

Abstract
Estradiol-17β and its metabolites which are sequentially synthesized by cytochrome P450s (CYP450s) and catechol-O-methyltransferase (COMT) to form 2 and 4-Hydroxyestradiol (2-OHE2 and 4-OHE2) and 2- and 4-Methoxestradiol (2-ME2, and 4-ME2) are elevated during pregnancy. We investigated whether CYP450s and COMT are expressed in uterine artery endothelial cells (UAECs) and if E2β and its metabolites modulate cell proliferation via ER-α and/or ER-β and play roles in physiologic uterine angiogenesis during pregnancy. Cultured ovine UAECs from pregnant (P-UAECs) and nonpregnant (NP-UAECs) ewes were treated with 0.1-100 nmol/L of E2β, 2-OHE2, 4-OHE2, 2-ME2, and 4-ME2. ER-α or ER-β specificity was tested using ICI 182,780, ER-α-specific MPP, ER-β –specific PHTPP antagonists and their respective agonists ER-α-specific PPT and ER-β –specific DPN. Angiogenesis was evaluated using BrdU Proliferation Assay. Utilizing confocal microscopy and Western analyses to determine enzyme location and levels, we observed CYP1A1, CYP1A2, CYP1B1, CYP3A4 and COMT expression in UAECs; however, expressions were similar between NP-UAECs and P-UAECs. E2β, 2-OHE2, 4-OHE2, and 4-ME2 treatments concentration-dependently stimulated proliferation in P-UAECs, but not NP-UAECs; 2-ME2 did not stimulate proliferation in either cell type. Proliferative responses of P-UAECs to E2β were solely mediated by ER-β, whereas responses to E2β metabolites were neither ER-α nor ER-β mediated. We demonstrate an important vascular role for E2β, its CYP450- and COMT-derived metabolites and ER-β in uterine angiogenesis regulation during pregnancy that may be dysfunctional in preeclampsia and other cardiovascular disorders.
Keywords: angiogenesis, hypertension, pregnancy, endothelium, estradiol metabolites, CYP450s
Introduction
Pregnancy is associated with dramatic uterine blood flow (UBF) rises resulting from vascular adaptations including vasodilatation and angiogenesis.1 These adaptations are critical in pregnancy since their dysfunctions are implicated in pathologic pregnancies such as preeclampsia which complicate 6-8% of all pregnancies in the USA and account for 50,000 maternal deaths per year worldwide.2,3,4
Regulation of vascular adaptations during pregnancy is mediated partly by estrogens, which are elevated during gestation.5 Estradiol-17β (E2β) infusion in sheep markedly reduces uterine and systemic vascular resistance causing rises in uterine and systemic blood flows.6 Uterine arterial administration of the nonselective estrogen receptor (ER) antagonist ICI 182,780 in pregnant sheep lowers UBF, demonstrating that endogenous estrogen via ERs helps maintain uterine perfusion.7 In human umbilical vein endothelial cells (HUVECs) 8 and myometrial microvascular ECs, E2β promotes proliferation, an index of angiogenesis.9
The effects of estrogen on uterine vascular adaptations may be further modulated by its biologically active metabolites. E2β metabolism catalyzed by cytochrome P450s (CYP450s) and catechol-O-methyltransferase (COMT) produces the catecholestradiols 2-Hydroxyestradiol (2-OHE2) and 4-Hydroxyestradiol (4-OHE2), and the methoxyestradiols 2-Methoxestradiol (2-ME2) and 4-Methoxyestradiol (4-ME2).10,11 Evidence supports the involvement of E2β-derived metabolites in pregnancy and in the regulation of angiogenesis; 2-ME2- and COMT-deficient mice exhibit preeclampsia-like symptoms including impaired angiogenesis and hypertension.12 Treatment with low concentration of 2-OHE2, 4-OHE2, 2-ME2, or 4-ME2 induces proliferation in cultured HUVECs whereas high concentration of 2-OHE2 or 2-ME2 inhibits proliferation.13
Thus, we tested the hypothesis that CYP450s and COMT may be expressed in the uterine vasculature and that E2β, its CYP450s and COMT-derived metabolites participate in the regulation of uterine angiogenesis during pregnancy. Late pregnant and nonpregnant ovine uterine artery endothelial cells (P-UAECs and NP-UAECs) consistently express ER-α and ER-β and exhibit pregnancy-specific responses to angiogenic ligands demonstrating that they are a good model to evaluate direct receptor-mediated actions of E2β and its metabolites.14,15 We investigated: 1) the expression and intracellular distribution of CYP450s and COMT in P-UAECs versus NP-UAECs; 2) whether E2β, 2-OHE2, 4-OHE2, 2-ME2, and 4-ME2 stimulate greater proliferation of P-UAECs than NP-UAECs; and 3) if E2β and its metabolites induce proliferative responses via ER-α and/or ER-β.
Methods
For complete details on specific materials and methodology, please see http://hyper.ahajournals.org.
Cell Preparation and Culture
Cell preparations were approved by the University of Wisconsin-Madison School of Medicine Research Animal Care Committee as previously described.14,15 UAECs were isolated and validated from late gestation (120-130 days; term= 147 days; n=6) and nonpregnant (luteal n=5 and follicular n=2) ewes.15 At passage 5, ~ 70% confluence, cells were transferred to slides, 96 well plates, or lysed for protein extraction as needed for respective experiments.
Protein Extraction and Western Immunoblotting
Western immunoblotting was performed as previously described.14 CYP1A1, CYP1A2, CYP1B1, CYP3A4, COMT, and ER-β expressions were detected using mouse anti-CYP1A1and rabbit anti-CYP1A2, anti-CYP1B1, anti-CYP3A4, anti-COMT or anti-ER-β antibodies. GAPDH was utilized as a loading control.
Immunofluorescence Confocal Microscopy
Immunofluorescence confocal microscopy was performed as previous described.14 UAECs were washed twice with ice cold PBS and fixed for 15 min with 3% paraformaldehyde. Fixed cells were rinsed with 50 mM glycine solution, permeabilized with 0.1% Triton-X for 3 min, blocked for 30 min with goat serum and incubated (20 min) with primary antibodies for CYP1A1, CYP1A2, CYP1B1, CYP3A4 and COMT. Subsequently, cells were incubated (30 min) with secondary antibodies Alexa Fluor 488 anti-mouse or anti-rabbit IgGs. Scanning was done with a radiance 2100 MP Rainbow confocal/multiphoton laser scan microscope system (Bio-Rad, Hercules, CA).
Experimental Treatments and Blockade and Activation of ER-α and ER-β
UAEC proliferation experiments were performed in quadruplicates and replicated in at least six NP-UAEC and P-UAEC preparations. For concentration response studies, UAECs in 96 well plates were serum starved (24 hrs) in endothelial basal medium (EBM) and medium was replaced with EBM or EBM containing 0.1, 1, 10 or 100 nmol/L E2β, 2-OHE2, 4-OHE2, 2-ME2 and 4-ME2 (24 hrs). ERs were blocked by pretreating UAECs for 1 hr with 1μmol/L of the ER antagonist ICI 182, 780 (ICI), or ER-α selective antagonist 1,3-Bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinyleth oxy)phenol]-1H-pyrazole dihydrochloride (MPP), or ER-β selective antagonist 4-[2-Phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrim idin-3-yl]phenol (PHTPP). Additional concentration response studies were performed using 0, 0.1, 1, 10 or 100 nmol/L of the ER-α selective agonist 4,4′,4″-(4-Propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT) or the ER-β selective agonist 2,3-bis(4-Hydroxyphenyl)-propionitrile (DPN). We also studied the effects of 0.1 nmol/L PPT + 0.1 nmol/L DPN and 1 μmol/L PHTPP + 0.1 nmol/L DPN to further evaluate receptor activation, additive effects, and specificity of ER-β selective agonist receptor activation.
BrdU Cell Proliferation Assays
BrdU was added for 16 hrs during the 24 hrs of steroid treatment and an in vitro index of proliferation was evaluated. Plates were read using Synergy HT Multi-Mode Microplate Reader (BioTek, Winooski, VT). Results are expressed as fold increases over untreated control after subtracting the “blank” (wells incubated without Brdu).
Statistical Analysis
Data (means ± SEM) were analyzed using a Two-way ANOVA with “Pregnancy” and “Concentration” as two “between” factors. Analyses of simple effects were performed using One-way ANOVA followed by post-hoc Student-Newman Keuls test. Pairwise comparisons were performed using Bonferroni or Student-Newman-Keuls test. Biphasic concentration response (deviation from the standard monotonic sigmoid shape) description was determined by nonlinear regression using the logarithm of agonist concentration against various responses. Level of significance was established a priori at P<0.05.
Results
CYP1A1, CYP1A2, CYP1B1, CYP3A4 and COMT are expressed in UAECs
Western analyses indicated the presence of CYP1A1, CYP1A2, CYP1B1, CYP3A4, and COMT in NP-UAECs and P-UAECs (Figure 1A). However, no differences were seen between NP-UAECs and P-UAECs in their levels of expression (Figure 1B). Confocal microscopy revealed no difference between NP-UAECs and P-UAECs in intracellular distribution patterns of these enzymes. Therefore, unless noted, P-UAECs images are shown. CYP1A1, CYP1A2, and CYP3A4 were localized in cytoplasmic and nuclear compartments of P-UAECs (Figure 2A, B and D). CYP1B1 was localized in the nuclear region, whereas COMT was localized in the cytoplasmic compartment (Figure 2C, 2E).
Figure 1.

(A) Immunoblots showing expression of CYP1A1, CYP1A2, CYP1B1, CYP3A4, COMT, and GAPDH in NP-UAECs and P-UAECs. (B) Densitometric analyses (Relative protein expression = enzyme expression OD /GAPDH OD) showed no difference between NP-UAECs (n=6) and P-UAECs (n=6); (P=0.949, One-Way ANOVA).
Figure 2.

Immunofluorescence microscopy showing intracellular localization of (A) CYP1A1, (B) CYP1A2, (C) CYP1B1, (D) CYP3A4, (E) COMT and (F) Negative Control in P-UAECs. Positive staining is green fluorescence with nuclei depicted in blue (DAPI). Pictures are representative of three experiments.
P-UAEC proliferation in response to E2β, 2-OHE2, 4-OHE2, 2-ME2 and 4-ME2
Biphasic concentration proliferative responses were observed in P-UAECs after E2β treatment with maximum responses observed at a concentration of 0.1 nmol/L (Figure 3A). In contrast, E2β did not induce NP-UAEC proliferation at any concentration. Similarly, P-UAECs but not NP-UAECs exhibited a biphasic proliferative response to 2-OHE2 and 4-OHE2 (Figure 3B, 3C). The magnitude of P-UAECs proliferation at 0.1 nmol/L of E2β, 2-OHE2 and 4-OHE2 were 2.07 ± 0.16, 1.79 ± 0.02 and 1.78 ± 0.02 fold of control, respectively.
Figure 3.

Concentration-dependent cell proliferation responses of NP-UAECs and P-UAECs to (A) E2β, (B) 2-OHE2, (C) 4-OHE2, (D) 2-ME2 and (E) 4-ME2. A biphasic proliferative response was observed in P-UAECs in response to E2β, 2-OHE2, and 4-OHE2 but not 4-ME2 compared to control with maximum responses at a physiologic concentration of 0.1 nmol/L (Two-Way ANOVA; Pregnancy × Concentration effect; E2β, F4,40=8.16, P<0.0001; 2-OHE2, F4,40=4.07, P=0.0073;, 4-OHE2, F4,40=3.69, P=0.0119; and 4-ME2, F4,40=5.05, P=0.002). NP-UAECs did not respond to E2β or its metabolites. No proliferation effect was observed with 2ME2. *Indicates an increase (P<0.05; n=6) in P-UAEC proliferation compared with both the respective NP-UAEC (n=7) group and untreated control.
2-ME2 did not stimulate proliferation of P-UAECs or NP-UAECs (Figure 3D). 4-ME2 at all concentrations induced proliferation in P-UAECs, but not in NP-UAECs (Figure 3E). Proliferation of P-UAECs at the physiologic concentration of 0.1 nmol/L of 4-ME2 was 1.50 ± 0.16 fold of control (Figure 3E). However, response to 4-ME2 was not biphasic and the maximum proliferation of 1.74 ± 0.04 fold was observed at 100 nmol/L. Additional validation of cell proliferation utilizing ViaLight Plus Kit (Lonza Inc., Rockland, ME) was performed and it confirmed increases in total viable cell numbers after treatment with E2β or its metabolites.
Proliferation of P-UAECs via classic ERs
Antagonism with ICI was tested at a physiologic concentration of 0.1 nmol/L E2β and its metabolites (Figure 4). ICI alone had no effect on P-UAEC proliferation, however, it totally abrogated proliferative responses to E2β indicating the requirement of ER-α and/or ER-β. In contrast, ICI did not have an effect of the proliferative responses of P-UAECs to 2-OHE2, 4-OHE2 and 4-ME2. Figure 4 also illustrates that at 0.1 nmol/L, E2β was more potent than 2-OHE2, 4-OHE2 and 4-ME2 which were equipotent in stimulating P-UAECs proliferation.
Figure 4.
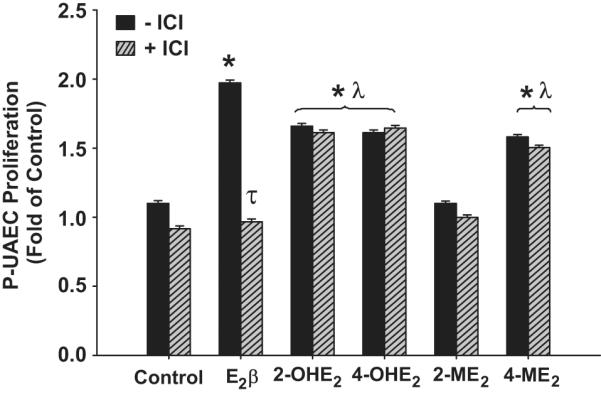
The effects of 1 μmol/L ICI on P-UAEC proliferative responses to 0.1 nmol/L of E2β, 2-OHE2, 4-OHE2, 2-ME2, and 4-ME2. ICI abrogated the response of P-UAECs to E2β but not in response to 2-OHE2, 4-OHE2 and 4-ME2 respectively (Two-Way ANOVA; Antagonist × Group effect; F5,60=25.272, P<0.001.*Indicates an increase (P<0.05, n=6) in P-UAEC proliferation compared to untreated control. τ Indicates inhibition (P<0.05) of P-UAEC proliferation with ICI; λ indicates lower P-UAEC proliferation (P<0.05) compared to E2β responses alone.
Proliferation of P-UAECs via ER-β not ER-α
In P-UAECs, ER-α blockade with 1 μmol/L MPP did not abolish the proliferative effects of E2β, 2-OHE2, 4-OHE2, or 4-ME2 (Figure 5). In contrast, E2β-induced proliferation was completely inhibited by 1 μmol/L of the ER-β selective antagonist PHTPP (Figure 6). However, PHTPP did not alter P-UAECs proliferative responses to the estrogen metabolites.
Figure 5.
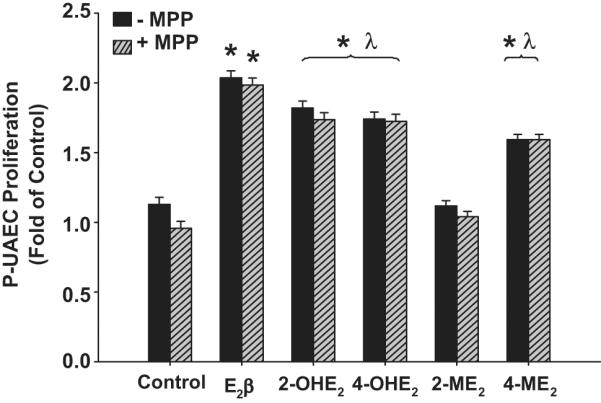
The effects of the ER-α antagonist MPP (1μmol/L) on P-UAEC proliferation responses to 0.1 nmol/L of E2β, 2-OHE2, 4-OHE2, 2-ME2 and 4-ME2. MPP had no effect on the proliferation responses of P-UAECs to 0.1 nmol/L of E2β, 2-OHE2, 4-OHE2, 2-ME2 and 4-ME2 (Two-Way ANOVA; Group effect, F5,60=14.315, P<0.001). Neither a main effect of MPP nor an interaction was noted. *Indicates an increase (P<0.05; n=6) in P-UAEC proliferation compared to untreated control; λ indicates lower P-UAEC proliferation (P<0.05) compared to E2β responses alone.
Figure 6.
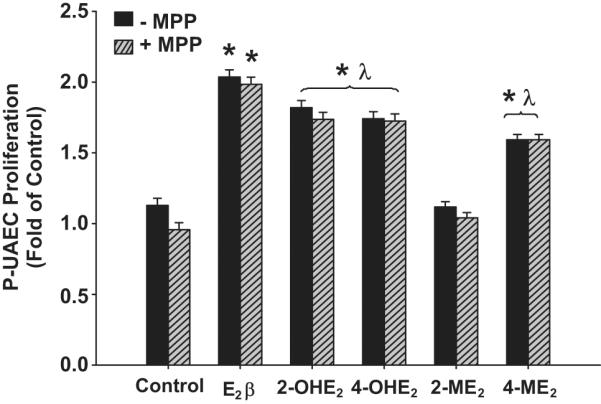
The effects of the ER-β antagonist PHTPP (1μmol/L) on P-UAEC proliferative responses to 0.1 nmol/L of E2β, 2-OHE2, 4-OHE2, 2-ME2, and 4-ME2 (Two-Way ANOVA; Antagonist × Group effect; F5,60=17.517, P<0.001. *Indicates an increase (P<0.05; n=6) in P-UAEC proliferation compared to untreated control. τ Indicates inhibition (P<0.05) of P-UAEC proliferation with PHTPP. λ Indicates lower P-UAEC proliferation (P<0.05) compared to E2β responses alone.
We further evaluated if the ER-β-mediated proliferative responses in P-UAECs were due to an increase in ER-β protein levels between NP-UAECs, P-UAECs and P-UAECs treated with 0.1 nmol/L E2β or its metabolites. Shown in Figure S1 (please see http://hyper.ahajournals.org), ER-β expressions were not different amongst these groups (P=0.943).
Treatment of P-UAECs with ER-α selective agonist PPT did not induce proliferation (Figure 7A). In contrast, all ER-β selective agonist DPN concentrations stimulated cell proliferation 1.50 ± 0.05 fold of control (Figure 7B). Because these P-UAEC responses were less than E2β alone and did not exhibit a concentration-dependent response, we examined combination of PPT (0.1 nmol/L) and DPN (0.1 nmol/L) (Figure 7C). No further increases in P-UAEC proliferative responses were observed. Moreover, pretreatment with 1 μmol/L PHTPP completely inhibited DPN-induced responses in P-UAECs; (Figure 7C).
Figure 7.

Concentration-dependent effects of (A) ER-α agonist PPT (B) ER-β agonist DPN and (C) their combination on cell proliferation responses of P-UAECs. Blockade of ER-β with PHTPP (1μmol/L) prior to treatment with ER-β agonist DNP is shown in (C). *Indicates an increase (P<0.05; n=7) in P-UAEC proliferation compared to untreated controls. λ Indicates a difference (P< 0.05) in P-UAEC proliferation in response to DPN or the combination of DNP and PPT compared to E2β only responses. τ Indicates inhibition (P<0.05) of P-UAEC proliferation with PHTPP.
Discussion
The key novel findings observed from this study are: 1) UAECs express CYP1A1, CYP1A2, CYP1B1, CYP3A4, and COMT; 2) E2β, 2-OHE2, 4-OHE2, and 4-ME2 stimulate P-UAEC, but not NP-UAEC, proliferation; and 3) E2β-induced cell proliferative responses are mediated primarily via ER-β, whereas E2β metabolites-induced proliferative responses are independent of ER-α and ER-β.
UAECs constitutively express enzymes that may metabolize E2β to its hydroxy-(CYP1A1, CYP1A2, CYP3A4, CYP1B1) and subsequently methoxy- (COMT) metabolites. Consistent with our findings, are reports showing that CYP450s and COMT are expressed in aortic, coronary artery and umbilical vein ECs.11, 16, 17,18 However, this is the first characterization of the localized intracellular expression of CYP450s in endothelial cells. Our data also confirm findings that COMT is primarily an intracellular cytosolic enzyme.19 Although little is known about the intracellular localization of these enzymes in endothelial cells, it is possible that intracellular compartmentalization is associated with enzymatic function.
The physiologic plasma E2β concentration in women ranges from 0.1-2.2 nmol/L and increase dramatically during pregnancy.5 We demonstrate that a physiologic concentration of E2β stimulates P-UAEC, but not NP-UAEC proliferation. The P-UAEC proliferative responses are similar to estrogenic stimulation of HUVECs and retinal microvascular ECs.8,13,20 Moreover, E2β promotes murine endometrial endothelial proliferation in vivo.21,22 These current data are also consistent with our previous findings23 that E2β increases P-UAEC [H3]-thymidine incorporation and tube formation; however maximum P-UAEC responses to E2β were seen at 1 nmol/L23 and not 0.1 nmol/L; NP-UAECs were not evaluated. These results demonstrate that P-UAEC proliferative responses are induced by gestational programming at the level of endothelial cell signaling, supporting reports that pregnancy-induced programming in P-UAECs leads to increased responsiveness to agonists and these effects are retained in cultured primary cell lines.15 Furthermore, the complete lack of mitogenic response of NP-UAECs may be specific to E2β since NP-UAECs show proliferation in response to ATP,VEGF, bFGF, and high (≥5%) serum.15, 24, 25,26 The mechanistic significance of pregnancy-induced estrogenic programming on physiologic angiogenesis in P-UAECs remains to be elucidated.
Plasma catecholestrogens levels in pregnancy are 10-fold greater than in nonpregnant women.27 Our finding that low levels of 2-OHE2 and 4-OHE2 stimulate P-UAEC, but not NP-UAEC proliferation, supports the proposal that CYP450s- and COMT-derived metabolites of E2β may play roles in the regulation of uterine angiogenesis during pregnancy. Low concentrations of 2-OHE2 and 4-OHE2 stimulate HUVEC proliferation and direct uterine arterial infusion of 2-OHE2 in nonpregnant sheep causes vasodilatation, whereas 4-OHE2 interacts directly with calcium channels to locally increase blood flow in gilts.13,28,29 These findings suggest that catecholestradiols play roles in pregnancy-induced vascular adaptations.
O-methylation of catecholestradiols produces less potent and antiproliferative metabolites of E2β.10 Interestingly, we demonstrate that 4-ME2 stimulated P-UAEC, but not NP-UAEC proliferation, consistent with observations that low 4-ME2concentrations stimulate HUVEC proliferation.13 However, 2-ME2 was not mitogenic on UAECs supporting numerous reports of its antiproliferative effects.30,31,32 The reason for divergent proliferative patterns between 2-ME2 and 4-ME2 is unclear. However, 2-ME2 disrupts tubulin polymeration and induces cell-cycle arrest in the mitotic phase in endothelial and smooth muscle cells.33,34,35 Thus, differences in association of 2-ME2 and 4-ME2 with regulators of mitosis, may likely account for their divergent responses.
Demonstrating a role for ER-α and ER-β, ICI abrogated E2β-induced P-UAEC proliferation, supporting previous observations that ICI blocks E2β-induced P-UAEC [H3]-thymidine incorporation23 and E2β-induced VEGF-mediated proliferation.9 Antagonism of ER-β with PHTPP abrogated E2β-induced P-UAEC proliferation and ER-β activation with DPN-induced proliferation demonstrating an ER-β only effect. However, although activation of ER-α with PPT did not alter P-UAEC proliferation, PPT stimulates proliferation of human myometrial microvascular endothelial cells.36 Therefore, the differences in P-UAECs proliferation in response to DPN, PPT, or E2β may be due to their distinct differences in affinity for ERs in association with the complex nature of ER-ligand complexes.37,38 Nevertheless, PHTPP inhibition of E2β- and DPN-induced P-UAEC proliferation validates that these E2β effects are solely ER-β mediated and independent of ER-α. Equally importantly, NP-UAECs, P-UAECs, and P-UAECs treated with E2β express similar levels of ER-β (Figure S1, please see http://hyper.ahajournals.org.) demonstrating that the ER-β-mediated E2β effects are not dependent on ER-β expression levels, but rather on other gestational-programming factors at the level of P-UAECs signaling.
E2β metabolites possess little affinity for classical ERs10,39 and unlike E2β, the effects of its metabolites on P-UAECs proliferation are not mediated via ER-α or ER-β. These results also indirectly suggest that CYP450s and COMT expressed in the UAECs do not possess high enough enzymatic activity under these conditions to metabolize E2β. However, E2β metabolites may induce proliferative effects via other estrogen associated receptors like GPR30 found in endothelial cells.40,41 Nonetheless, the exact mechanism of action of estrogen metabolites on uterine vascular ECs remains to be determined.
Perspectives
It is well established that E2β and its metabolites possess vascular protective effects on the cardiovascular system,42,43,44 but, little is known about estrogen metabolism and regulation of uterine angiogenesis during pregnancy. Therefore, understanding the biochemistry of E2β metabolism and the vascular physiology of E2β and its metabolites on the uterine endothelium may provide clues for understanding normal pregnancy-associated vascular adaptations and the dysfunction of endothelia in the pathophysiology of preeclampsia and other cardiovascular disorders.3,4
Supplementary Material
Acknowledgements
Terrance Phernetton, Jason Austin, Kreg Grindle, Gladys Lopez, Natasha Sean Fling, Kai Wang and Cindy Goss
Funding Sources Support: NIH Grants HL49210, HD38843, HL87144 (RRM), HL64703 (JZ), R25GM083252 (ML Carnes).
Footnotes
Disclosures None
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Magness RR. Maternal cardiovascular and other physiologic responses to the endocrinology of pregnancy. In: Bazer F, editor. The Endocrinology of Pregnancy. Humana press Inc; Totowa, NJ: 1998. pp. 507–539. [Google Scholar]
- 2.Samadi AR, Mayberry RM, Zaidi AA, Pleasant JC, McGhee N, Jr., Rice RJ. Maternal hypertension and associated pregnancy complications among African-American and other women in the United States. Obstet Gynecol. 1996;87:557–563. doi: 10.1016/0029-7844(95)00480-7. [DOI] [PubMed] [Google Scholar]
- 3.Pipkin FB. Risk Factors for Preeclampsia. N Engl J Med. 2001;344:925–926. doi: 10.1056/NEJM200103223441209. [DOI] [PubMed] [Google Scholar]
- 4.Luft FC. Pieces of the preeclampsia puzzle. Nephrol Dial Transplant. 2003 Nov;18:2209–2210. doi: 10.1093/ndt/gfg498. [DOI] [PubMed] [Google Scholar]
- 5.Albrecht ED, Pepe GJ. Placental steroid hormone biosynthesis in primate pregnancy. Endocr Rev. 1990;11:124–150. doi: 10.1210/edrv-11-1-124. [DOI] [PubMed] [Google Scholar]
- 6.Rosenfeld CR, Morriss FH, Jr., Battaglia FC, Makowski EL, Meschia G. Effect of estradiol-17beta on blood flow to reproductive and nonreproductive tissues in pregnant ewes. Am J Obstet Gynecol. 1976;124:618–629. doi: 10.1016/0002-9378(76)90064-8. [DOI] [PubMed] [Google Scholar]
- 7.Magness RR, Phernetton TM, Gibson TC, Chen DB. Uterine blood flow responses to ICI 182 780 in ovariectomized oestradiol-17beta-treated, intact follicular and pregnant sheep. J Physiol. 2005;565:71–83. doi: 10.1113/jphysiol.2005.086439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morales DE, McGowan KA, Grant DS, Maheshwari S, Bhartiya D, Kleinman HK, Schaper HW. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation. 1995;91:755–763. doi: 10.1161/01.cir.91.3.755. [DOI] [PubMed] [Google Scholar]
- 9.Gargett CE, Zaitseva M, Bucak K, Chu S, Fuller PJ, Rogers PA. 17Beta-estradiol up-regulates vascular endothelial growth factor receptor-2 expression in human myometrial microvascular endothelial cells: role of estrogen receptor-alpha and -beta. J Clin Endocrinol Metab. 2002;87:4341–4349. doi: 10.1210/jc.2001-010588. [DOI] [PubMed] [Google Scholar]
- 10.Zhu BT, Conney AH. Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis. 1998;19:1–27. doi: 10.1093/carcin/19.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Dubey RK, Tofovic SP, Jackson EK. Cardiovascular pharmacology of estradiol metabolites. J Pharmacol Exp Ther. 2004;308:403–409. doi: 10.1124/jpet.103.058057. [DOI] [PubMed] [Google Scholar]
- 12.Kanasaki K, Palmsten K, Sugimoto H, Ahmad S, Hamano Y, Xie L, Parry S, Augustin HG. Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature. 2008;453:1117–1121. doi: 10.1038/nature06951. [DOI] [PubMed] [Google Scholar]
- 13.Lippert C, Seeger H, Mueck AO, Lippert TH. The effects of A-ring and D-ring metabolites of estradiol on the proliferation of vascular endothelial cells. Life Sci. 2000;67:1653–1658. doi: 10.1016/s0024-3205(00)00747-5. [DOI] [PubMed] [Google Scholar]
- 14.Liao WX, Magness RR, Chen DB. Expression of estrogen receptors-alpha and -beta in the pregnant ovine uterine artery endothelial cells in vivo and in vitro. Biol Reprod. 2005;72:530–537. doi: 10.1095/biolreprod.104.035949. [DOI] [PubMed] [Google Scholar]
- 15.Bird IM, Sullivan JA, Di T, Cale JM, Zhang L, Zheng J, Magness RR. Pregnancy-dependent changes in cell signaling underlie changes in differential control of vasodilator production in uterine artery endothelial cells. Endocrinology. 2000;141:1107–1117. doi: 10.1210/endo.141.3.7367. [DOI] [PubMed] [Google Scholar]
- 16.Zacharia LC, Jackson EK, Gillespie DG, Dubey RK. Increased 2-methoxyestradiol production in human coronary versus aortic vascular cells. Hypertension. 2001;37:658–662. doi: 10.1161/01.hyp.37.2.658. [DOI] [PubMed] [Google Scholar]
- 17.Han Z, Miwa Y, Obikane H, Mitsumata M, Takahashi-Yanaga F, Morimoto M, Sasaguri T. Aryl hydrocarbon receptor mediates laminar fluid shear stress-induced CYP1A1 activation and cell cycle arrest in vascular endothelial cells. Cardiovasc Res. 2008;77:809–818. doi: 10.1093/cvr/cvm095. [DOI] [PubMed] [Google Scholar]
- 18.Conway DE, Sakurai Y, Weiss D, Vega JD, Taylor WR, Jo H, Eskin SG, Marcus CB, McIntire LV. Expression of CYP1A1 and CYP1B1 in human endothelial cells: regulation by fluid shear stress. Cardiovasc Res. 2009;81:669–677. doi: 10.1093/cvr/cvn360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mannisto PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51:593–628. [PubMed] [Google Scholar]
- 20.Suzuma I, Mandai M, Takagi H, Suzuma K, Otani A, Oh H, Kobayashi K, Honda Y. 17 Beta-estradiol increases VEGF receptor-2 and promotes DNA synthesis in retinal microvascular endothelial cells. Invest Ophthalmol Vis Sci. 1999;40:2122–2129. [PubMed] [Google Scholar]
- 21.Heryanto B, Rogers PA. Regulation of endometrial endothelial cell proliferation by oestrogen and progesterone in the ovariectomized mouse. Reproduction. 2002;123:107–113. doi: 10.1530/rep.0.1230107. [DOI] [PubMed] [Google Scholar]
- 22.Heryanto B, Lipson KE, Rogers PA. Effect of angiogenesis inhibitors on oestrogen-mediated endometrial endothelial cell proliferation in the ovariectomized mouse. Reproduction. 2003;125:337–346. doi: 10.1530/rep.0.1250337. [DOI] [PubMed] [Google Scholar]
- 23.Matsubara KMY, King AG, Zheng J, Abe E, Masaharu I, Magness RR. Regulation of endothelial cell proliferation by estrogen in reproductive organs. In: Kimura D, editor. Cell Growth Processes: New Research. Nova Science Publishers, Inc.; Hauppauge NY: 2008. pp. 159–182. [Google Scholar]
- 24.Yi FX, Boeldt DS, Gifford SM, Sullivan JA, Grummer MA, Magness RR, Bird IM. Pregnancy Enhances Sustained Ca2+ Bursts and Endothelial Nitric Oxide Synthase Activation in Ovine Uterine Artery Endothelial Cells Through Increased Connexin 43 Function. Biology of Reproduction. 2010;82:66–75. doi: 10.1095/biolreprod.109.078253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grummer MA, Sullivan JA, Magness RR, Bird IM. Vascular endothelial growth factor acts through novel, pregnancy-enhanced receptor signaling pathways to stimulate endothelial nitric oxide synthase activity in uterine artery endothelial cells. Biochem J. 2009;417:501–511. doi: 10.1042/BJ20081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sullivan JA, Grummer MA, Yi FX, Bird IM. Pregnancy-enhanced endothelial nitric oxide synthase (eNOS) activation in uterine artery endothelial cells shows altered sensitivity to Ca2+, U0126, and wortmannin but not LY294002--evidence that pregnancy adaptation of eNOS activation occurs at multiple levels of cell signaling. Endocrinology. 2006;147:2442–2457. doi: 10.1210/en.2005-0399. [DOI] [PubMed] [Google Scholar]
- 27.Ball P, Knuppen R. Catecholoestrogens (2-and 4-hydroxyoestrogens): chemistry, biogenesis, metabolism, occurrence and physiological significance. Acta Endocrinol Suppl (Copenh) 1980;232:1–127. [PubMed] [Google Scholar]
- 28.Rosenfeld CR, Jackson GM. Induction and inhibition of uterine vasodilation by catechol estrogen in oophorectomized, nonpregnant ewes. Endocrinology. 1982;110:1333–1339. doi: 10.1210/endo-110-4-1333. [DOI] [PubMed] [Google Scholar]
- 29.Stice SL, Ford SP, Rosazza JP, Van Orden DE. Interaction of 4-hydroxylated estradiol and potential-sensitive Ca2+ channels in altering uterine blood flow during the estrous cycle and early pregnancy in gilts. Biol Reprod. 1987;36:369–375. doi: 10.1095/biolreprod36.2.369. [DOI] [PubMed] [Google Scholar]
- 30.Fotsis T, Zhang Y, Pepper MS, Adlercreutz H, Montesano R, Nawroth PP, Schwelgerer L. The endogenous oestrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and suppresses tumour growth. Nature. 1994;368:237–239. doi: 10.1038/368237a0. [DOI] [PubMed] [Google Scholar]
- 31.Yue TL, Wang X, Louden CS, Gupta S, Pillarsetti K, Gu JL, Hart TK, Lysko PG. 2-Methoxyestradiol, an endogenous estrogen metabolite, induces apoptosis in endothelial cells and inhibits angiogenesis: possible role for stress-activated protein kinase signaling pathway and Fas expression. Mol Pharmacol. 1997;51:951–962. doi: 10.1124/mol.51.6.951. [DOI] [PubMed] [Google Scholar]
- 32.LaVallee TM, Zhan XH, Herbstritt CJ, Kough EC, Green SJ, Pribluda VS. 2-Methoxyestradiol inhibits proliferation and induces apoptosis independently of estrogen receptors alpha and beta. Cancer Res. 2002;62:3691–3697. [PubMed] [Google Scholar]
- 33.Ayumi T, Yoshiyasu K, Taku Y, Katsuken H, Masao I, Satoshi K. 2-Methoxyestradiol, an endogenous metabolite of estrogen, enhances apoptosis and β-Galactosidase expression in vascular endothelial cells. Biochemical and Biophysical Research Communications. 1998;248:9–12. doi: 10.1006/bbrc.1998.8902. [DOI] [PubMed] [Google Scholar]
- 34.Weirong S, Ioanna K, David SW. 2-Methoxyestradiol, an endogenous estradiol metabolite, differentially inhibits granulosa and endothelial cell mitosis: A potential follicular antiangiogenic regulator. Biology of Reproduction. 2001;65:622–627. doi: 10.1095/biolreprod65.2.622. [DOI] [PubMed] [Google Scholar]
- 35.Gui Y, Zheng XL. 2-Methoxyestradiol induces cell cycle arrest and mitotic cell apoptosis in human vascular smooth muscle cells. Hypertension. 2006;47:271–280. doi: 10.1161/01.HYP.0000199656.99448.dc. [DOI] [PubMed] [Google Scholar]
- 36.Zaitseva M, Yue DS, Katzenellenbogen JA, Rogers PA, Gargett CE. Estrogen receptor-alpha agonists promote angiogenesis in human myometrial microvascular endothelial cells. J Soc Gynecol Investig. 2004;11:529–535. doi: 10.1016/j.jsgi.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 37.Kraichely DM, Sun J, Katzenellenbogen JA, Katzenellenbogen BS. Conformational changes and coactivator recruitment by novel ligands for estrogen receptor-alpha and estrogen receptor-beta: correlations with biological character and distinct differences among SRC coactivator family members. Endocrinology. 2000;141:3534–3545. doi: 10.1210/endo.141.10.7698. [DOI] [PubMed] [Google Scholar]
- 38.Katzenellenbogen JA, Muthyala R, Katzenellenbogen BS. Nature of the ligand-binding pocket of estrogen receptor α and β: The search for subtype-selective ligands and implications for the prediction of estrogenic activity. Pure and Applied Chemistry. 2003;75:2397–2403. [Google Scholar]
- 39.Martucci C, Fishman J. Uterine estrogen receptor binding of catecholestrogens and of estetrol (1,3,5(10)-estratriene-3,15alpha,16alpha,17beta-tetrol) Steroids. 1976;27:325–333. doi: 10.1016/0039-128x(76)90054-4. [DOI] [PubMed] [Google Scholar]
- 40.Takada Y, Kato C, Kondo S, Korenaga R, Ando J. Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem Biophys Res Commun. 1997;240:737–741. doi: 10.1006/bbrc.1997.7734. [DOI] [PubMed] [Google Scholar]
- 41.Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol. 2008;70:165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 42.Rubanyi GM, Johns A, Kauser K. Effect of estrogen on endothelial function and angiogenesis. Vascul Pharmacol. 2002;38:89–98. doi: 10.1016/s0306-3623(02)00131-3. [DOI] [PubMed] [Google Scholar]
- 43.Dubey RK, Jackson EK. Cardiovascular protective effects of 17beta-estradiol metabolites. J Appl Physiol. 2001;91:1868–1883. doi: 10.1152/jappl.2001.91.4.1868. [DOI] [PubMed] [Google Scholar]
- 44.Miller VM, Duckles SP. Vascular actions of estrogens: functional implications. Pharmacol Rev. 2008;60:210–241. doi: 10.1124/pr.107.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.