

Abstract
Keratocytes of the corneal stroma secrete a unique population of proteoglycan molecules considered essential for corneal transparency. In healing corneal wounds, keratocytes exhibit a myofibroblastic phenotype in response to transforming growth factor β(TGF-β), characterized by expression of α-smooth muscle actin. This study examined proteoglycan and collagen expression by keratocytes in vitro during the TGF-β-induced keratocyte-myofibroblast transition. TGF-β-treated primary bovine keratocytes developed myofibroblastic features, including actin stress fibers anchored to paxillin-containing focal adhesions, cell-associated fibronectin, α5 integrin, and α-smooth muscle actin. Collagen I and III protein and mRNA increased in response to TGF-β. Secretion of [35S]sulfate-labeled keratan sulfate proteoglycans decreased markedly in response to TGF-β. Dermatan sulfate proteoglycans, however, increased in size and abundance. Protein and mRNA transcripts for normal stromal proteoglycans (lumican, keratocan, mimecan, and decorin) all decreased in response to TGF-β, but protein expression and mRNA for biglycan, a proteoglycan present in fibrotic tissue, was markedly up-regulated. These results show that TGF-β in vitro induces a proteoglycan expression pattern similar to that of corneal scars in vivo. This altered proteoglycan expression occurred coordinately with transdifferentiation of keratocytes to the myofibroblastic phenotype, implicating these cells as the source of fibrotic tissue in nontransparent corneal scars.
Transparency of the cornea to light is a function of the ultrastructure of the corneal stroma, a tissue composed of multiple lamellae of parallel, highly regular collagen fibrils. Trauma, inflammation, and several chronic pathological conditions lead to disruption of the stromal molecular architecture by the accumulation of a disorganized fibrotic extracellular matrix. This scarring is often associated with loss of corneal transparency and can contribute to permanent loss of vision. The essential role of corneal extracellular matrix in vision has led to an extensive characterization of the molecular components of the stroma of both normal and fibrotic corneas.
One specialized feature of the stroma is the composition of the hydrated matrix surrounding the collagen fibrils. This extrafibrillar matrix consists of proteoglycans and glycoproteins and includes a unique class of keratan sulfate-containing proteoglycans. Keratan sulfate glycosaminoglycan chains in the stroma modify three proteins, lumican, keratocan, and mimecan, that in other tissues occur as nonsulfated glycoproteins (1–3). The unique abundance and tissue-specific nature of the corneal keratan sulfate proteoglycans (KSPGs)1 suggests their role in corneal transparency. This hypothesis is supported by the occurrence of corneal opacity in diseases in which corneal keratan sulfate is absent and also in mice lacking lumican expression in which corneal keratan sulfate is reduced (4, 5). The lumican knockout mice develop corneal haze at 3–6 months of age, the same period of time during which corneal keratan sulfate accumulates in normal mouse corneas (6). In corneal scars resulting from trauma or from chronic pathologies, keratan sulfate is reduced and dermatan sulfate-containing proteoglycans are elevated (7–11). This characteristic alteration in stromal proteoglycans appears likely to be an important factor in the loss of corneal transparency.
Keratocytes are quiescent, neural crest-derived cells that populate the corneal stroma and secrete the molecular components of the stromal extracellular matrix. In healing corneal wounds, keratocytes become activated, migrate to the wound site, and begin mitosis (12). In later stages of healing, keratocytes develop F-actin stress fibers containing the muscle protein α-smooth muscle actin (13). These contractile cells, known as myofibroblasts, are hypothesized to be involved in wound closure (14). Keratocytes in vitro develop myofibroblastic characteristics in response to exogenous or autocrine TGF-β(15, 16). In vivo, antibodies blocking TGF-β eliminate the presence of myofibroblasts in healing wounds (17). These observations suggest that TGF-β plays an important role in the biology of wound healing and may be involved in the secretion of fibrotic tissue.
We recently demonstrated that bovine keratocytes maintained in vitro without serum or in reduced mitogen-horse serum, maintain stable secretion of proteoglycans similar to those produced in vivo (18). Growth of the cells in fetal bovine serum, however, rapidly reduced the KSPG synthesis by these cultures in a manner similar to that of healing wounds (19). This earlier study also presented preliminary data suggesting that KSPGs were down-regulated by exposure of keratocytes to TGF-β(19). In the current study, we have identified conditions under which bovine keratocytes develop myofibroblastic characteristics in response to TGF-β and examined the proteoglycans secreted during this transition. The results suggest that secretion of proteoglycans characteristic of fibrotic scars is coordinated with the development of a myofibroblastic phenotype as a response to TGF-β.
MATERIALS AND METHODS
Cell Culture
Primary cultures of bovine keratocytes were prepared and cultured at 4 ×104 cells/cm2 in Dulbecco’s modified Eagle’s medium/F-12 medium with antibiotics and 1% (v/v) platelet-poor horse serum as previously described (19). All protocols involved 6 days of culture. Recombinant human TGF-β1 (Sigma T7039) was added to a concentration of 1 ng/ml at different times during the 6-day culture to achieve the length of exposure noted in individual experiments.
Proteoglycan Analysis
Proteoglycans were metabolically labeled with [35S]sulfate (ICN Radiochemicals; catalog no. 64041), 100 μCi/ml or with a 35S-labeled mixture of methionine and cysteine (TransLabel; ICN Radiochemicals; catalog no. 51006) at 100 μCi/ml for 24 h before analysis. Proteoglycans were recovered from culture media by ion exchange chromatography on SPEC-NH2 microcolumns (Ansys Diagnostics, Inc.), and dermatan sulfate and keratan sulfate-containing proteoglycans were separated by fractional precipitation with 50 and 70% ethanol as described previously (19). The purified, labeled proteoglycans were dissolved in 0.1 M Tris acetate, 0.1% Triton X-100, pH 8, and radioactivity was determined by scintillation counting. The 70% (keratan sulfate) precipitate was digested for 2 h at 37 °C with 0.1 unit/ml affinity-pure chondroitinase ABC (Sigma) to remove residual dermatan sulfate. The intact proteoglycans were separated on 3–12% SDS-PAGE gels, transferred to Millipore Immobilon PVDF membranes, and detected by autoradiography using an Eastman Kodak Co. LE Transcreen and BIOMax MR film as previously described (19).
Western Blotting
Proteoglycan proteins were detected by immunoblotting after digestion of glycosaminoglycan chains from nonlabeled proteoglycans using a procedure similar to that described previously (20). Dermatan sulfate proteoglycans were treated with chondroitinase as described above, and keratan sulfate was digested with endo-β-galactosidase (Seikagaku Corp.; 4 milliunits/ml for 2 h at 37 °C). The core proteins were separated on 10% SDS-PAGE gels, transferred to polyvinylidene difluoride membranes, and detected with an affinity-purified antibody that detects all three KSPG proteins or with antisera against synthetic peptides of decorin (LF96) or biglycan (LF94) (10, 21, 22).
Collagen Analysis
Cultures exposed to TGF-β for different time periods were supplemented with sodium ascorbate (50 μg/ml) 24 h before harvesting and metabolically labeled with 50 μCi/ml [3H]proline for the final 18 h. The cell layer from each 35 mM dish was extracted in 0.5 ml of cold 0.5 M acetic acid, and cell debris was removed by centrifugation at 10,000 ×g. Noncollagenous proteins were digested with 20 μg/ml pepsin at 4 °C overnight followed by dialysis against 0.5 M acetic acid. Samples were lyophilized, proteins were separated on 12% SDS-PAGE gels and transferred to PVDF membranes, and proteins were detected by autoradiography or immunoblotting as described above using monoclonal antibody clone COL-I against collagen I and clone FH-7A against collagen III (Sigma).
Other cell-associated proteins were detected by scraping cells from a 35-mm culture well in 0.3 ml of SDS sample buffer, followed by heating at 70 °C for 20 min (19). Equal amounts of protein were separated by SDS-PAGE, transferred to PVDF membranes, and individual protein antigens were visualized using the following antibodies: for α5 integrin, clone P1D6 (Chemicon International MAB1965); for fibronectin, clone TV-1 (Chemicon International; MAB88904); and for α-smooth muscle actin, clone asm-1 (Roche Molecular Biochemicals catalog no. 1148818).
Northern Blotting
Total RNA isolated from cultured keratocytes was separated on 1% agarose-formaldehyde gels by electrophoresis. After electrophoresis, the gels were equilibrated for 10 min in 0.05 M NaOH, and the RNA was transferred under alkaline conditions to positively charged nylon membranes by downward flow for 2.5 h and fixed by ultraviolet exposure. Proteoglycan transcripts were detected with [32P]dATP-labeled DNA probes generated by random priming using modified dCTP (Strip-EZ DNA; Ambion Inc.) according to protocols provided by the manufacturer. Templates for keratan sulfate bovine proteins were generated by polymerase chain reaction from cDNA plasmids using the following primers: lumican, 508 base pairs, 5′-CATT-GACCTCCAGGTAATAG-3′(sense) and 5′-GCAATTGAAGAAGCTG-C-3′(antisense); keratocan, 953 base pairs, 5′-TTCAGCAATCTGGAG-AACCTG-3′(sense) and 5′-GTTAGATTGTTGTGTTGTCATGC-3′(antisense); mimecan, 608 base pairs, 5′-CAGCACCACCATCTCAGC-3′(sense) and 5′-CCAAGTAGAGGAAGGAGAGG-3′(antisense). Other cDNA probes were amplified from cDNA reverse transcribed from cultured keratocyte RNA using Superscript II (Life Technologies, Inc.) primed with oligo(dT) according to protocols provided by the manufacturer. Biglycan cDNA was amplified using 5′-TGCTGGACCTGCAGA-ATAATGAC-3′(sense) and 5′-AGGTTCAAAGCCGCTGTTCTC-3′(antisense), and decorin cDNA was amplified with 5′-GGATTGAACCAGA-TGATCGTCG-3′(sense), and TGTACTTATGATCGGCCACGC-3′(antisense). The amplified cDNA was cloned into a pGEM-T plasmid (Promega), and the identity of the inserted DNA was confirmed by sequence analysis. Bovine collagen α2(I) cDNA was amplified using 5′-GGTCGAAGTGGAGAGACAGG-3′(sense) and 5′-AGGTTCACCCA-CAGATCCAG-3′(antisense), and collagen α1(III) cDNA was amplified using 5′-TGGGTTGATCCTAACCAAGG-3′(sense) and 5-CCCAGTGT-GTTTTGTGCAAC-3′(antisense). Sequence of the collagen templates was confirmed by direct analysis of the amplified products. Murine ribosomal RNA and glyceraldhyde-3-phosphate dehydrogenase cDNA templates were obtained from Ambion Inc. Hybridization was carried out in 5 ml of Ultrahyb buffer (Ambion) at 45 °C overnight. The blots were washed twice with 2×SSC, 0.1% SDS at room temperature and twice with 0.2×SSC, 0.1% SDS at 45 °C for 30 min. After autoradiography, blots were stripped of labeled probes using proprietary buffers obtained from Ambion Inc. Autoradiograms in each figure were generated from a single blot, repeatedly stripped, and reprobed. Each result was repeated at least two times.
Immunohistochemistry
Primary keratocyte cultures in eight-well glass chamber slides (Nunc; catalog no. 177402) were rinsed briefly in room temperature PBS, fixed for 10–15 min in 3% paraformaldehyde in PBS at room temperature, rinsed in PBS, and stored at −4 °C in 50% glycerol in PBS until staining. Nonspecific binding was blocked with 1% bovine serum albumin in PBS. Paxillin was detected using monoclonal antibody clone PXC-10 (Sigma; catalog no. P1093) (1:400 dilution for 2 h) and then with Cy3-conjugated goat anti-mouse secondary (Accurate Chemical & Scientific; catalog no. JMG-165108) for 1 h. Filamentous actin was detected with fluorescein isothiocyanate-phalloidin (Sigma; catalog no. P5282) 2 μg/ml for 1 h. The double-labeled samples were photographed using a Zeiss LSM 410 confocal microscope with a 63×oil objective.
RESULTS
Cytoskeleton-Matrix Interactions in Response to TGF-β
We recently demonstrated that cultured primary bovine keratocytes maintained in low mitogen horse serum retain a morphology and proteoglycan expression characteristic of these cells in vivo (18, 19). Cultured rabbit keratocytes are also reported to maintain a stellate morphology in serum-free conditions and respond to TGF-β by reorganization of the actin cytoskeleton (15). In initial experiments, we found that cultures of primary bovine keratocytes exposed to TGF-β respond in a manner similar to that reported for rabbit keratocytes. Cultures maintained for 6 days in 1% platelet-poor horse serum exhibited a stellate morphology with extensive branched processes (Fig. 1A). Filamentous actin detected by phalloidin binding showed a limited cortical localization and was not organized into distinct stress fibers. Paxillin staining was diffuse. When cultured in the presence of 1 ng/ml TGF-β1 (Fig. 1B), the keratocytes showed marked alteration in morphology. Extensive actin stress fibers crossed the cell body and were anchored to the extracellular matrix via focal adhesions as demonstrated by basal accumulations of paxillin associated with the termini of the actin cables.
Fig. 1. Cytoskeletal reorganization in bovine keratocytes in response to TGF-β.
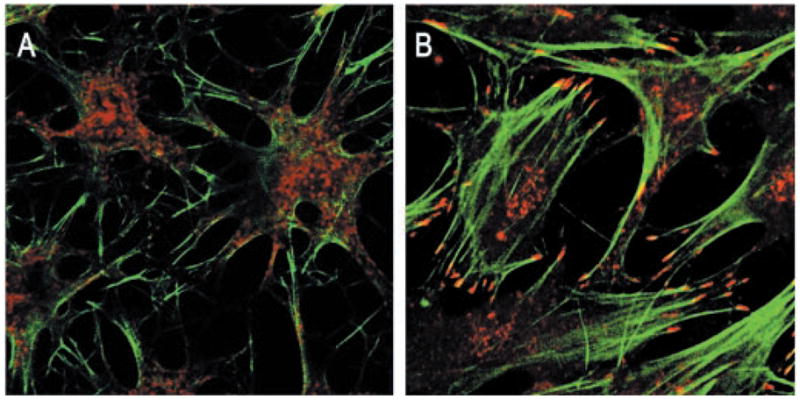
Primary keratocytes cultured 6 days in 1% platelet-poor horse serum (A) or cultured similarly with 1 ng/ml TGF-β(B) were fixed and stained with fluorescein isothiocyanate-phalloidin for filamentous actin (green) and anti-paxillin (red) to show focal adhesions as described under “Materials and Methods.”
The time course of the alteration in cytoskeletal/matrix interactions was documented by the data in Fig. 2. Fibronectin, initially not associated with the cell layer, begins to accumulate rapidly after 2 days of exposure to TGF-β(Fig. 2A). α5 integrin, a component of the fibronectin receptor (a5β1), like fibronectin, is not observed in the differentiated keratocyte but accumulates in the cells after 1 day of exposure to TGF-β(Fig. 2B). The appearance of α-smooth muscle actin (Fig. 2C) is concurrent with fibronectin and α5 integrin. The presence of this muscle-specific protein marks acquisition of a contractile myofibroblastic phenotype in response to TGF-β.
Fig. 2. Time course of matrix/cytoskeletal reorganization.
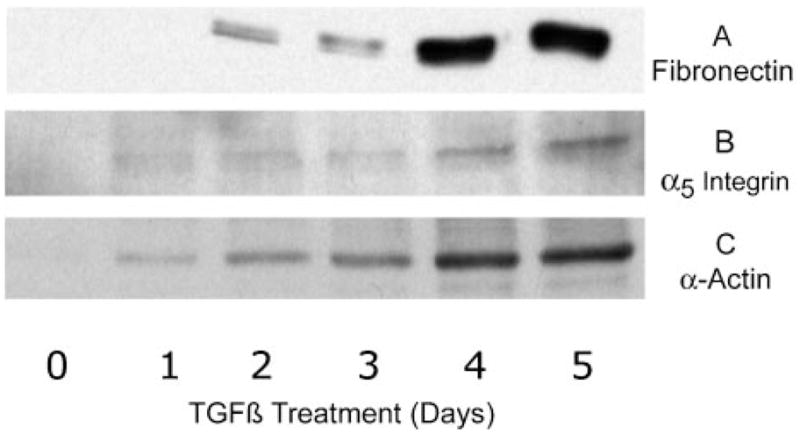
Six-day cultures of primary keratocytes were exposed to 1 ng/ml TGF-β for the length of time shown. The rinsed cell layer was solubilized in SDS sample buffer, and cell-associated proteins were detected by immunoblotting as described under “Materials and Methods” with anti-fibronectin (A), anti-α5 integrin (B), and anti-α smooth muscle actin antibodies (C).
Proteoglycans
In vivo keratocytes secrete keratan sulfate glycosaminoglycan chains modifying three proteins (lumican, keratocan, and mimecan) and dermatan sulfate chains primarily attached to decorin. Alteration of this proteoglycan secretion profile in response to TGF-β was examined by isolating keratan and dermatan sulfate-containing proteoglycans, metabolically labeled in the glycosaminoglycan moieties with [35S]sulfate or in the protein component with [35S]methionine. As shown in Fig. 3A, incorporation of [35S]sulfate into keratan sulfate decreased dramatically in response to TGF-β treatment, whereas incorporation into dermatan sulfate (Fig. 3B) was increased. Interestingly, labeling with [35S]methionine documented a moderate decrease for both proteoglycan fractions in response to TGF-β. The ratio of sulfate (glycosaminoglycan) to methionine (protein) in the secreted proteoglycans consequently responded in an opposite fashion for the two proteoglycan fractions (Fig. 3C). Keratan sulfate/protein ratios dropped to 10 –20% of the control value over a period of 2–3 days of TGF-β exposure, whereas dermatan sulfate proteoglycans increased by about 2-fold over the same time period. Qualitative SDS-PAGE gel analysis of the [35S]sulfate-labeled proteoglycans (Fig. 4) was consistent with the results of the quantitative analysis of Fig. 3. Keratan sulfate proteoglycans (Fig. 4A) were markedly decreased in abundance and simultaneously underwent a decrease in molecular size in response to TGF-β. Sulfate-labeled dermatan sulfate proteoglycans (Fig. 4B), conversely, increased in abundance and in size during TGF-β treatment. A minor component of high molecular size was observed after 3 days of treatment.
Fig. 3. Proteoglycan biosynthesis in response to TGF-β.

Six-day cultures of primary keratocytes were exposed to 1 ng/ml TGF-β for the length of time shown and labeled with [35S]sulfate (filled symbols) or [35S]methionine (open symbols) in the final 18 h of culture. Proteoglycans isolated from culture media were separated into pools containing keratan sulfate (A) and dermatan sulfate (B) as described under “Materials and Methods.” C, the ratio of sulfate/methionine incorporation was calculated for KSPG (circles) and for dermatan sulfate proteoglycans (triangles). Data are expressed as a percentage of control cultures. Error bars show standard deviation among triplicate samples.
Fig. 4. SDS-PAGE analysis of proteoglycans.
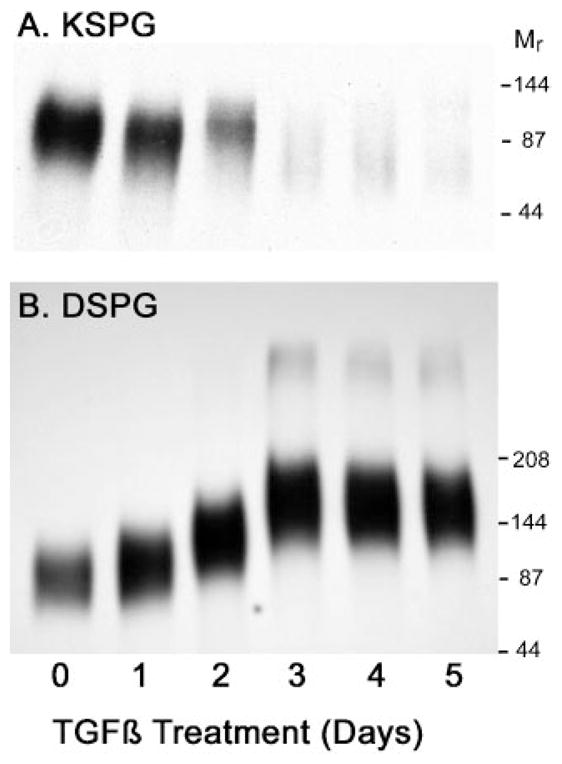
Secreted proteoglycans labeled with [35S]sulfate as described in Fig. 3 were separated by electrophoresis on 3–8% SDS-PAGE gels and detected by autoradiography as described under “Materials and Methods.” A, KSPG, keratan sulfate containing proteoglycans; B, DSPG, dermatan sulfate proteoglycans; Mr; molecular weight of prestained protein standards.
The primary dermatan sulfate-containing stromal proteoglycan is decorin (23), but biglycan, a similar proteoglycan that is not present in normal stroma, has been observed to accumulate in corneas with chronic pathological conditions (10). Biglycan protein contains two glycosaminoglycan attachment sites and in most tissues exhibits a higher molecular size than decorin (10, 24). Immunoprecipitation of the sulfate-labeled dermatan sulfate-containing proteoglycans from TGF-β-treated cultures with monospecific antibodies to decorin and biglycan (Fig. 5) showed the high molecular weight dermatan sulfate proteoglycan accumulating in response to TGF-β to precipitate with the anti-biglycan antibody but not the antibody to decorin.
Fig. 5. Immunoprecipitation of decorin and biglycan proteoglycans.
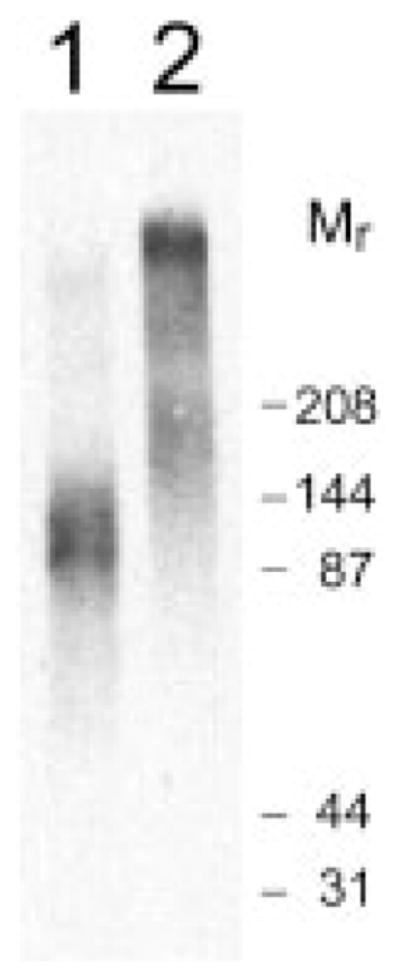
Sulfate-labeled dermatan sulfate proteoglycans from keratocytes treated with TGF-β for 6 days (as described in Fig. 3) were isolated by immunoprecipitation with peptide antibodies LF96 to decorin (lane 1) or LF94 to biglycan (lane 2), separated by SDS-PAGE, and detected by autoradiography as described under “Materials and Methods.” Mr, molecular weight of prestained protein standards.
Detection by immunoblotting of core proteins released from nonlabeled secreted proteoglycans (Fig. 6) supported the conclusions of Fig. 3, suggesting that KSPG proteins undergo a modest overall decrease after 5–6 days of exposure to TGF-β. Secreted decorin also decreased somewhat in abundance over the 6-day treatment period. Biglycan, on the other hand, was not detected in untreated cultures but appeared in the pool of secreted proteoglycan within 2 days of TGF-β treatment and increased over several days.
Fig. 6. Proteoglycan protein response to TGF-β.
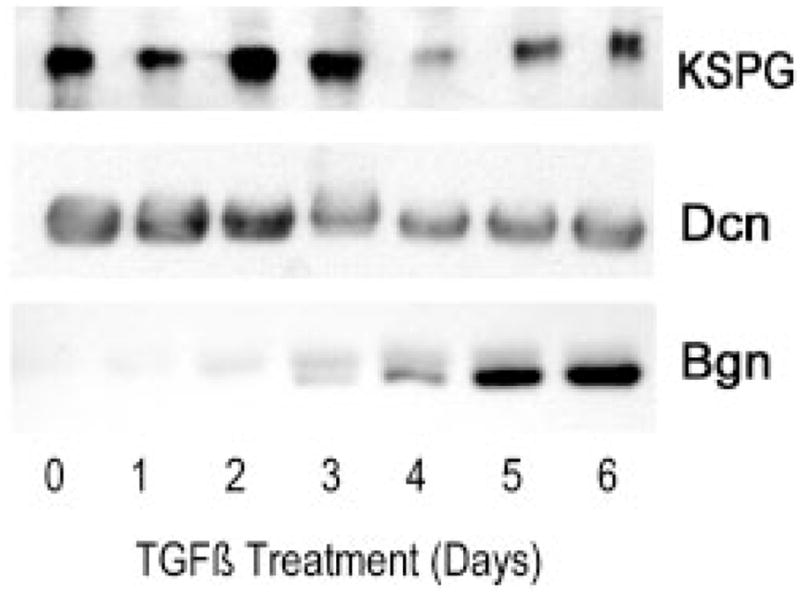
KSPG proteins from nonlabeled culture medium of a TGF-β time course similar to that in Fig. 3 were isolated and treated with endo-β-galactosidase to release keratan sulfate-linked proteins. These were detected by immunoblotting from a 10% SDS-PAGE gel using an affinity-purified antibody that recognizes all three bovine corneal keratan sulfate-linked proteins as described under “Materials and Methods.” Dermatan sulfate-linked proteins were released by chondroitinase ABC digestion. Decorin (Dcn) and biglycan (Bgn) were detected by immunoblotting as described under “Materials and Methods.”
Pools of mRNA for the KSPG proteins, detected by Northern blotting in Fig. 7, exhibited a pattern of alterations similar to that seen in the secreted proteoglycan proteins. Lumican transcripts underwent a moderate decrease, whereas those for mimecan exhibited a more marked reduction. Mimecan transcripts occur in several alternately spliced forms, and it appeared that TGF-β treatment shifted the ratio of these in addition to decreasing their overall abundance. Keratocan transcripts were present at the limit of detection and did not exhibit much apparent alteration during TGF-β treatment. RNA transcripts of decorin, similar to the protein levels, were reduced somewhat by TGF-β, and expression of biglycan mRNA, almost undetectable in untreated cultures, was rapidly up-regulated (Fig. 8).
Fig. 7. Messenger RNA pools of KSPG-linked proteins.
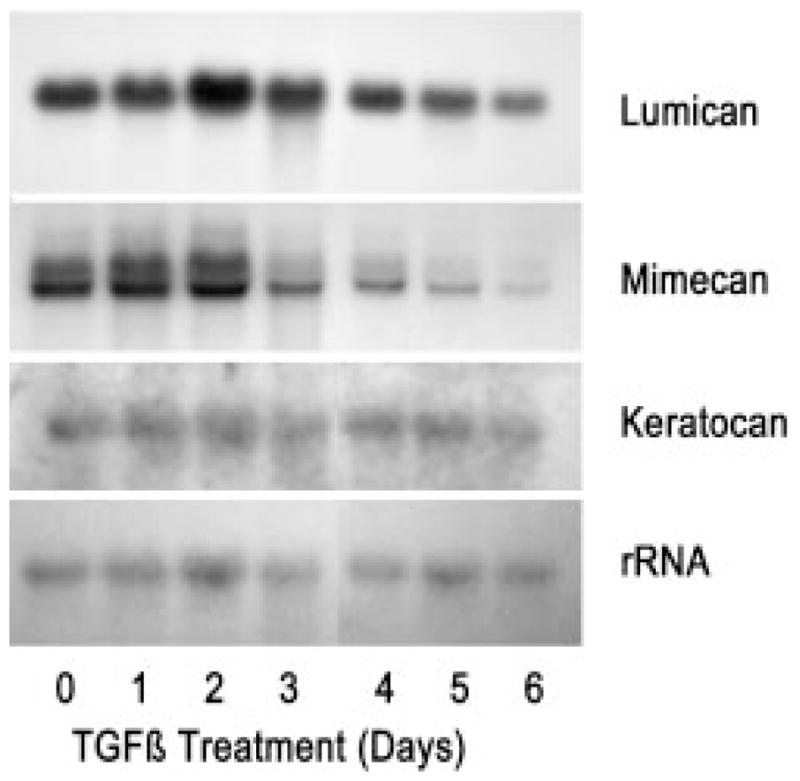
Five-μg samples of total RNA from TGF-β-treated cultures similar to those in Fig. 3 were separated by gel electrophoresis and subjected to sequential blotting for lumican, mimecan, keratocan, and rRNA, as described under “Materials and Methods.”
Fig. 8. Messenger RNA pools for dermatan sulfate-linked proteins.
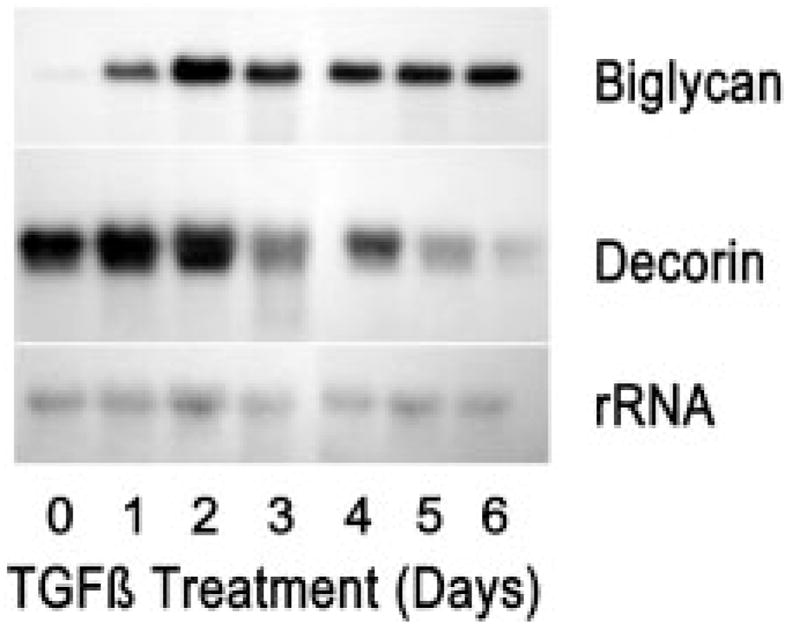
Five-μg samples of total RNA from 6-day primary cultures of bovine keratocytes exposed to 1 ng/ml TGF-β for differing times were separated and probed for decorin and biglycan transcripts as described under “Materials and Methods.”
Collagen
In most tissues, fibrosis is associated with marked accumulation of collagen. In cornea, type I and particularly type III collagens are more abundant in healing wounds and in fibrotic regions of chronic pathological corneas and mRNA pools for these proteins are elevated (25–31). We observed that [3H]proline incorporation into pepsin-resistant protein was dramatically increased in response to TGF-β (Fig. 9A). This pepsin-resistant protein band showed reactivity with both collagen types I and type III antibodies in Western blotting. Increased pools of collagen α2(I) and α1(III) mRNA were observed in the cells by Northern blotting (Fig. 9B). This up-regulation of fibrotic collagen expression in response to TGF-β correlates with biglycan expression implicating myofibroblasts in secretion of fibrotic tissue.
Fig. 9. Up-regulation of collagen in response to TGF-β.

A, 6-day keratocyte cultures exposed to TGF-β for various lengths of time were metabolically labeled with [3H]proline, and cell layer-associated pepsin-resistant proteins were extracted and separated by SDS-PAGE as described under “Materials and Methods.” A, a single major labeled protein band was detected by autoradiography ([3H]proline). The same band was immunologically reactive by immunoblotting with antibodies to collagen I and III. B, 5-μg samples of total RNA from 6-day cultures of bovine keratocytes exposed to 1 ng/ml TGF-β for differing times were separated and probed sequentially for collagen α2(I) and collagen α1(III) transcripts and ribosomal RNA as described under “Materials and Methods.”
DISCUSSION
In this study, we found that bovine keratocytes respond to TGF-β by the development of extensive F-actin stress fibers terminating at paxillin-containing focal adhesions. The formation of new cytoskeletal elements correlates with accumulation of cell-associated fibronectin and its receptor, α5 integrin. Such a fibronectin matrix and associated cytoskeleton is not observed in keratocytes in quiescent cultures (Fig. 1A), nor is it characteristic of keratocytes in vivo (13). Expression of and interaction with a fibronectin matrix, however, represents a classic marker of the fibroblastic phenotype and is exhibited by most adherent cells in culture as well as by keratocytes that populate healing corneal wounds (13, 15, 25). Stromal fibronectin accumulation is also observed in human corneas from several chronic pathological conditions as well as in acute healing wounds (29, 32).
α-Smooth muscle actin is a protein originally thought to be restricted to muscle cells, and its appearance in fibroblastic cells has given rise to the term “myofibroblast.” Corneal myofibroblasts are contractile and have been suggested to play a role in wound closure (14). Accumulation of α-smooth muscle actin in rabbit keratocytes has been shown to depend on interaction of the fibronectin receptor with its ligand (the amino acid sequence RGD) (33). Consequently, it is thought that the corneal myofibroblast represents a phenotype derived from fibroblasts, not directly from the differentiated keratocytes, which lack cell-associated fibronectin (34). Myofibroblastic cells are widely observed in normal and pathological tissue environments, and in lung and liver there is an association of myofibroblastic cell population with tissue fibrosis (35–37).
In healing corneal wounds, increased collagen deposition is associated with the presence of keratocytes containing α-smooth actin, suggesting that the myofibroblastic phenotype identifies cells responsible for deposition of fibrotic tissue (38). Only low level collagen expression is observed in normal adult cornea, but in the late phase of healing wounds, a marked increase in collagen biosynthesis and mRNA pools has been demonstrated (39). Type III collagen, not a component of normal stroma, is detected in stromal fibrotic tissue (26, 27, 29, 30) and as such represents a good marker for fibrosis. Our demonstration that up-regulation of type I and III collagen expression (Fig. 9) correlates with α-smooth actin accumulation in vitro strengthens the hypothesized link between myofibroblastic cells and fibrotic tissue deposition in the cornea.
Numerous earlier reports have documented lack of keratan sulfate biosynthesis by fibroblastic cultures of keratocytes maintained in fetal bovine serum and subcultured by trypsinization (40). Our more recent studies found that keratocytes in serum-free or reduced serum media secrete keratan sulfate for extended periods (18, 19). Cell proliferation induced by basic fibroblast growth factor does not down-regulate keratan sulfate biosynthesis, suggesting that cell division per se is not involved. Our current data show a dramatic and rapid effect of TGF-β on keratan sulfate biosynthesis. Stromal fibroblast cultures both produce and respond to TGF-β in an auto-crine fashion (16, 41). TGF-β is also present in wounded and pathological corneas (42). Considered together, these results suggest that TGF-β may be the primary agent responsible for keratan sulfate loss in fibroblastic cultures and in the cornea during wound healing and inflammation. The loss of keratan sulfate must precede or be independent of myofibroblast formation, because although not all cultures of stromal fibroblasts exhibit α-smooth actin expression, none have been found to express keratan sulfate (16).
In the current experiments, exposure to TGF-β caused a moderate decrease in mRNA pools for keratan sulfate-linked proteins and a similar decrease in KSPG core proteins labeled with methionine or detected by immunoblotting. Incorporation of sulfate into keratan sulfate on these proteoglycans, on the other hand, was rapidly and dramatically reduced by TGF-β, resulting in a change in the KS/protein ratio of 10-fold compared with untreated cells (Fig. 1). This result suggests a change in the size and/or sulfation of the KS chains modifying the KSPG proteins. The decrease in KSPG molecular size (Fig. 4) is also consistent with an alteration in the glycosylation of the KSPG proteins. Altered keratan sulfate glycoforms were identified in cultures of stromal fibroblasts (40). In these cells, lumican, keratocan, and mimecan were modified with oligolac-tosamine chains of low sulfation (40). These data suggest that reduction of keratan sulfate biosynthesis comes as a result of changes in glycosyl- or sulfotransferase activity in the keratocytes. A report by Nakazawa et al. (43) documented loss of N-acetylglucosamine-6-O-sulfotransferase activity in chicken keratocytes exposed to serum in vitro, providing a candidate enzyme for this modulation.
Dermatan sulfate-linked proteoglycans secreted by keratocytes respond in a reciprocal manner to the keratan sulfate proteoglycans (Fig. 3). Overall, the biosynthesis of dermatan sulfate-linked proteins decreased in response to TGF-β, whereas the intact proteoglycans increased in size and sulfation. These results suggest an increase in the length and/or sulfation of the dermatan sulfate chains modifying these proteins. Such alterations in corneal dermatan sulfate have been observed in healing wounds and in human corneas undergoing rejection, suggesting that this response in vitro models fibrosis in vivo (8 –11, 44, 45). The exact nature of the structural changes in the dermatan sulfate and the enzymes that effect these changes remain to be elucidated.
The most marked change in proteoglycan proteins secreted in response to TGF-β was in biglycan. Biglycan is not present in the normal stroma and neither biglycan mRNA nor biglycan protein secretion was detected in untreated cultured keratocytes. Within 24 h of TGF-β treatment, however, biglycan mRNA was detected and protein was detected in 48 h (Figs. 6 and 8). Biglycan transcripts and protein increased during several days of exposure to TGF-β. We previously found biglycan to be markedly increased in the stromas of human corneas with chronic pathological conditions (10). The large molecular size of the biglycan secreted by keratocytes in response to TGF-β is similar to a large molecular weight dermatan sulfate proteoglycan observed in healing rabbit corneal wounds (11). Biglycan accumulation is also associated with fibrosis in lung and hepatic tissues (37, 46). These data together with our current observations suggest biglycan as a sensitive marker of fibrotic tissue deposition in the stroma.
The up-regulation of biglycan in the keratocyte appears to be transcriptional; i.e. biglycan mRNA was not detected in the untreated cells. The biglycan gene promoter is reported to contain TGF-β response elements; however, biglycan transcription does not necessarily respond directly to TGF-β (47). In the keratocyte system, we observed that biglycan expression in primary cultures was not altered by TGF-β in the complete absence of serum (data not shown). Consequently, the current experiments were carried out in 1% platelet-poor horse serum, a medium that maintains much of the keratocyte differentiated phenotype (19). Rabbit keratocytes do become myofibroblastic in response to TGF-β in serum-free conditions (15), a transition dependent on interaction with extracellular RGD-containing ligands (33). Thus, the response to TGF-β in both systems appears to require signals in addition to TGF-β.
Acknowledgments
We thank Dr. Larry Fisher for donation of antibodies to decorin and biglycan and Dr. Gary W. Conrad for support in several aspects of this work.
Footnotes
This work was supported by National Institutes of Health Grants EY09368 (to J. L. F.), P30-EY08098 (University of Pittsburgh Department of Ophthalmology Core Grant), and EY00952 (to Gary W. Conrad); Research to Prevent Blindness; and the Eye and Ear Foundation of Pittsburgh.
The abbreviations used are: KSPG, keratan sulfate proteoglycan; TGF-β, transforming growth factor β; PAGE, polyacrylamide gel electrophoresis; PVDF, polyvinylidene difluoride.
References
- 1.Corpuz LM, Funderburgh JL, Funderburgh ML, Bottomley GW, Prakash S, Conrad GW. J Biol Chem. 1996;271:9759–9763. doi: 10.1074/jbc.271.16.9759. [DOI] [PubMed] [Google Scholar]
- 2.Funderburgh JL, Funderburgh ML, Brown SJ, Vergnes JP, Hassell JR, Mann MM, Conrad GW. J Biol Chem. 1993;268:11874–11880. [PubMed] [Google Scholar]
- 3.Funderburgh JL, Corpuz LM, Roth MR, Funderburgh ML, Tasheva ES, Conrad GW. J Biol Chem. 1997;272:28089–28095. doi: 10.1074/jbc.272.44.28089. [DOI] [PubMed] [Google Scholar]
- 4.Chakravarti S, Magnuson T, Lass JH, Jepson KJ, LaMantia C, Carroll H. J Cell Biol. 1998;141:1277–1286. doi: 10.1083/jcb.141.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hassell JR, Newsome DA, Krachmer JH, Rodrigues MM. Proc Natl Acad Sci U S A. 1980;77:3705–3709. doi: 10.1073/pnas.77.6.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ying S, Shiraishi A, Kao CW, Converse RL, Funderburgh JL, Swiergiel J, Roth MR, Conrad GW, Kao WW. J Biol Chem. 1997;272:30306–30313. doi: 10.1074/jbc.272.48.30306. [DOI] [PubMed] [Google Scholar]
- 7.Cintron C, Kublin CL. Dev Biol. 1977;61:346–357. doi: 10.1016/0012-1606(77)90304-9. [DOI] [PubMed] [Google Scholar]
- 8.Cintron C, Gregory JD, Damle SP, Kublin CL. Invest Ophthalmol Vis Sci. 1990;31:1975–1981. [PubMed] [Google Scholar]
- 9.Funderburgh JL, Chandler JW. Invest Ophthalmol Vis Sci. 1989;30:435–442. [PubMed] [Google Scholar]
- 10.Funderburgh JL, Hevelone ND, Roth MR, Funderburgh ML, Rodrigues MR, Nirankari VS, Conrad GW. Invest Ophthalmol Vis Sci. 1998;39:1957–1964. [PubMed] [Google Scholar]
- 11.Hassell JR, Cintron C, Kublin C, Newsome DA. Arch Biochem Biophys. 1983;222:362–369. doi: 10.1016/0003-9861(83)90532-5. [DOI] [PubMed] [Google Scholar]
- 12.Matsuda H, Smelser GK. Exp Eye Res. 1973;16:427–442. doi: 10.1016/0014-4835(73)90100-0. [DOI] [PubMed] [Google Scholar]
- 13.Jester JV, Barry PA, Lind GJ, Petroll WM, Garana R, Cavanagh HD. Invest Ophthalmol Vis Sci. 1994;35:730–743. [PubMed] [Google Scholar]
- 14.Garana RM, Petroll WM, Chen WT, Herman IM, Barry P, Andrews P, Cavanagh HD, Jester JV. Invest Ophthalmol Vis Sci. 1992;33:3271–3282. [PubMed] [Google Scholar]
- 15.Jester JV, Barry-Lane PA, Cavanagh HD, Petroll WM. Cornea. 1996;15:505–516. [PubMed] [Google Scholar]
- 16.Masur SK, Dewal HS, Dinh TT, Erenburg I, Petridou S. Proc Natl Acad Sci U S A. 1996;93:4219–4223. doi: 10.1073/pnas.93.9.4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moller-Pedersen T, Cavanagh HD, Petroll WM, Jester JV. Curr Eye Res. 1998;17:736–747. [PubMed] [Google Scholar]
- 18.Beales MP, Funderburgh JL, Jester JV, Hassell HR. Invest Ophthalmol Vis Sci. 1999;40:1658–1663. [PubMed] [Google Scholar]
- 19.Long CJ, Roth MR, Tasheva ES, Funderburgh M, Smit R, Conrad GW, Funderburgh JL. J Biol Chem. 2000;275:13918–13923. doi: 10.1074/jbc.275.18.13918. [DOI] [PubMed] [Google Scholar]
- 20.Jost CJ, Funderburgh JL, Mann M, Hassell JR, Conrad GW. J Biol Chem. 1991;266:13336–13341. [PubMed] [Google Scholar]
- 21.Funderburgh JL, Funderburgh ML, Mann MM, Conrad GW. J Biol Chem. 1991;266:14226–14231. [PubMed] [Google Scholar]
- 22.Fisher LW, Stubbs JT, 3rd, Young MF. Acta Orthop Scand Suppl. 1995;266:61–65. [PubMed] [Google Scholar]
- 23.Blochberger TC, Vergnes JP, Hempel J, Hassell JR. J Biol Chem. 1992;267:347–352. [PubMed] [Google Scholar]
- 24.Fisher LW, Termine JD, Young MF. J Biol Chem. 1989;264:4571–4576. [PubMed] [Google Scholar]
- 25.Maguen E, Alba SA, Burgeson RE, Butkowski RJ, Michael AF, Kenney MC, Nesburn AB, Ljubimov AV. Cornea. 1997;16:675–682. [PubMed] [Google Scholar]
- 26.Kato T, Nakayasu K, Kanai A. Jpn J Ophthalmol. 2000;44:334–341. doi: 10.1016/s0021-5155(00)00168-4. [DOI] [PubMed] [Google Scholar]
- 27.Power WJ, Kaufman AH, Merayo-Lloves J, Arrunategui-Correa V, Foster CS. Curr Eye Res. 1995;14:879–886. doi: 10.3109/02713689508995127. [DOI] [PubMed] [Google Scholar]
- 28.Dutt JE, Ledoux D, Baer H, Foster CS. Cornea. 1996;15:606–611. [PubMed] [Google Scholar]
- 29.Ljubimov AV, Burgeson RE, Butkowski RJ, Couchman JR, Wu RR, Ninomiya Y, Sado Y, Maguen E, Nesburn AB, Kenney MC. Invest Ophthalmol Vis Sci. 1996;37:997–1007. [PubMed] [Google Scholar]
- 30.Ljubimov AV, Alba SA, Burgeson RE, Ninomiya Y, Sado Y, Sun TT, Nesburn AB, Kenney MC, Maguen E. Exp Eye Res. 1998;67:265–272. doi: 10.1006/exer.1998.0511. [DOI] [PubMed] [Google Scholar]
- 31.Delaigue O, Arbeille B, Rossazza C, Lemesle M, Roingeard P. Graefes Arch Clin Exp Ophthalmol. 1995;233:331–338. doi: 10.1007/BF00200481. [DOI] [PubMed] [Google Scholar]
- 32.Jester JV, Petroll WM, Barry PA, Cavanagh HD. Invest Ophthalmol Vis Sci. 1995;36:809–819. [PubMed] [Google Scholar]
- 33.Jester JV, Huang J, Barry-Lane PA, Kao WW, Petroll WM, Cavanagh HD. Invest Ophthalmol Vis Sci. 1999;40:1959–1967. [PubMed] [Google Scholar]
- 34.Fini ME. Prog Retin Eye Res. 1999;18:529–551. doi: 10.1016/s1350-9462(98)00033-0. [DOI] [PubMed] [Google Scholar]
- 35.Badid C, Mounier N, Costa AM, Desmouliere A. Histol Histopathol. 2000;15:269–280. doi: 10.14670/HH-15.269. [DOI] [PubMed] [Google Scholar]
- 36.Stokes MB, Holler S, Cui Y, Hudkins KL, Eitner F, Fogo A, Alpers CE. Kidney Int. 2000;57:487–498. doi: 10.1046/j.1523-1755.2000.00868.x. [DOI] [PubMed] [Google Scholar]
- 37.Venkatesan N, Ebihara T, Roughley PJ, Ludwig MS. Am J Respir Crit Care Med. 2000;161:2066–2073. doi: 10.1164/ajrccm.161.6.9909098. [DOI] [PubMed] [Google Scholar]
- 38.Ishizaki M, Zhu G, Haseba T, Shafer SS, Kao WW. Invest Ophthalmol Vis Sci. 1993;34:3320–3328. [PubMed] [Google Scholar]
- 39.Cionni RJ, Katakami C, Lavrich JB, Kao WW. Curr Eye Res. 1986;5:549–558. doi: 10.3109/02713688609015118. [DOI] [PubMed] [Google Scholar]
- 40.Funderburgh JL, Funderburgh ML, Mann MM, Prakash S, Conrad GW. J Biol Chem. 1996;271:31431–31436. doi: 10.1074/jbc.271.49.31431. [DOI] [PubMed] [Google Scholar]
- 41.Song QH, Singh RP, Richardson TP, Nugent MA, Trinkaus-Randall V. J Cell Biochem. 2000;77:186–199. doi: 10.1002/(sici)1097-4644(20000501)77:2<186::aid-jcb3>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 42.Vesaluoma MH, Tervo TT. J Refract Surg. 1998;14:447–454. doi: 10.3928/1081-597X-19980701-11. [DOI] [PubMed] [Google Scholar]
- 43.Nakazawa K, Takahashi I, Yamamoto Y. Arch Biochem Bio-phys. 1998;359:269–282. doi: 10.1006/abbi.1998.0897. [DOI] [PubMed] [Google Scholar]
- 44.Anseth A, Fransson LA. Exp Eye Res. 1969;8:302–309. doi: 10.1016/s0014-4835(69)80043-6. [DOI] [PubMed] [Google Scholar]
- 45.Sawaguchi S, Yue BY, Chang I, Sugar J, Robin J. Invest Ophthalmol Vis Sci. 1991;32:1846–1853. [PubMed] [Google Scholar]
- 46.Hogemann B, Edel G, Schwarz K, Krech R, Kresse H. Pathol Res Pract. 1997;193:747–751. doi: 10.1016/S0344-0338(97)80052-0. [DOI] [PubMed] [Google Scholar]
- 47.Ungefroren H, Krull NB. J Biol Chem. 1996;271:15787–15795. doi: 10.1074/jbc.271.26.15787. [DOI] [PubMed] [Google Scholar]