

Abstract
Small-molecule hepatitis C virus (HCV) NS3 protease inhibitors such as boceprevir (SCH 503034) have been shown to have antiviral activity when they are used as monotherapy and in combination with pegylated alpha interferon and ribavirin in clinical trials. Improvements in inhibitor potency and pharmacokinetic properties offer opportunities to increase drug exposure and to further increase the sustained virological response. Exploration of the structure-activity relationships of ketoamide inhibitors related to boceprevir has led to the discovery of SCH 900518, a novel ketoamide protease inhibitor which forms a reversible covalent bond with the active-site serine. It has an overall inhibition constant (K*i) of 7 nM and a dissociation half-life of 1 to 2 h. SCH 900518 inhibited replicon RNA at a 90% effective concentration (EC90) of 40 nM. In biochemical assays, SCH 900518 was active against proteases of genotypes 1 to 3. A 2-week treatment with 5× EC90 of the inhibitor reduced the replicon RNA level by 3 log units. Selection of replicon cells with SCH 900518 resulted in the outgrowth of several resistant mutants (with the T54A/S and A156S/T/V mutations). Cross-resistance studies demonstrated that the majority of mutations for resistance to boceprevir and telaprevir caused similar fold losses of activity against all three inhibitors; however, SCH 900518 retained more activity against these mutants due to its higher intrinsic potency. Combination treatment with alpha interferon enhanced the inhibition of replicon RNA and suppressed the emergence of resistant replicon colonies, supporting the use of SCH 900518-pegylated alpha interferon combination therapy in the clinic. In summary, the results of the preclinical characterization of the antiviral activity of SCH 900518 support its evaluation in clinical studies.
Hepatitis C virus (HCV) is a major cause of chronic hepatitis worldwide. Despite recent advances, the eradication of chronic HCV infection remains challenging. Approximately 50% of patients infected with the most prevalent form of the virus (genotype 1) fail to respond to treatment with the current standard of care of pegylated alpha interferon in combination with ribavirin (4, 13). Thus, there is a great unmet medical need for the development of new agents with activity against HCV to improve the sustained virological response (SVR). The HCV genome is translated as a single polyprotein precursor which is processed by cellular and viral proteases into structural proteins (the C, E1, and E2 proteins), followed by nonstructural (NS) proteins (the p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B proteins). The NS3 protein is multifunctional, with the N-terminal region (amino acids 1 to 181) encoding a serine protease and the remaining polypeptide encoding a helicase. The NS3 protease noncovalently binds to the cofactor NS4A, which contributes to the formation and stability of the active site and helps to anchor the NS3/NS4A complex to the membrane. The NS3 protease is responsible for the processing of the HCV polyprotein at the NS3-NS4A, NS4A-NS4B, NS4B-NS5A, and NS5A-NS5B junctions (for a review, see reference 19).
The NS3 protease is essential for viral replication (6) and represents an important target for antiviral therapy. The clinical efficacies of NS3 protease inhibitors were first demonstrated with compounds targeting the enzyme active site in monotherapy and in combination with pegylated interferon: BILN 2061, a macrocyclic tripeptide in a short monotherapy trial (8), and boceprevir (SCH 503034) and telaprevir (VX-950), both of which are ketoamide derivatives (21, 22, 27, 28). Mutations conferring resistance to these small-molecule inhibitors have been identified in the NS3 protease domain by selection of replicon cells in the presence of compounds; many of the mutations selected in vitro have also been detected in patients during clinical trials of boceprevir and telaprevir (9, 11, 21, 25, 27, 28). Although encouraging clinical results have been achieved with these first-generation protease inhibitors, additional improvements in inhibitor potency and pharmacokinetic properties offers opportunities to increase drug coverage and to further increase the SVR.
The combination of medicinal chemistry and structure-based design has led to the synthesis of a new compound, SCH 900518 (narlaprevir; Fig. 1) (1), with improved antiviral activity in the replicon system compared with the activities of boceprevir (12) and telaprevir (16, 17). The characterization of its inhibition mechanism and in vitro resistance profile is presented in this report.
FIG. 1.
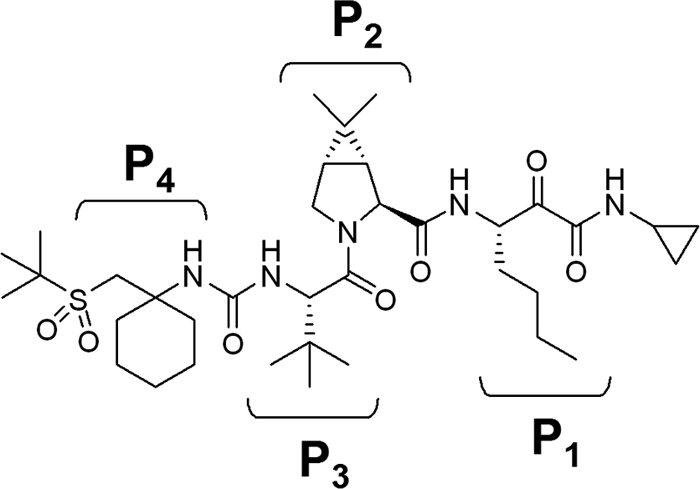
Structure of SCH 900518. P1 to P4 refer to different side chains.
MATERIALS AND METHODS
Continuous spectrophotometric assay.
Recombinant single-chain HCV NS3 and NS4A proteases from genotypes 1 to 3 were prepared as described previously (2, 23, 26). HCV protease assays were performed by use of a 200-μl reaction mixture, as described previously (29). Typically, 100 μl protease was added to 100 μl of assay buffer containing the chromogenic substrate acetyl-DTEDVVP(norvaline)-O-4-phenylazophenol. The change in the absorbance was monitored over 60 min with a Spectromax Plus microtiter plate reader (Molecular Devices).
Progress curve analysis.
Curves of the time-dependent progress of product formation in the presence of various concentrations of inhibitor were first fit to the modified Morrison equation (equation 1) (14) to obtain the initial velocity (vi), the steady-state velocity (vs), and the constant for the apparent rate of reaching the steady state (kobs). No-inhibitor controls yielded the uninhibited initial velocity (vo). The parameters obtained were fit to a two-step, postbinding slow isomerization model, according to the following procedure.
A plot of vi/vo versus the inhibitor concentration was fit to equation 2 to derive the apparent inhibition constant Kiapp for the initial, fast binding step. Similarly, a plot of vs/vo versus the inhibitor concentration was fit to equation 3 to derive the overall apparent inhibition constant K*appi for the subsequent slow isomerization step. Finally, a plot of kobs versus the inhibitor concentration was fit to equation 4 to derive the two isomerization rate constants k3 (from EI [initial enzyme-inhibitor complex] to EI* [final enzyme-inhibitor complex], where E represents the enzyme and I represents the inhibitor) and k4 (from EI* to EI) while fixing or floating Kiapp. The Kiapp and K*appi values are converted into Ki and K*i values by using equations 5a and 5b, respectively, with input of the substrate concentration and its predetermined Michaelis-Menten constant (Km). The inhibition constant, K*i, was used as a measure of inhibitor potency and should be distinguished from the term Ki, which is used for rapid-binding noncovalent competitive inhibitors.
To calculate koff, the overall rate constant of dissociation of the enzyme-inhibitor complex, we first estimated the magnitude of k2 (the rate of dissociation of EI to E and I) from Ki and reasonable ranges of k1 (105 to 108 M−1 s−1, the rate of association of E and I to form EI) using equation 6. The resultant estimate, necessarily an upper estimate for koff, is calculated with equation 7. The lower limit of the half-life of dissociation (t1/2) is calculated by using equation 8.
The two-step, postbinding slow isomerization model is E + I ↔ EI ↔ EI*.
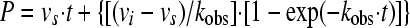 |
(1) |
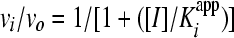 |
(2) |
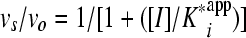 |
(3) |
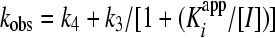 |
(4) |
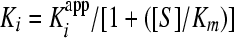 |
(5a) |
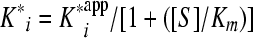 |
(5b) |
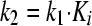 |
(6) |
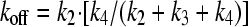 |
(7) |
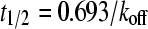 |
(8) |
where t is time, [I] is the inhibitor concentration, and [S] is the substrate concentration.
Replicon assay.
Replicon (genotype 1b) construction was performed as described previously (12, 25). To measure antireplicon activity, replicon cells were seeded in 96-well collagen I-coated plates, SCH 900518 was added at 24 h postseeding, and the plates were incubated for 3 days, at which point the cells were lysed and the replicon RNA level was measured by real-time PCR. The 50% effective concentration (EC50) was the drug concentration necessary to achieve an increase in the cycle threshold (CT) of 1 over the projected baseline CT. The EC90 was the drug concentration necessary to achieve an increase in CT of 3.2 over the projected baseline CT.
For combination treatment with alpha interferon, replicon cells were treated for 3 days with SCH 900518, which was serially diluted 1:2.5 for a 5-point titration. At each concentration of SCH 900518, alpha interferon was titrated in. The replicon RNA level was measured by real-time PCR (TaqMan assay) with GAPDH RNA as an endogenous control. Relative replicon RNA inhibition (dCT) was calculated as follows:
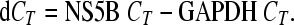 |
Combination index (CI) values were calculated as described previously (3) for alpha interferon in combination with multiple fixed concentrations of SCH 900518 that were approximately equal to or below its EC50, EC75, and EC90 by using the following nonexclusive formula:
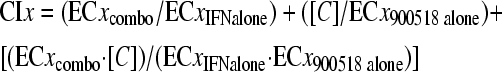 |
where x refers to either the 50, 75, or 90% inhibition parameter and ECxIFN, alone and ECxcombo represent the concentrations of alpha interferon alone and in combination with the fixed concentration of SCH 900518 ([C]) needed to inhibit viral replication by 50, 75, or 90%. ECx900518, alone represents the ECx of SCH 900518 alone. A CI of 1 is considered additive, a CI of <1 is considered synergistic, and a CI of >1 is considered antagonistic.
Replicon resistance studies.
To select replicon cells resistant to SCH 900518, subconfluent monolayers of genotype 1b replicon cells and parental line Huh-7 were cultured with various concentrations of the compound (in the resistance study, the EC90 of SCH 900518 was 65 nM) with or without alpha interferon. All cells were passed at a 1:10 ratio when they reached 95% confluence. The colonies that survived selection were pooled and expanded for further analysis.
To identify mutations in the NS3 protease domain which conferred resistance to SCH 900518, total cellular RNA was isolated from pooled colonies and amplified by reverse transcription-PCR (RT-PCR), as described previously (25). The RT-PCR products were purified with a QIAquick PCR purification kit (Qiagen) and sequenced with 3130xl genetic analyzer (Applied Biosystems). Alternatively, the RT-PCR products were cloned into the TOPO TA vector (Invitrogen), and the plasmid DNA from five to six bacterial colonies was sequenced.
X-ray structural analysis.
The crystal structure of SCH 900518 complexed with the NS3-NS4A protease was solved in-house (1). To illustrate the key resistance-conferring loci, the structure of SCH 900518 is presented as a stick model, and the side chains of residues are shown by using CPK models on the Connolly surface of the NS3 protease.
Protein structure accession number.
The crystal structure of SCH 900518 complexed with the NS3-NS4A protease solved in-house can be found in PDB under code 3LON.
RESULTS
Inhibition of HCV NS3 protease in vitro.
Exploration of the structure-activity relationships of ketoamide inhibitors related to boceprevir has resulted in the development of the novel ketoamide SCH 900518 (Fig. 1). X-ray structural analysis has shown that SCH 900518 forms a covalent bond with the active-site Ser139 (1). In the continuous assay, the time required for stable covalent adduct formation is typified by a slow onset of inhibition, as shown in Fig. 2. Progress curve analysis estimated the overall inhibition constant (K*i) of SCH 900518 for genotype 1b NS3 protease to be 7 ± 1 nM (n = 12 experiments). SCH 900518 was estimated to dissociate from the enzyme-inhibitor complex with a lower limit of the half-life of 1 to 2 h (for details, see Materials and Methods). SCH 900518 was also active against proteases from genotypes 1a, 2a, and 3a (K*is, 0.7 nM, 3 nM, and 7 nM, respectively).
FIG. 2.

(A) Inhibition of HCV genotype 1b NS3 protease by SCH 900518. The progress curve for SCH 900518 shows the slow binding inhibition. The term K*i is derived from the velocity at steady state and is used to distinguish the overall inhibition constant obtained for the slow (e.g., covalent)-binding inhibitors from the term Ki used for rapid-binding noncovalent competitive inhibitors. (B) Inhibition of HCV replicon by SCH 900518. Replicon cells were treated with SCH 900518 for 3 days, and replicon RNA levels were measured by TaqMan assay. The average (with SD) of the relative reduction in the replicon RNA level (dCT) from triplicate measurements was plotted against the log compound concentration.
Replicon activity.
The antiviral activity of SCH 900518 was evaluated in the genotype 1b HCV replicon system. The EC50 and EC90 values for SCH 900518 were 20 ± 6 nM (n = 49 experiments) and 40 ± 10 nM (n = 63 experiments), respectively, in a 72-h assay (Fig. 2B). Prolonged exposure to 1.2× EC90 SCH 900518 resulted in a 2-log-unit decrease in RNA levels by day 14 (Fig. 3). Dosing with 5× EC90 resulted in a 3-log-unit decrease in RNA levels by day 14. No cytotoxicity was observed with SCH 900518 in replicon cells by the MTS assay (50% cytotoxic concentration > 25 μM).
FIG. 3.

Effect of treatment duration and inhibitor concentration on replicon RNA level. Replicon cells were treated with various concentrations of SCH 900518, as indicated. The replicon cells were refreshed with the compound every 2 to 3 days, and the cells were split when they became confluent. The log RNA reduction at day 0 is set equal to 0. The model used to fit the curve is one-phase exponential decay. The reduction in the amount of replicon RNA is calculated as follows: dCT = NS5B CT − GAPDH CT, where ddCT = dCT of treated samples − dCT for the vehicle control and where the log RNA reduction = log(1/2ddCT). IC90, 90% inhibitory concentration.
Combination studies with interferon alfa-2b.
When interferon alfa-2b was coadministered with SCH 900518, there was a concentration-dependent, enhanced inhibition of replicon RNA compared to that achieved with treatment with SCH 900518 alone (Fig. 4A). The activity of the combination of alpha interferon and SCH 900518 was shown to be at least additive. The CIs at 50%, 75%, and 90% inhibition levels (± standard deviations [SDs] from two to four experiments) were calculated to be 0.3 ± 0.1, 0.6 ± 0.2, and 0.6 ± 0.1, respectively. Importantly, the use of SCH 900518 in combination with interferon alfa-2b also suppressed the emergence of resistant replicon colonies (Fig. 4B). Culturing of replicon-bearing cells in the presence of 3× EC90 SCH 900518 for 3 weeks resulted in the emergence of resistant colonies at a level of approximately 0.13%. Increases in the concentration of SCH 900518 to 6× EC90 and 15× EC90 reduced the number of resistant colonies that emerged by about 14- and 65-fold, respectively. The emergence of resistant colonies was reduced about 10-fold when 3× EC90 SCH 900518 was used with interferon alfa-2b (30 IU/ml, which is ∼10× EC90) compared to the rate of emergence of resistant colonies when SCH 900518 was used alone. This suggests that SCH 900518 could be used in combination with interferon-based therapy to minimize the emergence of resistant mutants.
FIG. 4.

(A) Combination treatment with interferon alfa-2b enhances the inhibition of replicon RNA. Replicon cells were treated for 3 days with SCH 900518, which was serially diluted. At each concentration of SCH 900518, alpha interferon was titrated in. The replicon RNA level was measured by real-time PCR (TaqMan assay) with GAPDH RNA as an endogenous control. Relative replicon RNA inhibition (dCT) is calculated as dCT = NS5B CT − GAPDH CT. (B) Combination treatment with interferon alfa-2b suppresses the emergence of replicon resistance. Replicon cells were cultured with various concentrations of the compound with or without alpha interferon. All cells were passed at a 1:10 ratio when they reached confluence. The colonies that survived selection were stained and counted. IC90, 90% inhibitory concentration.
Characterization of resistant cells.
Sequence analysis of replicon clones recovered from the SCH 900518 selection identified mutations at two different loci (amino acids 54 and 156) in the protease domain (Table 1). After selection with 6× EC90 of SCH 900518, T54A/S mutations, which conferred an approximately 10-fold increase in the EC90 values, were the predominant changes observed in resistant replicon cells. Increasing the selection concentration to 15× EC90 led to the emergence and outgrowth of mutations at the A156 locus, which conferred higher levels of resistance to the compound. A double mutation (T54A and A156T) was observed in one clone at the higher selecting concentration. Replicon cells carrying the double mutation were resistant to SCH 900518 even when it was used at 40 μM. The locations of T54 and A156 and the structure of SCH 900518 complexed with the NS3-NS4A protease are shown in Fig. 5. Importantly, replicon cells resistant to SCH 900518 remained sensitive to interferon alfa-2b (Table 2).
TABLE 1.
Resistance-conferring mutations selected by SCH 900518
| Mutation(s) | Fold increase in EC90 | Frequencya at the following drug level: |
|
|---|---|---|---|
| 6× EC90 | 15× EC90 | ||
| T54A/S | 10 | 4/6 | 0/5 |
| A156S | 30-60 | 1/6 | 3/5 |
| A156T/V | ∼500 | 1/6 | 1/5 |
| T54A, A156T | >600 | 0/6 | 1/5 |
Frequency is represented as the number of clones carrying the mutation/total number of clones sequenced.
FIG. 5.

Structural view of resistance loci of SCH 900518 identified from replicon studies. The structure of SCH 900518 is presented as a stick model. Side chains of key residues are shown by using CPK models on the Connolly surface of the NS3 protease. The colors of the residues are defined as follows: red, active-site Ser139; magenta, Ala156; yellow, Thr54.
TABLE 2.
Sensitivity to alpha interferon in replicon cells selected by SCH 900518
| SCH 900518 treatment | EC50 (U/ml) | EC90 (U/ml) |
|---|---|---|
| No selection | 0.1 | 3 |
| Selection with 6× EC90 | 0.3 | 4.5 |
| Selection with 15× EC90 | 0.1 | 3.5 |
Further studies showed that SCH 900518 was cross-resistant to mutations against boceprevir (24) in enzymatic and replicon assays (Table 3). The potency of SCH 900518 was moderately reduced (approximately 10-fold or less) by the following resistance mutations: V36M/A, F43C, T54A/S, and V170A. SCH 900518 lost significantly more activity against mutations at locus 155 (R155K; 15-fold by the replicon assay) and locus 156 (A156S/T; 70- to >1,000-fold by the replicon assay). As previously shown with boceprevir, A156T conferred the highest level of resistance to SCH 900518. The fold increase in resistance for mutants with double mutations was approximately equal to the product of the individual mutations (i.e., the increase was multiplicative rather than additive). However, SCH 900518 remained fully active against mutants with the D168V mutation, which confers resistance to BILN 2061, a nonketoamide NS3 protease inhibitor (11).
TABLE 3.
Enzyme and replicon activity of SCH 900518 against mutations conferring resistance to boceprevira
| Parameter | Mutation(s) | Fold increaseb |
|---|---|---|
| K*i | V36M | 3 |
| V36A | 5 | |
| F43C | 8 | |
| T54A | 8 | |
| T54Sc | 3 | |
| R155K | 8 | |
| A156S | 100 | |
| A156T | 3,000 | |
| D168V | 1 | |
| V170A | 6 | |
| V36 M + R155K | 160 | |
| T54S + R155Kc | 58 | |
| EC50 | V36M | 8 |
| T54A | 11 | |
| R155K | 15 | |
| A156S | 70 | |
| A156T | >1,000 | |
| D168V | 1 | |
| V170A | 10 | |
| V36 M + R155K | 130 |
Data are the means of two or more experiments, unless indicated otherwise.
Fold increase in parameter value with SCH 900518 treatment relative to that for the wild type.
Data are from a single experiment.
DISCUSSION
SCH 900518 is a potent, novel serine protease inhibitor designed to inhibit HCV NS3 protease and, therefore, inhibit viral replication in infected host cells. Structural and kinetic analyses have shown that the mechanism of inhibition involves the covalent yet reversible binding of SCH 900518 to the NS3 protease active-site serine (Ser139) through a ketoamide functional group (1; this study). SCH 900518 inhibits the NS3 protease in two steps. In the first step, it binds to the enzyme noncovalently. In the second, slower step, the α-ketoamide group is rearranged to form a covalent bond with the active-site Ser139. The overall inhibition constant K*i, derived from the velocity at steady state, is used to measure the potency of slow-binding inhibitors such as SCH 900518, boceprevir, and telaprevir and is distinguished from the term Ki used for rapid-binding noncovalent competitive inhibitors such as BILN 2061. The covalent enzyme-inhibitor complex is usually very stable. Progress curve analysis showed that SCH 900518 dissociates from the stable complex with a half-life of 1 to 2 h, which is similar to that of telaprevir (1 to >5 h) (16-18) and slightly shorter than that of boceprevir (10 to 20 h) (12) but significantly longer than that previously reported for BILN 2061 (on the order of minutes) (8, 18). Estimates of the value of the dissociation constant can vary by the different types of assays used to measure the stability of enzyme-inhibitor complexes (e.g., progress curve analysis, reactivation assay, and affinity binding assay). Assay conditions such as the enzyme and substrate sequences can also affect measurements of kinetics. Nevertheless, our results are consistent with the slow dissociation kinetics of ketoamide inhibitors, and this mechanism of an increased residence time may be responsible, at least in part, for the observed potent antiviral activities of ketoamide inhibitors.
Alignment of the sequences of over 250 isolates of HCV encompassing all genotypes suggested that the proximal subsite residues of the protease are completely conserved (2). Consistent with the prediction based on sequence conservation, SCH 900518 is equally active against proteases from genotype 1b, 2a, and 3a isolates but is 5- to 10-fold more active against the genotype 1a protease in the enzymatic assay. Two other ketoamide inhibitors, boceprevir and telaprevir, have also demonstrated activity against a broad spectrum of genotypes (10, 16, 26), although differences in relative activity across genotypes have been observed in different assay systems. Studies with proteases from multiple isolates of each genotype are required to fully assess variations across genotypes. In the replicon assay, the EC50 and EC90 values for SCH 900518 were 20 nM and 40 nM, respectively, which makes it approximately 10-fold more potent than boceprevir and telaprevir, the two ketoamide inhibitors currently in late-stage clinical development. Extended treatment with SCH 900518 demonstrated inhibition of replicon RNA in concentration- and time-dependent manners, which resulted in up to a 3-log-unit reduction in the replicon RNA level without a rebound. When interferon alfa-2b was coadministered with SCH 900518, there was a concentration-dependent enhanced inhibition of replicon RNA. Calculation of the combination index indicated no antagonism between the two agents (CI < 1). It has recently been recommended that a more conservative criterion be used to report drug-drug interactions (15). Using this more stringent cutoff, there is modest synergy between alpha interferon and SCH 900518 at low concentration (CI50 < 0.5 ); at higher concentrations, the two agents appear to be additive (CI75 and CI90 > 0.5 to 4). Importantly, the emergence of resistant mutants was significantly reduced when SCH 900518 was used in combination with interferon alfa-2b. The enhanced inhibition of replicon RNA and the suppressed emergence of resistant colonies suggest the potential for the clinical use of SCH 900518 in combination with pegylated alpha interferon therapy.
In replicon cells selected with SCH 900518, mutations at amino acids 54 and 156 in the protease domain were observed, and both of these have been identified in resistance studies with boceprevir (25). An increase in the selection concentration reduced the number of resistant colonies and resulted in a shift in the predominant mutations toward those conferring higher levels of resistance (from T54A/S at 6× EC90 to A156S/T/V at 15× EC90). Replicons carrying the A156T mutation were significantly less fit than replicons carrying other resistance mutations (25), consistent with the observed reduction in the resistance frequency at higher selection concentrations. Of note, replicon cells resistant to SCH 900518 remained sensitive to interferon alfa-2b. X-ray structural analysis (1) showed that the A156 side chain is in van der Waals contact with the P2 and P4 side chains of SCH 900518. Mutation of A156 to bulkier residues (Ser/Thr/Val) causes stereo hindrance in the region. To avoid crowding, the P2 and P4 backbone would shift away from T156, resulting in weaker hydrogen-bonding interactions, as previously observed with boceprevir (5). Although T54 is not in direct contact with the inhibitor, it is important in keeping F43 (which is part of the S1′ substrate-binding pocket) in its proper conformation through hydrogen bonding with L44, as proposed by Zhou et al. (30). A reduced interaction or the loss of such an interaction would increase the flexibility of F43 and weaken the SCH 900518 binding. Consistent with the hypothesis, change of F43 to C43 resulted in an 8-fold loss in binding affinity (Table 3).
Further characterization of the SCH 900518 resistance profile showed that SCH 900518 was cross-resistant to mutations conferring resistance to boceprevir and telaprevir. A majority of mutations (e.g., V36M/A, F43C, T54A/S, and V170A) caused similar fold losses of activity against all three inhibitors (Table 3) (24). However, SCH 900518 was more potent against these mutants due to its high intrinsic level of potency. The exceptions are mutations at R155 and A156, which appeared to affect the activity of SCH 900518 more than that of boceprevir. The clinical implications of the differences in the in vitro resistance profiles of boceprevir and SCH 900518 remain to be determined.
Evaluations of HCV NS3 protease inhibitors have shown encouraging results in improving SVR rate, validating the NS3 protease as a promising drug target. In a phase II trial with treatment-naïve patients (the SPRINT-1 trial), use of the combination of boceprevir (800 mg three times a day) and the standard of care (SOC) for 24 to 48 weeks achieved an SVR rate of 55 to 75%, whereas the SVR for the control arm (which consisted of the SOC for 48 weeks) was 38% (7). Boceprevir is currently in a phase III clinical trial to further evaluate its efficacy and safety profile. The results obtained in early clinical trials of SCH 900518 have also been very encouraging (20). With its more potent in vitro antiviral activity as well as improved pharmacokinetic profile and physicochemical characteristics (1), SCH 900518 has the potential to provide HCV-infected patients with another treatment option in the future.
Acknowledgments
We thank Rachael Steiner and Julie Strizki for helpful discussions.
This work is dedicated to the memory of our beloved coauthor and colleague Angela Skelton.
Footnotes
Published ahead of print on 2 March 2010.
REFERENCES
- 1.Arasappan, A., F. Bennett, S. L. Bogen, S. Venkatraman, M. Blackman, K. Chen, S. Hendrata, Y. Huang, R. M. Huelgas, L. Nair, A. I. Padilla, W. Pan, R. Pike, P. Pinto, S. Ruan, M. Sannigrahi, F. Velazquez, B. Vibulbhan, W. Wu, W. Yang, A. K. Saksena, V. Girijavallabhan, N. Y. Shih, J. Kong, T. Meng, Y. Jin, J. Wong, P. McNamara, A. Prongay, V. Madison, J. J. Piwinski, K. C. Cheng, R. Morrison, B. Malcolm, X. Tong, R. Ralston, and F. G. Njoroge. Discovery of narlaprevir (SCH 900518): a potent, second generation HCV NS3 serine protease inhibitor. ACS Med. Chem. Lett. doi: 10.1021/ml9000276. [DOI] [PMC free article] [PubMed]
- 2.Beyer, B. M., R. Zhang, Z. Hong, V. Madison, and B. A. Malcolm. 2001. Effect of naturally occurring active site mutations on hepatitis C virus NS3 protease specificity. Proteins 43:82-88. [PubMed] [Google Scholar]
- 3.Chou, T. C., and P. Talalay. 1984. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22:27-55. [DOI] [PubMed] [Google Scholar]
- 4.Fried, M. W., M. L. Shiffman, K. R. Reddy, C. Smith, G. Marinos, F. L. Goncales, Jr., D. Haussinger, M. Diago, G. Carosi, D. Dhumeaux, A. Craxi, A. Lin, J. Hoffman, and J. Yu. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347:975-982. [DOI] [PubMed] [Google Scholar]
- 5.Guo, Z., A. Prongay, X. Tong, T. Fischmann, S. Bogen, F. Velazquez, S. Venkatraman, F. G. Njoroge, and V. Madison. 2006. Computational study of the effects of mutations A156T, D168V, and D168Q on the binding of HCV protease inhibitors. J. Chem. Theory Comput. 2:1657-1663. [DOI] [PubMed] [Google Scholar]
- 6.Kolykhalov, A. A., K. Mihalik, S. M. Feinstone, and C. M. Rice. 2000. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 74:2046-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwo, P., E. J. Lawitz, J. McCone, E. Schiff, J. Vierling, D. Pound, M. Davis, J. Galati, S. Gordon, N. Ravendhran, L. Rossaro, F. Anderson, I. Jacobson, R. Rubin, K. Koury, C. Brass, E. Chaudhri, and J. Albrecht. 2009. HCV SPRINT-1 final results: SVR 24 from a phase 2 study of boceprevir plus PegIntron (peginterferon alfa-2b)/ribavirin in treatment-naive subjects with genotype-1 chronic hepatitis C. J. Hepatol. 50:S4. [Google Scholar]
- 8.Lamarre, D., P. C. Anderson, M. Bailey, P. Beaulieu, G. Bolger, P. Bonneau, M. Bos, D. R. Cameron, M. Cartier, M. G. Cordingley, A. M. Faucher, N. Goudreau, S. H. Kawai, G. Kukolj, L. Lagace, S. R. LaPlante, H. Narjes, M. A. Poupart, J. Rancourt, R. E. Sentjens, R. St. George, B. Simoneau, G. Steinmann, D. Thibeault, Y. S. Tsantrizos, S. M. Weldon, C. L. Yong, and M. Llinas-Brunet. 2003. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426:186-189. [DOI] [PubMed] [Google Scholar]
- 9.Lin, C., K. Lin, Y. P. Luong, B. G. Rao, Y. Y. Wei, D. L. Brennan, J. R. Fulghum, H. M. Hsiao, S. Ma, J. P. Maxwell, K. M. Cottrell, R. B. Perni, C. A. Gates, and A. D. Kwong. 2004. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061: structural analysis indicates different resistance mechanisms. J. Biol. Chem. 279:17508-17514. [DOI] [PubMed] [Google Scholar]
- 10.Liverton, N. J., S. S. Carroll, J. Dimuzio, C. Fandozzi, D. J. Graham, D. Hazuda, M. K. Holloway, S. W. Ludmerer, J. A. McCauley, C. J. McIntyre, D. B. Olsen, M. T. Rudd, M. Stahlhut, and J. P. Vacca. 2009. MK-7009, a potent and selective inhibitor of hepatitis C virus NS3/4A protease. Antimicrob. Agents Chemother. 54:305-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu, L., T. J. Pilot-Matias, K. D. Stewart, J. T. Randolph, R. Pithawalla, W. He, P. P. Huang, L. L. Klein, H. Mo, and A. Molla. 2004. Mutations conferring resistance to a potent hepatitis C virus serine protease inhibitor in vitro. Antimicrob. Agents Chemother. 48:2260-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malcolm, B. A., R. Liu, F. Lahser, S. Agrawal, B. Belanger, N. Butkiewicz, R. Chase, F. Gheyas, A. Hart, D. Hesk, P. Ingravallo, C. Jiang, R. Kong, J. Lu, J. Pichardo, A. Prongay, A. Skelton, X. Tong, S. Venkatraman, E. Xia, V. Girijavallabhan, and F. G. Njoroge. 2006. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob. Agents Chemother. 50:1013-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manns, M. P., J. G. McHutchison, S. C. Gordon, V. K. Rustgi, M. Shiffman, R. Reindollar, Z. D. Goodman, K. Koury, M. Ling, and J. K. Albrecht. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958-965. [DOI] [PubMed] [Google Scholar]
- 14.Morrison, J. F., and C. T. Walsh. 1988. The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 61:201-301. [DOI] [PubMed] [Google Scholar]
- 15.Odds, F. C. 2003. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 52:1. [DOI] [PubMed] [Google Scholar]
- 16.Perni, R. B., S. J. Almquist, R. A. Byrn, G. Chandorkar, P. R. Chaturvedi, L. F. Courtney, C. J. Decker, K. Dinehart, C. A. Gates, S. L. Harbeson, A. Heiser, G. Kalkeri, E. Kolaczkowski, K. Lin, Y. P. Luong, B. G. Rao, W. P. Taylor, J. A. Thomson, R. D. Tung, Y. Wei, A. D. Kwong, and C. Lin. 2006. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease. Antimicrob. Agents Chemother. 50:899-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perni, R. B., G. Chandorkar, P. R. Chaturvedi, L. F. Courtney, C. J. Decker, C. A. Gates, S. L. Harbeson, A. D. Kwong, C. Lin, K. Lin, Y. P. Luong, W. Markland, B. G. Rao, R. D. Tung, and J. A. Thompson. 2003. VX-950: the discovery of an inhibitor of the hepatitis C NS3 4A protease and a potential hepatitis C virus therapeutic. Hepatology 38:972A. [Google Scholar]
- 18.Rajagopalan, R., S. Misialek, S. Stevens, D. Myszka, B. Brandhuber, J. Ballard, S. Andrews, S. Seiwert, and K. Kossen. 2009. Inhibition and binding kinetics of the hepatitis C virus NS3 protease inhibitor ITMN-191 reveals tight binding and slow dissociative behavior. Biochemistry 11:11. [DOI] [PubMed] [Google Scholar]
- 19.Reed, K. E., and C. M. Rice. 2000. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr. Top. Microbiol. Immunol. 242:55-84. [DOI] [PubMed] [Google Scholar]
- 20.Reesink, H. W., J. F. Bergmann, J. de Bruijne, C. J. Weegink, J. J. van Lier, A. van Vliet, A. Keung, J. Li, J. O'Mara, M. A. Treitel, E. A. Hughes, H. L. A. Janssen, and R. J. de Knegt. 2009. Safety and antiviral activity of SCH 900518 administered as monotherapy and in combination with peginterferon alfa-2b to naive and treatment-experienced HCV-1 infected patients. J. Hepatol. 50(Suppl. 1):S35. [Google Scholar]
- 21.Sarrazin, C., T. L. Kieffer, D. Bartels, B. Hanzelka, U. Muh, M. Welker, D. Wincheringer, Y. Zhou, H. M. Chu, C. Lin, C. Weegink, H. Reesink, S. Zeuzem, and A. D. Kwong. 2007. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132:1767-1777. [DOI] [PubMed] [Google Scholar]
- 22.Sarrazin, C., R. Rouzier, F. Wagner, N. Forestier, D. Larrey, S. K. Gupta, M. Hussain, A. Shah, D. Cutler, J. Zhang, and S. Zeuzem. 2007. SCH 503034, a novel hepatitis C virus protease inhibitor, plus pegylated interferon alpha-2b for genotype 1 nonresponders. Gastroenterology 132:1270-1278. [DOI] [PubMed] [Google Scholar]
- 23.Taremi, S. S., B. Beyer, M. Maher, N. Yao, W. Prosise, P. C. Weber, and B. A. Malcolm. 1998. Construction, expression, and characterization of a novel fully activated recombinant single-chain hepatitis C virus protease. Protein Sci. 7:2143-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tong, X., S. Bogen, R. Chase, V. Girijavallabhan, Z. Guo, F. G. Njoroge, A. Prongay, A. Saksena, A. Skelton, E. Xia, and R. Ralston. 2008. Characterization of resistance mutations against HCV ketoamide protease inhibitors. Antiviral Res. 77:177-185. [DOI] [PubMed] [Google Scholar]
- 25.Tong, X., R. Chase, A. Skelton, T. Chen, J. Wright-Minogue, and B. A. Malcolm. 2006. Identification and analysis of fitness of resistance mutations against the HCV protease inhibitor SCH 503034. Antiviral Res. 70:28-38. [DOI] [PubMed] [Google Scholar]
- 26.Tong, X., Z. Guo, J. Wright-Minogue, E. Xia, A. Prongay, V. Madison, P. Qiu, S. Venkatraman, F. Velazquez, F. G. Njoroge, and B. A. Malcolm. 2006. Impact of naturally occurring variants of HCV protease on the binding of different classes of protease inhibitors. Biochemistry 45:1353-1361. [DOI] [PubMed] [Google Scholar]
- 27.Zeuzem, S., C. Sarrarin, R. Rouzier, A. Tarral, N. Brion, N. Forestier, S. Gupta, D. Deckman, K. Fellows, M. Hussain, D. Cutler, and J. Zhang. 2005. Anti-viral activity of SCH 503034, a HCV protease inhibitor, administered as monotherapy in HCV-1 patients refractory to pegylated interferon (PEG-IFN-α). Hepatology 42:233A. [Google Scholar]
- 28.Zeuzem, S., C. Sarrarin, F. Wagner, R. Rouzier, N. Forestier, S. Gupta, M. Hussain, A. Shah, D. Cutler, and J. Zhang. 2005. Combination therapy with HCV protease inhibitor SCH 503034, plus PEG-Intron in hepatitis C genotype-1 PEG-Intron non-responders. Hepatology 42:276A. [Google Scholar]
- 29.Zhang, R., B. M. Beyer, J. Durkin, R. Ingram, F. G. Njoroge, W. T. Windsor, and B. A. Malcolm. 1999. A continuous spectrophotometric assay for the hepatitis C virus serine protease. Anal. Biochem. 270:268-275. [DOI] [PubMed] [Google Scholar]
- 30.Zhou, Y., D. J. Bartels, B. L. Hanzelka, U. Muh, Y. Wei, H. M. Chu, A. M. Tigges, D. L. Brennan, B. G. Rao, L. Swenson, A. D. Kwong, and C. Lin. 2008. Phenotypic characterization of resistant Val36 variants of hepatitis C virus NS3-4A serine protease. Antimicrob. Agents Chemother. 52:110-120. [DOI] [PMC free article] [PubMed] [Google Scholar]