

Abstract
It is well established that the glutamate decarboxylase (GAD) system is central to the survival of Listeria monocytogenes at low pH, both in acidic foods and within the mammalian stomach. The accepted model proposes that under acidic conditions extracellular glutamate is transported into the cell in exchange for an intracellular γ-aminobutyrate (GABAi). The glutamate is then decarboxylated to GABAi, a reaction that consumes a proton, thereby helping to prevent acidification of the cytoplasm. In this study, we show that glutamate supplementation had no influence on either growth rate at pH 5.0 or survival at pH 2.5 when L. monocytogenes 10403S was grown in a chemically defined medium (DM). In response to acidification, cells grown in DM failed to efflux GABA, even when glutamate was added to the medium. In contrast, in brain heart infusion (BHI), the same strain produced significant extracellular GABA (GABAe) in response to acidification. In addition, high levels of GABAi (>80 mM) were found in the cytoplasm in response to low pH in both growth media. Medium-swap and medium-mixing experiments revealed that the GABA efflux apparatus was nonfunctional in DM, even when glutamate was present. It was also found that the GadT2D2 antiporter/decarboxylase system was transcribed poorly in DM-grown cultures while overexpression of gadD1T1 and gadD3 occurred in response to pH 3.5. Interestingly, BHI-grown cells did not respond with upregulation of any of the GAD system genes when challenged at pH 3.5. The accumulation of GABAi in cells grown in DM in the absence of extracellular glutamate indicates that intracellular glutamate is the source of the GABAi. These results demonstrate that GABA production can be uncoupled from GABA efflux, a finding that alters the way we should view the operation of bacterial GAD systems.
The capacity to produce γ-aminobutyric acid (GABA) through glutamate decarboxylation is commonly found in both Gram-negative and Gram-positive bacterial genera (10, 12). In several cases, this reaction has been shown to be critical for bacteria to survive potentially lethal acidic environments (15, 18, 20). It is generally held that the hydrogen ion consumed during the decarboxylation reaction helps to prevent excessive acidification of the cytoplasm, thereby protecting the cells against acidic environments. The GABA produced in the reaction is removed from the cell through the activity of an antiporter that exchanges a GABA molecule for an extracellular glutamate (Glu) molecule (6, 12).
In Listeria monocytogenes, the Gram-positive food-borne pathogen that was the focus of the present study, the glutamate decarboxylase (GAD) system has been shown to play an essential role in acid tolerance (8, 9). Mutants compromised in their ability to catalyze this decarboxylation reaction survive poorly both in acidic foods (8) and gastric juice (9). The GAD system in most L. monocytogenes strains is encoded by a total of five genes. There are three genes, designated gadD1, gadD2, and gadD3, that encode distinct glutamate decarboxylase enzymes and two genes, designated gadT1 and gadT2, that encode two Glu-GABA antiporters. These genes are organized at three separate genetic loci: gadD1T1, gadT2D2, and gadD3 (11). The decarboxylase/antiporter system encoded by gadT2D2 plays a central role in allowing survival under extreme acidic conditions; mutants lacking either the GadT2 antiporter or the GadD2 decarboxylase are highly sensitive to low pH (9, 11). In contrast, the GadD1/GadT1 decarboxylase/antiporter system appears to be more important for growth under moderately acidic conditions (11). The genes encoding this system are absent from most serotype 4 strains, and this generally correlates with a reduced ability of these strains to grow well at low pH (11). The role of GadD3 is less clear since it has not been possible to generate a deletion mutant lacking the corresponding gene (9).
Although the activity of the decarboxylase is generally thought to be coupled directly to the antiporter activity (i.e., the efflux of GABA is coupled to the supply of Glu) there is little direct evidence for this, even in bacteria where the system has been very well characterized. Most studies of the bacterial GAD system have used complex growth media when studying acid tolerance and GABA production (7, 8, 15). In the present study, we sought to determine whether extracellular Glu is a requirement for the production of GABA in L. monocytogenes. To do this, we have used a chemically defined growth medium (DM) that supports the growth of L. monocytogenes but does not include Glu. The results indicate that cells cultured in this medium do not produce extracellular GABA (GABAe) in response to low pH but are capable of accumulating substantial pools of intracellular GABA (GABAi). We establish that some component of complex medium is indispensable for efficient efflux of GABA. Surprisingly, supplementation of the DM with Glu failed to stimulate the extracellular release of GABA. We show that the GadD2/GadT2 decarboxylase/antiporter system is not transcribed when cells are grown in DM and suggest that this accounts for much of the difference in GABA production between cells cultured in DM and complex growth medium.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Wild-type Listeria monocytogenes strain 10403S (serotype 1/2a) was used throughout this study. The stock culture was stored at −80°C in 15% (vol/vol) glycerol. Prior to the experiments, the stock culture was streaked onto brain heart infusion (BHI) agar (LAB, Lancashire, United Kingdom) and incubated at 37°C overnight. A single colony from this medium was transferred to 2 ml of sterile BHI broth (LAB, Lancashire, United Kingdom) or DM and incubated overnight at 37°C with shaking (160 rpm). No additional buffering agents were added to either growth medium. Subsequently, a portion of these overnight cultures served as the inoculum (0.3% [vol/vol]) to prepare the cultures that were used in the experiments. These cultures were prepared in 250-ml conical flasks containing 20 ml of the same medium as the one used for the inoculum and incubated overnight at 37°C with shaking (160 rpm). DM was prepared according to Amezaga et al. (3).
Growth under acidic conditions.
Growth experiments were performed in BHI and DM or in the same media supplemented with 1 mM or 10 mM l-glutamate (Glu; Sigma-Aldrich, Steinheim, Germany). The pH of the growth medium was adjusted to 5.0 or 6.4 with the addition of 3 M HCl. These media were used to prepare 20-ml cultures in conical flasks as described above and to monitor growth under acidic conditions at 37°C with shaking (160 rpm). Growth was monitored by taking samples at regular time intervals and measuring the optical density at 600 nm (OD600).
Survival under acidic conditions.
Survival experiments were performed in BHI and DM. In these experiments, the pH of the overnight-grown cultures was adjusted to 2.5 with the addition of 3 M HCl. In experiments assessing the effect of Glu on acid resistance, Glu was added to the DM growth medium at a final concentration of 10 mM and the pH was adjusted to 2.5. Samples were obtained prior to the pH adjustment, and thereafter every 20 min up to 60 min, and 10-fold serial dilutions were prepared and plated onto BHI agar in triplicate. These plates were incubated at 37°C overnight, and subsequently colonies were counted to assess survival under lethal acidic conditions.
GABase assay.
A commercial preparation known as GABase was used to determine the intracellular and extracellular concentration of GABA. This assay, originally described by Tsukatani et al. (19), uses both γ-aminobutyric acid aminotransferase (GABGT) and succinic semialdehyde dehydrogenase (SSDH) from Pseudomonas fluorescens (Sigma-Aldrich, Steinheim, Germany). Prior to experiments, overnight cultures had their pH, optical density, and cell concentration measured. With control cultures, we proceeded with the measurement of GABA without any pH adjustment, while the rest of the cultures had their pH adjusted to various values and maintained for a certain length of time at 37°C before GABA measurement (see Fig. 2, 3, and 5). In other experiments (see Fig. 6), cells from overnight cultures were harvested by centrifugation at 7,690 × g and resuspended in acidified (pH 3.5) BHI, DM, or various mixtures of BHI with DM (plus 10 mM Glu) and BHI with water (plus 10 mM Glu) in various proportions. All acidified media were supplemented with 10 μg ml−1 chloramphenicol, which is a protein synthesis inhibitor, and its addition allowed us to study the function of the GAD system proteins in different media.
FIG. 2.

Monitoring of the concentration of extracellular GABA through time (min) following the adjustment of pH of overnight cultures of L. monocytogenes 10403S grown until the stationary phase in BHI and in DM supplemented with 10 mM Glu. The data presented here were obtained from one representative experiment out of a series of three with comparable results.
FIG. 3.

Concentration of extracellular GABA in a pH range (2.5 to 7.6) for L. monocytogenes 10403S in BHI, DM, and DM plus 30 mM Glu. The time of the exposure was 80 min. The data presented here were obtained from one representative experiment out of a series of three with comparable results.
FIG. 5.

Accumulation of GABAi over time in stationary-phase cells of L. monocytogenes 10403S in BHI or DM at pH 3.5. The concentration of GABAe is demonstrated on the secondary axis (dashed lines). Values are means of experiments performed in triplicate. Error bars represent standard deviation (n = 3).
FIG. 6.

(A) Concentration of GABAe (mM) exported following the resuspension of the cells of an overnight culture of L. monocytogenes 10403S grown in 20 ml BHI in a similar volume of various mixes of BHI with DM (plus 10 mM Glu) and BHI with water (plus 10 mM Glu) at pH 3.5 supplemented with 10 μg ml−1 chloramphenicol. Values are means of experiments performed in triplicate. Error bars represent standard deviation (n = 3). (B) Concentration of GABAi (mM) accumulated in the cells of an overnight culture of L. monocytogenes 10403S grown in 20 ml BHI following its resuspension in a similar volume of various mixes of BHI with DM (plus 10 mM Glu) and BHI with sterile water (plus 10 mM Glu) at pH 3.5 supplemented with 10 μg ml−1 chloramphenicol. The concentration of intracellular GABA represents amounts of GABA released by cells contained in 20 ml of culture following their resuspension in 0.5 ml of water and boiling for 10 min. Values are means of experiments performed in triplicate. Error bars represent standard deviation (n = 3). (C) Concentration of GABAe following the transfer of cells contained in a culture of L. monocytogenes 10403S grown overnight in DM or BHI to an equivalent volume of acidified DM or BHI (pH 3.5) containing 10 μg ml−1 chloramphenicol. Following the transfer of cells in the acidified medium, they remained at low pH for 45 min. Values are means of experiments performed in triplicate. Error bars represent standard deviation (n = 3). (D) Concentration of GABAi following the transfer of cells contained in a culture of L. monocytogenes 10403S grown overnight in DM or BHI to an equivalent volume of DM or BHI (pH 3.5). Cells remained at low pH for 45 min. Values are means of experiments performed in triplicate. Error bars represent standard deviation (n = 3).
Subsequently, a sample of 500 μl from each culture was centrifuged at 8,000 × g for 10 min, and 10 μl from the supernatant was incubated for 300 min at 37°C with 90 μl of the assay mixture containing 80 mM Tris buffer (pH 9), 750 mM sodium sulfate, 10 mM dithiothreitol, 1.4 mM NADP+, 2 mM α-ketoglutarate, and 0.3 g liter−1 GABase in each well of a 96-well microtiter plate. Previously, standard solutions containing 0, 1, 2, or up to 10 mM GABA were prepared in the same sterile medium used to grow the cultures. Ten microliters from each standard solution was included in the measurement to construct a standard curve for GABA measurement. Subsequently, the microtiter plate containing the samples and the standards was placed in a Sunrise spectrophotometer (Tecan, Männedorf, Switzerland) operated by Magellan software (Tecan, Männedorf, Switzerland) and the optical density at 340 nm was measured every 1 min for 3 h at 37°C. The GABA contained in the samples or the standards in the presence of α-ketoglutarate is converted to succinic semialdehyde (SS), through the activity of GABGT (equation 1). The SS is in turn transformed to succinate in the presence of NADP+ by the activity of SSDH (equation 2). In the last reaction, the NADP+ is transformed into NADPH, which absorbs at 340 nm, and since the reaction is stoichiometric, the levels of NADPH directly reflect the levels of GABA in the mix. The concentration of GABA in the samples was calculated using calibration curves generated with the standard solutions:
 |
(1) |
and
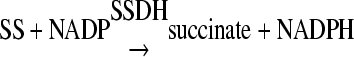 |
(2) |
One limitation of the GABase assay was that it was not possible to discriminate between GABA and SS in samples that might have contained both intermediates. This is because the assay relied on two enzymes that were supplied as a mixture with the assay kit, resulting in measurements that reflect the sum of the GABA and SS concentrations. To overcome this difficulty, the levels of SS present in samples from cells grown either in DM or BHI were measured. To do this, we used 2-aminoethyl hydrogen sulfate at a concentration of 80 mM to inhibit the activity of GABGT without affecting the activity of SSDH in the assay (13). Therefore, the use of the inhibitor results in the measurement of the SS concentration only. In all cases tested, the SS levels were found to be very low and represented less than 5% of the total GABA measurement. All reagents used for the GABase assay were obtained from Sigma-Aldrich (Steinheim, Germany).
Determination of GABAi concentration.
A modification of the GABase assay was used to measure GABAi in all cell lysates. Prior to experiments, overnight cultures had their pH, optical density, and cell concentration measured. With control cultures, we proceeded with the measurement of GABAi without any pH adjustment, while the rest of the cultures had their pH adjusted to 3.5 and maintained for a certain length of time at 37°C before GABAi measurement (Fig. 5). Medium-swap experiments with supplementation of 10 μg ml−1 chloramphenicol (Fig. 6) were performed as described earlier in the “GABase assay” section.
Subsequently, 20 ml of culture grown in BHI overnight was centrifuged at 9,000 × g for 10 min, and following the complete removal of the supernatant, the pelleted cells were retrieved. Subsequently, the pellet was resuspended in 500 μl of sterile water contained in a sterile plastic tube and placed in boiling water for 10 min. As determined experimentally (data not shown), this process is sufficient to kill all of the cells. Furthermore, we confirmed that the cell lysates contained all of the GABAi by demonstrating that an increase in the boiling time did not result in any increase in the levels of GABAi measured in similar samples. Neither did the boiling lead to a loss of GABA through heat inactivation since control samples with known GABA concentrations were found to have constant levels of GABA regardless of the boiling time. The lysate containing all of the GABAi was centrifuged at 8,000 × g for 10 min on a benchtop centrifuge, and the pellet, which consisted of cell debris, was discarded while the supernatant was placed in a sterile 1.5-ml tube. Ten microliters of the supernatant was used for the measurement of the concentration of GABAi.
Real-time PCR determination of GAD gene transcription.
Real-time PCR was used to determine the relative expression of the genes encoding the GAD system, using the 16S rRNA gene as a reference gene. Cells were grown overnight (16 h) in triplicate (biological replicates) in 20-ml flasks with BHI and DM at 37°C, and prior to RNA isolation, they were mixed with 4 ml ice-cold 5% phenol solution in ethanol to stop the RNA synthesis and the degradation of the RNA already synthesized. Subsequently cells were harvested by centrifugation at 7,690 × g. Total RNA was obtained in triplicate from each one of the three biological replicates using the RNeasy kit (Qiagen, Hilden, Germany), quantified with the use of the NanoDrop (Thermo Scientific, Wilmington, DE), treated with DNase (Turbo DNA-free; Ambion, Austin, TX), and used to synthesize cDNA. To obtain cDNA, 50 ng of mRNA was mixed with 1 μl of random primers (Invitrogen, Carlsbad, CA), 1 μl deoxynucleoside triphosphate (dNTP) mix (Invitrogen, Carlsbad, CA), and sterile water (nuclease-free water, Ambion, Austin, TX) up to 12 μl. The mixture was heated up to 65°C for 5 min, transferred on ice, and centrifuged briefly before 4 μl of First Strand buffer and 2 μl dithiothreitol (DTT) was added. Following an incubation of 2 min at 25°C, 1 μl of Superscript II and 1 μl of sterile water were added. Subsequently, the mixture was incubated at 25°C for 10 min, at 42°C for 50 min, and finally at 70°C for 15 min before cDNA was stored at −20°C. Relative quantification of the expression of the genes was carried out by fluorometric real-time PCR using the SYBR green master mix and the Quantitect SYBR green PCR kit (Qiagen, Hilden, Germany). Following the instructions of the kit, 5 μl of cDNA was mixed with 10 μl QuantiTect SYBR green PCR master mix (containing fast-start Taq DNA polymerase, SYBR green dye, buffer, and MgCl2), and the appropriate set of primers (Table 1) was added to a final concentration of 0.2 μM each. Each set of primers was designed with specificity toward the genome of L. monocytogenes strain 10403S (accession no. AARZ02000000), producing amplicons in the range of 207 to 268 bp (Table 1). Finally, molecular-biology-grade water was added to the mixture, up to a final volume of 20 μl, and placed in a well of the 96-microtiter plate (LightCycler 480 multiwell plate 384; Roche Diagnostic GmbH, Manheim, Germany). Once the other PCRs were set up in the other wells, the plate was placed in the LightCycler 480 instrument (Roche Diagnostic GmbH, Manheim, Germany), and the PCR was programmed starting with an initial denaturation at 95°C for 10 min, followed by amplification for 40 cycles at 95°C for 1 s; 5 s at various annealing temperatures, depending on the melting temperature of the set of primers (Table 1); and 72°C for 9 to 11 s.
TABLE 1.
Primers used in this studya
| Primer | Sequence (5′→3′) | Position (bp) in gene | Tm (°C)b | Amplicon length (bp) |
|---|---|---|---|---|
| gadT1F | CGCAATAACGGCATCTCTTT | 48-67 | 63.8 | 249 |
| gadT1R | ACCGACTGTGATTTGGAACC | 278-297 | 63.7 | |
| gadD1F | CGCTTCGGATATGAGGGTTA | 988-1007 | 63.6 | 226 |
| gadD1R | AGTGGATACGCCGGTACTTG | 1195-1214 | 63.7 | |
| gadD3F | GAAACGCTCGAGAAAAATGC | 61-80 | 63.6 | 237 |
| gadD3R | AGTTTGGTCGTTTTGCCTGT | 279-298 | 63.3 | |
| gadT2F | CACGGCTAAAATCGCAAAAT | 438-457 | 63.5 | 207 |
| gadT2R | GGAAGCTTCAACACCCATGT | 626-645 | 63.8 | |
| gadD2F | AATACCTTGCCCATGCAGTC | 1049-1068 | 63.7 | 268 |
| gadD2R | GGCTTGGAAATCTTGGATGA | 1298-1317 | 63.8 | |
| 16SF | TGGGGAGCAAACAGGATTAG | 914-933 | 63.8 | 213 |
| 16SR | TAAGGTTCTTCGCGTTGCTT | 1107-1127 | 63.6 |
This study was the source of all primers shown.
Tm, melting temperature.
The specificity of amplification for each product was determined by a melting curve analysis at 95°C for 1 s and 65°C for 15 s, followed by a progressive increase of the temperature to 95°C with a ramp rate of 0.11°C s−1, with continued measurement of fluorescence, and finally cooling of the plate at 40°C for 30 s. Alongside each real-time PCR assay, a control reaction without added cDNA was run as a negative control. The efficiencies of the real-time PCR assays were estimated through the use of five decimal dilutions of cDNA and genomic DNA samples using the LightCycler 480 software program (Roche Diagnostic GmbH, Manheim, Germany). The PCR efficiencies for each set of the primers were determined as 2.27 (16S rRNA gene), 1.94 (gadT1), 2.12 (gadD1), 2.03 (gadD3), 2.07 (gadT2), and 2.09 (gadD2).
Relative expression was calculated as a ratio between expression of target genes (gadD1, gadT1, gadD2, gadT2, and gadD3) and the expression of the 16S rRNA gene, which served as reference gene in each cDNA sample. Calculations were carried out following the “advanced relative quantification” settings of the LightCycler 480 software program with PCR efficiency correction. Relative expression of a gene in BHI compared to that in DM was calculated by comparison of its relative expression to the 16S rRNA gene in each medium and expressed as fold change.
Statistical analysis of RT-PCR results.
For each reverse transcription-PCR (RT-PCR) experiment, measurements were performed on three independent biological samples, with three technical replicates performed on each. The data were normalized to the 16S rRNA level using advanced relative quantification with PCR efficiency correction (using Roche Lightcylcer 480 software). Student's t test was performed on the biological replicates to determine the statistical significance of the differences in expression between BHI and DM for each gene at each time point. Differences with P values of <0.05 were considered statistically significant, while P values of <0.01 were considered highly statistically significant.
RESULTS
Glu does not influence growth or survival at low pH in DM.
The finding that the growth of L. monocytogenes at low pH relies on the GAD system (11) suggested that the presence of extracellular Glu might help to relieve the growth inhibition caused by acidic conditions. To investigate this hypothesis, L. monocytogenes 10403S was grown in DM adjusted to either pH 5.0 or 6.4 in the presence or absence of Glu. In the absence of any added Glu, the growth rate of the cultures was reduced by approximately 70% ± 1.28% at pH 5.0 compared to pH 6.4 (Fig. 1A and B). The presence of 1 mM Glu in the DM resulted in statistically significant increases of 4.6% ±0.76% and 25% ± 9% in the specific growth rate for pH 6.4 and pH 5.0, respectively. However, the presence of 10 mM Glu in the DM had no significant impact on the specific growth rates of cultures growing at either pH (Fig. 1A and B). These data suggest that extracellular Glu can not be used by cells growing under these conditions to mitigate the inhibitory effects of acidic pH.
FIG. 1.

(A) Glu has no effect on growth of L. monocytogenes 10403S cells in DM (± 1 mM or 10 mM Glu) with pH 5 and 6.41 at 37°C with shaking. The data presented here were obtained from one representative experiment out of a series of three with comparable results. (B) Glu has no effect on growth of L. monocytogenes 10403S in DM. The specific growth rate (μ) is shown during the exponential phase of growth in DM (± 1 mM or 10 mM Glu) at pH 5 or 6.41 at 37°C with shaking. Values are means of experiments performed in triplicate. Error bars represent standard deviation (n = 3). (C) Glu has no effect on the acid resistance of L. monocytogenes 10403S in DM. Cells were grown overnight in DM (± 10 mM Glu) up to stationary phase and challenged in pH 2.5. Values are means of experiments performed in triplicate. Error bars represent standard deviation (n = 3). Asterisks indicate the presence of a small but significant (P < 0.05) difference.
Since earlier studies had shown a dramatic effect of Glu on the ability to survive potentially lethal acid challenges (8, 9), we investigated whether the presence of Glu in DM could protect L. monocytogenes 10403S against a challenge at pH 2.5. Surprisingly, when stationary-phase cultures were challenged at pH 2.5, cells grown in the presence of 10 mM Glu lost viability at the same rate as the untreated control (Fig. 1C). The addition of Glu (10 mM) immediately prior to acid challenge also had no impact on survival (data not shown). These data suggested the possibility that the GAD system may not be functional when cells are grown in DM.
L. monocytogenes fails to export GABA at low pH when grown in DM.
The ability to utilize Glu for acid tolerance is coupled to the efflux of GABA through an antiport system. We investigated whether L. monocytogenes cells grown in DM could efflux GABA in response to low pH. After overnight growth, the concentrations of cells were similar in DM and BHI (approximately 6.0 × 109 CFU ml−1), and therefore differences in GABA levels could not be attributed to differences in cell numbers. The pH values of the overnight cultures were recorded as pH 6.1 to 6.2 for the BHI cultures and pH 6.5 to 6.6 for the DM-grown cultures. First, the concentration of GABAe was monitored over time at various pH values in both BHI and in DM supplemented with 10 mM Glu. In BHI, the levels of GABAe were undetectable at pH 7.0, while significant GABA export was detected at pH 4.0 and at pH 3.5 (Fig. 2). In contrast, in DM with Glu, very low concentrations of GABAe (0.24 to 0.30 mM) were detected regardless of the pH value in the cultures. These barely detectable GABAe levels in DM suggest that GABAe export does not take place in this medium, despite the presence of extracellular Glu and the acidic conditions.
It was possible that GABA efflux might only occur outside the range of pH values tested. To address this, the production of GABAe was investigated over a full range of acidic pH values from 2.5 to 7.6 both in BHI and in DM with or without 30 mM Glu. Stationary-phase cultures were acidified with HCl to each pH in this range, and GABAe was measured after 80 min. Strikingly, GABAe levels remained consistently low in DM regardless of the pH of the medium and regardless of whether Glu was present (Fig. 3). These data are consistent with the growth and survival data, which show that extracellular Glu fails to protect DM-grown L. monocytogenes from acidic conditions. In contrast, cells cultured in BHI produced significant levels of GABAe, but only when the pH of the medium was reduced below 4.5. The highest concentrations of GABAe were detected when the pH was between 3.5 and 2.5, where levels of approximately 5 mM were reached (Fig. 3). This is probably close to the maximum GABAe level achievable since loss of cell viability below this pH will curtail the decarboxylation reaction.
When the rates of survival of 10403S at pH 2.67 in BHI and DM were compared, the tolerance to acid was found to reflect the capacity to produce GABAe. At pH 2.67 in BHI, the numbers of CFU remained unaffected during the course of the experiment, while in DM there was a reduction of greater than 5 log cycles in the number of viable cells detected (Fig. 4). As described above, the presence of Glu in DM did not affect the acid resistance of the cells (Fig. 1C). These results indicate that cells from the stationary phase grown in BHI are much more resistant to the acidic conditions than cells from the same phase of growth grown in DM, and this difference in survival correlates well with the export of GABA.
FIG. 4.

Acid resistance of L. monocytogenes 10403S at pH 2.67 in BHI and DM. Values are means of experiments performed in triplicate. Error bars represent standard deviation (n = 3).
Significant accumulation of GABAi during acid stress.
Most studies investigating the role of GABA in acid resistance have focused on the export of GABA, which results in the accumulation of GABAe in the medium. In this study, we considered the possibility that GABA might accumulate intracellularly. GABAi was measured in boiled cell lysates, as described in Materials and Methods. Control experiments performed with nonboiled cell pellets confirmed that the GABA detected in these assays could only have been derived from lysed cells and could not have occurred by GABA efflux during the course of the assay (data not shown). By using this assay, cells grown in BHI or DM at pH 7.0 were found to contain no detectable GABAi (data not shown). However, when the media were acidified to pH 3.5 with HCl, both cultures were found to rapidly accumulate GABAi (Fig. 5). Within 10 min at pH 3.5, the BHI-grown culture had reached a steady-state level of GABAi corresponding to a value of 10 mM. A similar amount was produced in cultures grown in DM, although a steady level was not reached until approximately 40 min after acidification. GABAe production was also determined in parallel, by measuring the GABA level present in culture supernatants. As before, almost no GABAe was produced by cultures grown in DM, while significant levels were produced by BHI-grown cells (Fig. 5). The appearance of GABAe lagged significantly behind the accumulation of GABAi, with GABAe levels continuing to increase steadily even 2 h after acidification, long after the intracellular steady-state level had been reached. Importantly, these data demonstrate that GABA can be produced at high levels in the cell without the simultaneous occurrence of GABA efflux into the medium. They also indicate that GABAi can be produced without a supply of extracellular Glu.
A component of BHI medium is required for efficient GABA efflux.
The data presented above suggested that DM-grown cells could synthesize but not export GABA. Two possible explanations for this effect were considered: (i) DM may contain some component that inhibits the transport of GABA out of the cell, and (ii) some component present in BHI may be required to allow normal GABA efflux.
In order to distinguish between these possibilities, an experiment was performed in which cells grown in BHI were transferred to an acidified mixture of BHI with DM or water in a range of proportions (50%/50%, 75%/25%, 90%/10%, and 100%/0%). The acidified mixtures were supplemented with 10 μg ml−1 chloramphenicol to prevent de novo protein synthesis from influencing the outcome of the experiment. When BHI was diluted with either DM or water, there was a significant reduction in the levels of GABAe produced. Dilution to 50% with either water or DM almost completely abolished GABAe production (Fig. 6A). The effect of diluting the medium with water was essentially the same as dilution with DM, indicating that it was the dilution of BHI per se that had a negative impact on GABAe production and not some inhibitory component present in DM (Fig. 6A). In contrast, the dilution of the medium had a much more limited impact on GABAi production. Even when the BHI medium was diluted 50% with DM or water, the activity remained greater than 60% of that of the undiluted control (Fig. 6B). Since all of the diluted media were supplemented with 10 mM Glu, it was not possible that Glu limitation caused the effect on GABA production. Together, these data suggest that some component of the BHI medium other than Glu is required for efficient efflux of GABA from the cell.
Differential induction of GAD system genes in BHI and DM.
If the presence of some component in BHI was required for efficient GABA efflux, then it might be expected that cells grown in DM but acidified in BHI might be capable of GABA efflux. To test this, cells grown overnight in either DM or BHI were pelleted by centrifugation and resuspended in either acidified DM or BHI (pH 3.5) supplemented with 10 μg ml−1 chloramphenicol. The levels of GABAe and GABAi were then assayed after a 45-min incubation at this pH. Cells grown in BHI but then assayed in DM produced no detectable GABAe, while the control culture transferred to acidified BHI produced GABAe as expected. Cells grown in DM failed to produce GABAe, regardless of which acidified medium they were incubated in prior to the GABA measurement (Fig. 6C). This result suggests that cells grown in DM are not capable of producing GABAe, even when the assay is performed in BHI, perhaps indicating that GAD decarboxylase/antiporter is not expressed when cells are grown in this medium. The ability to produce GABAi, however, was not compromised by growing cells in DM, provided that the assay was performed with cells acidified in BHI (Fig. 6D).
To investigate whether growth in DM influenced the transcription of the GAD system, RT-PCR was used to measure the relative levels of the GAD gene transcripts in cultures grown overnight in BHI and DM and subsequently challenged with HCl (pH 3.5) in the same medium. In the absence of acid challenge (0 min), the transcription of the genes encoding the GadD1T1 and GadD3 systems was not significantly affected by the growth medium. In contrast, the genes encoding the GadT2D2 system were transcribed at very high levels in BHI compared to DM, and once acid challenge was introduced, transcription dropped rapidly (<10 min) by approximately 80% in BHI-grown cells, reaching levels similar to those of the acid-treated DM-grown cells (Fig. 7). Furthermore, in BHI-grown cells, acid challenge resulted in downregulation of gadD1T1, while transcription of gadD3 remained stable. In contrast, in DM-grown cells transcription of all GAD genes was induced upon acid challenge and that of gadD1T1 and gadD3 (the highest transcript levels) was rapid (<10 min) and reached significantly higher levels than that of the corresponding genes in acid-treated BHI-grown cells. Induction of transcription of gadT2D2 in DM-grown cells upon acid treatment was relatively slow (>18 min), and it always remained lower than that in acid-treated BHI-grown cells (Fig. 7).
FIG. 7.

Transcription of the gad genes in response to acidification. Relative normalized (based on the 16S rRNA gene) expression of all genes of the GAD system (gadD1, gadT1, gadT2, gadD2, and gadD3) in BHI (•) and DM (○) before (0 min) and after (10, 18, and 20 min) acidification (pH 3.5 with 3 M HCl). For each comparison, the data are expressed as a percentage of the maximal level detected for that transcript. In order to allow comparisons between the expression of the different gad genes the actual relative normalized (based on the 16S rRNA gene) expression level for each maximum value is also indicated on each graph. Following advanced relative quantification and normalization based on the expression of the 16S rRNA gene, a Student's t test was performed to estimate the statistical significance of the differences in expression between BHI and DM for each gene at each time point. Differences that were found to be statistically significant (P < 0.05) or highly significant (P < 0.01) are indicated on the graphs by * and **, respectively.
DISCUSSION
It is well established that the GAD system plays an important role in survival of L. monocytogenes under acidic conditions both in acidic foods and in the stomach (9, 11). The protective effect of the GAD system is believed to arise through the consumption of a proton that occurs during the decarboxylation reaction, which helps to maintain the cytoplasmic pH when the external pH drops. The Glu for this reaction is provided by the activity of an antiporter that exchanges extracellular Glu for GABAi (9, 11). In this study, we show that Glu added to DM is not used by L. monocytogenes for Glu decarboxylation, since no significant GABAe was detected under any condition involving this growth medium. The consequence of this is that the presence of Glu in this medium influences neither the growth rate under mildly acidic conditions nor the survival of cells when they are exposed to a lethal pH (Fig. 1). This result was surprising since GABAe is readily produced when the same strain of L. monocytogenes is found in the acidified complex medium, BHI (Fig. 2). Indeed there is a good correlation between the ability to produce GABAe and acid tolerance, since cultures challenged with acid in BHI have a significant survival advantage compared to DM-grown cultures (Fig. 4).
The GABAi values detected (Fig. 5) represented the GABA that was released during the boiling step of the assay, as described in Materials and Methods. If it is assumed that all of the GABAi is released during boiling, then it is possible to estimate the true GABAi concentration, given that the number of cells present in the assay is known (1.3 × 1011 ± 2.3 × 1010 CFU ml−1) and the cell volume for this specific strain has recently been estimated to be in the range 0.264 to 0.443 μm3 (14). Using these values, the steady-state GABAi concentration after acidification is estimated to be at least between 85 ± 3 mM and 142 ± 5 mM. Since this high concentration of GABAi occurs in DM-grown cells in the absence of extracellular Glu, the synthesis of GABAi must rely on the intracellular pools of Glu. As in most bacteria, L. monocytogenes maintains a substantial pool of Glu in the cytoplasm, where it is believed to act as a counter ion to potassium and plays a role in the osmotic stress response (3, 17). Such a high intracellular concentration of GABA is rather surprising since it has been described as a toxic metabolite, both in Bacillus subtilis (5) and in the fungus Apergillus nidulans (4) and as previously suggested for L. monocytogenes (1, 2). The intracellular pool of GABA in L. monocytogenes is probably constrained at a tolerable level by a catabolic pathway that is predicted to convert GABA to succinate via the intermediate SS (1, 9, 11). Importantly the catabolism of GABAi to succinate would also regenerate Glu, as a by-product of the transamination reaction that converts GABA to SS. Although there is no direct biochemical evidence that this pathway exists, it has been shown that a mutant lacking the putative SSDH (encoded by lmo0913) is sensitive to low pH, and this has been suggested to be due to an inability to catabolize GABAi (1). The data presented here indicate that at least under acidic conditions the accumulation of high GABAi levels does occur. Presumably the production of GABAi is beneficial to the cell under these conditions because the removal of protons that occurs during Glu decarboxylation helps to prevent excessive acidification of the cytoplasm, and this outweighs any toxic effects that GABAi might have on the cell.
It is interesting to observe that the steady-state GABAi concentrations achieved in acidified cells were similar in both growth media. However, the rate of GABAi production was significantly slower in DM-grown cells, only reaching a maximum level after 40 min, compared to less than 10 min for cells grown in BHI (Fig. 5). The significantly higher level of expression of the gadT2D2 genes in BHI-grown cells prior to the acid challenge (Fig. 7) presumably accounts for the difference in the rates of GABAi synthesis. Cells exposed to acid in BHI fail to induce the transcription of any of the gad genes, suggesting that the decarboxylases already present in the cell are sufficient to mediate GABA production. In contrast, cells exposed to acid in DM rapidly induce the transcription of all three gad operons (Fig. 7), with the gadD3 transcript being produced at the highest level after exposure to low pH. Thus, there is a striking difference in the transcriptional response to acidification observed between cells grown in BHI and DM. However, it is not only the transcription that is affected by the medium. Overall, the levels of GABAi produced in cells acidified in DM with chloramphenicol are lower than those in cells acidified in BHI with chloramphenicol, regardless of the medium they are first grown in (Fig. 6D). The inclusion of chloramphenicol in this experiment prevents de novo protein synthesis and therefore gives an indication of the existing state of the cells prior to acidification. The results therefore suggest that the decarboxylase reaction itself may also be influenced by the growth medium, independently of effects on transcription of the gad genes.
It is perhaps surprising that transcription of gadD1T1 is found to be rapidly induced at pH 3.5 in DM, since earlier studies indicate that GadD1 does not play a significant role in extreme acid survival (9). Indeed the gadD1T1 operon is not found in all strains of L. monocytogenes (e.g., it is absent in serotype 4b strains), which means that the findings outlined in the present study for strain 10403S (serotype 1/2a) may not necessarily pertain in other strains. The rapid induction of gadD3 suggests that GadD3 might also be responsible for contributing to GABAi synthesis under these conditions. It has not yet been possible to directly study the role of GadD3 in acid tolerance since generating a gadD3 gene deletion has proved difficult (9). However, it is interesting to note that an insertion mutation in the gadD3 gene (lmo2434) was found to impair the intracellular growth of L. monocytogenes in a colonic epithelial cell line, suggesting that it may have some role in establishing infections within the host (16).
The finding that DM-grown cells fail to efflux GABA even when Glu is present in the growth medium was somewhat unexpected. This result is apparently caused by two separate factors. First, the genes encoding the GadT2D2 system are transcribed at a very low level in DM-grown cultures. This system has previously been shown to be essential for acid tolerance in L. monocytogenes (9, 11). Therefore, the absence of this system from cells grown in DM is likely to contribute significantly to the acid sensitivity seen in this medium (Fig. 4). Furthermore, this finding suggests that GadT2 antiporter is likely to be the primary mediator of GABA-Glu exchange under extreme acid conditions. Second, it appears that even when cells are grown in BHI (and therefore express the GadT2D2 system normally), they are still not capable of GABA efflux when they are transferred to acidified DM immediately before the assay (Fig. 6C). Dilution of the BHI medium, either with water or with DM, also has a negative impact on GABA efflux (Fig. 6A). These results suggest that some component or property of BHI medium, other than Glu, is essential for the normal functioning of the GABA/Glu antiport system. It is interesting to speculate that L. monocytogenes may have evolved a regulatory system that only allows GABA efflux to occur when nutrients are plentiful; when resources are less plentiful, it may make metabolic sense to retain the GABA within the cell, perhaps for recycling to succinate.
The precise role that GABA efflux plays in the acid tolerance of L. monocytogenes remains uncertain. The results presented here show that cells grown in DM survive poorly when subjected to lethal acid conditions compared to cells grown in BHI, and this correlates well with the pattern of GABA efflux observed. However, the rates of GABAi synthesis are also different between the two growth media (Fig. 5), and it is possible that this difference plays an important role in determining survival rates after acid challenge. If the sole beneficial effect of the GAD system is to help maintain the cytoplasmic pH through the decarboxylation of Glu, then the synthesis of GABAi could be the critical step in determining the acid tolerance, with GABA efflux merely facilitating the uptake of new substrate for the decarboxylation reaction. The speed of GABAi synthesis is impressive, with >80 mM GABAi produced within 10 min of acidification, when cells are grown in BHI. In contrast GABAe levels reach only 5 mM even 2 h after the acidification of BHI-grown cells (Fig. 5). Such a dramatic difference in both the concentrations and synthesis rates of GABAi and GABAe suggests that the most important and most immediate impact of GABA production will be on the cytoplasmic pH. It remains unclear at present whether GABAe might also act to provide additional protection against acidification, perhaps by acting as a buffer outside the cell, as has been proposed previously (6). Alternatively, GABAe may simply be produced to prevent the intracellular concentration of GABA from reaching toxic levels, although this idea presents some difficulties since the concomitant Glu uptake would presumably be channeled into the decarboxylation reaction. Ongoing research in this laboratory is currently trying to distinguish between these two possibilities.
In summary, this study has shown that significant differences exist in the operation of the GAD system in L. monocytogenes when cells are grown in different growth media. Specifically, the responses to a lethal acid challenge are markedly different, with GABA efflux only detected when cells are grown in a complex medium (BHI). These differences derive from differences in both the transcription of the GAD genes as well as the functioning of the system in different media. This study provides the first intracellular measurements of GABA in this pathogen, and the data reveal that very high intracellular levels of GABA are produced in response to an acid challenge. Together, these findings show that in L. monocytogenes the synthesis of GABA can be uncoupled from its efflux and suggest an important role for the intracellular pool of Glu in allowing this pathogen to cope with sudden changes in the pH of its environment.
Acknowledgments
The authors are grateful to members of the Bacterial Stress Response Group for helpful discussions and to the Environmental Change Institute at National University of Ireland, Galway, for administrative support.
This work was supported by a Food Institutional Research Measure grant from the Department of Agriculture, Fisheries and Food (grant no. 06/RD/C/459) and by an EU Marie Curie Transfer of Knowledge program grant (GAMIDI).
Footnotes
Published ahead of print on 16 April 2010.
REFERENCES
- 1.Abram, F., E. Starr, K. A. G. Karatzas, K. Matlawska-Wasowska, A. Boyd, M. Wiedmann, K. J. Boor, D. Connally, and C. P. O'Byrne. 2008. Identification of components of the SigB regulon in Listeria monocytogenes that contribute to acid and salt tolerance. Appl. Environ. Microbiol. 74:6848-6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abram, F., W.-L. Su, M. Wiedmann, K. J. Boor, P. Coote, C. Botting, K. A. G. Karatzas, and C. P. O'Byrne. 2008. Proteomic analyses of a Listeria monocytogenes mutant lacking sigB identify new components of the SigB regulon and highlight a role for SigB in the utilization of glycerol. Appl. Environ. Microbiol. 74:594-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amezaga, M.-R., I. Davidson, M. Debra, A. Verheul, T. Abee, and I. R. Booth. 1995. The role of peptide metabolism in the growth of Listeria monocytogenes ATCC 23074 at high osmolarity. Microbiology 141:41-49. [DOI] [PubMed] [Google Scholar]
- 4.Arst, H. N., Jr. 1976. Integrator gene in Aspergillus nidulans. Nature 262:231-234. [DOI] [PubMed] [Google Scholar]
- 5.Belitsky, B. R., and A. L. Sonenshein. 2002. GabR, a member of a novel protein family, regulates the utilization of γ-aminobutyrate in Bacillus subtilis. Mol. Microbiol. 45:569-583. [DOI] [PubMed] [Google Scholar]
- 6.Booth, I., P. Cash, and C. O'Byrne. 2002. Sensing and adapting to acid stress. Antonie Van Leeuwenhoek 81:33-42. [DOI] [PubMed] [Google Scholar]
- 7.Castanie-Cornet, M.-P., T. A. Penfound, D. Smith, J. F. Elliott, and J. W. Foster. 1999. Control of acid resistance in Escherichia coli. J. Bacteriol. 181:3525-3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cotter, P. D., K. O'Reilly, and H. Colin. 2001. Role of the glutamate decarboxylase acid resistance system in the survival of Listeria monocytogenes LO28 in low pH foods. J. Food Prot. 64:1362-1368. [DOI] [PubMed] [Google Scholar]
- 9.Cotter, P. D., C. G. M. Gahan, and C. Hill. 2001. A glutamate decarboxylase system protects Listeria monocytogenes in gastric fluid. Mol. Microbiol. 40:465-475. [DOI] [PubMed] [Google Scholar]
- 10.Cotter, P. D., and C. Hill. 2003. Surviving the acid test: responses of Gram-positive bacteria to low pH. Microbiol. Mol. Biol. Rev. 67:429-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cotter, P. D., S. Ryan, C. G. M. Gahan, and C. Hill. 2005. Presence of GadD1 glutamate decarboxylase in selected Listeria monocytogenes strains is associated with an ability to grow at low pH. Appl. Environ. Microbiol. 71:2832-2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foster, J. W. 2004. Escherichia coli acid resistance: tales of an amateur acidophile. Nat. Rev. Microbiol. 2:898-907. [DOI] [PubMed] [Google Scholar]
- 13.Fowler, L. J., and R. A. John. 1972. Active-site-directed irreversible inhibition of rat brain 4-aminobutyrate aminotransferase by ethanolamine O-sulphate in vitro and in vivo. Biochem. J. 130:569-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giotis, E. S., I. S. Blair, and D. A. McDowell. 2007. Morphological changes in Listeria monocytogenes subjected to sublethal alkaline stress. Int. J. Food Microbiol. 120:250-258. [DOI] [PubMed] [Google Scholar]
- 15.Hersh, B., F. Farooq, D. Barstad, D. Blankenhorn, and J. Slonczewski. 1996. A glutamate-dependent acid resistance gene in Escherichia coli. J. Bacteriol. 178:3978-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joseph, B., K. Przybilla, C. Stuhler, K. Schauer, J. Slaghuis, T. M. Fuchs, and W. Goebel. 2006. Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J. Bacteriol. 188:556-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patchett, R. A., A. F. Kelly, and R. G. Kroll. 1992. Effect of sodium chloride on the intracellular solute pools of Listeria monocytogenes. Appl. Environ. Microbiol. 58:3959-3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanders, J. W., K. Leenhouts, J. Burghoorn, J. R. Brands, G. Venema, and J. Kok. 1998. A chloride-inducible acid resistance mechanism in Lactococcus lactis and its regulation. Mol. Microbiol. 27:299-310. [DOI] [PubMed] [Google Scholar]
- 19.Tsukatani, T., T. Higuchi, and K. Matsumoto. 2005. Enzyme-based microtiter plate assay for γ-aminobutyric acid: application to the screening of γ-aminobutyric acid-producing lactic acid bacteria. Anal. Chim. Acta 540:293-297. [Google Scholar]
- 20.Waterman, S., and P. Small. 1996. Identification of sigma S-dependent genes associated with the stationary-phase acid-resistance phenotype of Shigella flexneri. Mol. Microbiol. 21:925-940. [DOI] [PubMed] [Google Scholar]