarding and addictive nature of nicotine is mediated by cholinergic action in subcortical structures (Maskos et al., 2005), and learning and memory functions of ACh are subserved by cholinergic projections to the hippocampal formation (Everitt and Robbins, 1997) with a lesser role for the neocortical cholinergic projection (Hasselmo and McGaughy, 2004). However, based on work in rodents, the cholinergic projection from the basal forebrain area (BFA) to neocortex is believed to be primarily involved in processes that underlie arousal and attentional modulation (Everitt and Robbins, 1997; Hasselmo and McGaughy, 2004; Sarter et al., 2005).
In addition to (or perhaps as part of) its role in higher functions, cholinergic neuromodulation in the neocortex is also involved in normal sensory processing; after lesions of the BFA, many visual cortical neurons no longer respond to visual stimuli (Sato et al., 1987a). The role of ACh in the processing of sensory stimuli has been extensively studied, particularly in the visual cortex (Muller and Singer, 1989; Murphy and Sillito, 1991; Roberts et al., 2005; Sato et al., 1987a, 1987b; Sillito and Kemp, 1983; Zinke et al., 2006), but the reported effects of ACh are diverse and in some cases contradictory. In order to clarify the role of ACh in the sensory cortices, we pursued a detailed anatomical and pharmacological dissection of neocortical cholinergic neuromodulation, beginning with the role of nicotinic ACh receptors (nAChRs) in a well-studied cortical model system, the primary visual cortex (V1) of the macaque monkey.
In macaques, V1 receives its cholinergic innervation from the nucleus basalis (Pearson et al., 1983). In the neocortex, ACh is usually released from varicosities that are not apposed to a synaptic specialization (Aoki and Kabak, 1992; Beaulieu and Somogyi, 1991; but see Turrini et al., 2001). This mode of release, known as volume transmission, means that receptor subtype and localization critically determine signaling specificity. There are multiple ionotropic and metabotropic subclasses of AChRs (Brown et al., 1997; Metherate, 2004), and in V1, both muscarinic AChRs (mAChRs) (metabotropic, primarily the m1 and m2 subtypes; Tigges et al., 1997) and nicotinic (ionotropic) AChRs (Han et al., 2000) are expressed at significant levels.
Although previous anatomical studies exist suggesting a presynaptic localization for nAChRs on thalamic axons arriving in primary sensory areas (Parkinson et al., 1988), and although in vitro studies also indicate a nicotinic modulation of ascending pathways into cortex (Gil et al., 1997; Hsieh et al., 2000; Kimura et al., 1999), the precise type and localization of the nAChRs responsible for these effects is still not known. We conducted a tract-tracing and immunocytochemical electron microscopic (immuno-EM) study to localize mAChRs and nAChRs within layer 4c, the principal thalamic recipient zone in macaque V1. To provide a more complete description of the cholinergic modulation of V1, we also used dual immunofluorescence to determine the extent to which AChRs, both nicotinic (this study) and muscarinic (Disney et al., 2006), are expressed by inhibitory versus excitatory neurons. Finally, to confirm predictions made by these anatomical data, we conducted in vivo experiments that combined iontophoresis of cholinergic agonists with extracellular recordings of the functional properties of neurons in all layers of V1.
Here we show that presynaptic high-affinity nAChRs are found on afferent fibers arriving from the principal visual thalamic relay, the lateral geniculate nucleus (LGN), in layer 4c of V1 in the macaque monkey. We also demonstrate that iontophoretic application of nicotine enhances the response gain (i.e., increases the spiking response to a given stimulus contrast) of neurons in layer 4c of V1, but not in other layers. These effects are dose dependent, transient, and in most neurons not due to changes in maintained discharge.
RESULTS
AChR Expression at Thalamocortical Synapses in Layer 4c
First we determined the subcellular distribution of nAChRs and mAChRs in V1 using tract-tracing and electron microscopy (EM). We injected an anterograde tracer into the primary visual thalamic relay, the LGN, of three macaques in order to identify thalamic terminals in layer 4c of V1 (Figures 1A and 1B). Thalamocortical (TC) synapses in layer 4c were identified and the AChR distribution (presynaptic and postsynaptic) at synapses onto spines and shafts was determined by EM. We compared tracer-positive (thalamic) synapses with tracer-negative (presumed nonthalamic) excitatory (i.e., asymmetric, Figure 1B) synapses and found β2 subunits (nAChR) expressed presynaptically at 76% of thalamic synapses onto spines (Figure 1C, left bar; 29/38 synapses), but at 0% of thalamic synapses onto dendritic shafts (Figure 1C, second bar from left; 0/9 synapses). Because glutamatergic (i.e., spiny) neurons receive most of their excitatory synapses at spines, while GABAergic (aspiny or sparsely spiny) neurons receive excitatory synapses directly to the dendritic shaft, this nAChR localization at synapses onto spines and not shafts indicates a targeted modulation of the thalamic input to cortical excitatory cells. In contrast to the strong expression at thalamic synapses, nAChRs were present at only 12% (29/242) of other excitatory synapses onto shafts and spines in layer 4c (Figure 1C, right pair of bars).
Figure 1. Localization of AChRs by EM-Immunocytochemistry.
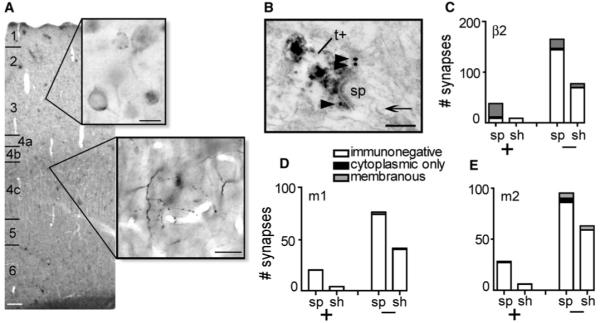
(A) Labeling for the β2-nAChR subunit by silver-intensified gold (upper inset; scale bar, 10 μm) and anterogradely traced LGN terminals by ABC-DAB (middle inset; scale bar, 20 μm). (B) An EM image of a tracer-labeled terminal (t+) synapsing onto a spine (sp; note the spine apparatus, arrow), showing that tract-tracing allows identification of thalamic terminals in single planes of section in which the presynaptic element is otherwise difficult to distinguish. Silver particles (arrowheads) indicate presynaptic β2-nAChRs. Scale bar, 200 nm. (C) The quantification of β2 labeling at layer 4c synapses (x axis: sp, spine; sh, shaft; +, labeled terminals; −, unlabeled terminals). 76% of thalamic synapses onto spines in layer 4c are presynaptically immunoreactive for β2 subunits. No thalamic synapses onto dendrites are β2-nAChR-ir. Layer 4c thalamic synapses are immunonegative for m1 (D) and m2 (E) muscarinic receptors.
In contrast to their strong expression of nicotinic receptors, no thalamic axons or terminals expressed membrane-associated m1AChRs (Figure 1D, left pair of bars; 0/24), and only one thalamic terminal was immunoreactive for the m2AChR (Figure 1E, left pair of bars; 1/34). m2AChRs were, however, observed on nonterminal membranes of 27% (6/22) of thalamic axons in the lower half of layer 4c (4cβ). No axon segments in upper layer 4c (4cα) were m2 immunoreactive. Finally, a small population of nonthalamic excitatory synapses expressed mAChRs (Figures 1D and 1E, right pair of bars; m1: 3/117, m2: 13/158). The predominant effect of ACh acting on thalamic axons in layer 4c is thus expected to be a presynaptic, nAChR-mediated modulation of the thalamic input to excitatory neurons.
nAChR Expression by Interneurons in Macaque V1
To further elucidate how ACh acts on the cortical circuit (i.e., beyond the TC synapse) we compared β2-nAChR subunit expression between excitatory and inhibitory neurons. We have shown previously that mAChRs are strongly expressed by inhibitory neurons in macaque V1 (Disney et al., 2006). Inhibitory neurons can be identified using antibodies directed at the neurotransmitter γ-aminobutyric acid (GABA)—the remaining neurons can be presumed to be excitatory. We examined coexpression of β2-nAChR subunits with GABA using dual-label immunofluorescence microscopy and found that few V1 neurons, whether excitatory or inhibitory, express β2 subunits. Of those that do, most are GABAergic (Figures 2A and 2B). Across layers, 75% (644/861) of β2-nAChR-immunoreactive (nAChR-ir) somata were GABA immunoreactive. However, this represented only 25% (644/2577) of the GABAergic population. GABAergic neurons comprise ~20% of all V1 neurons (Beaulieu et al., 1992). Thus, only ~7% of V1 neurons express β2-nAChR subunits, of which >70% (i.e., 5% of the total V1 population) are β2 subunit-expressing inhibitory neurons. This is qualitatively supported by the observation that dendrites which, in the above EM study, expressed β2 subunits received asymmetric synapses directly onto the dendritic shaft (i.e., they were aspinous and putatively inhibitory;Figure 2C).
Figure 2. Expression of Nicotinic Receptors by V1 Interneurons.
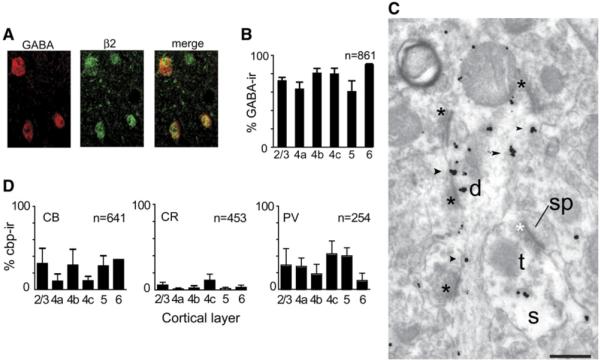
(A) Dual immunofluorescence from layer 3. The red channel (left) shows GABA immunoreactivity; the green channel (middle) shows β2-nAChR-immunoreactivity. In the merged image (right), dual-labeling appears yellow. In every layer the proportion of β2-nAChR-ir somata that are also GABA-ir exceeds 60% (B). By EM there is prominent β2-nAChR immunolabeling of putatively inhibitory dendrites—in (C), a layer 4cβ shaft (d) receives direct asymmetric synapses (black asterisks), suggesting that it is the dendrite of a GABAergic (aspiny) neuron. The dendrite is densely β2-nAChR immunoreactive (black arrowheads). Also visible is an asymmetric synapse onto a spine (sp) that has presynaptic β2-nAChR immunoreactivity in the terminal (t) cytoplasm. The empty space inside the terminal (s) is the tissue surface—data were collected very close to the tissue/EPON interface. Scale bar, 200 nm. (D) Some β2-nAChR-ir neurons express the calcium-binding proteins (CBP) calbindin (CB) (left; 111 of 641 neurons, 17%) and parvalbumin (PV) (right; 76 of 254 neurons, 30%). In contrast, very few β2-nAChR-ir neurons in any layer express calretinin (CR) (middle; 25 of 453 neurons, 6%). All error bars = SEM.
Inhibitory neurons can be subdivided based upon calcium-binding protein (CBP) expression, a classification scheme that may predict a neuron’s spiking properties (Kawaguchi and Kubota, 1993; Toledo-Rodriguez et al., 2004). The three CBPs, parvalbumin (PV), calbindin D-28k (CB), and calretinin (CR) are expressed in macaque V1 (DeFelipe et al., 1999; Meskenaite, 1997; Van Brederode et al., 1990). We examined the coexpression of the β2-nAChR subunit with each of these markers (Figure 2D) and found that most β2-nAChR-ir neurons in macaque V1 express PV (which in rodents is a marker for the fast-spiking [FS] neuronal phenotype; Kawaguchi and Kubota, 1993; Toledo-Rodriguez et al., 2004). β2 expression was moderate (approximately one-third of cells) in the CB neurons of layers 2, 3, 4b, 5, and 6, but very weak in CB neurons within the thalamic recipient layers 4a and 4c. PV neurons showed low to moderate β2 subunit expression across all layers. It should be noted, however, that while both PV and CB neurons express nicotinic receptors, PV neurons represent 75% of GABAergic neurons in macaque V1 and greatly outnumber CB neurons, particularly in layers 4a–4c (Van Brederode et al., 1990). CR neurons rarely expressed β2 subunits.
The Effect of Nicotine on V1 Responses In Vivo
Our fluorescence and EM data together show that β2 subunit-containing nAChRs are expressed by most TC terminals in layer 4c and by very few intrinsic V1 neurons. Additionally, mAChRs are not expressed by TC terminals. Consequently, the dominant cholinergic effect in layer 4c of V1 should be a nicotinic modulation of TC transmission. nAChRs are ionotropic cation channels; they produce a fast membrane depolarization which, at an axon terminal, would increase the probability of vesicle release. Thus, combining iontophoretically applied nicotine with visual stimulation should result in elevated neuronal responses in layer 4c. Altering the relationship between stimulus contrast and spike rate in this way would represent a form of visual gain change.
A sensitive measure of visual gain used in humans and macaques is responsiveness to contrast (see Figure S1 in the Supplemental Data available with this article online; Carrasco et al., 2000; Ling and Carrasco, 2006; McAdams and Reid, 2005; Reynolds et al., 2000; Williford and Maunsell, 2006). We measured the extracellular spiking response to optimized drifting grating stimuli (see Experimental Procedures) of increasing luminance contrast at 72 sites across the layers of V1 with and without nicotine application. In approaching these data, first we analyzed the population response in each cortical layer using a nonparametric (model-free) analysis. Second, we used a common parametric (model-based) analysis of the contrast-response relationship to determine the nature of the gain change. Gain was increased at 18 of 27 layer 4c sites by the nonparametric analysis and at 13 of 27 sites by the parametric analysis.
Nonparametric Analysis
To quantify the gain effect, we used a nonparametric measure, taking the average spike rate (across two to five repeats, with one repeat being one sequence of 12 contrast steps spanning 2%–96%, with interleaved blanks) at each stimulus contrast and summing across all 12 contrasts tested. This measure is equivalent to the area under the contrast curve and does not depend on fitting a model function to the data. The area under the contrast-response curve (or response area) at the maximum applied iontophoretic current (usually 120 nA; range 40–160 nA) was normalized to the area before nicotine ejection (baseline response area) and the results averaged by layer. By this analysis, nicotine strongly enhanced responses in layer 4c (Figure 3A) but not in other layers.
Figure 3. The Effect of Nicotine on Visual Gain in V1.

(A) The average, across neurons, of the summed contrast responses (area under the contrast-response curve) obtained at the maximum applied iontophoretic current. Data are normalized to the response obtained before any nicotine was applied. Nicotine produces an increase in gain in layer 4c but not in any other layer (number of sites in parentheses above each bar).
(B) The development of the nicotine effect in 4c across repeats of contrast-response measurement.
(C) Normalized contrast-response area was stationary in the absence of nicotine. Only data for sites enhanced by nicotine are shown in (C) (i.e., not all 4c sites as in [A] and [B]; see main text).
y axis, all graphs: normalized area. All error bars = SEM.
To identify significant effects we compared the response area at the maximum applied iontophoretic current with the response area of the immediately preceding control (no drug) condition. If the mean area with nicotine was 3 standard deviations (SD) above (or below) the mean across the control repeats, the site was deemed significantly enhanced (or suppressed). By this categorization, 22 of 72 sites (18 in 4c, 3 in unknown layers, 1 in layer 3) were enhanced and 7 sites (3 in layer 2/3, 3 in layer 5, 1 in an unknown layer) were suppressed. The remaining units showed no significant changes (43/72). Enhanced cells did not differ from unaffected layer 4c cells on any receptive field or baseline response property we measured (simple/complex, breadth of orientation tuning, direction selectivity, surround suppression). We included in our baseline measures a metric that captures the mixing of thalamic inputs carrying luminance (magnocellular pathway) and color (parvocellular pathway) information (Johnson et al., 2001). Based on a null response to an isoluminant stimulus and on responsiveness to very-low-contrast stimuli, 9 of the 18 enhanced layer 4c sites reflected a dominance of magnocellular (luminance) inputs. The nicotine effect was observed in both the magnocellular (4cα) and parvocellular (4cβ) subdivisions of layer 4c and was dynamic: there was a significant (p < 0.001; paired t test) increase in the area under the contrast curve across the first two repeats with a plateau after 48–96 s of nicotine exposure (Figure 3B). With iontophoretic currents below 100 nA, the response area increased across all repeats, suggesting that currents >100 nA may have released saturating amounts of nicotine. Contrast-response area was stationary before nicotine application (Figure 3C).
The effect of nicotine appeared at 20 nA iontophoretic current in the enhanced neurons and at 40 nA iontophoretic current in neurons that were suppressed. In both groups the effect was monotonic with increasing current, up to 120 nA (Figure 4A). Most neurons that were enhanced at the maximum current level were also significantly enhanced at lower current levels, and the population shows enhancement at the lowest levels tested (1–39 nA, usually 20 nA; Figure 4A). Neurons recovered rapidly, returning to baseline early in the first recovery run (i.e., <45 s) after each nicotine application (shown for the enhanced group in Figure 4B).
Figure 4. Nicotine Effects Were Dose Responsive and Recovered Rapidly.

(A) The averaged, normalized response area as a function of ejection current for enhanced, suppressed, and unaffected neurons.
(B) Nicotine iontophoresis was always followed by recovery runs in which the contrast-response function was measured without iontophoresis of any substance. These data come from the first recovery run after nicotine was delivered. At enhanced sites, high ejection currents often produced a recovery overshoot (below baseline), but enhancement was still seen in subsequent nicotine runs. (Note: first open square is labeled “recovery” because the order of delivery of nicotine doses varied from neuron to neuron).
All error bars = SEM.
Because our nicotine solution was at ~pH 3, control experiments were run to eliminate the possibility that the observed enhancement was a pH effect. At 38 sites (including 12 enhanced sites), combined current/pH controls were run using saline adjusted to pH 3. While there was a very weak excitatory effect of this solution on some neurons (it was suppressive in others), it could account for no more than 10% of the magnitude of the nicotine effect (Figure 5A). Enhanced neurons were not different from other neurons in their responses to pH or high-ejection currents (Figure 5B). Additionally, our preliminary data on the effects of ACh (pH 5) and carbachol (pH 6.5) on contrast responses in layer 4c reveal a similar enhancement with these closer-to-neutral pH solutions (Figure 5C). Finally, in preliminary antagonist experiments, mecamylamine (an nAChR antagonist) blocked the nicotine effect at six of eight enhanced sites (Figures 5D and 5E).
Figure 5. The Gain Enhancement in Layer 4c Is Not a pH Effect.
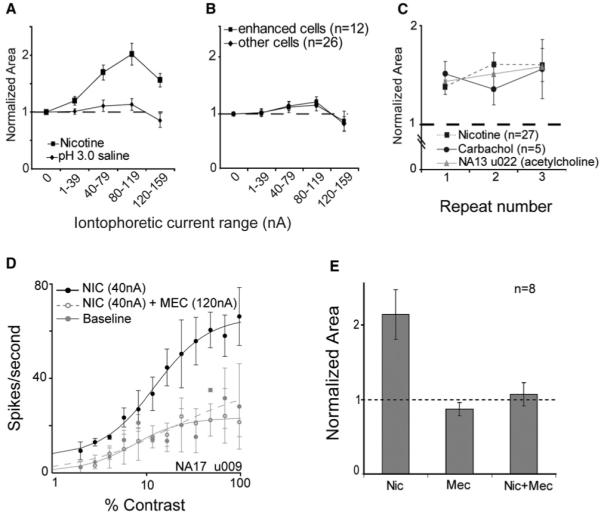
Combined current and pH controls were run for 38 (of 72) neurons, including 12 (of 22) enhanced sites. A saline solution, adjusted to pH 3.0, was ejected using the same range of currents as was used to eject nicotine. In this way the effects of pH and current are both examined. (A) For the 12 enhanced sites, the pH 3.0 saline was weakly excitatory (diamonds), but the magnitude of the effect was 10-fold smaller than that observed for nicotine (squares). The peak normalized response areas (both obtained at 80 nA) were as follows: pH 3.0 saline = 1.096, nicotine = 2.020. At very high ejection currents (>80 nA), the pH 3.0 solution was actually slightly suppressive. n = 8 for the 120–159 nA data point. In (B), it can be seen that the effect of the pH 3.0 solution on normalized response area did not differ between enhanced (squares) and unaffected (diamonds) sites. In addition, in preliminary experiments using the broad cholinergic agonists ACh (gray triangles; pH 5.0) and carbachol (circles; pH 6.5), we observed a similar enhancement in layer 4c with nonacidic solutions (C). Our preliminary antagonist studies also show that the effect of nicotine is eliminated following receptor blockade. (D) Data for an example cell in which mecamylamine was used as an nAChR antagonist. Fits were obtained for contrast-response functions under baseline conditions (filled gray circles/solid lines) and during iontophoresis of nicotine (black circles/lines; 40 nA) or nicotine (40 nA) plus mecamylamine (120 nA; open gray circles/dashed lines). The effect of nicotine was completely blocked by mecamylamine in this cell, as it was for five other cells ([E]; effect was blocked in six of eight enhanced cells; data shown is the average across all eight cells tested with mecamylamine).
Error bars = SEM for (A)–(C) and (E), SD in (D).
Increased spike rates during nicotine application could be due to changes in visually driven or spontaneous activity. To determine the level of spontaneous discharge, we summed the spike rate across the blank (no stimulus) periods that were interleaved between stimulus presentations. The difference in spike rate during blank stimuli between the nicotine and baseline conditions (before any drug was applied) was used to estimate the change in spontaneous discharge due to the drug, adopting the same mean ±3 SD criterion described above. Using this measure, 8 of 22 enhanced sites showed significant increases in background firing, and 1 showed suppression. For the eight sites with increased background firing, we subtracted the summed spike rate across the blanks from the spike rate across the stimulus presentations for both conditions to determine the effect of nicotine on the visually driven response alone. Effects on background activity, assessed in this manner, accounted for the entire nicotine enhancement in 2 (of 22) cases. Thus, 7 of 22 enhanced sites showed mixed background and driven spike rate changes (6 with increased background, 1 suppressed) and 13 of 22 showed a pure enhancement of driven activity.
Parametric Analysis
Changes in visual gain, as measured by the contrast-response function, are often assessed by a parametric (i.e., model-based) analysis, which can give insights into the mechanisms that underlie gain changes (e.g., Reynolds et al., 2000; Williford and Maunsell, 2006). To allow comparison with earlier studies, we fit the contrast responses with a model function that provides values for the slope, asymptote (maximum response rate; Rmax), half-maximum (contrast giving the half-maximum response; c50) and y-offset (spontaneous activity; sFR) of a hyperbolic ratio function (Naka-Rushton, see Experimental Procedures and Figure S1) describing the relationship between increasing contrast and spike rate. We then performed 1000 bootstrapped fits on data from the nicotine level that produced the maximum effect (see Experimental Procedures) and the immediately preceding no-drug control condition (Figure 6A) to obtain estimates of the parameters in each condition. The distributions of the parameter estimates from the nicotine and baseline conditions were compared to determine statistical significance (Figures 6B and 6C).
Figure 6. Parametric Analysis: Single-Cell Example.
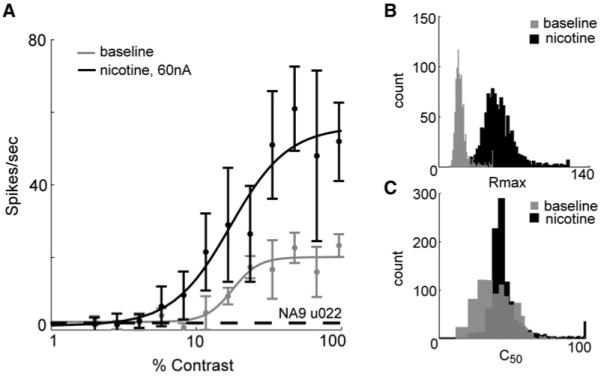
Spike responses were averaged and fit with a Naka-Rushton function (see Experimental Procedures). The parameters Rmax, c50, and n are the maximum firing rate and the contrast and slope at half-maximum, respectively; the parameter sFR captures offset attributable to background activity. (A) The fits obtained for a representative cell from layer 4c. The nicotine curve (black) is shifted to the left—indicating increased sensitivity—and shows a higher maximum response than baseline (gray). Error bars = SD. A bootstrap analysis (1000 samples) performed on data for this cell (see Experimental Procedures) shows that nicotine alters Rmax (B), but not c50 (C).
The fit value for the Rmax parameter was significantly increased at 10 sites, all in layer 4c (Figure 7A), indicating a response (or activity) gain effect of nicotine (see Figure S1). A further three sites in 4c and five sites for which histological confirmation of layer was unavailable approached significance (p < 0.10). Nicotine did not alter contrast gain; significant (p < 0.05) changes were very rare in the c50 parameter (Figure 7B; 2 of 72 sites; 1 in layer 2/3, the other in 4b). The sFR (3 of 72 sites; 2 in layer 4c, 1 in 2/3) and slope (1 layer 4c site) parameters were also unchanged by nicotine in most cases. Ten of the thirteen layer 4c sites identified by bootstrap as showing increases (p < 0.1) in Rmax were among those identified as enhanced by the nonparametric analysis above.
Figure 7. Population Data on Rmax Changes from Parametric Analysis.
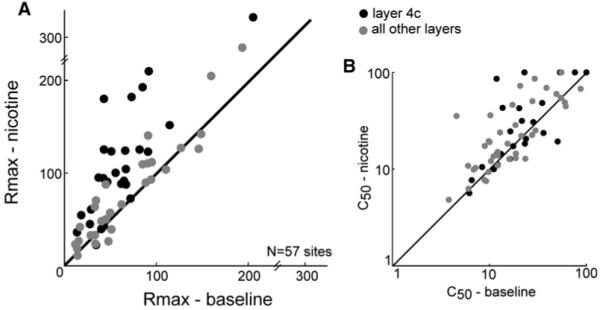
(A) A bootstrap analysis on the Rmax parameter of fits from the baseline (x axis) and nicotine (y axis) conditions shows that Rmax is elevated by nicotine at most layer 4c sites (black circles). In contrast, the data from other layers (gray circles) is clustered around the unity line. Nicotine did not alter c50 in any layer (B). Data from 15 recording sites for which histological reconstruction was not possible is not shown. None of the omitted sites showed a significant change in Rmax or c50.
Reliable Enhancement of Low-Contrast Responses with Nicotine
A feature of the effect of nicotine that is not captured by the above analyses is the emergence of reliable responses to low-contrast stimuli, indicating a reduction in contrast threshold. Figures 8A–8C show three example cells from layer 4c (all significantly altered by nicotine by both parametric and nonparametric analysis). The contrast value at which each cell’s driven rate reliably rises above background (i.e., when the error bar for that data point clears the horizontal line representing the background firing rate) is lower with nicotine for all three cells (as it is for a fourth cell, shown in Figure 6A). This is representative of the effect of nicotine across the population of enhanced cells and indicates that low-contrast detection is improved with nicotine. Figures 8A–8C also illustrate that this gain effect would best be described as a multiplicative (as opposed to an additive) response gain for most cells, supported by the lack of changes in the c50 or sFR parameters. A small number of cells showed evidence of an additive activity gain or a mixed activity/response gain. InFigure 8D, data for neurons showing a significant change in Rmax (all in layer 4c) have been normalized, averaged, and fit to show the population response to nicotine versus control—the increased sensitivity and responsiveness are clearly evident.
Figure 8. Nicotine Lowers Detection Thresholds for Neurons in Layer 4c.
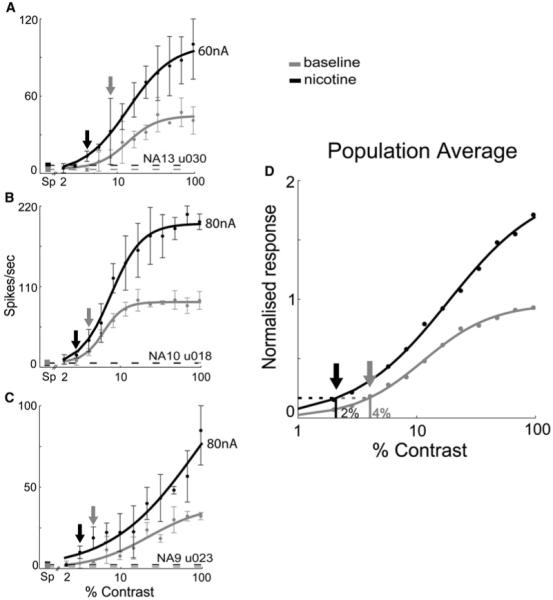
Fits are shown for three cells in layer 4c (A–C) and for the population of cells that showed Rmax changes (D) with nicotine data in black and baseline data in gray. Spontaneous firing rates in (A)–(C) are indicated by the filled squares at the far left of each graph (“Sp” on the x axis) and by the dashed lines extending from the squares. For all three cells, the contrast at which the nicotine response reliably rises above the threshold set by the background activity (black arrows, set to the first point for which the error bars clear the dashed line) is lower than the corresponding threshold contrast under baseline conditions. Error bars = SD. In (D), data from the neurons that showed changes in Rmax (p < 0.1) have been normalized to the maximum firing rate obtained under baseline conditions, averaged, and fit. Spontaneous activity was not significantly altered in these neurons and the normalized spontaneous rate has thus been averaged across both conditions—the value shown is this mean plus 3 SD (dashed line). The threshold contrast defined by this level of background activity is lower (~2% contrast) with nicotine than under baseline conditions (~4%). Closer inspection reveals that this is the case regardless of the chosen value for threshold contrast.
DISCUSSION
We have shown that in macaque V1, β2-nAChRs are found presynaptically at TC synapses onto spines, but not at TC synapses onto dendritic shafts. Furthermore, very few neurons intrinsic to V1 express β2-nAChR subunits. Of the V1 neurons that do express β2-nAChRs, most are GABAergic neurons expressing either CB or PV. Consistent with our anatomical data, we have also demonstrated in vivo that nicotine reliably enhances the gain of responses to visual stimuli in layer 4c, but not in other layers. As a consequence, through enhanced TC transmission, nicotine enhances detection of visual stimuli.
Nicotinic Modulation of TC Synapses and Inhibition
Our anatomical data are consistent with and extend those from previous studies. While there is no prior quantitative data, it has been suggested previously that among the neurons in the rodent cortex, high-affinity nAChRs are selectively expressed by inhibitory interneurons (Metherate, 2004). We show that in layers 2–6 of macaque V1, 75% of the cells expressing nAChRs also express GABA. This would predict an excitatory effect of nicotine on some inhibitory neurons, particularly those expressing CB or PV. In rodents, inhibitory interneurons are often subdivided into two classes, FS and non-FS neurons. FS cells express PV, and some non-FS cells express CB (Kawaguchi and Kubota, 1993). Non-FS interneurons recorded in vitro have been shown to be exited by nicotine (Christophe et al., 2002; Gulledge et al., 2007; Xiang et al., 1998). These data are consistent with our observation that CB-ir neurons express β2-nAChRs. We also found that a distinct subset of PV-ir neurons expresses β2-nAChRs. This is interesting, given previous studies of FS (PV-ir) neurons in rodents, which report either no cholinergic modulation (Gulledge et al., 2007; Kawaguchi, 1997) or a solely muscarinic modulation (Xiang et al., 1998). It may be that FS cells as a group respond heterogeneously to cholinergic agonists, but also that an as yet unidentified, anatomically specified subgroup of FS cells express β2-nAChRs. It will be a goal of future research to determine the firing pattern and neuronal geometry of our dually labeled β2-nAChR/PV interneurons.
While the localization of high-affinity nAChRs to thalamic afferents in cortical input layers has been suggested previously based on receptor autoradiography combined with thalamic lesions (Lavine et al., 1997; Parkinson et al., 1988), these studies could not resolve the localization or numerosity of layer 4 nAChRs. Our EM study directly verifies the localization of β2-nAChRs on afferent terminals in monkeys. Importantly, by using EM we were also able to show that β2-nAChRs are found at the thalamic input onto spines, but not onto dendritic shafts. Inhibitory neurons are sparsely spiny or aspiny and receive excitatory inputs directly to the dendritic shaft. Thus, the population of TC synapses onto dendritic shafts observed in this study probably contains within it (if it is not entirely comprised of) thalamic input to inhibitory neurons. This selective expression indicates the potential for nicotine to strengthen the ascending drive to excitatory over inhibitory thalamorecipient neurons.
We also found that terminal segments of thalamic axons did not express mAChRs. We did, however, observe a small population of TC axons (i.e., nonterminal segments) in layer 4cβ expressing m2 AChRs. In this subdivision of layer 4c, the effect of cholinergic modulation on TC transmission may not be completely captured due to our use of a selective nicotinic agonist. The presence of m2 AChRs in layer 4cβ has been reported previously (Mrzljak et al., 1996), in an EM study that did not employ a method for distinguishing TC from cortical axons. Our data support the conclusion by Mrzljak et al. that ACh may suppress activity in the parvocellular visual channel (which arrives in layer 4cβ).
Nicotinic Gain Control in V1
Our physiology experiments show that nicotine enhances visual gain in layer 4c in a manner that decreases contrast threshold and increases overall responsiveness, and that nicotine produces suppressive effects outside layer 4c. A putatively presynaptic nicotinic mechanism that protects ascending input to cortex from cholinergic suppression has been shown previously in slice preparations of rodent somatosensory (Gil et al., 1997), visual (Kimura et al., 1999), auditory (Hsieh et al., 2000) and prefrontal (Lambe et al., 2005) cortices. These studies demonstrate that nicotine increases the amplitude of EPSPs arising from stimulation of ascending pathways, but has no effect on EPSPs in corticocortical pathways. What cannot be determined from in vitro intracellular recordings is how (or whether) this increased EPSP amplitude, demonstrated for a brief train of electrical stimuli, will translate into altered spiking responses to a sensory stimulus. Our data thus represent a step forward in understanding the effect of nicotinic modulation on sensory processing. We show that activating nAChRs in layer 4c of V1 strengthens the input from the visual world sufficiently to allow neurons to reliably respond to low-contrast stimuli, which can support better stimulus detection.
From layer 4c, these effects should propagate throughout the visual cortex, because the major ascending visual input in primates is via layer 4c. Furthermore, effects within single neurons in 4c may become more pronounced where the receptive fields of multiple V1 neurons are summed. A first step toward understanding the nature of that propagation would involve separate recording and iontophoresis electrodes to allow activation of nicotinic receptors in layer 4c while recordings are made in other layers.
Nicotinic Suppression outside Layer 4c
The nicotinic suppression we demonstrate in layers 2, 3, and 5 is also consistent with the anatomical data we have presented. In the abovementioned in vitro slice studies, cholinergic suppression of corticocortical transmission was reported, but was shown to be a muscarinic effect. The effect we observe is most likely due to activation of inhibitory neurons, as our anatomical data show that very few principal cells in V1 express nAChRs.
Contribution of Low-Affinity nAChRs
While most cortical high-affinity nAChRs incorporate β2 subunits, there is also a population of α-bungarotoxin-sensitive nAChRs in macaque V1. These receptors are homomers of α7 nicotinic subunits and are interesting because they pass calcium ions with a high affinity (relative to sodium ions). Receptor autoradiography has shown that α7-nAChRs are present as a band in layer 4c of macaque V1 (Han et al., 2003). It is not known whether they are expressed on TC afferents or by intrinsic V1 neurons. Unfortunately, available antibodies directed against the α7 subunit did not pass our control experiments for use in macaque. The extent to which α7-nAChRs contribute to the effects we obtained using nicotine as an agonist in V1 remains unresolved. They have a 5-fold lower affinity for (and desensitize more rapidly to) nicotine, compared with β2-nAChRs (in heterologous expression systems; Fenster et al., 1997). However, this lower affinity may mean that only a subset of α7-nAChRs is in a desensitized state at any one time, and so these receptors may have contributed—alongside the β2 subunit-containing nAChRs—to the increased visual gain we observed. The strong expression of α7-nAChRs in layer 4c, however, reinforces the importance of this layer as a site for nAChR-mediated cholinergic modulation.
Desensitization of Nicotinic Receptors
High-affinity nAChRs also desensitize to agonist exposure, although variation in the extent to which native nAChRs desensitize (including nondesensitizing responses) has been reported (Dani et al., 2000; Lambe et al., 2005). It is interesting that such a process is not apparent in our data. It is unlikely that the enhancement we observed is itself a result of desensitization; elevated spiking due to receptor desensitization would require the loss of a normally tonic inhibitory effect. As nAChRs are cation channels, such tonic inhibition would have to be mediated by inhibitory neurons. Our immunofluorescence data show that inhibitory neurons express nAChRs in all layers of V1, but enhancement was seen only in layer 4c. That an effect specific to layer 4c (nicotinic enhancement) is the result of an anatomical feature also specific to 4c (thalamic terminal expression of nAChRs) is the more parsimonious explanation. Additionally, recovery from desensitization to agonist exposure of >1 min takes place over minutes (Reitstetter et al., 1999), while the responses we observed recovered in under 45 s.
Natural Cholinergic Modulation
The nicotinic gain effects we demonstrate physiologically arise from local agonist application on a background of what may be unusually low basal ACh release (given the anaesthetized state). Natural release in the awake animal probably differs from iontophoretic application in two regards: ACh is released continuously (Jimenez-Capdeville and Dykes, 1996) and more broadly. The extent of cortical neuropil covered by a single basal forebrain afferent in the macaque is not known, but in the rodent each axon innervates a region approximately 1–1.5 mm in diameter (Price and Stern, 1983).
ACh will also activate both nAChRs and mAChRs. nAChRs are ionotropic, so for a brief window (<200 ms) after an increase in local ACh, nicotinic effects will probably dominate over the slower metabotropic effects mediated by muscarinic receptors. In rodent somatosensory cortex in vitro, the net effect of the nicotinic enhancement at TC synapses and muscarinic suppression at cortico-cortical synapses is that ACh leaves TC inputs relatively enhanced on a background of suppressed intracortical transmission (Gil et al., 1997).
In vivo, locally applied ACh has mixed effects on visual responses in cat area 17 (Sato et al., 1987b; Sillito and Kemp, 1983) and in marmoset and macaque V1 (Roberts et al., 2005; Zinke et al., 2006). Previous studies do report neurons that show an enhanced response with ACh, with, interestingly, a larger proportion of neurons being enhanced in cat versus monkey. The only previous attempt at pharmacological dissection of cholinergic effects in visual cortex (Sato et al., 1987b) concluded that the facilitatory effects of ACh are mediated by mAChRs and not nAChRs. However, this study was performed using halothane anesthesia, which antagonizes nAChRs (Mori et al., 2001), making interpretation of their results difficult.
It seems likely that natural ACh release would produce a more modest increase in spike rate than what we have observed, due to the rapid and effective nature of acetylcholinesterase, the higher tonic ACh release in the awake animal (above which phasic release may represent a smaller increment), and concurrent suppression via activation of m2 AChRs on some thalamic afferents and on some synapses between pyramidal neurons (Levy et al., 2006). Interneurons in macaque V1 also strongly express mAChRs (Disney et al., 2006), but whether the net effect of ACh acting via mAChRs on inhibitory neurons will be an increase or decrease in activity will depend on the effectors for the activated G-proteins within each interneuron population.
ACh and Attention
In rodent, the cholinergic system is believed to be central to the mechanisms underlying attention (Sarter et al., 2005). Interestingly, cholinergic neuromodulation is—to our knowledge—not invoked as a candidate mechanism for visual attention in primates, although similarities in the effects of ACh and of visual attention have been noted by another group working with V1 (Roberts et al., 2005; Zinke et al., 2006). Generally, glutamatergic feedback from higher visual areas, visuomotor areas, or both is the preferred underlying model circuit for visual spatial attention in the primate (Desimone and Duncan, 1995; Reynolds and Chelazzi, 2004). Jack et al. (2006) extend this model and conclude that while glutamatergic feedback probably underlies spatial attention, modulation by ACh, norepinephrine, or both may play a role in sensory gating.
Just how restricted the region of cortex influenced by ACh can be will determine whether cholinergic effects finally fall in a domain more properly called “arousal,” or whether they will be found capable of subserving more specific processes. At some point the distinction between arousal and attention becomes difficult to operationally define. Too little is currently known about the physiology of the BFA or about the anatomy of the BFA projection to cortex to resolve these questions.
Data often cited in favor of a glutamatergic mechanism for attention is the finding that frontal eye field (FEF) stimulation alters visual receptive field properties and improves attentional performance in monkeys (Armstrong et al., 2006; Moore and Fallah, 2004). Interestingly, the FEF, in addition to projecting down through the visual areas, also sends a direct projection to the BFA (Russchen et al., 1985). It has also been shown in rodents that activating glutamate receptors in the prefrontal cortex causes ACh release in the posterior parietal cortex (Nelson et al., 2005). Perhaps the FEF (and other frontal and prefrontal areas) coactivates both glutamatergic and cholinergic pathways leading back to the sensory cortices, which act in concert to generate attentional effects.
Overall, attentional effects on single units recorded in visual cortex (McAdams and Reid, 2005; Reynolds et al., 2000; Williford and Maunsell, 2006) and those induced by nicotine in layer 4c are remarkably similar, although the effects in the awake animal, particularly in V1, are far more modest—which we would expect with endogenous ACh release (see above subsection, “Natural Cholinergic Modulation”). Our data clearly demonstrate that ACh, acting through nAChRs, serves to enhance the sensory drive to early visual cortex in the macaque. Given the demonstrated role for ACh in mechanisms of attention in rodents, a role for cholinergic neuromodulation in models of spatial attention in the visual system of the primate—subserved in part by nicotinic gain control in layer 4c of V1—should be considered and investigated further.
EXPERIMENTAL PROCEDURES
Brief methods are outlined below. For details see Supplementary Methods.
Animals
Adult male macaca fascicularis were used in these experiments. Procedures were approved by the Institutional Care and Use Committee for NYU, in accordance with NIH guidelines.
Immuno-EM
Tracer Injection Surgery
Tracer injections were made in three anaesthetized animals. Three injection tracks (three deposits/track; nine total) were made per animal, at 8 mm antero-posterior (AP)/11 mm medio-lateral (ML) (with respect to interaural zero), 9 mm AP/11 mm ML, and posterior to the central sulcus at 11 mm ML. Following craniotomy and dural reflection, 0.25 μl deposits of 10% biotinylated dextran-amine (3000 MW, BioDesign) and 10% micro-ruby (BioDesign) in sterile saline were made by Hamilton syringe at depths of 27, 26, and 25 mm below the pial surface. The dura and bone plug were then replaced and “sealed” using agar. After ~96 hr of unrelated physiological recording (Solomon et al., 2004), a dose of 60 mg/kg pentobarbital was delivered, resulting in a flat EEG.
Histological Preparation
Animals were exsanguinated and then transcardially perfused with 4 liters of 4% paraformaldehyde (PFA) with 0.25% glutaraldehyde (GLA) in 0.1 M PB. V1 and the LGN were removed and post-fixed at 4°C in 4% PFA. Within 48 hr, the blocks were sectioned at 40 μm and reacted for 30 min in 1% sodium borohydride in PB. Every third V1 section was set aside as a cytochrome-oxidase (CO) reference for determining layers. CO histochemistry was commenced <72 hr after perfusion, as described elsewhere (Disney et al., 2006).
‘Double DAB’ Procedure for Light Microscopic Identification of Thalamic Injection Sites and V1 Terminal Fields
All LGN sections and every second V1 section were incubated in 1% hydrogen peroxide in 0.01 M PBS for 30 min to block endogenous peroxidases. They were then placed into an avidin-HRP complex (Vectastain Elite ABC Kit, Vector Laboratories) with 0.3% Triton x-100 overnight at room temperature (RT). Sections were then reacted for 15 min using the ABC-DAB technique (Hsu et al., 1981). After rinsing, the V1 set was placed into fresh avidin-HRP solution for 3 hr and the ABC-DAB reaction was repeated. All sections were mounted and dried overnight before a 10 min, on-slide exposure to 0.1% osmium tetroxide in PB to enhance staining.
Single-Label Immunocytochemistry for EM
Using the above V1 ABC-DAB reference set, sequential sections with labeled thalamic terminal fields were selected. A dimethylsulfoxide “freeze-thaw” technique (Wouterlood and Jorritsma-Byham, 1993) was used to improve antibody penetration. After blocking endogenous peroxidases (see preceding subsection), sections were incubated in 1% IgG-free bovine serum albumin (BSA, Molecular Probes) with 0.05% sodium azide (Sigma), 0.04% Triton x-100 (Triton) and 0.1% Photoflo (Kodak) in PBS for 30 min.
Primary antibodies were diluted in PBS with 1% BSA and 0.05% sodium azide (Standard Blocking Solution). Added to this buffer were a rabbit anti-β2 nicotinic subunit (45 μg/mL; BioDesign), a rabbit anti-m1AChR (1:200; Chemicon) or a rat anti-m2AChR (1:250; Chemicon). Free-floating sections were incubated in one primary antibody for 72 hr at RT.
Silver-intensified immunogold was used for visualization. After the primary antibody incubation, sections were incubated overnight at RT in a 0.8 nm gold-conjugated secondary antibody (goat anti-rabbit IgG or goat anti-mouse IgG; Aurion), diluted 1:100 in Standard Blocking Solution. Sections were then post-fixed with 2% GLA in PBS for 10 min and, after brief washes in 0.2 M citrate buffer (pH 6.0) to remove phosphate buffer salts, the gold particles were enhanced for 5 to 12 min using the Amersham IntenSE silver kit.
Following autometallography, sections were processed to visualize the tract-tracer using the double DAB procedure described above, omitting Triton x-100 in the overnight incubation. The now dually labeled sections were fixed with 0.5% osmium tetroxide in PB for 30 min, followed by dehydration in 50% ethanol and an overnight incubation in 4% uranyl acetate in 70% ethanol at 4°C. The next day the dehydration series proceeded through 70%, 90%, and 100% ethanol and three 30 min acetone rinses before overnight infiltration of 1:1 EMBED 812 (EM Sciences)/acetone and then 100% EMBED 812. Following embedding and curing in EPON capsules, the tissue was resectioned at 80–90 nm, mounted on formvar-coated nickel grids, and counterstained with Reynold’s lead citrate before inspection under a JEOL 1200 XL transmission electron microscope.
Data Collection
Using tissue maps drawn by camera lucida, data were collected from thalamic terminal fields near the tissue/EPON interface. Images were taken at 20,000–40,000× magnification using either film or a Hamamatsu CCD camera controlled by ATM software. Analysis was undertaken offline.
Immunofluorescence
Tissue Preparation
Seven animals were perfused, and visual cortex was blocked, removed, post-fixed, sectioned, and stored as described above, following unrelated physiology experiments.
GABA/β2 Subunit Dual Immunofluorescence
Three animals were used for this experiment. Following freeze-thawing, random sections were blocked in Standard Blocking Solution and exposed to 2 antibodies in a single 72 hr, coincubation step at RT. The polyclonal rabbit anti-β2 nicotinic subunit antibody (described above) was combined with a monoclonal mouse anti-GABA (1:100; Sigma).
Secondary antibodies were diluted 1:50 in Standard Blocking Solution. GABA-ir sites were visualized using Alexa 594 chicken anti-mouse IgG (Molecular Probes) and nAChR-ir sites with Alexa 488 chicken anti-rabbit IgG (Molecular Probes). This incubation proceeded in the dark, at RT, for 4–6 hr. Sections were then rinsed, mounted, coverslipped, and stored in the dark at 4°C.
Confocal Microscopy
Image montages from pia to white matter were taken of the opercular surface of V1 (2°–8° parafoveal visual field representation) using a Zeiss LSM 310 confocal microscope. Counting was undertaken offline.
CBP/nAChR Dual Immunofluorescence
Four animals were used for this experiment. The protocol followed was identical to that described for GABA/nAChR dual-labeling, with the exception that 0.3% Triton x-100 (Triton) was added to the blocking solution in place of the freeze-thaw process. CBPs were detected using antibodies directed against PV (goat anti-PV 1:5000; Swant), CB (mouse anti-CB D-28K 1:5000; Swant) and CR (goat anti-CR, 1:1000; Swant).
Physiological Recordings
Recordings of single or multiple units (generally <3 cells) were made in anaesthetized (sufentanil citrate; 6–24 μg), paralyzed (vencuronium bromide) macaques with a carbon fiber iontophoresis/physiology electrode (Carbostar, Kation Scientific). The signal from the fiber was amplified differentially (Dagan, Minnesota) with band-pass filtering (300 Hz to 10 kHz) and digitized using an A/D board (SGI). Spikes were discriminated and time-stamped by custom software running on a Silicon Graphics computer. Spike waveforms were selected using a discriminator window and stored for offline analysis.
Nicotine (0.25 M), sodium chloride (0.25 M), mecamylamine (0.1 M), and Chicago Sky Blue 6B (2%), all dissolved in water, were delivered via multibarrel pipettes (tip diameter 1–2 μm) connected to a Dagan multichannel pump. Ten nano-Amp holding currents were used to prevent leak.
After quantitative measures of orientation, spatial frequency, temporal frequency, and area tuning using drifting grating stimuli were made (Xing et al., 2005), a drifting grating optimized for these parameters was selected and contrast responses were measured in 12 logarithmic steps from 2%–96% contrast, with or without ejection of drug solutions. Drug ejection preceded visual stimulation by 10 s and never exceeded 90 s without recovery (minimum 5 min). A baseline contrast response was recorded before each ejection period and 2 or 3 “recovery” contrast responses were recorded after drug application. This series of recordings was repeated with varying ejection currents up to a limit of 160 nA.
After physiological recording, animals were sacrificed and perfused, and blocks of V1 were removed and sectioned. Laminar track reconstruction was aided by CO histochemistry with Nissl counterstaining.
Parametric Analysis
For each condition (baseline, nicotine and/or mecamylamine at varying ejection currents, recovery and controls), responses (R) were averaged across 2–5 repeats of the sequence of stimulus contrasts (C). A Naka-Rushton function,
was fit to these data, and the parameters Rmax, c50, and n were obtained, along with a parameter (sFR) capturing the offset attributable to spontaneous firing.
The nicotine ejection current at which the maximal change in these fit parameters was observed was determined and a parametric boot-strap was performed by sampling randomly, with replacement, from a Gaussian distribution defined by the mean and standard deviation of the response to each contrast. Each set of sample responses was fit with a Naka-Rushton function. One thousand iterations were performed to obtain estimates of the parameter distributions. Statistical analyses of the difference between the nicotine condition and its baseline were conducted for each fit parameter.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Lennie for animals and lab facilities for tracer injections; V. Mahadomrongkul, C. Farb, C. Henry, S. Joshi, P. Williams, D. Xing, and A. Henrie for assistance with experiments; and E. Goldberg and M. Schiff for helpful comments on the manuscript. This work was supported by R01-NS41091 and R01-EY13145 to C.A.; EY8300 and EY15549 to M.J.H.; and core grant P30-EY13079.
Footnotes
Supplemental Data
The Supplemental Data for this article can be found online at http://www.neuron.org/cgi/content/full/56/4/701/DC1/.
REFERENCES
- Aoki C, Kabak S. Cholinergic terminals in the cat visual cortex: ultrastructural basis for interaction with glutamate-immunoreactive neurons and other cells. Vis. Neurosci. 1992;8:177–191. doi: 10.1017/s0952523800002832. [DOI] [PubMed] [Google Scholar]
- Armstrong KM, Fitzgerald JK, Moore T. Changes in visual receptive fields with microstimulation of frontal cortex. Neuron. 2006;50:791–798. doi: 10.1016/j.neuron.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Beaulieu C, Somogyi P. Enrichment of cholinergic synaptic terminals on GABAergic neurons and coexistence of immunoreactive GABA and choline acetyltransferase in the same synaptic terminals in the striate cortex of the cat. J. Comp. Neurol. 1991;304:666–680. doi: 10.1002/cne.903040412. [DOI] [PubMed] [Google Scholar]
- Beaulieu C, Kisvarday Z, Somogyi P, Cynader M, Cowey A. Quantitative distribution of GABA-immunopositive and -immunonegative neurons and synapses in the monkey striate cortex (area 17) Cereb. Cortex. 1992;2:295–309. doi: 10.1093/cercor/2.4.295. [DOI] [PubMed] [Google Scholar]
- Brown DA, Abogadie FC, Allen TG, Buckley NJ, Caulfield MP, Delmas P, Haley JE, Lamas JA, Selyanko AA. Muscarinic mechanisms in nerve cells. Life Sci. 1997;60:1137–1144. doi: 10.1016/s0024-3205(97)00058-1. [DOI] [PubMed] [Google Scholar]
- Carrasco M, Penpeci-Talgar C, Eckstein M. Spatial covert attention increases contrast sensitivity across the CSF: support for signal enhancement. Vision Res. 2000;40:1203–1215. doi: 10.1016/s0042-6989(00)00024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophe E, Roebuck A, Staiger JF, Lavery DJ, Charpak S, Audinat E. Two types of nicotinic receptors mediate an excitation of neocortical layer I interneurons. J. Neurophysiol. 2002;88:1318–1327. doi: 10.1152/jn.2002.88.3.1318. [DOI] [PubMed] [Google Scholar]
- Dani JA, Radcliffe KA, Pidoplichko VI. Variations in desensitization of nicotinic acetylcholine receptors from hippocampus and midbrain dopamine areas. Eur. J. Pharmacol. 2000;393:31–38. doi: 10.1016/s0014-2999(00)00003-0. [DOI] [PubMed] [Google Scholar]
- DeFelipe J, Gonzalez-Albo MC, Del Rio MR, Elston GN. Distribution and patterns of connectivity of interneurons containing calbindin, calretinin, and parvalbumin in visual areas of the occipital and temporal lobes of the macaque monkey. J. Comp. Neurol. 1999;412:515–526. doi: 10.1002/(sici)1096-9861(19990927)412:3<515::aid-cne10>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Desimone R, Duncan J. Neural mechanisms of selective visual attention. Annu. Rev. Neurosci. 1995;18:193–222. doi: 10.1146/annurev.ne.18.030195.001205. [DOI] [PubMed] [Google Scholar]
- Disney AA, Domakonda K, Aoki C. Differential expression of muscarinic acetylcholine receptors across excitatory and inhibitory cells in visual cortical areas V1 and V2 of the macaque monkey. J. Comp. Neurol. 2006;499:49–63. doi: 10.1002/cne.21096. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Central cholinergic systems and cognition. Annu. Rev. Psychol. 1997;48:649–684. doi: 10.1146/annurev.psych.48.1.649. [DOI] [PubMed] [Google Scholar]
- Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA. Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J. Neurosci. 1997;17:5747–5759. doi: 10.1523/JNEUROSCI.17-15-05747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher M, Colombo PJ. Ageing: the cholinergic hypothesis of cognitive decline. Curr. Opin. Neurobiol. 1995;5:161–168. doi: 10.1016/0959-4388(95)80022-0. [DOI] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Park SB, Kawaguchi Y, Stuart GJ. Heterogeneity of phasic cholinergic signaling in neocortical neurons. J. Neurophysiol. 2007;97:2215–2229. doi: 10.1152/jn.00493.2006. [DOI] [PubMed] [Google Scholar]
- Han ZY, Le Novere N, Zoli M, Hill JA, Champtiaux N, Changeux JP. Localization of nAChR subunit mRNAs in the brain of Macaca mulatta. Eur. J. Neurosci. 2000;12:3664–3674. doi: 10.1046/j.1460-9568.2000.00262.x. [DOI] [PubMed] [Google Scholar]
- Han ZY, Zoli M, Cardona A, Bourgeois JP, Changeux JP, Le Novere N. Localization of [3H]nicotine, [3H]cytisine, [3H]epibatidine, and [125I]alpha-bungarotoxin binding sites in the brain of Macaca mulatta. J. Comp. Neurol. 2003;461:49–60. doi: 10.1002/cne.10659. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, McGaughy J. High acetylcholine levels set circuit dynamics for attention and encoding and low acetylcholine levels set dynamics for consolidation. Prog. Brain Res. 2004;145:207–231. doi: 10.1016/S0079-6123(03)45015-2. [DOI] [PubMed] [Google Scholar]
- Hsieh CY, Cruikshank SJ, Metherate R. Differential modulation of auditory thalamocortical and intracortical synaptic transmission by cholinergic agonist. Brain Res. 2000;880:51–64. doi: 10.1016/s0006-8993(00)02766-9. [DOI] [PubMed] [Google Scholar]
- Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem. Cytochem. 1981;29:577–580. doi: 10.1177/29.4.6166661. [DOI] [PubMed] [Google Scholar]
- Jack AI, Shulman GL, Snyder AZ, McAvoy M, Corbetta M. Separate modulations of human V1 associated with spatial attention and task structure. Neuron. 2006;51:135–147. doi: 10.1016/j.neuron.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Jasper HH, Tessier J. Acetylcholine liberation from cerebral cortex during paradoxical (REM) sleep. Science. 1971;172:601–602. doi: 10.1126/science.172.3983.601. [DOI] [PubMed] [Google Scholar]
- Jimenez-Capdeville ME, Dykes RW. Changes in cortical acetylcholine release in the rat during day and night: differences between motor and sensory areas. Neuroscience. 1996;71:567–579. doi: 10.1016/0306-4522(95)00439-4. [DOI] [PubMed] [Google Scholar]
- Johnson EN, Hawken MJ, Shapley R. The spatial transformation of color in the primary visual cortex of the macaque monkey. Nat. Neurosci. 2001;4:409–416. doi: 10.1038/86061. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Selective cholinergic modulation of cortical GABAergic cell subtypes. J. Neurophysiol. 1997;78:1743–1747. doi: 10.1152/jn.1997.78.3.1743. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. Correlation of physiological subgroupings of nonpyramidal cells with parvalbumin- and calbindinD28k-immunoreactive neurons in layer V of rat frontal cortex. J. Neurophysiol. 1993;70:387–396. doi: 10.1152/jn.1993.70.1.387. [DOI] [PubMed] [Google Scholar]
- Kimura F, Fukuda M, Tsumoto T. Acetylcholine suppresses the spread of excitation in the visual cortex revealed by optical recording: possible differential effect depending on the source of input. Eur. J. Neurosci. 1999;11:3597–3609. doi: 10.1046/j.1460-9568.1999.00779.x. [DOI] [PubMed] [Google Scholar]
- Lambe EK, Olausson P, Horst NK, Taylor JR, Aghajanian GK. Hypocretin and nicotine excite the same thalamocortical synapses in prefrontal cortex: correlation with improved attention in rat. J. Neurosci. 2005;25:5225–5229. doi: 10.1523/JNEUROSCI.0719-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine N, Reuben M, Clarke PB. A population of nicotinic receptors is associated with thalamocortical afferents in the adult rat: laminal and areal analysis. J. Comp. Neurol. 1997;380:175–190. doi: 10.1002/(sici)1096-9861(19970407)380:2<175::aid-cne3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Levy RB, Reyes AD, Aoki C. Nicotinic and muscarinic reduction of unitary excitatory postsynaptic potentials in sensory cortex; dual intracellular recording in vitro. J. Neurophysiol. 2006;95:2155–2166. doi: 10.1152/jn.00603.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S, Carrasco M. Sustained and transient covert attention enhance the signal via different contrast response functions. Vision Res. 2006;46:1210–1220. doi: 10.1016/j.visres.2005.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, Evrard A, Cazala P, Cormier A, Mameli-Engvall M, et al. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–107. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- McAdams CJ, Reid RC. Attention modulates the responses of simple cells in monkey primary visual cortex. J. Neurosci. 2005;25:11023–11033. doi: 10.1523/JNEUROSCI.2904-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meskenaite V. Calretinin-immunoreactive local circuit neurons in area 17 of the cynomolgus monkey, Macaca fascicularis. J. Comp. Neurol. 1997;379:113–132. [PubMed] [Google Scholar]
- Metherate R. Nicotinic acetylcholine receptors in sensory cortex. Learn. Mem. 2004;11:50–59. doi: 10.1101/lm.69904. [DOI] [PubMed] [Google Scholar]
- Moore T, Fallah M. Microstimulation of the frontal eye field and its effects on covert spatial attention. J. Neurophysiol. 2004;91:152–162. doi: 10.1152/jn.00741.2002. [DOI] [PubMed] [Google Scholar]
- Mori T, Zhao X, Zuo Y, Aistrup GL, Nishikawa K, Marszalec W, Yeh JZ, Narahashi T. Modulation of neuronal nicotinic acetylcholine receptors by halothane in rat cortical neurons. Mol. Pharmacol. 2001;59:732–743. doi: 10.1124/mol.59.4.732. [DOI] [PubMed] [Google Scholar]
- Mrzljak L, Levey AI, Rakic P. Selective expression of m2 muscarinic receptor in the parvocellular channel of the primate visual cortex. Proc. Natl. Acad. Sci. USA. 1996;93:7337–7340. doi: 10.1073/pnas.93.14.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller CM, Singer W. Acetylcholine-induced inhibition in the cat visual cortex is mediated by a GABAergic mechanism. Brain Res. 1989;487:335–342. doi: 10.1016/0006-8993(89)90837-8. [DOI] [PubMed] [Google Scholar]
- Murphy PC, Sillito AM. Cholinergic enhancement of direction selectivity in the visual cortex of the cat. Neuroscience. 1991;40:13–20. doi: 10.1016/0306-4522(91)90170-s. [DOI] [PubMed] [Google Scholar]
- Nelson CL, Sarter M, Bruno JP. Prefrontal cortical modulation of acetylcholine release in posterior parietal cortex. Neuroscience. 2005;132:347–359. doi: 10.1016/j.neuroscience.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Parkinson D, Kratz KE, Daw NW. Evidence for a nicotinic component to the actions of acetylcholine in cat visual cortex. Exp. Brain Res. 1988;73:553–568. doi: 10.1007/BF00406614. [DOI] [PubMed] [Google Scholar]
- Pearson RC, Gatter KC, Brodal P, Powell TP. The projection of the basal nucleus of Meynert upon the neocortex in the monkey. Brain Res. 1983;259:132–136. doi: 10.1016/0006-8993(83)91075-2. [DOI] [PubMed] [Google Scholar]
- Price JL, Stern R. Individual cells in the nucleus basalis–diagonal band complex have restricted axonal projections to the cerebral cortex in the rat. Brain Res. 1983;269:352–356. doi: 10.1016/0006-8993(83)90145-2. [DOI] [PubMed] [Google Scholar]
- Reitstetter R, Lukas RJ, Gruener R. Dependence of nicotinic acetylcholine receptor recovery from desensitization on the duration of agonist exposure. J. Pharmacol. Exp. Ther. 1999;289:656–660. [PubMed] [Google Scholar]
- Reynolds JH, Chelazzi L. Attentional modulation of visual processing. Annu. Rev. Neurosci. 2004;27:611–647. doi: 10.1146/annurev.neuro.26.041002.131039. [DOI] [PubMed] [Google Scholar]
- Reynolds JH, Pasternak T, Desimone R. Attention increases sensitivity of V4 neurons. Neuron. 2000;26:703–714. doi: 10.1016/s0896-6273(00)81206-4. [DOI] [PubMed] [Google Scholar]
- Rezvani AH, Levin ED. Cognitive effects of nicotine. Biol. Psychiatry. 2001;49:258–267. doi: 10.1016/s0006-3223(00)01094-5. [DOI] [PubMed] [Google Scholar]
- Roberts MJ, Zinke W, Guo K, Robertson R, McDonald JS, Thiele A. Acetylcholine dynamically controls spatial integration in marmoset primary visual cortex. J. Neurophysiol. 2005;93:2062–2072. doi: 10.1152/jn.00911.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russchen FT, Amaral DG, Price JL. The afferent connections of the substantia innominata in the monkey, Macaca fascicularis. J. Comp. Neurol. 1985;242:1–27. doi: 10.1002/cne.902420102. [DOI] [PubMed] [Google Scholar]
- Sarter M, Hasselmo ME, Bruno JP, Givens B. Unraveling the attentional functions of cortical cholinergic inputs: interactions between signal-driven and cognitive modulation of signal detection. Brain Res. Brain Res. Rev. 2005;48:98–111. doi: 10.1016/j.brainresrev.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Sato H, Hata Y, Hagihara K, Tsumoto T. Effects of cholinergic depletion on neuron activities in the cat visual cortex. J. Neurophysiol. 1987a;58:781–794. doi: 10.1152/jn.1987.58.4.781. [DOI] [PubMed] [Google Scholar]
- Sato H, Hata Y, Masui H, Tsumoto T. A functional role of cholinergic innervation to neurons in the cat visual cortex. J. Neurophysiol. 1987b;58:765–780. doi: 10.1152/jn.1987.58.4.765. [DOI] [PubMed] [Google Scholar]
- Sillito AM, Kemp JA. Cholinergic modulation of the functional organization of the cat visual cortex. Brain Res. 1983;289:143–155. doi: 10.1016/0006-8993(83)90015-x. [DOI] [PubMed] [Google Scholar]
- Solomon SG, Peirce JW, Lennie P. The impact of suppressive surrounds on chromatic properties of cortical neurons. J. Neurosci. 2004;24:148–160. doi: 10.1523/JNEUROSCI.3036-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigges M, Tigges J, Rees H, Rye D, Levey AI. Distribution of muscarinic cholinergic receptor proteins m1 to m4 in area 17 of normal and monocularly deprived rhesus monkeys. J. Comp. Neurol. 1997;388:130–145. [PubMed] [Google Scholar]
- Toledo-Rodriguez M, Blumenfeld B, Wu C, Luo J, Attali B, Goodman P, Markram H. Correlation maps allow neuronal electrical properties to be predicted from single-cell gene expression profiles in rat neocortex. Cereb. Cortex. 2004;14:1310–1327. doi: 10.1093/cercor/bhh092. [DOI] [PubMed] [Google Scholar]
- Turrini P, Casu MA, Wong TP, De Koninck Y, Ribeiro-da-Silva A, Cuello AC. Cholinergic nerve terminals establish classical synapses in the rat cerebral cortex: synaptic pattern and age-related atrophy. Neuroscience. 2001;105:277–285. doi: 10.1016/s0306-4522(01)00172-5. [DOI] [PubMed] [Google Scholar]
- Van Brederode JF, Mulligan KA, Hendrickson AE. Calcium-binding proteins as markers for subpopulations of GABAergic neurons in monkey striate cortex. J. Comp. Neurol. 1990;298:1–22. doi: 10.1002/cne.902980102. [DOI] [PubMed] [Google Scholar]
- Vazquez J, Baghdoyan HA. Basal forebrain acetylcholine release during REM sleep is significantly greater than during waking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001;280:R598–R601. doi: 10.1152/ajpregu.2001.280.2.R598. [DOI] [PubMed] [Google Scholar]
- Williford T, Maunsell JH. Effects of spatial attention on contrast response functions in macaque area V4. J. Neurophysiol. 2006;96:40–54. doi: 10.1152/jn.01207.2005. [DOI] [PubMed] [Google Scholar]
- Wouterlood FG, Jorritsma-Byham B. The anterograde neuroanatomical tracer biotinylated dextran-amine: comparison with the tracer Phaseolus vulgaris-leucoagglutinin in preparations for electron microscopy. J. Neurosci. Methods. 1993;48:75–87. doi: 10.1016/s0165-0270(05)80009-3. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Huguenard JR, Prince DA. Cholinergic switching within neocortical inhibitory networks. Science. 1998;281:985–988. doi: 10.1126/science.281.5379.985. [DOI] [PubMed] [Google Scholar]
- Xing D, Shapley RM, Hawken MH, Ringach DL. Effect of stimulus size on the dynamics of orientation selectivity in Macaque V1. J Neurophysiol. 2005;94:799–812. doi: 10.1152/jn.01139.2004. [DOI] [PubMed] [Google Scholar]
- Zinke W, Roberts MJ, Guo K, McDonald JS, Robertson R, Thiele A. Cholinergic modulation of response properties and orientation tuning of neurons in primary visual cortex of anaesthetized Marmoset monkeys. Eur. J. Neurosci. 2006;24:314–328. doi: 10.1111/j.1460-9568.2006.04882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.