

Abstract
The present review focuses on a unique bio-molecular construction kit based on surface-layer (S-layer) proteins as building blocks and patterning elements, but also major classes of biological molecules such as lipids, membrane-active peptides and membrane proteins, and glycans for the design of functional supported lipid membranes. The biomimetic approach copying the supramolecular building principle of most archaeal cell envelopes merely composed of a plasma membrane and a closely associated S-layer lattice has resulted in robust and fluid lipid membranes. Most importantly, S-layer supported lipid membranes spanning an aperture or generated on solid and porous substrates constitute highly interesting model membranes for the reconstitution of responsive transmembrane proteins and membrane-active peptides. This is of particular challenge as one-third of all proteins are membrane proteins such as pore-forming proteins, ion channels, and receptors. S-layer supported lipid membranes are seen as one of the most innovative strategies in membrane protein-based nanobiotechnology with potential applications that range from pharmaceutical (high-throughput) drug screening over lipid chips to the detection of biological warfare agents.
I. INTRODUCTION
Model membranes (lipid mono- or bilayers with associated or integral proteins) have attracted lively interest in recent years as the advances in genome mapping revealed that approximately one-third of the genes of an organism encode for membrane proteins like pores, ion channels, receptors, and membrane-anchored enzymes.1,2 These proteins are key factors in the cell’s metabolism, for example in cell-cell interaction, signal transduction, and transport of ions and nutrients, and, thus, in health and disease.3 Due to this important function, membrane proteins are a preferred target for pharmaceuticals (at present more than 60% of consumed drugs)4 and have received widespread recognition for their application in drug discovery, protein-ligand screening, and biosensors.
Contrary to this fundamental role in biology, accessibility of membrane proteins by experimental techniques remains challenging. Although more than 47 000 3-D structures of proteins have been resolved up to date,5 only approximately 250 of those are membrane proteins.6 Membrane proteins exhibit an amphiphilic character and, thus, require a lipid matrix to adopt their proper structure and function.7,8 Biological systems are exceedingly complex and, in order to avoid uncontrollable interactions and to gain understanding of their basic mechanism, it is often necessary to reduce the number of parameters like the quantity and broad variety of lipid species, peripheral and integral objectionable membrane proteins, or glycosylated biomolecules. One approach to elucidate the functioning of ion channels or receptor proteins is their reconstitution into planar lipid membranes.
The present review intends to give a summary on a particular biomimetic planar lipid membrane, which consists of, besides the lipid matrix, a closely associated proteinaceous surface (S-) layer lattice as a stabilizing and tethering structure. S-layers are crystalline bacterial cell surface layers9,10 and constitute one of the most common outermost cell envelope components of prokaryotic organisms (archaea and bacteria, Fig. 1).11 The template for such a composite membrane is the cell envelope structure of gram-negative archaea, which is composed of a plasma membrane, a closely attached or even integrated or penetrating S-layer lattice, and embedded and integral membrane proteins. Mimicking this building principle, membranes composed of artificial or isolated lipid molecules can be stabilized by the closely attached S-layer lattice or, the other way round, on S-layer lattices, membranes combining stability and fluidity can be generated.12-15 Finally, model membrane proteins but also membrane-active peptides can be incorporated into these composite structures and their biological function could be demonstrated on the single functional unit level.
FIG.1.

In (a), freeze-etching preparation of a whole cell of Bacillus sphaericus with a square S-layer lattice is shown. Bar corresponds to 200 nm. Schematic illustration of the supramolecular architecture of the three major classes of prokaryotic cell envelopes containing crystalline bacterial cell surface layers (S-layers). (b) Cell envelope structure of gram-negative archaea with S-layers as the only component external to the cytoplasmic membrane. (c) Cell envelope as observed in gram-positive archaea and bacteria. In bacteria the rigid wall component is primarily composed of peptidoglycan. In archaea other wall polymers (e.g., pseudomurein) are found. (d) Cell envelope profile of gram-negative bacteria composed of a thin peptidoglycan layer and an outer membrane. If present, the S-layer is closely associated with the lipopolysaccharide of the outer membrane. Modified after Ref. 15. Copyright 1999 Reprinted with permission from Wiley-VCH.
II. DESCRIPTION OF S-LAYER PROTEINS AND SCWP
While considerable variation exists in the complexity and structure of prokaryotic cell envelopes, it is possible to classify cell envelope profiles into their main groups on the basis of structure, biochemistry, and function [Figs. 1(b)–1(d)]. It is now evident that one of the most common surface structures on archaea and bacteria are monomolecular crystalline arrays of proteinaceous subunits [Fig. 1(a)] termed S-layers.10,16 Chemical analysis and genetic studies on a variety of S-layers have shown that with few exceptions they are composed of a single homogeneous protein or glycoprotein species with molecular masses ranging from 40 to 200 kD.17,18 These are often weakly acid proteins with isoelectric points in the range of 4 to 6. A few post-translational modifications are known to occur in S-layer proteins, including protein phosphorylation and protein glycosylation. While most archaeal S-layer proteins appear to be glycosylated, this posttranslational modification is much less common among bacteria.17,19
S-layer subunits can be aligned in lattices with oblique, square, or hexagonal symmetry (Fig. 2) with a center-to-center spacing of the morphological units of approximately 3–35 nm. Hexagonal lattice symmetry is predominantly observed at archaea.20-22 High resolution electron microscopy and scanning probe microscopy revealed that most S-layers are 5 to 25 nm thick and have a rather smooth outer surface and a more corrugated inner surface. Among S-layer lattices of archaea, pillarlike extensions on the inner surface may even penetrate into the plasma membrane [Fig. 1(b)].23,24 Since S-layers are monomolecular assemblies of identical protein subunits, they exhibit pores of identical size and morphology. In numerous S-layer lattices more than one distinct type of pore (generally in the 2 to 8 nm range) has been identified.22,25-27
FIG. 2.
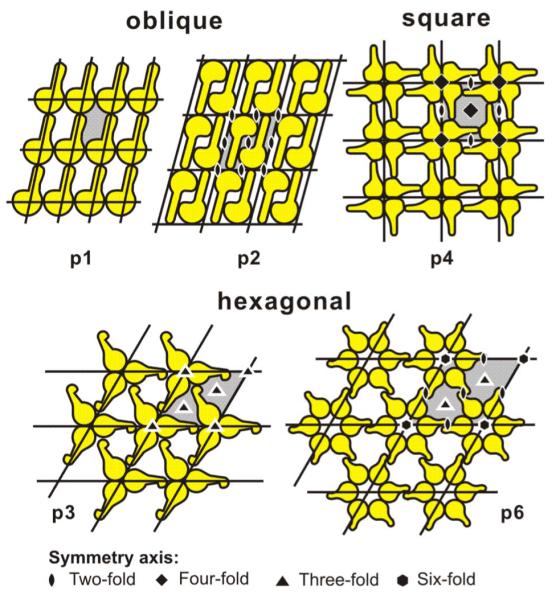
Schematic drawings of possible S-layer lattice types. Owning to the chirality of proteins, space group symmetries with mirror-reflection lines or glide-reflection lines are not possible in S-layer lattices. Modified after Ref. 27 (Bacterial surface layer proteins: a simple versatile biological self-assembly system in nature; Fig. 2). Copyright 2006 with kind permission of Springer Science and Business Media.
Labeling experiments with differently charged marker molecules and recrystallization of isolated S-layer proteins on differently charged solid supports revealed a charge-neutral outer and a net negative or net positive inner S-layer face for many Bacillaceae with respect to their native orientation on the cell. This characteristic of S-layer subunits appears to be essential for the proper orientation during local insertion in the course of lattice growth. From a general point of view, it is now evident that S-layers are dynamic closed surface crystals with the intrinsic ability to continuously assume a structure of low free energy during cell growth and cell division.
Different methods have been developed for the detachment of S-layers and for their disintegration into protomeric units.17,22,28,29 Since S-layer proteins of most bacteria interact with each other through non-covalent forces they can be solubilized with high concentrations of agents that break hydrogen bonds (e.g., guanidine hydrochloride, urea). Isolated S-layer subunits frequently maintain the ability to recrystallize upon removal (e.g., dialysis) of the disintegrating agent. Reassembly of isolated subunits at the air/water interface, on Langmuir–Blodgett (LB) films, liposomes, or a great variety of solid supports (e.g., polymers, silicon wafers, metals), has proven to be an easy and reproducible way for generating extended closed S-layer lattices.15,17,30-32 In accordance with S-layer proteins recrystallized on solid surfaces, the orientation of the protein arrays at liquid interfaces and lipid films was determined by anisotropy in the physicochemical surface properties of the protein lattice. For example, electron microscopy and scanning force microscopy examination revealed that recrystallized S-layer protein from selected gram-positive Bacillaceae [Fig. 1(c)] were oriented with their outer charged less hydrophilic surface against the air/water interface and with their negatively charged, more hydrophilic inner face against positively charged or zwitterionic head groups of phospholipid or tetraetherlipid films.33,34
In gram-positive bacteria with S-layer lattices associated with the rigid wall component [Fig. 1(c)] secondary cell wall polymers (SCWPs) have been recognized as components that facilitate a specific interaction between S-layer monomers and the peptidoglycan sacculus. In bacilli, the N-terminal region was found to be responsible for anchoring the S-layer subunits to the rigid cell envelope layer whereas in many lactobacilli binding occurs via the C-terminal portion in a defined orientation to the SCWPs. Structurally, the SCWPs resemble to some extent teichoic acids of gram-positive organisms, but in addition to the common negative charge, they can also be uncharged.35 It is now evident that the highly specific lectin-type binding between S-layer proteins and SCWPs is an important mechanism for generating and maintaining a dynamic protein crystal on a bacterial cell surface during all stages of cell growth and division.35
For many nanobiotechnological applications recrystallization of S-layer proteins in a defined orientation (e.g., on solid supports, liposomes, and lipid films) is most relevant. The biomimetic approach of functionalizing surfaces and interfaces with SCWP now enables binding and crystallization of the S-layer (fusion) proteins in a reproducible way.36
III. CONCEPT OF S-LAYER/LIPID MEMBRANES
The concept for S-layer stabilized lipid membranes (Ss-LMs) is based on a biomimetic approach.37 Biomimetics in general is the application of methods and systems found in nature to the study and design of engineering systems and modern technology. These human-made processes, substances, devices, or systems that imitate nature are of special interest to researchers in nanotechnology, robotics, artificial intelligence, the medical industry, and the military.
On closer examination of the cell envelope structure of archaea a fairly simple but highly robust building principle has been observed (Fig. 3).38,39 Concerning the robustness, it is interesting to note that some archaea dwell under very harsh conditions like temperatures up to 121 °C, pH-value even below 0, very high pressures (up to 1200 Pa), and high salt concentrations (>2 M salt).38,40 Due to this exceptional stability of archaea, a biomimetic approach utilizing the biological building principle of these cell envelope structures has been applied and resulted in the fabrication of SsLMs.12,41,42 In this architecture either a tetraetherlipid monolayer or an artificial phospholipid mono- or bilayer replaces the cytoplasmic membrane, and isolated bacterial S-layer proteins are attached either on one or both sides of the lipid film (Fig. 3).
FIG. 3.

Schematic illustration of (A) an archaeal cell envelope structure composed of the cytoplasmic membrane with integral membrane proteins and a S-layer lattice, integrated into the cytoplasmic membrane. Using this supramolecular construction principle, biomimetic membranes can be generated (B,C). The cytoplasmic membrane is replaced by a phospholipid bilayer (B) or a tetraetherlipid monolayer (C) and S-layer proteins derived from Bacillaceae are recrystallized to form a closed lattice on the lipid film. Subsequently integral model membrane proteins can be reconstituted into the S-layer-supported lipid membrane. As indicated in (B) and (C), a second S-layer lattice may be recrystallized on the top to stabilize the layered architecture and provide a nanoporous filter function. Modified after Ref. 17. Copyright (2003) with permission from Wiley-VCH.
Tetraetherlipids are bipolar, membrane-spanning lipids (Fig. 4). The hydrophobic part of the molecule consists of a 72 member macro-cycle, which is formed by two glycerol units that are bridged by two biphytanoyl chains.43 Instead of ester groups connecting in phospholipids, the alkyl chains, and the glycerol moieties, ether groups are existent in these lipids40 providing an enhanced robustness under acidic conditions.45,46
FIG. 4.
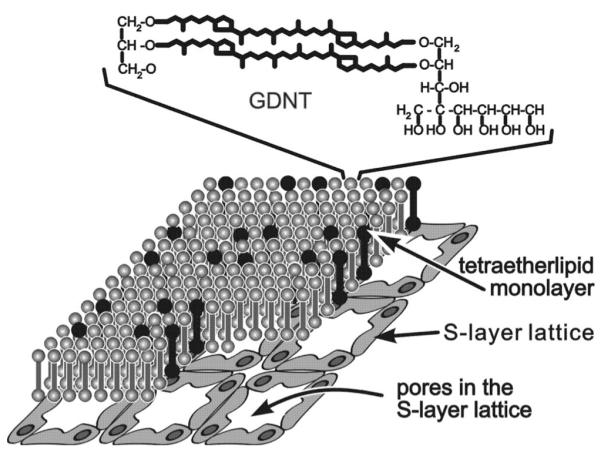
Schematic illustration of the composite S-layer/GDNT-monolayer structure. The chemical structure of the glycerol dialkyl nonitol tetraetherlipid (GDNT) molecules is shown in the top of the figure. The black colored GDNT molecules represent the most favored lipids in the GDNT monolayer whose associated head groups may interact with defined domains in the S-layer lattice (not drawn to scale). Reprinted from Ref. 44. Copyright 1998 Reprinted with permission from Elsevier.
For bacterial S-layer proteins it has been demonstrated that protein domains or functional groups on the S-layer lattice particularly interact via electrostatic forces with some head groups of the lipid molecules (Fig. 4). Primary and secondary binding sites for lipid molecules on the inner and outer face, respectively, have been postulated.33 In contrast to solid surfaces, lipid films in a phase-separated state get slightly modulated during the recrystallization process of S-layer proteins as alkane chains of the fluid lipid were driven into a state of higher order.47 However, although peptide side groups of the S-layer protein interpenetrate the phospholipid head groups almost in its entire depth, no increase of the conductivity of the lipid membrane has been observed.41 In contrast, mostly even a decrease in conductivity could be measured. Thus, S-layer lattices constitute unique supporting scaffolding for lipid membranes.14,41
IV. CHARACTERIZATION TOOLS
A. S-layer protein self-assembly to form crystalline lattices
1. Nanometer-scale microscopic tools
Transmission electron microscopy (TEM) is the classical method of imaging structures at atomic or molecular level. In most biological applications the sample has to be prepared by appropriate techniques prior to the transfer in the electron microscope in order to preserve the structure of the biological sample at molecular resolution.48 The most critical step concerns the drying of the wet sample. Negative staining techniques often go hand in hand with a chemical fixation and thus stabilization of the biological structure. With this technique, S-layer protein monolayer formation at a liquid-air interface was studied by TEM.49 Cryo techniques, however, make use of the less destructive drying when ice is directly sublimated into the vapor phase.50 But a successful cryo preparation is only possible when water contained in the sample had been so rapidly frozen initially that only amorphous and not crystalline ice was formed. This process is known as “vitrification” of ice. Freezing rates of approximately 10 000 K s−1 are mandatory. Subsequently, either freeze-etching [see Fig. 1(a)] or freeze-drying in combination with high resolution shadowing is used to make replicas of the surface topography of the frozen samples. In particular, freeze-drying is used for planar samples such as biomembranes. While the described preparation techniques are used to image the surface of the samples, ultra-thin sectioning is the only method that allows obtaining a cross section of the specimen. The sample is immersed in a polymer after substituting the water in the biological material by an appropriate organic solvent. After hardening of the polymer the sample is cut into 70–90 nm thick slices, stained, and transferred into the microscope.
An alternative approach to electron microscopical investigations is provided by scanning force microscopy (SFM).48 In SFM an ultra-fine tip is scanned over the sample surface. This can be done by either scanning in contact mode where the tip follows the corrugations of the sample directly or in noncontact mode where the tip is brought into oscillations leading to an intermittent contact with the sample. The contact mode is the high resolution imaging mode (Fig. 5) but requires extremely low forces (typically 1 nN to 100 pN) in order to avoid damaging the soft biological material. Although the noncontact mode does not yield such high resolution it is a most gentle imaging mode since the tip is only tapping over the surface. Nevertheless, both SFM imaging modes allow the investigation of the sample in its native most often wet state. Thus, SFM is the most appropriate method for studying biological materials and systems in their native environments. In addition, SFM allows us to image dynamic processes such as two-dimensional crystal growth of S-layer proteins on solid surfaces in real-time.51
FIG. 5.

SFM images (deflection mode) of (a) the S-layer protein SbpA from Bacillus sphaericus CCM 2177 recrystallized on a QCM-D gold-coated sensor surface (image size 800×800 nm2) and (b) on a SCWP-covered gold-coated sensor surface (image size 700×700 nm2).
2. Real-time analysis of molecular self-assembly
Two powerful surface-sensitive techniques are quartz crystal microbalance (QCM) with dissipation monitoring (QCM-D) and surface plasmon resonance (SPR) spectroscopy.52-54 SPR and QCM are ultra-sensitive mass sensors that monitor the real-time change in an adsorbed amount of material. In contrast to SPR, QCM senses in addition to the adsorbed amount of material also the mass of trapped and coupled water.55,56 By presenting different surfaces to a biomolecule, QCM can also differentiate between varying affinities of adsorption in response to specific surface chemistry.52,57 QCM techniques have also advanced to incorporate simultaneous measurements of energy dissipation changes during adsorption processes, which has been coined QCM-D, where D signifies the energy lost over energy stored.58 QCM-D measurements not only yield adsorbed quantity (and affinity) of hydrated material at the liquid/solid interface but directly provide the kinetics of the process and the system energy losses during the adsorption. SPR spectroscopy is an optical technique measuring optical pathlength changes and by careful calibration it can be used to detect and characterize the adsorbed mass and to investigate the kinetics of adsorption processes at a gold surface.53,54 Using QCM-D and SPR in parallel is a powerful approach enabling interesting insights in the hydration of (bio)materials and the understanding of complex self-assembly processes.
The S-layer protein SbpA from Bacillus sphaericus CCM 2177 has been recrystallized on gold-coated sensor surfaces with and without chemisorbed thiolated SCWP. The increase in mass per time has been investigated by QCM-D (E4, Q-Sense) and SPR (Biacore 2000, Biacore). Furthermore, the morphology of the S-layer protein on sensor surfaces has been investigated by SFM measurements (Nanoscope III, Digital Instruments).
The mass of the chemisorbed SCWP was determined by SPR to 380–445 ng/cm2.59 This is in good agreement with QCM-D measurements revealing a calculated protein mass of 440±50 ng/cm2 (n=13). SFM studies revealed a soft surface texture on the gold surface without any structural details of the SCWP (data not shown).
The recrystallization of SbpA on gold surfaces gave raise to an increase in mass of 620 to 650 ng/cm2 as measured by SPR.59 As at QCM-D measurements, the value for the dissipation factor D only moderately increased upon SbpA self-assembly; the frequency shift could be analyzed according to the Sauerbrey equation60 and resulted in a SbpA mass of 1360±160 ng/cm2 (n=6) (Fig. 6). The difference in mass measured by QCM-D and SPR, respectively, can be attributed to trapped water inside and coupled water to the S-layer lattice. The effective density ρeff of the self-assembled S-layer could be calculated from the SPR and QCM-D data61 by assuming a density for the protein and water of 1.35 g/cm3 (Ref. 62) and 1.0 g/cm3, respectively, and was determined to ρeff=1.14 g/cm3. These results are in good agreement with the data and calculations based on a previous study, where ~40% of the volume of the S-layer lattice is occupied by protein and ~60% consists of water.34 With this calculated ρeff it was possible to estimate the effective thickness of the S-layer on the gold surface, which was found to be 12 nm. This thickness is higher than that found for SbpA recrystallized on a phospholipid monolayer (~9 nm) as determined by x-ray reflectivity and grazing incidence diffraction studies.34 However, preliminary SFM studies investigating the thickness of the S-layer on a gold-coated surface also suggested a value of up to 12.5 nm. The SFM image showed a crystalline protein layer consisting of small S-layer patches on the gold-coated QCM-D sensor.
FIG. 6.
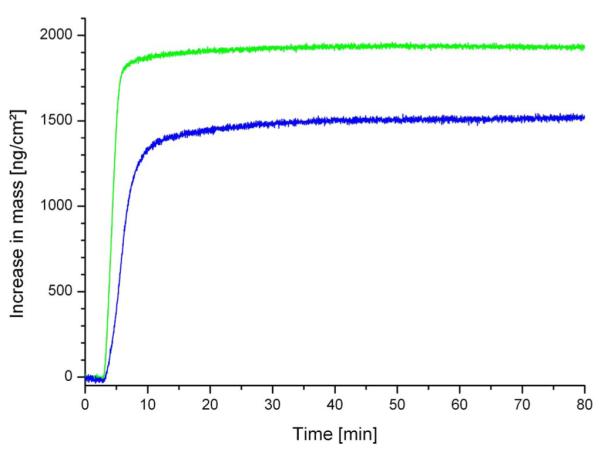
Increase in mass vs time upon the adsorption/binding and recrystallization of the S-layer protein SbpA on a gold-covered QCM-D sensor (blue line) and on a SCWP-coated gold-covered QCM-D sensor (green line).
High-resolution electron microscopical and SFM studies have shown that the protein monolayers are not monocrystalline over the entire surface, but consist of a mosaic of randomly aligned crystalline domains (Fig. 5).51 Analysis of the crystallization process revealed that crystal growth of S-layers at interfaces is initiated at randomly distributed nucleation sites composed of proteins or small protein assemblies from the bulk solution. Subsequently, the crystalline domains rapidly grow laterally in all directions until neighboring areas meet and a closed coherent monolayer is formed (Figs. 5 and 6).51 This polycrystalline layer consisted of domains 100–200 nm in diameter. No three-dimensional growth occurred at the solid support.
As determined by QCM-D, the recrystallization of SbpA on a SCWP-coated gold surface gave raise to a higher frequency shift compared to SbpA on a plain gold sensor surface and resulted in a mass of 1960±30 ng/cm2 (Fig. 6). As expected, a significantly lower amount of mass, close to the amount observed for SbpA on a plain sensor surface, was determined by SPR measurements.59 Since at QCM-D measurements the dissipation factor D was close to zero, the mass could be inferred by the Sauerbrey equation with a ρeff of 1.14 g/cm3 to a thickness of 17.2 nm. Further studies are currently in progress to elucidate the physicochemical background for the difference in thickness of approximately 5 nm between the hydrated lattice of SbpA on gold with and without a SCWP coating in more detail. The SFM image, however, showed a closed, very flat S-layer with a good long-range order of the SbpA lattice [Fig. 5(b)].
The recrystallization process was finished after approximately 40 and 25 min for SbpA on gold and SCWP-coated gold, respectively, as determined by SPR (Ref. 59) and QCM-D (Fig. 6). The maximal increase in mass per area and time was determined by both techniques and was found to be more than two times faster on the SCWP-coated compared to the plain gold sensor surface. Thus, the recrystallization of SbpA on the “natural” biomimetic surface occurred much faster (Fig. 6), most probably due to the higher flexibility of the underlying SCWP cushion allowing a fast arrangement of the S-layer subunits to form a closed S-layer lattice [Fig. 5(b)].
B. S-layer stabilized lipid membranes
1. Biosurface-sensitive techniques
The simplest biomimetic SsLMs were generated by the recrystallization of isolated bacterial S-layer proteins on a phospholipid monolayer [Fig. 7(a)].37,49,63 These SsLMs have been characterized by TEM,64,65 dual-label fluorescence microscopy, Fourier-transform infrared spectroscopy,47 and x-ray and neutron reflectivity measurements.34,66,67 Closed S-layer lattices covering the entire area of the lipid film have been observed with lipid films composed of zwitterionic phospholipids in the liquid-condensed phase. Most S-layer subunits are weakly acidic proteins and therefore show a charge-neutral outer and a net-negative inner face at a neutral pH. The S-layer lattice orientates toward the phospholipid films with its inner, net-negatively charged face with respect to its native orientation on the bacterial cell wall.49 The S-layer recrystallization process has been demonstrated to be facilitated by addition of a small portion of positively charged surfactants (e.g., hexadecylamine)68,69 or lipid derivatives.70 From this observation it has been concluded that electrostatic interactions between exposed carboxyl groups on the inner face of the S-layer lattice (primary binding sites)29 and the zwitterionic lipid head groups are primarily responsible for the defined orientation of the subunits. For such an alignment, it has been suggested that there are at least two to three contact points between the lipid film and the attached S-layer protein.33,71 In other words less than 5% of the lipid molecules are anchored to protein domains on the S-layer lattice whereas the remaining ≥95% lipid molecules may diffuse freely in the membrane between pillars consisting of anchored lipid molecules.12,41 Because of its widely retained fluid characteristic this nanopatterned lipid membrane is also referred to as “semifluid membrane” (Fig. 4).37
FIG. 7.
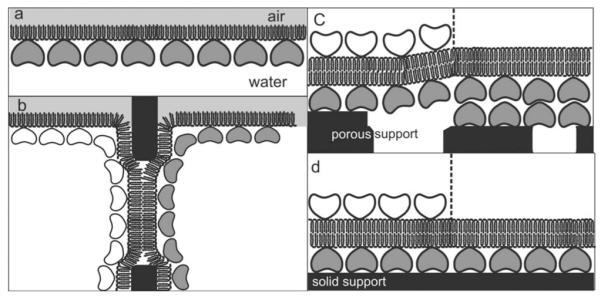
Schematic illustrations of various S-layer-supported lipid membranes. (a) S-layer protein has been recrystallized from the aqueous phase on a phospholipid monolayer. In (b), a folded or painted membrane has been generated to span a Teflon aperture. Subsequently S-layer protein can be injected into one or both compartments whereby the protein self-assembles to form closely attached S-layer lattices on the BLMs. (c) On a SUM a BLM can be generated by a modified LB technique. As a further option, a closed S-layer lattice can be attached on the external side of the SUM-supported BLM (left part). (d) Solid supports can be covered by a closed S-layer lattice and subsequently BLMs can be generated using combinations of the LB and Langmuir–Schaefer techniques and vesicle fusion. As shown in (c), a closed S-layer lattice can be recrystallized on the external side of the solid supported BLM (left part). Modified after Ref. 71. Copyright 2004 Reprinted with permission from Wiley-VCH.
The membrane tension of bilayer lipid membranes (BLMs) upon the attachment of S-layer proteins has been determined by dynamic light scattering.70 For BLMs, the collective motions of the lipid molecules are dominated by membrane tension rather than by membrane curvature energy. S-layer lattices recrystallized at both faces of the BLM [Fig. 7(b)] resulted in a considerable reduction of the membrane tension, whereas the membrane bending energy increased by three orders of magnitude. This result indicated that the attached S-layer lattice has facilitated the transverse shear motions of the lipid molecules.70 In accordance with voltage pulse experiments,72 a significant increase of the previously negligible surface viscosity of the membrane has been observed as a consequence of S-layer protein attachment.70
The lateral diffusion of lipid molecules in different solid supported lipid membranes has been studied by the fluorescence recovery after photobleaching technique.73 SsLMs have been fabricated in several ways. S-layer proteins have been self-assembled with their outer, more hydrophobic, face on silanized silicon surfaces before generating a BLM by the LB and Langmuir–Schaefer techniques.73 For this purpose the S-layer covered silicon wafer was placed in a trough filled with subphase and on the air-water interphase a lipid monolayer with a certain surface pressure was generated. Subsequently, the S-layer covered silicon was vertically pulled out of the subphase (LB technique), leading to a first lipid monolayer on the S-layer lattice. Then, after adjusting the surface pressure to the desired value, the lipid-S-layersilicon architecture was placed horizontally on the lipid monolayer and carefully dipped into the subphase to generate a lipid bilayer (Langmuir–Schaefer technique).73 This structure was kept under water during all measurements. The mobility of labeled lipid molecules within this composite structure has been compared with silane- and dextran-supported phospholipid membranes. The lateral diffusion of lipids was highest in the SsLMs compared to silane- or dextran-supported lipid structures.73 This finding may be explained by the low amount of lipid molecules immobilized on the S-layer lattice and by the repetitive local interactions of the S-layer lattice with the lipid head groups.
2. Bioelectrochemical methods
In general, lipid membranes are characterized by the resistance and capacitance. The membrane resistance provides information on the impermeability of the lipid membrane, in particular for ions, and is for defectless membranes in the giga-ohm range. The membrane capacitance is a measure for the average thickness of the isolating part of the lipid membrane and may vary, depending on the lipid molecules and preparation method, between 0.4 and 1.0 μF/cm2 (Table I). The mechanical properties of SsLMs have been studied by relaxation experiments and by capacitance measurements during the application of a hydrostatic pressure. To summarize, relaxation experiments reveal a considerably longer delay time between the voltage pulse and the appearance of the initial defect at SsLMs.69 Hydrostatic pressure applied across painted BLMs causes them to bulge, resulting in an increase of capacitance. A significantly higher pressure compared to BLMs is required for SsLMs from the S-layer-faced side to observe any change in capacitance. This result indicates in the latter case an enhanced mechanical stability of the SsLMs.69,74
Table I.
Electrophysical parameters and stability of plain and S-layer-supported lipid membranes. Rm=membrane resistance, Rs=specific membrane resistance, Cs=specific membrane capacitance, and SUM =S-layer ultrafiltration membrane.
|
Rm (GΩ) |
Rs (MΩ cm2) |
Cs (μF/cm2) |
Stability (h) |
|
|---|---|---|---|---|
| Painted BLM | 7.6 | 33.3 | 0.40 | - |
| Painted BLM | 9.4 | 50 | 0.41 | - |
| + S-layer | ||||
| Folded BLM | 8.7±0.5 | 1.15±0.07 | 0.84±0.05 | 6–7 |
| Folded BLM1 | 3.9±2.3 | 1.85±0.31 | 0.83±0.05 | - |
| + S-layer | ||||
| Bilayer on SUM |
15.4±1.9 | 11.6±1.45 | 0.61±0.1 | 8±3 |
| Tetraetherlipid monolayer on SUM |
116.3 | 100 | 0.76±0.01 | - |
| Bilayer on S-layer covered gold electrode |
≤80 (5–80) | ≤80 (5–80) | 0.53±0.1 | 5–46 |
| Tetraetherlipid monolayer on S-layer covered gold electrode |
≤60 | ≤60 | 0.68±0.03 | 30–46 |
| ≤60 (1–60) | ≤60 (1–60) | 41–106a |
With an additional SbpA cover.
Lipid membranes on porous and solid substrates were generated by a modified LB technique. A polymer foil with an aperture approximately 10−3 cm2 in area was placed on the substrates, which have previously been covered by a closed S-layer lattice. This assembly was mounted vertically in the measuring chamber.75 A first electrode was placed behind the porous substrate or the solid one was directly used as electrode. A second electrode was placed in the chamber, which was filled with electrolyte above the aperture. Then a lipid film was generated on the air-electrolyte interphase and by means of a syringe electrolyte was sucked out of the chamber so that the lipid film covered, among others, the polymer foil and the S-layer lattice accessible through the aperture. To generate the second lipid layer, the electrolyte was carefully injected back into the chamber and in most cases an electrically tight and stable bilayer could be generated by this technique.
In general, lipid membranes generated on a porous support [Fig. 7(c)] combine the advantage of possessing an essentially unlimited ionic reservoir on each side of the bilayer lipid membrane and of easy manual handling. However, the surface properties of porous supports, like roughness or great differences in pore size, have significantly impaired the stability of attached BLMs. Thus, a strategy to use an S-layer ultrafiltration membrane (SUM) with the S-layer as the stabilizing and biomimetic layer between the BLM and the porous support was introduced.69 SUMs are isoporous structures with very sharp molecular exclusion limits and are manufactured by depositing S-layer-carrying cell wall fragments on commercial microfiltration membranes (MFMs) with an average pore size of approximately 0.4 μm.76-78 SFM measurements reveal approximately three times lower roughness for SUMs compared to untreated microfiltration membranes.79
Composite SUM-supported phospholipid bilayers are tight structures with breakdown voltages well above 500 mV during their whole lifetime of approximately 8 h.69 For a comparison, lipid membranes on a plain nylon MFM reveal a lifetime of about 3 h. Specific capacitance measurements and reconstitution experiments demonstrate that the lipid membrane on the SUM consists of two phospholipid layers as the pore-forming protein α-hemolysin (αHL) can be reconstituted into lytic channels. In contrast, no pore formation is observed with BLMs generated on the MFM.79
The main phospholipid isolated from Thermoplasma acidophilum (MPL), a membrane-spanning tetraetherlipid, and also mixtures of MPL with 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC) have been spread at the air/water interface.79 Monomolecular films have been transferred by one (at MPL and mixtures) or two (at DPhPC) steps on the SUM. Beside the convincing data for its resistance and capacitance, SUM-supported MPL membranes showed a longevity of 8.3±2.9 h (Table I). An additional monomolecular S-layer protein lattice recrystallized on the lipid-faced side, forming a S-layer–lipid membrane–S-layer sandwichlike structure, increases the lifetime significantly to about 1 day.79 Phospholipid bilayers and tetraetherlipid monolayers have also been generated on S-layer covered gold electrodes [Fig. 7(d)] and exhibited a remarkable long-term robustness of up to approximately 5 days in the sandwichlike structure described earlier (Table I).
Membrane-active peptides like alamethicin, gramicidin A, or valinomycin have been incorporated in SsLMs. In a first study, a tetraetherlipid monolayer was clamped on the tip of a micropipette (tip-dip technique).44 Valinomycin could not only be incorporated in S-layer supported tetraetherlipid monolayers, but a tenfold increase of the lifetime was also observed for the latter one compared to a tetraetherlipid monolayer without an attached S-layer lattice.40 In a further study, gramicidin A was incorporated in tetraetherlipid monolayers and phospholipid bilayers that were deposited on SUMs.79 These membranes revealed not only a remarkable stability, particularly with an S-layer cover, but the most striking result was that high-resolution conductance measurements on single gramicidin pores were feasible. The functionality of lipid membranes resting on S-layer covered gold electrodes has been demonstrated by the reconstitution of alamethicin, gramicidin A, and valinomycin.75 Due to the formation of conductive alamethicin channels, the membrane resistance dropped from 80 to ~1 MΩ cm2, whereas the capacitance was not altered. Partial inhibition of the alamethicin channels with amiloride and analogs has been demonstrated as increasing amounts of inhibitor gave rise to an increased membrane resistance.75 Furthermore, a SsLM with incorporated valinomycin, a potassium-selective ion carrier, revealed in a potassium buffer a 410-fold lower resistance than bathed in a sodium buffer.75
In reconstitution experiments, the pore formation of the staphylococcal αHL (Ref. 80) has been examined at plain and SsLMs.81 αHL added to the lipid-exposed side of the S-layer-supported BLM resulted in pore formation as determined by the increase in conductance. No assembly has been detected upon adding αHL monomers to the S-layer face of the composite membrane. Therefore, it is concluded that the intrinsic molecular sieving properties of the S-layer lattice did not allow passage of αHL monomers through the S-layer lattice. In comparison to plain BLMs, SsLMs have a decreased tendency to rupture in the presence of αHL, indicating an enhanced stability due to the attached S-layer lattice.81 Most interestingly, even single pore recordings have been performed with αHL reconstituted in SsLMs (Ref. 82) and also with BLMs resting on a SUM.69
V. SUMMARY AND CONCLUSION
S-layer stabilized lipid membranes mimic the supramolecular building principles of archaeal cell envelopes, which have been optimized for billions of years of evolution in most extreme habitats. In general, the most commonly observed outermost prokaryotic cell envelope structures are S-layers. Isolated S-layer subunits, recombinantly produced or isolated from S-layer carrying bacteria, have the intrinsic feature to self-assemble into two-dimensional arrays on lipid films and at various surfaces like gold-covered sensor surfaces, silicon wafers, or glass slides.83,84 S-layer lattices are highly porous structures at the nanometer scale. Few domains on the S-layer proteins, which are spatially separated according to the lattice type, interact via non-covalent forces with the lipid head groups of the adjacent lipid monolayer. This interaction gives rise to nanopatterned lipid membranes where few lipid molecules are immobilized, whereas all the remaining lipids possess very low diffusional constraints. Since the fluidity of the membrane is confined in nanopatterned domains, a so-called “semifluidity” can be observed for the whole S-layer supported lipid membrane. The strategy to use lipid membranes in-between two S-layers is of particular interest and has already led to a remarkable stability of many days. In addition, the water-containing S-layer lattices act as tethering structures providing an ionic reservoir and, most important, enough space for protein domains of incorporated membrane proteins, that protrude from the lipid membrane. Thus, S-layers are an auspicious alternative to tether molecules and polymer-cushioned supports and constitute highly interesting model membrane systems.
ACKNOWLEDGMENTS
Financial support from the Austrian Science Fund (FWF, Project No. 20256-B11), the Erwin-Schrödinger Society for Nanosciences, the FP6 EC STREP NASSAP (Project No. 13352), and the Air Force Office of Scientific Research, USA (AFOSR, Project Nos. FA9550-06-1-0208 and FA9550-07-1-0313) is gratefully acknowledged. The authors thank Jacqueline Friedmann for her skillful assistance.
References
- 1.Gerstein M, Hegyi H. FEMS Microbiol. Rev. 1998;22:277. doi: 10.1111/j.1574-6976.1998.tb00371.x. [DOI] [PubMed] [Google Scholar]
- 2.Galdiero S, Galdiero M, Pedone C. Curr. Protein Pept. Sci. 2007;8:63. doi: 10.2174/138920307779941541. [DOI] [PubMed] [Google Scholar]
- 3.Viviani B, Gardoni F, Marinovich M. Int. Rev. Neurobiol. 2007;82:247. doi: 10.1016/S0074-7742(07)82013-7. [DOI] [PubMed] [Google Scholar]
- 4.Ellis C, Smith A. Nat. Rev. Drug Discovery. 2004;3:237. doi: 10.1038/nrd1332. [DOI] [PubMed] [Google Scholar]
- 5.Henrick K, Feng Z, Bluhm WF, Dimitropoulos D, Doreleijers JF, Dutta S, Flippen-Anderson JL, Ionides J, Kamada C, Krissinel E, Lawson CL, Markley JL, Nakamura H, Newman R, Shimizu Y, Swaminathan J, Velankar S, Ory J, Ulrich EL, Vranken W, Westbrook J, Yamashita R, Yang H, Young J, Yousufuddin M, Berman HM. Nucleic Acids Res. 2007;36:D426. doi: 10.1093/nar/gkm937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raman P, Cherezov V, Caffrey M. Cell. Mol. Life Sci. 2006;63:36. doi: 10.1007/s00018-005-5350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurley JH. Chem. Biol. 2003;10:2. doi: 10.1016/s1074-5521(03)00006-1. [DOI] [PubMed] [Google Scholar]
- 8.Torres J, Stevens TJ, Samso M. Trends Biochem. Sci. 2003;28:137. doi: 10.1016/S0968-0004(03)00026-4. [DOI] [PubMed] [Google Scholar]
- 9.Sleytr UB. Nature (London) 1975;257:400. doi: 10.1038/257400a0. [DOI] [PubMed] [Google Scholar]
- 10.Sleytr UB. Int. Rev. Cytol. 1978;53:1. doi: 10.1016/s0074-7696(08)62240-8. [DOI] [PubMed] [Google Scholar]
- 11.Sleytr UB, Beveridge TJ. Trends Microbiol. 1999;7:253. doi: 10.1016/s0966-842x(99)01513-9. [DOI] [PubMed] [Google Scholar]
- 12.Schuster B, Sleytr UB. Curr. Nanosci. 2006;2:143. [Google Scholar]
- 13.Schuster B, Sleytr UB. Rev. Mol. Biotechnol. 2000;74:233. doi: 10.1016/s1389-0352(00)00014-3. [DOI] [PubMed] [Google Scholar]
- 14.Schuster B, Gufler PC, Pum D, Sleytr UB. IEEE Trans. Nanobiosci. 2004;3:16. doi: 10.1109/tnb.2004.824267. [DOI] [PubMed] [Google Scholar]
- 15.Sleytr UB, Messner P, Pum D, Sára M. Angew. Chem. Int. Ed. 1999;38:1034. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1034::AID-ANIE1034>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 16.Sleytr UB, Messner P, Pum D, Sára M, editors. Crystalline Bacterial Cell Surface Layers. Springer-Verlag; Berlin: 1988. pp. 1–193. [DOI] [PubMed] [Google Scholar]
- 17.Sleytr UB, Sára M, Pum D, Schuster B, Messner P, Schäffer C. Biopolymers. Vol. 7. Wiley-VCH; Weinheim: 2003. pp. 285–338. [Google Scholar]
- 18.Sára M, Sleytr UB. J. Bacteriol. 2000;182:859. doi: 10.1128/jb.182.4.859-868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schäffer C, Messner P. Glycobiology. 2004;14:31R. doi: 10.1093/glycob/cwh064. [DOI] [PubMed] [Google Scholar]
- 20.König H. Can. J. Microbiol. 1988;34:395. [Google Scholar]
- 21.Kandler O. Zbl. Bakt. Hyg. I. Abt. Orig. C. 1982;3:149. [Google Scholar]
- 22.Sleytr UB, Messner P, Pum D, Sára M, editors. Molecular Biology Intelligence Unit. Academic; Landes, Austin, TX: 1996. pp. 1–230. [Google Scholar]
- 23.Baumeister W, Lembcke G. J. Bioenerg. Biomembr. 1992;24:567. doi: 10.1007/BF00762349. [DOI] [PubMed] [Google Scholar]
- 24.Mayr J, Lupas A, Kellermann J, Eckerskorn C, Baumeister W, Peters J. Curr. Biol. 1996;6:739. doi: 10.1016/s0960-9822(09)00455-2. [DOI] [PubMed] [Google Scholar]
- 25.Hovmöller S, Sjögren A, Wang DN. Prog. Biophys. Mol. Biol. 1988;51:131. doi: 10.1016/0079-6107(88)90012-0. [DOI] [PubMed] [Google Scholar]
- 26.Baumeister W, Wildhaber I, Phipps BM. J. Can. Microbiol. 1989;35:215. doi: 10.1139/m89-034. [DOI] [PubMed] [Google Scholar]
- 27.Pum D, Sára M, Schuster B, Sleytr UB. Nanotechnology: Science and Computation. Springer-Verlag; Berlin: 2006. pp. 277–290. [Google Scholar]
- 28.Beveridge TJ. Curr. Opin. Struct. Biol. 1994;4:202. [Google Scholar]
- 29.Messner P, Sleytr UB. Adv. Microb. Physiol. 1992;33:213. doi: 10.1016/s0065-2911(08)60218-0. [DOI] [PubMed] [Google Scholar]
- 30.Sleytr UB, Sára M, Pum D, Schuster B. Supramolecular Polymers. 2nd ed CRC, Taylor & Francis; Boca Raton, FL: 2005. pp. 583–616. [Google Scholar]
- 31.Sleytr UB, Egelseer E, Ilk N, Pum D, Schuster B. FEBS J. 2007;274:323. doi: 10.1111/j.1742-4658.2006.05606.x. [DOI] [PubMed] [Google Scholar]
- 32.Sleytr UB, Huber C, Ilk N, Pum D, Schuster B, Egelseer E. FEMS Microbiol. Lett. 2007;267:131. doi: 10.1111/j.1574-6968.2006.00573.x. [DOI] [PubMed] [Google Scholar]
- 33.Wetzer B, Pfandler A, Györvary E, Pum D, Lösche M, Sleytr UB. Langmuir. 1998;14:6899. [Google Scholar]
- 34.Weygand M, Wetzer B, Pum D, Sleytr UB, Cuvillier N, Kjaer K, Howes PB, Lösche M. Biophys. J. 1999;76:458. doi: 10.1016/S0006-3495(99)77213-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferner-Ortner J, Mader C, Ilk N, Sleytr UB, Egelseer EM. J. Bacteriol. 2007;189:7154. doi: 10.1128/JB.00294-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sleytr UB, Sára M, Mader C, Schuster B, Unger FM. No. A 732/2000 Patent. 2000 Apr 26;
- 37.Pum D, Sleytr UB. Thin Solid Films. 1994;244:882. [Google Scholar]
- 38.Stetter KO. Extremophiles. 2006;10:357. doi: 10.1007/s00792-006-0012-7. [DOI] [PubMed] [Google Scholar]
- 39.Sleytr UB, Messner P. Encyclopedia of Microbiology. Vol. 1. Academic; San Diego, CA: 2000. pp. 889–906. [Google Scholar]
- 40.Kashefi K, Lovely DR. Science. 2003;301:934. doi: 10.1126/science.1086823. [DOI] [PubMed] [Google Scholar]
- 41.Schuster B. NanoBiotechnology. 2005;1:153. [Google Scholar]
- 42.Schuster B, Pum D, Sàra M, Sleytr UB. Mini Rev. Med. Chem. 2006;6:909. doi: 10.2174/138955706777935026. [DOI] [PubMed] [Google Scholar]
- 43.De Rosa M. Thin Solid Films. 1996;284–285:13. [Google Scholar]
- 44.Schuster B, Pum D, Sleytr UB. Biochim. Biophys. Acta. 1998;1369:51. doi: 10.1016/s0005-2736(97)00206-x. [DOI] [PubMed] [Google Scholar]
- 45.Hanford MJ, Peeples TL. Appl. Biochem. Biotechnol. 2002;97:45. doi: 10.1385/abab:97:1:45. [DOI] [PubMed] [Google Scholar]
- 46.Strobl C, Six L, Heckmann K, Henkel B, Ring K. Z. Naturforsch. C. 1984;40c:219. [Google Scholar]
- 47.Diederich A, Hödl C, Pum D, Sleytr UB, Lösche M. Colloids Surf., B. 1996;6:335. [Google Scholar]
- 48.Hoppert M. Microscopic Techniques in Biotechnology. Wiley-VCH; Weinheim: 2003. [Google Scholar]
- 49.Pum D, Weinhandl M, Hödl C, Sleytr UB. J. Bacteriol. 1993;175:2762. doi: 10.1128/jb.175.9.2762-2766.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robards AW, Sleytr UB. Practical Methods in Electron Microscopy. Vol. 10. Elsevier; Amsterdam: 1985. pp. 293–300. [Google Scholar]
- 51.Györvary E, Stein O, Pum D, Sleytr UB. J. Microsc. 2003;212:300. doi: 10.1111/j.1365-2818.2003.01270.x. [DOI] [PubMed] [Google Scholar]
- 52.Cooper MA, Singleton VT. J. Mol. Recognit. 2007;20:154. doi: 10.1002/jmr.826. [DOI] [PubMed] [Google Scholar]
- 53.Nguyen B, Tanious FA, Wilson WD. Methods. 2007;42:150. doi: 10.1016/j.ymeth.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 54.Phillips KS, Cheng Q. Anal. Bioanal. Chem. 2007;387:1831. doi: 10.1007/s00216-006-1052-7. [DOI] [PubMed] [Google Scholar]
- 55.Rodahl M, Höök F, Krozer A, Brzezinski P, Kasemo B. Rev. Sci. Instrum. 1995;66:3924. [Google Scholar]
- 56.Höök F, Rodahl M, Kasemo B, Brzezinski P. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12271. doi: 10.1073/pnas.95.21.12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Höök F, Kasemo B. Piezoelectric Sensors. Springer Verlag; Berlin: 2006. pp. 425–448. [Google Scholar]
- 58.Johannsmann D. Piezoelectric Sensors. Springer Verlag; Berlin: 2006. pp. 49–110. [Google Scholar]
- 59.Pisecker M. Master’s thesis. University for Natural Resources and Applied Life Sciences; Vienna: 2005. [Google Scholar]
- 60.Sauerbrey G. Z. Phys. 1959;155:206. [Google Scholar]
- 61.Larsson C, Rodahl M, Höök F. Anal. Chem. 2003;75:5080. doi: 10.1021/ac034269n. [DOI] [PubMed] [Google Scholar]
- 62.Fischer H, Polikarpov I, Craievich AF. Protein Sci. 2004;13:2825. doi: 10.1110/ps.04688204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schuster B, Gufler PC, Pum D, Sleytr UB. Langmuir. 2003;19:3393. [Google Scholar]
- 64.Nomellini FJ, Küpcü S, Sleytr UB, Smit J. J. Bacteriol. 1997;179:6349. doi: 10.1128/jb.179.20.6349-6354.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smit E, Oling F, Demel R, Martinez B, Pouwels PH. J. Mol. Biol. 2001;305:245. doi: 10.1006/jmbi.2000.4258. [DOI] [PubMed] [Google Scholar]
- 66.Weygand M, Schalke M, Howes PB, Kjaer K, Friedmann J, Wetzer B, Pum D, Sleytr UB, Lösche M. J. Mater. Chem. 2000;10:141. [Google Scholar]
- 67.Weygand M, Kjaer K, Howes PB, Wetzer B, Pum D, Sleytr UB, Lösche M. J. Phys. Chem. B. 2002;106:5793. [Google Scholar]
- 68.Küpcü S, Sára M, Sleytr UB. Biochim. Biophys. Acta. 1995;1235:263. doi: 10.1016/0005-2736(95)80013-6. [DOI] [PubMed] [Google Scholar]
- 69.Schuster B, Pum D, Sara M, Braha O, Bayley H, Sleytr UB. Langmuir. 2001;17:499. [Google Scholar]
- 70.Hirn R, Schuster B, Sleytr UB, Bayerl TM. Biophys. J. 1999;77:2066. doi: 10.1016/S0006-3495(99)77048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sleytr UB, Egelseer E, Pum D, Schuster B. NanoBiotechnology: Concepts, Methods and Perspectives. Wiley-VCH; Weinheim: 2003. pp. 77–92. [Google Scholar]
- 72.Schuster B, Diederich A, Bähr G, Sleytr UB, Winterhalter M. Eur. Biophys. J. 1999;28:583. doi: 10.1007/s002490050240. [DOI] [PubMed] [Google Scholar]
- 73.Györvary E, Wetzer B, Sleytr UB, Sinner A, Offenhäusser A, Knoll W. Langmuir. 1999;15:1337. [Google Scholar]
- 74.Schuster B, Sleytr UB. Biochim. Biophys. Acta. 2002;1563:29. doi: 10.1016/s0005-2736(02)00370-x. [DOI] [PubMed] [Google Scholar]
- 75.Gufler PC, Pum D, Sleytr UB, Schuster B. Biochim. Biophys. Acta. 2004;1661:154. doi: 10.1016/j.bbamem.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 76.Sára M, Sleytr UB. J. Bacteriol. 1987;169:2804. doi: 10.1128/jb.169.6.2804-2809.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weigert S, Sára M. J. Membr. Sci. 1995;106:147. [Google Scholar]
- 78.Weigert S, Sára M. J. Membr. Sci. 1996;121:185. [Google Scholar]
- 79.Schuster B, Weigert S, Pum D, Sára M, Sleytr UB. Langmuir. 2003;19:2392. [Google Scholar]
- 80.Bhakdi S, Tranum-Jensen J. Microbiol. Rev. 1991;55:733. doi: 10.1128/mr.55.4.733-751.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schuster B, Pum D, Braha O, Bayley H, Sleytr UB. Biochim. Biophys. Acta. 1998;1370:280. doi: 10.1016/s0005-2736(97)00274-5. [DOI] [PubMed] [Google Scholar]
- 82.Schuster B, Sleytr UB. Bioelectrochemistry. 2002;55:5. doi: 10.1016/s1567-5394(01)00148-7. [DOI] [PubMed] [Google Scholar]
- 83.Sleytr UB, Sára M, Pum D, Schuster B. Prog. Surf. Sci. 2001;68:231. [Google Scholar]
- 84.Sleytr UB, Györvary E, Pum D. Prog. Org. Coat. 2003;47:279. [Google Scholar]