

Abstract
Prostate adenocarcinoma is the second leading cause of cancer death among men, due primarily to the fact that the majority of prostate cancers will eventually spread to the skeleton. Metastatic dissemination requires a complex series of coordinated events that result in cells that escape from the primary tumor into the circulation and eventually colonize a distant organ. The ability of these cells to evolve into macroscopic metastases depends strongly on their compatibility with, and ability to utilize, this new microenvironment. We previously showed that bone-metastatic prostate cancer cells exposed to human bone marrow respond by activation of cell survival pathways, such as PI3K/Akt, and that these events are mediated by the alpha-receptor for platelet-derived growth factor (PDGFRα). Our studies and others have shown that PDGFRα may be activated by mechanisms independent of PDGF ligand binding.
Here we provide conclusive evidence that soluble components of human bone marrow can activate PDGFRα through a mechanism that does not require the canonical binding of PDGF ligand(s) to the receptor. In particular, we found dimerization of PDGFRα monomers is not induced by human bone marrow, but this does not prevent both receptor phosphorylation and downstream signaling from occurring. To establish the relevance of this phenomenon in vivo, we employed a PDGFRα mutant lacking the extracellular ligand-binding domain. Our studies show that this truncated PDGFRα was able to restore bone-metastatic potential of prostate cancer cells as effectively as the full-length form of the receptor.
INTRODUCTION
Prostate adenocarcinoma is the second leading cause of cancer related death among men, despite a largely successful treatment of the primary tumor following early detection [1]. The main problem for therapy is that the majority of prostate cancers will eventually disseminate to the skeleton. This complication leads to a significant decline in quality of life and is currently untreatable, representing the main cause of death in patients with advanced disease [2].
Metastatic dissemination is a concerted multi-step process in which cancer cells spread from the primary tumor into the vasculature, survive in the circulation and reach distant organs [3]. Cancer cells that spread to secondary sites must initially adhere to the luminal surface of endothelial cells, and subsequently migrate in response to chemo-attractant cues produced by the tissue microenvironment [4] [5]. Further progression into clinically significant metastases depends strongly on the ability of cancer cells to support their survival and proliferation in the parenchyma of the secondary organ. In fact, it is widely agreed that cancer cells that fail to adapt to a specific organ microenvironment will either perish or remain dormant; where they are incapable of causing harm to the patient unless growth is resumed [6] [7].
Cancer cells that disseminate to the skeleton through the circulatory system encounter the bone marrow immediately following extravasation from the vascular sinusoids. Their propensity to grow into macroscopic secondary tumors is most likely dictated by favorable conditions offered by this tissue, such as compatible trophic factors [8] [9]. Thus, interference with these symbiotic interactions has been proposed as a powerful means to counteract skeletal metastases [10].
Effective therapies against skeletal metastases might be achieved by identification and blockade of molecular targets that result in disruption of the host/tumor relationship. To this end, we exposed bone-metastatic prostate cancer cells to bone marrow aspirates from human donors, and found that they responded by activation of downstream cell survival pathways, such as PI3K/Akt [11]. We also determined that these events are mediated by the alpha-receptor for platelet-derived growth factor (PDGFRα), a receptor tyrosine kinase expressed at higher levels in bone-metastatic cells than in cells that lack bone tropism [12]. In addition, we demonstrated that targeting PDGFRα with a humanized monoclonal antibody dramatically prevents the growth of skeletal lesions in an animal model of disseminated prostate cancer [13]. When PDGFRα was over-expressed in prostate cancer cells that lacked bone-metastatic potential, it restored their ability to survive during the first days following extravasation in the bone marrow. This allowed these cells to progress into secondary bone tumors that were comparable in number and size to those produced by metastatic cancer cells that endogenously express higher levels of the receptor [13]. The emphasis we place on the role of PDGFRα in prostate cancer progression and dissemination is also based on studies by other groups which reported detection of PDGFRα in human samples from both primitive prostate adenocarcinoma [14] [15] and bone secondary tumors [16]. While the contribution of PDGFRα in the promotion of skeletal metastases from prostate and breast cancers is progressively emerging [17] [18], studies focusing on the mechanisms of receptor activation and recruitment of downstream signaling pathways have provided some exciting results. In particular, recent evidence suggests that PDGFRα may be activated by novel mechanisms, alternative to the conventional binding of PDGF ligand(s) to the receptor. For instance, Lei and Kazlauskas have shown that generation of reactive oxygen species (ROS) is sufficient for Src kinase-mediated activation of PDGFRα [19]. Additional evidence demonstrates that PDGFRα associates with, and is activated in response to, infection by human cytomegalovirus [20]. In agreement with these studies, we have found that activation of PDGFRα by human bone marrow could occur despite blockade of the extracellular ligand-binding domain [11].
Here we provide conclusive evidence that the acellular fraction of human bone marrow can activate PDGFRα through a mechanism that does not require the canonical binding of PDGF ligand(s) to the receptor. In particular, we found that human bone marrow does not induce dimerization of PDGFRα monomers, but this does not prevent both receptor phosphorylation and downstream signaling from occurring. To establish the relevance of this phenomenon in vivo, we employed a PDGFRα mutant lacking the extracellular ligand-binding domain. Our studies show that this truncated PDGFRα was able to restore bone-metastatic potential of prostate cancer cells as effectively as the full-length form of the receptor.
MATERIALS AND METHODS
Reagents
Both PP2 (4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo []pyrimidine) and NAC (N-acetyl-l-cysteine) were obtained from Calbiochem (San Diego, CA).
Cell Lines and Cell Culture
PC3-ML and PC3-N cell lines were derived from the parental PC-3 cell line (ATCC, Manassas, VA) as previously described [21]. DU145 cells were purchased from ATCC. All cell lines were cultured at 37°C and 5% CO2 in DMEM supplemented with 10% fetal bovine serum (Hyclone, Logan, UT) and 0.1% gentamicin (Invitrogen, Carlsbad, CA). Cells were engineered to stably-express EGFP as described below.
Ectopic PDGFRα Expression
Human full-length PDGFRα (NM_006206) and its truncated form (αΔX) were expressed in the identical lentiviral vector (America Pharma Source) used to obtain stably fluorescent cells as previously described [13]. The truncated PDGFR(αΔX) lacks amino acids 45-524, corresponding to the entire extracellular domain with the exception of the signal peptide (AA 1-44). Amino acid 45 in this truncated receptor corresponds to leucine 525 of the full-length form, which is the beginning of the transmembrane domain [22]. Cells were transduced using an MOI of 50 ifu/cell. As the lentiviral vector contains an EGFP-IRES site, the cells expressing PDGFRα were isolated and purified by flow cytometry and sorting based on their fluorescence intensity.
Human Bone Marrow Acquisition and Processing
Bone marrow samples from healthy male donors (ages 18–45) were supplied by Lonza Biosciences (Poeitics™ Donor Program). Samples were shipped and maintained at 4° C throughout processing. Briefly, samples were centrifuged (1,500 rpm for 20 minutes) in order to separate the acellular and cellular phases. Supernatant containing the acellular phase was removed and filtered using 0.8µm and 0.22µm filters in succession followed by storage at −80°C.
In Vitro Experimental Protocols
Cells were starved from serum for four hours prior to being exposed to bone marrow or PDGF-AA (30ng/ml). Fifty micro-liters of processed bone marrow were administered to cells in 1ml of experimental medium for a final 1:20 dilution. PP2 was used at a concentration of 10µM and IMC-3G3 was used at a concentration of 20µg/ml. Receptor crosslinking experiments were carried out on ice and/or at 4°C using the chemical crosslinker BS3 (Pierce) at a concentration of 2mM. Briefly, cells were incubated with either ligand or bone marrow for the time periods indicated, followed by incubation with BS3 for 30m at 4°C. Crosslinking was then quenched by the addition of 20 mM Tris for five minutes at room temperature.
SDS-PAGE and Western Blotting
Cell lysates were obtained and SDS-PAGE and Western Blot analysis performed as previously described [23], with few modifications. Membranes were blotted with antibodies targeting phospho-Akt (Ser-473, Cell Signaling Technology), PDGFRα (R&D Systems) and total Akt (Cell Signaling). Primary antibody binding was detected using an HRP-conjugated secondary antibody (Pierce). Chemiluminescent signals were obtained using SuperSignal West Femto reagents (Pierce) and detected with the Fluorochem 8900 imaging system and relative software (Alpha Innotech). Densitometry analysis was performed using the UN-SCAN IT software (Silk Scientific). Samples were run on the same gels for effective comparison of intensity levels. Each experiment was repeated at least three times and provided similar results.
Detection of PDGFRα phosphorylation
Cells were washed twice with ice cold PBS and lysed with immunoprecipitation buffer (50mM Tris, 150mM NaCl, 10mM NaF, 10mM sodium pyrophosphate, 1% NP40) supplemented with protease and phosphatase inhibitors (Protease Inhibitor Cocktail Set III, Phosphatase Inhibitor Cocktail Set II, Calbiochem). 750µg of cell lysates were incubated with agarose-conjugated anti-PDGFRα primary antibody (Santa Cruz) for one hour at 4°C. Immunoprecipitation was carried out according to the manufacturer’s protocol, with immunoprecipitated protein run on a 7.5% polyacrylamide gel. Western blotting was carried out as previously described using an antibody directed against phospho-Tyrosine (Cell Signaling) or phospho-PDGFRα (Tyr-742, Cell Signaling). Equal loading was confirmed by stripping the membrane and blotting for total PDGFRα.
Animal model of metastasis
Five week-old male immuno-compromised mice (CB17-SCRF) were obtained from Taconic and housed in a germ-free barrier. At six weeks of age, mice were anesthetized with 100mg/kg ketamine and 20mg/kg xylazine and successively inoculated in the left cardiac ventricle with cancer cells (5×104 in 100µl of serum-free DMEM/F12). All cancer cells used in our in vivo studies were stably transduced using a lentiviral vector (America Pharma Source) expressing enhanced Green Fluorescent Protein (eGFP), either with an exogenous gene insert (full-length PDGFRα or its αΔX truncated form) or as an empty-vector. PC3-ML and PC3-N cells expressing the empty-vector showed metastatic abilities identical to their wild-type counterparts, as established in our previous studies [11], [13]. As also previously described, the only soft-tissue organs that we find are harboring cancer cells at four weeks post-inoculation are the adrenal glands. However, these tumors remain contained in size and never produce tumor burden in the inoculated animals [13].
Mice were sacrificed at specified time-points following inoculation and tissues prepared as described below. All experiments were conducted in accordance with NIH guidelines for the humane use of animals. All protocols involving the use of animals were approved by the Drexel University College of Medicine Committee for the Use and Care of Animals.
Tissue Preparation
Bones and soft-tissue organs were collected and fixed in 4% formaldehyde solution for 24 hours and then transferred into fresh formaldehyde for additional 24 hours. Soft tissues were then placed either in 30% sucrose for cryoprotection or 1% formaldehyde for long-term storage. Bones were decalcified in 0.5M EDTA for 7 days followed by incubation in 30% sucrose. Tissues were maintained at 4°C for all aforementioned steps and frozen in O.C.T. medium (Electron Microscopy Sciences, Hatfield, PA) by placement over dry-ice chilled 2-methylbutane (Fisher, Pittsburgh, PA). Serial sections of 80µm in thickness were obtained using a Microm HM550 cryostat (Mikron, San Marcos, CA).
Fluorescence stereomicroscopy and morphometric analysis of metastases
Bright-field and fluorescent images of skeletal metastases were acquired using a SZX12 Olympus stereomicroscope coupled to an Olympus DT70 CCD color camera. Digital images were analyzed with ImageJ software (http://rsb.info.nih.gov/ij/) and calibrated for measurement by obtaining a pixel to millimeter ratio. Morphometric evaluation of skeletal tumors was conducted by analysis of serial cryosections, in which the largest representative tumor section for each metastasis was identified. A freehand tool was used to outline the border of each metastatic lesion, and the area was computed using the ImageJ 'measure area' function.
Statistics
Data shown in figures are representative of at least two experiments providing similar results. We analyzed number and size of skeletal metastases between groups using a two-tailed Student’s t-test. A value of P ≤ 0.05 was considered statistically significant.
RESULTS AND DISCUSSION
We have previously shown that the activation of the PI3K/Akt pathway in bone-metastatic prostate cancer cells exposed to human bone marrow aspirates depends mostly on PDGFRα signaling [11]. Furthermore, prostate cancer cells that lack bone-metastatic potential - and were less responsive to bone marrow – could activate Akt to the same extent of bone-metastatic cells following PDGFRα overexpression [13]. Therefore, our first series of experiments sought to ascertain whether the acellular fraction of human bone marrow would induce phosphorylation of PDGFRα - as is the case when this receptor binds, and is activated by, its proper PDGF ligands [24]. As done for our previous studies, we employed PC3-ML cells, a sub-population of the human PC3 cell line originally derived from a skeletal metastasis in a patient affected by prostate adenocarcinoma [25]. PC3-ML cells were originally selected for their invasiveness in vitro in concert with their bone-metastatic potential in animal models [21]. When these cells were exposed to bone marrow, PDGFRα phosphorylation was clearly detected and lasted at least 30 minutes (Fig.1A). The ability of bone marrow to phosphorylate PDGFRα could be consistently observed using aspirates withdrawn from different donors (Fig.1B, top).
FIGURE 1. Human bone marrow induce phosphorylation of PDGFRα and activation of downstream Akt pathway in prostate cancer cells.
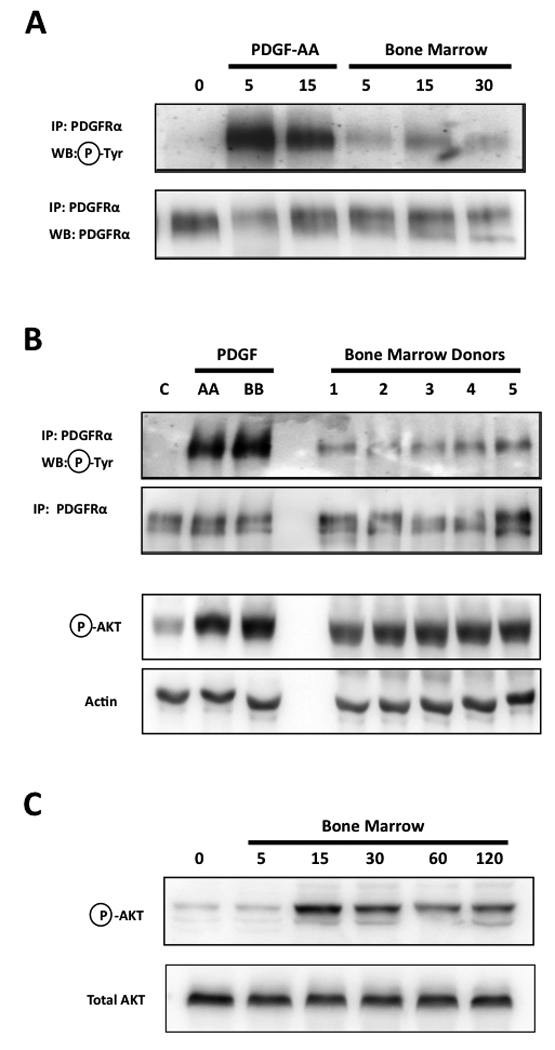
The exposure of PC3-ML prostate cancer cells to acellular human bone marrow induces phosphorylation of PDGFRα, although of a lower magnitude compared to that produced by the proper ligand PDGF-AA (30ng/ml) (A); this effect was consistently reproduced by all human samples tested (B, top); despite producing a lower extent of PDGFRα phosphorylation than PDGF-AA, bone marrow activated the downstream signaling kinase Akt to an extent almost equivalent to that of the growth factor (B, bottom) and in a time-dependent fashion (C).
Structural and functional studies have shown that PDGF binding induces dimerization of PDGFR; this allows the trans-phosphorylation of several conserved tyrosine residues on the juxtaposed intracellular portion of each receptor [26]. In addition to enhancing the catalytic activity of the kinase domain, this event creates docking sites for the binding and activation of signal transduction mediators containing SH2 domains, including members of the PI3K family [27]. At least eight tyrosine residues on PDGFRα become phosphorylated upon binding of PDGF ligand(s) [28], likely contributing to the strong signal observed in our experiments (Fig.1 A–B).
Interestingly, the considerably lower magnitude of PDGFRα phosphorylation induced by bone marrow - as compared to that induced by PDGF ligand(s) – did not translate to a significantly lower activation of the downstream Akt pathway. In fact, remarkably close levels of Akt phopshorylation were observed after exposing PC3-ML cells to either PDGF or bone marrow (Fig. 1B, bottom) and as evidenced by a time-dependent kinetic (Fig. 1C). In addition, we had previously demonstrated that the majority of this observed Akt activation is due solely to PDGFRα signaling [11]. This suggests that the activation of PI3K, which is functionally upstream of, and mainly responsible for, Akt phosphorylation, must be almost equivalent in these two conditions. Thus, we speculate that the majority of PDGFRα phosphorylation caused by bone marrow corresponds to Tyr-731 and Tyr-742 residues, which specifically bind and activate PI3K [24]. This would explain the lower magnitude of receptor phosphorylation as compared to stimulation by PDGF ligands. However, the phosphorylation of only select tyrosine residues could hardly be justified in the event that bone marrow induces PDGFRα dimerization. In fact, whereas dimers could be observed upon exposure of PC3-ML cells to PDGF-AA, human bone marrow consistently lacked the ability to induce dimerization of PDGFRα in these cells (Fig. 2A–B).
FIGURE 2. Recruitment of PDGFRα by human bone marrow occurs in the absence of receptor dimerization.
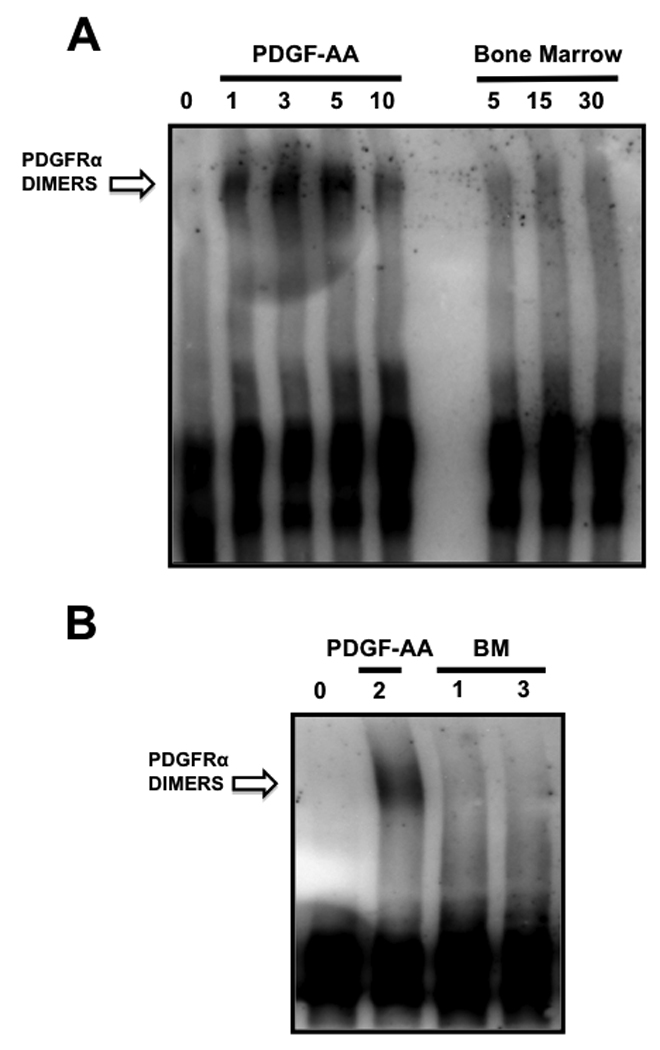
Phosphorylation of PDGFRα by PDGF-AA was preceded by receptor dimerization, observed as soon as 1 minute after exposure of cells to the ligand. However, bone marrow did not induce PDGFRα dimerization even in cells that were stimulated for up to 30 minutes.
This initial evidence of an unorthodox mechanism used by bone marrow to initiate PDGFRα signaling was further confirmed by a second set of experiments in which dimerization was blocked using a monoclonal antibody directed against the extracellular portion of the receptor. The humanized antibody IMC-3G3 blocks the ligand-binding domain of PDGFRα [11], [29], thereby inhibiting receptor dimerization following treatment with PDGF-AA (Fig.3A). By blocking ligand-induced dimerization, IMC-3G3 also prevented PDGFRα trans-phosphorylation (Fig.3B), which normally occurs by juxtaposition of intracellular kinase domains between receptor monomers [27] [30]. However, no differences in PDGFRα phosphorylation were observed when cells were exposed to bone marrow alone or in the presence of IMC-3G3 (Fig.3B). This result provides compelling evidence that dimerization is not a pre-requisite for the activation of PDGFRα by the soluble fraction of human bone marrow in PC3-ML prostate cancer cells. If this was indeed the case, an active kinase must still phosphorylate the tyrosine residues responsible for recruitment of PI3K to the receptor, as this step should precede downstream activation of Akt by PDGFRα exposed to bone marrow. One likely possibility was the hetero-dimerization of PDGFRα with other tyrosine-kinase receptors. However, this event could be excluded by the absence of any dimer formation following treatment of cells with bone marrow and subsequent cell-surface receptor crosslinking (Fig.2).
FIGURE 3. Bone marrow-mediated activation of PDGFRα occurs upon blockade of ligand-induced receptor dimerization.
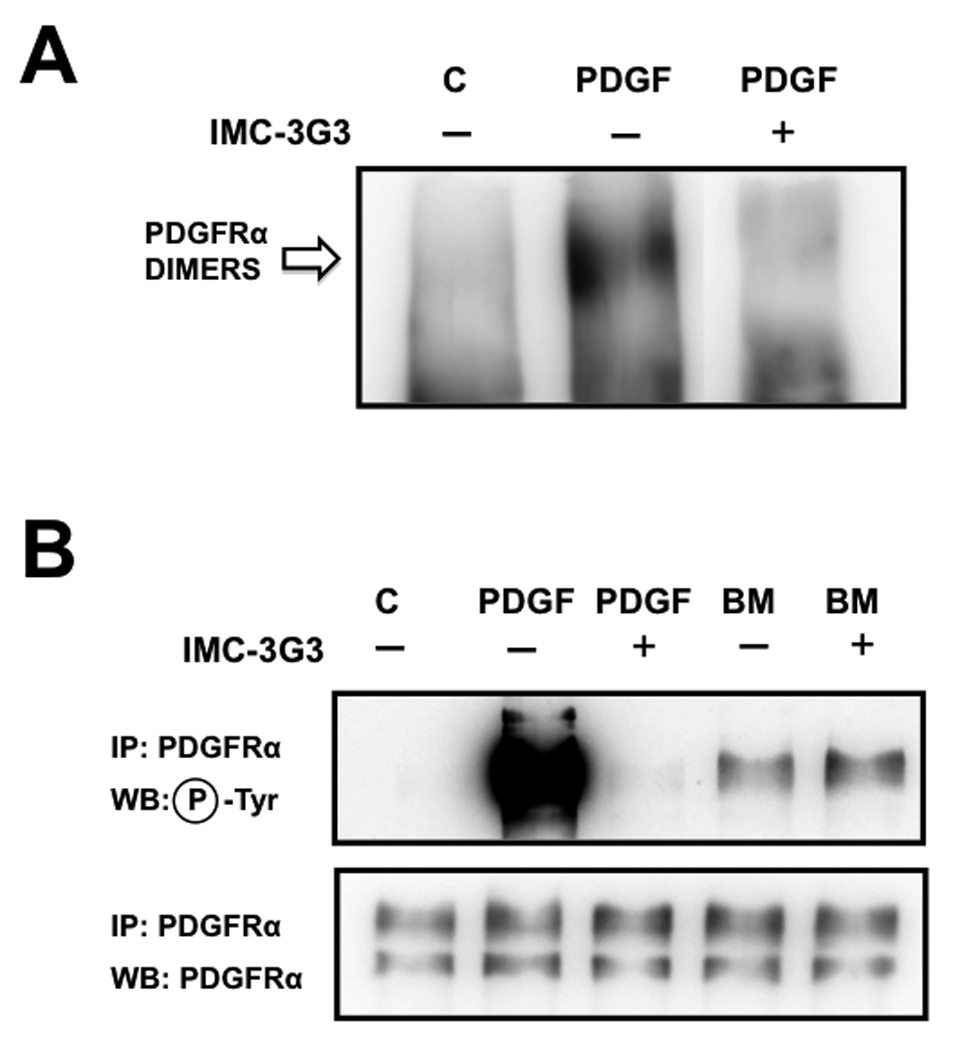
Blockade of the ligand binding domain of PDGFRα using the monoclonal antibody IMC-3G3 (20µg/ml) prevents receptor dimerization from occurring in response to PDGF-AA ligand (A); As expected, blockade of receptor dimerization prevented receptor phosphorylation in cells exposed to PDGF-AA. However, bone marrow was able to activate PDGFRα despite blockade of PDGFRα dimerization (B).
Thus, we hypothesize that PDGFRα is activated without recruitment of its ligand-binding domain and consequent dimerization, possibly by a soluble tyrosine-kinase that phosphorylates the monomeric form of the receptor. In fact, this idea is strongly supported by a recent study of proliferative vitreoretinopathy showing that activated Src family kinases (SFKs) phosphorylate PDGFRα in mouse embryo fibroblasts exposed to rabbit vitreous humor, the clear viscous fluid that occupies the space between the retina and lens of vertebrates’ ocular bulb [31].
In these cells, growth factors outside of the PDGF family and present in the vitreous can activate their specific receptor(s), thereby increasing the levels of intracellular reactive oxygen species (ROS). This event led to the direct activation of SFKs, which then mediated the phosphorylation of PDGFRα [19]. We have previously shown that human bone marrow activates downstream Akt despite containing negligible levels of PDGF ligands (measured at 150pg/ml by ELISA [11]), and also induces evident PDGFRα phosphorylation (Fig. 4A). Thus, if bone marrow were engaging PDGFRα in prostate cancer cells with a mechanism analogous to vitreous humor in mouse embryo fibroblasts, this would similarly require an increase in intracellular ROS. In this case, either anti-oxidants or inhibitors of SFKs should then block the activation of PDGFRα. To test this possibility, we treated PC3-ML cells with either 5mM hydrogen peroxide alone or bone marrow in the presence of the anti-oxidant N-Acetylcysteine (NAC, 10mM). The first condition – adopted to induce intracellular ROS production -– was unable to induce PDGFRα phosphorylation. Analogously, NAC did not prevent the activation of the receptor and downstream PI3K/Akt signaling caused by bone marrow (data not shown).
FIGURE 4. PDGF-independent but receptor kinase-dependent phosphorylation of PDGFRα induced by human bone marrow.
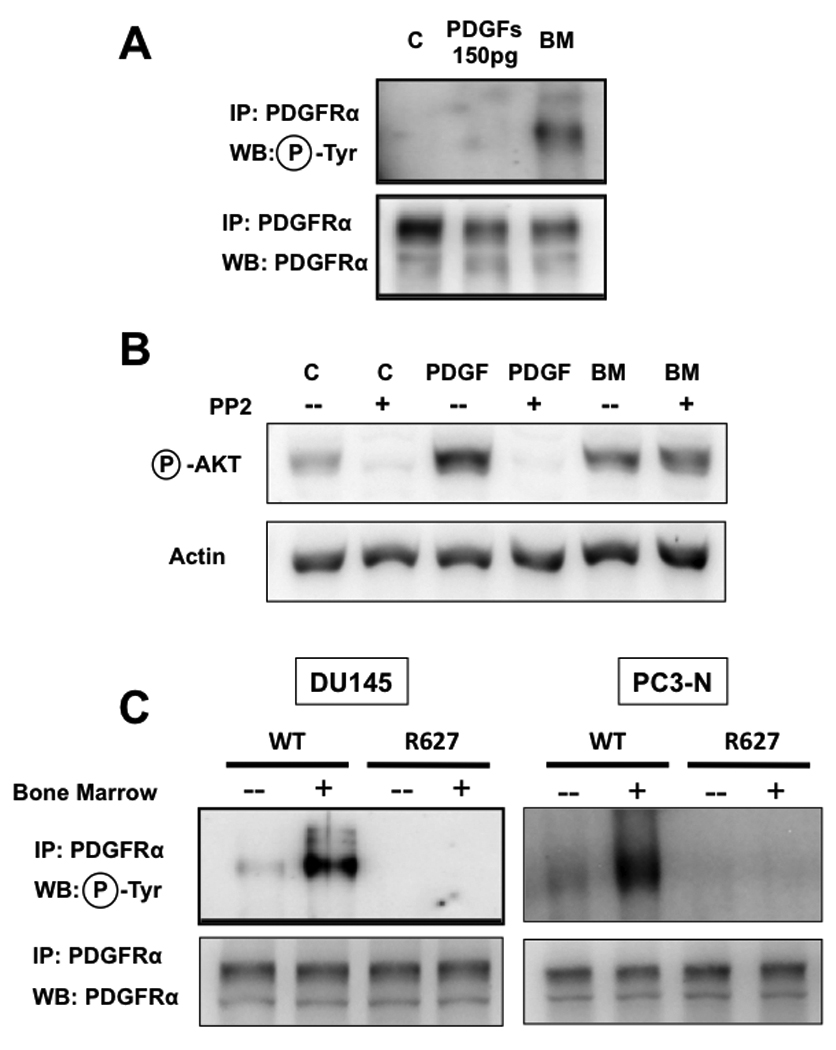
Exposure of PC3-ML cells to the same concentrations of PDGF ligands detected in human bone marrow fails to achieve receptor phosphorylation (A) or downstream Akt signaling [11]. Signaling through PDGFRα in PC3-ML cells exposed to PDGF ligands depends on the activation of SFKs and was blocked with PP2, an inhibitor of this family of kinases. In contrast, bone marrow-induced signaling through PDGFRα was insensitive to PP2; (B); The R627 kinase-dead mutant of PDGFRα was over-expressed in both DU-145 cells (C, left) and PC3-N (C, right) and demonstrates that bone marrow-mediated activation of PDGFRα requires the receptor kinase-domain for its phosphorylation..
We could also exclude the involvement of SFKs in the activation and signaling of PDGFRα induced by bone marrow by using PP2, a specific inhibitor of this family of kinases [32], [33]. In PC3-ML cells, PP2 did not prevent Akt phosphorylation by bone marrow (Fig. 4B), whereas it completely blocked stimulation of this pathway by PDGF-AA. This finding is in agreement with a described role of SFKs in coordinating ligand-induced recruitment of signaling pathways by tyrosine-kinase receptors, by acting upstream of PI3K [34] [35].
Taken together, these results lead us to conclude that human bone marrow activates PDGFRα on prostate cancer cells in a manner that is distinct from the factors found in vitreous humor, supporting the idea that there may be multiple mechanisms whereby this receptor can be activated without involvement of its canonical PDGF ligands.
It is widely recognized that the activity of the kinase domain in tyrosine kinase receptors is significantly augmented upon dimerization and trans-phosphorylation [30]. Because we observed a lack of PDGFRα dimerization upon exposure of prostate cancer cells to human bone marrow, one would predict that the kinase domain is neither necessary nor responsible for the phosphorylation of monomeric PDGFRα. To effectively investigate this idea, we used two different prostate cancer cell lines that show low levels of this receptor (PC3-N) or completely fail to express it (DU-145) [12] and engineered them to stably express a kinase-inactive PDGFRα mutant (R627)[36]. Surprisingly, both R627-expressing cell types failed to show receptor phosphorylation upon bone marrow exposure, in contrast to when the same cell types over-express the wild-type form of PDGFRα (Fig. 4C). Thus, these results implicate the kinase domain in the phosphorylation and signaling of monomeric PDGFRα induced by bone marrow. This observation suggests the possibility of a receptor autophosphorylation event, independent of ligand-induced dimerization, and most likely affecting the tyrosine residues responsible for PI3K binding and activation. The identification of the mechanism and signaling mediators responsible for this event are the focus of an ongoing investigation in our laboratory.
In previous animal studies we have shown that PDGFRα can confer bone-metastatic potential to prostate cancer cells [13]. Intriguingly, the role exerted by this receptor in the metastatic process may require its indirect activation – likely through the activity of yet unidentified factor. To further investigate this peculiarity of PDGFRα signaling, we stably transfected cancer cells with a truncated receptor mutant (αΔX) that lacks the extracellular ligand-binding domain (see Materials and Methods for details) and is therefore unable to bind or be activated by proper PDGF ligand(s) or additional molecules that could activate the receptor is a spurious fashion [22].
A first set of experiments was conducted using DU-145 prostate cancer cells, which normally do not express PDGFRα [12] and would therefore not interfere by providing additional signaling through its wild-type full form. The αΔX expressed in these cells (Fig. 5A) was clearly insensitive to PDGF-AA, as expected due to its lack of binding capability and shown by the absence of any receptor phosphorylation (Fig. 5B, top). However, the exposure of DU-145(αΔX) cells to human bone marrow aspirates induced strong phosphorylation of the truncated receptor, conclusively demonstrating its ligand-independent activation (Fig. 5B, top). This phosphorylation involved the Tyr-742 residue (Fig. 5B, bottom), which on PDGFRα specifically corresponds to the PI3K binding site through its SH-2 domain [30] [24], unequivocally linking the soluble fraction of human bone marrow to the downstream activation of the Akt signaling pathway in prostate cancer cells. This event could be observed in cells either constitutively expressing PDGFRα (Fig.1 and refs. [11] [13]) or engineered to express the exogenous form of this receptor (Fig.5C).
FIGURE 5. PDGFRα activation by human bone marrow is independent of the extracellular ligand-binding domain.
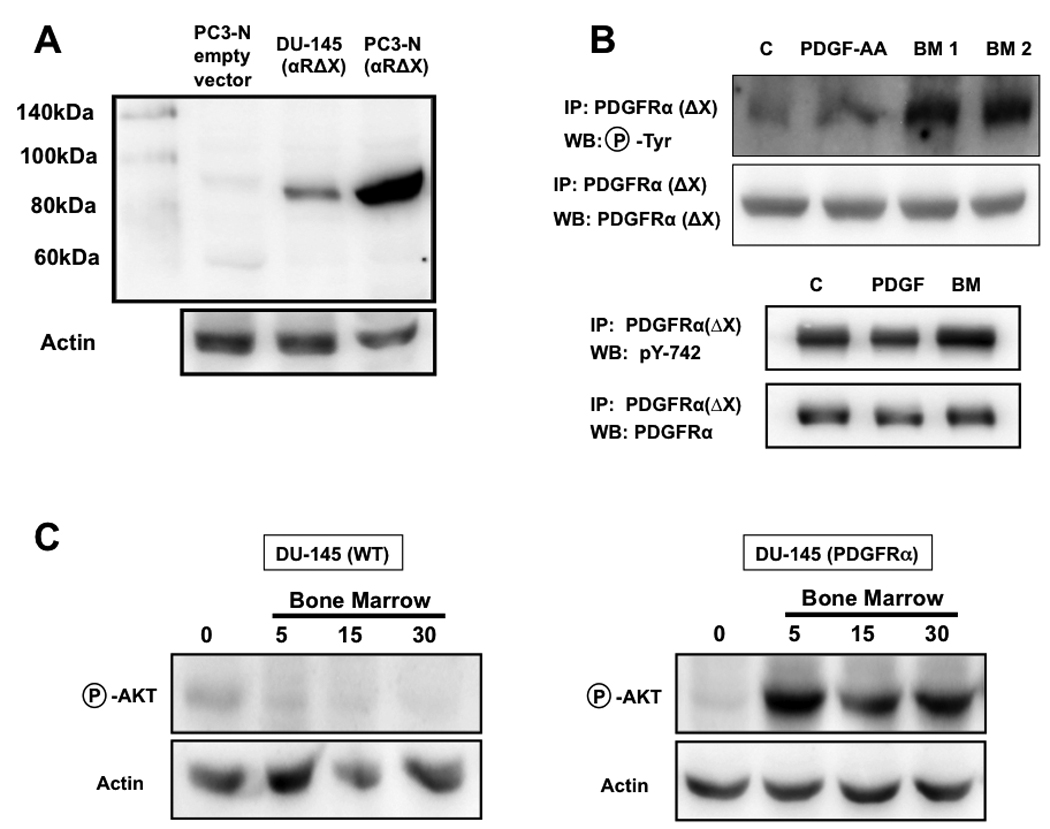
A truncated PDGFRα missing the extracellular binding domain (αΔX) was stably expressed in DU-145 cells, which normally lack PDGFRα expression (A) [12]. As expected, this mutated PDGFRα could not be phosphorylated in cells exposed to PDGF-AA; however, two different samples of human bone marrow elicited a strong receptor phosphorylation (B, top), which involved the Tyr-742 residue, a specific binding site for PI3K (B, bottom); the exposure of wild-type DU-145 cells to bone marrow did not produce an appreciable activation of the PI3K/Akt pathway, which was conferred by the stable expression of PDGFRα, (C).
A second series of experiments aimed to ascertain whether the αΔX receptor could reproduce the enhanced capacity of skeletal metastases from prostate cancer cells, as we had previously shown for its full-length form [13]. A confirmatory result would convincingly demonstrate that PDGFRα could induce a pro-metastatic phenotype, with pronounced bone-tropism, through a ligand-independent transactivation caused by the acellular fraction of bone marrow. PC3-N cells were originally selected from the parental PC3 cell line for their lack of invasiveness in vitro [21]. We have previously shown that PC3-N cells express lower levels of PDGFRα and respond weakly to human bone marrow as compared to their bone-metastatic counterpart PC3-ML cells [12] [13]. Most importantly, PC3-N cells disseminate to the bone when inoculated in the blood circulation of SCID mice, but fail to grow into macroscopic skeletal metastases unless they are engineered to over-express full-length PDGFRα [13]. When PC3-N cells were stably transduced with a lentiviral vector expressing PDGFR(αΔX) (Fig. 5A) and exposed to bone marrow, Akt was phosphorylated to the same extent observed in either PC3-ML cells (Fig.6A) or PC3-N cells expressing full-length PDGFRα treated in the same fashion.
FIGURE 6. PDGFRα(ΔX) induces a bone metastatic phenotype that is indistinguishable from cells expressing full-length PDGFRα.
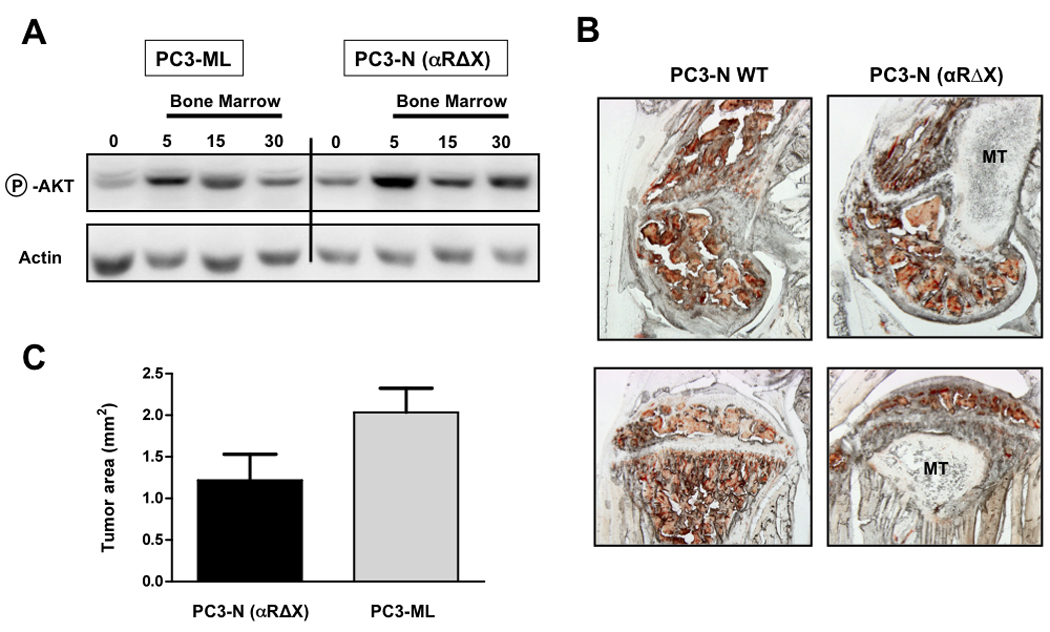
Expression of PDGFRα(Δx) in non-bone-metastatic PC3-N cells results in an increased activation of downstream Akt upon exposure to human bone marrow, to an extent comparable to that observed in bone-metastatic PC3-ML cells (A); PC3-N(αΔx) were able to successfully form skeletal lesions four weeks after their intracardiac inoculation in SCID mice (B; top is femur, bottom is tibia); Importantly, the size of metastases produced by PC3-N(αΔx) cells did not differ significantly from lesions formed by the metastatic PC3-ML cells, which express substantially higher levels of endogenous, full-length PDGFRα (C); (MT = metastatic tumor). Seven mice were inoculated with PC3-N(αΔx) cells in two different experiments and presented 13 skeletal tumors in total. Eight mice were inoculated with PC3-ML-(empty vector) cells in two different experiments and presented 22 skeletal tumors in total.
In order to test their bone-metastatic potential, PC3-N(αΔX) cells were then inoculated via an intracardiac route into SCID mice and the skeletal metastases observed at 4-week post-inoculation were compared to those detected in mice receiving the highly bone-metastatic PC3-ML cells. When the bone tumors in these two groups of mice were compared, no significant differences in size were observed and the location and bone-destructive properties induced by the αΔX and full-length PDGFRα were nearly identical (Fig.6B,C). These results demonstrate that PDGFRα can both increase the responsiveness of human prostate cancer cells to acellular bone marrow in vitro and promote their progression in the bone microenvironment in the absence of any involvement of its ligand-binding domain.
Recognizing that PDGFRα could contribute to dissemination and metastatic progression of prostate adenocarcinoma independently of direct ligand stimulation has significant translational implications. It could be inferred that anti-cancer therapeutics that are designed to block PDGF ligand binding to PDGFRα would not prevent the activation of downstream signaling pathways in cells that have spread to the bone marrow. In contrast, therapeutic approaches that induce internalization of PDGFRα may provide a better option to inhibit downstream signaling elicited by this receptor in disseminated prostate cancer cells when exposed to the bone marrow microenvironment.
ACKNOWLEDGMENTS
The authors wish to thank Dr. Olimpia Meucci (Drexel College of Medicine) for helpful discussion, Dr. Gregg Johannes and Mr. Jeff Thomas (Department of Pathology, Drexel College of Medicine) for help with the PDGFRα expressing vector, Dr. Nick Loizos (ImClone Systems Inc., NY) for providing the IMC-3G3 antibody. The cDNA for full-length human PDGFRα was a kind gift of Dr. Carl-Henrik Heldin (Ludwig Institute for Cancer Research, Uppsala, Sweden).
GRANT SUPPORT
This work was funded by the W.W. Smith Charitable Trust and Department of Defense Prostate Cancer Program grant PC080987 (AF) and by The National Institutes of Health EY012509 (AK).
REFERENCES
- 1.Denmeade SR, Isaacs JT. A history of prostate cancer treatment. Nat Rev Cancer. 2002;2:389–396. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mohammad KS, Fournier PG, Guise TA, Chirgwin JM. Agents Targeting Prostate Cancer Bone Metastasis. Anti-cancer agents in medicinal chemistry. 2009 doi: 10.2174/187152009789735008. [DOI] [PubMed] [Google Scholar]
- 3.Fidler I. Critical determinants of metastasis. Semin Cancer Biol. 2002;12:89–96. doi: 10.1006/scbi.2001.0416. [DOI] [PubMed] [Google Scholar]
- 4.Fidler I. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 5.Chambers AF, Groom A, Macdonald I. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 6.Fehm T, Mueller V, Marches R, et al. Tumor cell dormancy: implications for the biology and treatment of breast cancer. APMIS. 2008;116:742–753. doi: 10.1111/j.1600-0463.2008.01047.x. [DOI] [PubMed] [Google Scholar]
- 7.Horak CE, Lee JH, Marshall JC, et al. The role of metastasis suppressor genes in metastatic dormancy. APMIS. 2008;116:586–601. doi: 10.1111/j.1600-0463.2008.01213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bussard K, Gay C, Mastro A. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2008;27:41–55. doi: 10.1007/s10555-007-9109-4. [DOI] [PubMed] [Google Scholar]
- 9.Buijs JT, van der Pluijm G. Osteotropic cancers: from primary tumor to bone. Cancer Lett. 2009;273:177–193. doi: 10.1016/j.canlet.2008.05.044. [DOI] [PubMed] [Google Scholar]
- 10.Mundy G. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 11.Dolloff N, Russell MR, Loizos N, Fatatis A. Human bone marrow activates the Akt pathway in metastatic prostate cells through transactivation of the alpha-platelet-derived growth factor receptor. Cancer Res. 2007;67:555–562. doi: 10.1158/0008-5472.CAN-06-2593. [DOI] [PubMed] [Google Scholar]
- 12.Dolloff N, Shulby S, Nelson A, et al. Bone-metastatic potential of human prostate cancer cells correlates with Akt/PKB activation by alpha platelet-derived growth factor receptor. Oncogene. 2005;24:6848–6854. doi: 10.1038/sj.onc.1208815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russell M, Jamieson W, Dolloff N, Fatatis A. The alpha-receptor for platelet-derived growth factor as a target for antibody-mediated inhibition of skeletal metastases from prostate cancer cells. Oncogene. 2009;28:412–421. doi: 10.1038/onc.2008.390. [DOI] [PubMed] [Google Scholar]
- 14.Fudge K, Wang CY, Stearns ME. Immunohistochemistry analysis of platelet-derived growth factor A and B chains and platelet-derived growth factor alpha and beta receptor expression in benign prostatic hyperplasias and Gleason-graded human prostate adenocarcinomas. Mod Pathol. 1994;7:549–554. [PubMed] [Google Scholar]
- 15.Hofer M, Fecko A, Shen R, et al. Expression of the platelet-derived growth factor receptor in prostate cancer and treatment implications with tyrosine kinase inhibitors. Neoplasia. 2004;6:503–512. doi: 10.1593/neo.04157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chott A, Sun Z, Morganstern D, et al. Tyrosine kinases expressed in vivo by human prostate cancer bone marrow metastases and loss of the type 1 insulin-like growth factor receptor. Am J Pathol. 1999;155:1271–1279. doi: 10.1016/S0002-9440(10)65229-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.George DJ. Receptor tyrosine kinases as rational targets for prostate cancer treatment: platelet-derived growth factor receptor and imatinib mesylate. Urology. 2002;60:115–121. doi: 10.1016/s0090-4295(02)01589-3. discussion 122. [DOI] [PubMed] [Google Scholar]
- 18.Carvalho I, Milanezi F, Martins A, et al. Overexpression of platelet-derived growth factor receptor alpha in breast cancer is associated with tumour progression. Breast Cancer Res. 2005;7:R788–R795. doi: 10.1186/bcr1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lei H, Kazlauskas A. Growth factors outside of the platelet-derived growth factor (PDGF) family employ reactive oxygen species/Src family kinases to activate PDGF receptor alpha and thereby promote proliferation and survival of cells. J Biol Chem. 2009;284:6329–6336. doi: 10.1074/jbc.M808426200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soroceanu L, Akhavan A, Cobbs CS. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature. 2008;455:391–395. doi: 10.1038/nature07209. [DOI] [PubMed] [Google Scholar]
- 21.Wang M, Stearns ME. Isolation and characterization of PC-3 human prostatic tumor sublines which preferentially metastasize to select organs in S.C.I.D. mice. Differentiation. 1991;48:115–125. doi: 10.1111/j.1432-0436.1991.tb00250.x. [DOI] [PubMed] [Google Scholar]
- 22.Lei H, Velez G, Hovland P, et al. Growth factors outside the PDGF family drive experimental PVR. Invest Ophthalmol Vis Sci. 2009;50:3394–3403. doi: 10.1167/iovs.08-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shulby SA, Dolloff N, Stearns M, et al. CX3CR1-fractalkine expression regulates cellular mechanisms involved in adhesion, migration, and survival of human prostate cancer cells. Cancer Res. 2004;64:4693–4698. doi: 10.1158/0008-5472.CAN-03-3437. [DOI] [PubMed] [Google Scholar]
- 24.Heldin CH, Ostman A, Rönnstrand L. Signal transduction via platelet-derived growth factor receptors. Biochim Biophys Acta. 1998;1378:F79–F113. doi: 10.1016/s0304-419x(98)00015-8. [DOI] [PubMed] [Google Scholar]
- 25.Kaighn ME, Narayan KS, Ohnuki Y, et al. Establishment and characterization of a human prostatic carcinoma cell line (PC-3) Investigative urology. 1979;17:16–23. [PubMed] [Google Scholar]
- 26.Heldin CH. Structural and functional studies on platelet-derived growth factor. EMBO J. 1992;11:4251–4259. doi: 10.1002/j.1460-2075.1992.tb05523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heldin CH, Ostman A. Ligand-induced dimerization of growth factor receptors: variations on the theme. Cytokine & Growth Factor Reviews. 1996;7:3–10. doi: 10.1016/1359-6101(96)00002-0. [DOI] [PubMed] [Google Scholar]
- 28.Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 29.Loizos N, Xu Y, Huber J, et al. Targeting the platelet-derived growth factor receptor alpha with a neutralizing human monoclonal antibody inhibits the growth of tumor xenografts: implications as a potential therapeutic target. Molecular Cancer Therapeutics. 2005;4:369–379. doi: 10.1158/1535-7163.MCT-04-0114. [DOI] [PubMed] [Google Scholar]
- 30.Kazlauskas A, Cooper JA. Autophosphorylation of the PDGF receptor in the kinase insert region regulates interactions with cell proteins. Cell. 1989;58:1121–1133. doi: 10.1016/0092-8674(89)90510-2. [DOI] [PubMed] [Google Scholar]
- 31.Holekamp NM. The vitreous gel: more than meets the eye. Am J Ophthalmol. 2010;149:32–36. doi: 10.1016/j.ajo.2009.07.036. [DOI] [PubMed] [Google Scholar]
- 32.Saito Y, Haendeler J, Hojo Y, et al. Receptor heterodimerization: essential mechanism for platelet-derived growth factor-induced epidermal growth factor receptor transactivation. Mol Cell Biol. 2001;21:6387–6394. doi: 10.1128/MCB.21.19.6387-6394.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emaduddin M, Bicknell DC, Bodmer WF, Feller SM. Cell growth, global phosphotyrosine elevation, and c-Met phosphorylation through Src family kinases in colorectal cancer cells. Proc Natl Acad Sci USA. 2008;105:2358–2362. doi: 10.1073/pnas.0712176105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koga F, Xu W, Karpova TS, et al. Hsp90 inhibition transiently activates Src kinase and promotes Src-dependent Akt and Erk activation. Proc Natl Acad Sci USA. 2006;103:11318–11322. doi: 10.1073/pnas.0604705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan PC, Chen YL, Cheng CH, et al. Src phosphorylates Grb2-associated binder 1 upon hepatocyte growth factor stimulation. J Biol Chem. 2003;278:44075–44082. doi: 10.1074/jbc.M305745200. [DOI] [PubMed] [Google Scholar]
- 36.Rosenkranz S, DeMali KA, Gelderloos JA, et al. Identification of the receptor-associated signaling enzymes that are required for platelet-derived growth factor-AA-dependent chemotaxis and DNA synthesis. J Biol Chem. 1999;274:28335–28343. doi: 10.1074/jbc.274.40.28335. [DOI] [PubMed] [Google Scholar]