

Abstract
XylS protein, a member of the AraC family of transcriptional regulators, comprises a C-terminal domain (CTD) involved in DNA binding and an N-terminal domain required for effector binding and protein dimerization. In the absence of benzoate effectors, the N-terminal domain behaves as an intramolecular repressor of the DNA binding domain. To date, the poor solubility properties of the full-length protein have restricted XylS analysis to genetic approaches in vivo. To characterize the molecular consequences of XylS binding to its operator, we used a recombinant XylS-CTD variant devoid of the N-terminal domain. The resulting protein was soluble and monomeric in solution and activated transcription from its cognate promoter in an effector-independent manner. XylS binding sites in the Pm promoter present an intrinsic curvature of 35° centered at position −42 within the proximal site. Gel retardation and DNase footprint analysis showed XylS-CTD binding to Pm occurred sequentially: first a XylS-CTD monomer binds to the proximal site overlapping the RNA polymerase binding sequence to form complex I. This first event increased Pm bending to 50° and was followed by the binding of the second monomer, which further increased the observed global curvature to 98°. This generated a concomitant shift in the bending center to a region centered at position −51 when the two sites were occupied (complex II). We propose a model in which DNA structure and binding sequences strongly influence XylS binding events previous to transcription activation.
XylS, the transcriptional regulator of the meta-cleavage pathway for alkylbenzoate catabolism in Pseudomonas putida TOL plasmid, senses the presence of 3-methylbenzoate (3MB), and activates transcription from the meta-cleavage operon Pm promoter (15), a process that requires σ32 in the exponential phase and σ38 in the stationary phase (11). XylS overproduction through a toluene-induced regulatory cascade also leads to transcription activation in a 3MB-independent manner (15, 20). XylS protein is a member of the AraC family of transcriptional regulators (17, 36, 62). The proteins of this family share significant homology over a 100-amino-acid segment that constitutes the DNA binding domain. Most family members contain an additional domain involved in effector recognition and oligomerization which modulates their transcriptional activity (6, 37, 55).
The AraC family members studied in detail so far use different mechanisms to regulate transcription. Most AraC class proteins related to stress response (e.g., MarA and SoxS) are active as monomers. These two proteins are composed of a single domain, the C-terminal DNA binding domain (22, 35), and they activate transcription from both class I and class II promoters in a process exclusively depending upon their concentrations in the cell (22, 34). In contrast, AraC family members involved in carbon metabolism (e.g., AraC, RhaS, and MelR) are usually active as dimers and appear to mainly activate class II promoters (17). Activation by this second group of AraC family proteins responds to the presence of their specific effectors. Effector recognition by the N-terminal domain (NTD) triggers a conformational change in the regulator that leads to its correct positioning at its binding sites (21, 32, 55, 56).
XylS is composed of two separate and independently functional domains. Genetic analysis located its DNA binding domain in the C-terminal end of the protein, connected by a short linker domain (amino acids 205 to 213) to the 204-amino-acid N-terminal end involved in 3MB recognition and dimerization (12, 28, 49, 50). We have shown in electrophoresis mobility shift assays (EMSA) and in vivo that the XylS N-terminal domain repressed XylS-C-terminal domain (CTD) DNA binding and activation capacity. In both cases, 3MB had a releasing effect on this repression (12). In fact, early work showed a tagged XylS C-terminal domain was able to activate transcription without the contribution of any additional determinant present in the N-terminal domain and in a 3MB-independent manner (28).
X-ray crystallography of MarA-DNA and Rob-DNA complexes showed that the conserved DNA binding domain was composed of seven α-helices folding into two helix-turn-helix (HTH) motifs. A similar arrangement is observed in the recent high-resolution structure of ToxT, the regulator of the two primary virulence factors of Vibrio cholerae (33). The structure of the two domains of this protein is most similar to the structure of the N-terminal and C-terminal domains of AraC (48). Nuclear magnetic resonance (NMR) techniques revealed an inherent flexibility in MarA that accounted for its ability to bind different marbox sequences present in E. coli chromosome. In MarA interaction with its binding sites, the recognition helix of each HTH motif was inserted into adjacent major grooves of the DNA (10, 47). This is in contrast to the structure observed in Rob interaction with the micF promoter, which occurs through only one of the HTH motifs, the second helix making contacts with the phosphate backbone (31). In XylS, a thorough genetic analysis allowed us to determine XylS binding geometry. This protein recognizes two 15-bp direct repeats (TGCA-N6-GGNTA) in the Pm promoter, each consisting of two highly conserved sequences, the 5′-box A (TGCA) and the 3′-box B (GGNTA) separated by 6 bp, spanning one DNA helical turn (19, 27) (Fig. 1). This organization parallels the promoter organization of the AraC-controlled PBAD promoter (40). Previous results showed that XylS binds the direct repeats in a head-to-tail organization, with α-helix 3 of the first HTH motif (residues Arg242 and Asn246) recognizing specific bases of the A boxes and α-helix 6 of the second HTH motif interacting with the B boxes (residues Arg296 and Glu299) (13).
FIG. 1.

Pm sequence and DNA probes used in this work. The sequences of oligonucleotides containing one or two XylS binding sites used for gel shift experiments are shown. A1B1A2B2 sequence corresponds to wild-type Pm. Letters correspond to the A and B boxes at each binding site, which are indicated by arrows below the sequence (D, distal; P, proximal).
Like most members of the AraC family, XylS is poorly soluble, hampering the possibility to obtain active protein preparations and making it unamenable to biochemical analysis (1, 26, 37, 39, 61). In the work reported here, we have integrated different approaches to explore XylS DNA binding properties. We overexpressed and purified a His-tagged XylS C-terminal domain fusion protein and obtained active protein preparations. We have found that XylS-CTD monomers bind to tandem sites and that their sequential binding provokes a gradual bending of DNA. Our analysis suggests that XylS-CTD DNA recognition not only depended on specific amino acid base contacts (13) but also was influenced by intrinsic DNA conformation features, especially by binding-induced DNA bending. In the end, this repositioning of the regulator-DNA complex would enable to establish the contacts with RNA polymerase (RNAP) required for transcription activation.
MATERIALS AND METHODS
Bacterial strains, culture media, and plasmids.
The bacterial strains and plasmids used in this study are listed in Table 1. Bacterial strains were grown at 30°C in Luria-Bertani medium supplemented, when required, with 100 μg/ml ampicillin, 25 μg/ml kanamycin, or 20 μg/ml rifampin. Escherichia coli BL21(DE3) was grown on 2×YT medium (1.6% [wt/vol] Bacto tryptone, 1% [wt/vol] yeast extract, and 0.5% [wt/vol] sodium chloride) for protein production.
TABLE 1.
E. coli strains and plasmids used in this work
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| Strains | ||
| CC118 λ Pm::lacZ | Rifr; CC118 with chromosomal mini-Tn5::Pm::lacZ insertion | 29 |
| BL21(DE3) | Carries T7 RNA polymerase under control of lacUV5 promoter | Novagen |
| DH5α | supE44 lacU169(φ80lacZΔM15) hsdR17 (rK− mK−) recA1 endA1 gyrA96 thi-1 relA1 | 23 |
| Plasmids | ||
| pJLR100 | Apr; Pm cloned in pEMBL9 | 46 |
| pGP1-2 | Kmr; contains an inducible T7 RNA polymerase genea | 58 |
| pERD103 | xylS IncQ Kmr | 45 |
| pET16b | Apr; protein expression vector | Novagen |
| pET-XylS-C | Apr; pET16b derivative used to produce His-tagged XylS C-terminal domain | 12 |
| pET-XylS | Apr; pET16b derivative used to produce His-tagged XylS | 12 |
| pET-XylS-N | Apr; pET16b derivative used to produce His-tagged XylS N-terminal domain | 12 |
| pBend2::Pm80 | Apr; pBend2 derivative carrying 86-bp DNA fragment containing Pm promoter | 19 |
pGP1-2 contains gene I (RNA polymerase) of phage T7 under the control of the inducible PL promoter of phage λ; pGP1-2 also contains the Plac promoter and the gene for the heat-sensitive λ repressor, cI857.
Cloning, overexpression, and purification of His-tagged XylS derivatives.
The 968-bp DNA fragment covering wild-type XylS gene was PCR amplified from the TOL plasmid DNA, digested with the NdeI and XhoI enzymes, and cloned into the pET16b vector (Novagen). The resulting clones were sequenced to confirm XylS sequence. The 400-bp DNA fragment covering the XylS C-terminal domain (XylS residues 196 to 321) was PCR amplified from the TOL plasmid DNA, digested with NdeI and XhoI enzymes, and cloned into the pET16b vector (Novagen). The resulting clones were sequenced to confirm XylS-CTD sequence. To purify the truncated derivative XylS-CTD protein (XylS C-terminal domain), freshly transformed BL21(DE3) cells harboring pET-XylS-C plasmid were grown in 500 ml 2× YT medium (53) at 30°C until turbidity at 660 nm reached 0.5. Culture was then transferred to 16°C and incubated with 0.1 mM isopropyl-beta-d-thiogalactopyranoside (IPTG) for 3 to 4 h, and cells were pelleted and frozen. The cell pellet was resuspended in 50 ml of lysis buffer (20 mM sodium phosphate, pH 7.5, 300 mM NaCl, 0.1 mM EDTA, 2.5 mM 2-mercaptoethanol, 10% [vol/vol] glycerol, 10 mM imidazole, and 1 mM Complete [Roche] protease inhibitor cocktail) and disrupted in a French pressure cell. The crude extract was centrifuged at 30,000 × g for 1 h, filtered through a 0.45-μm pore-size filter and loaded onto a 5 ml Ni-agarose column (Amersham Biosciences) preequilibrated with buffer A-XylS-C (lysis buffer without the Complete cocktail). The column was washed with buffer A-XylS-C until nonspecifically bound material had been removed. A 40-ml imidazole gradient in buffer A-XylS-C (from 0 to 1 M) was then applied. XylS-CTD eluted at about 500 mM imidazole. Eluted fractions containing XylS-CTD protein were dialyzed against storage buffer (20 mM sodium phosphate, pH 7.5, 300 mM NaCl, 2 mM dithiothreitol [DTT], 10% glycerol) and stored at −70°C until use. The 643-bp DNA fragment covering the XylS N-terminal domain (XylS residues 1 to 207) was amplified by PCR from pERD103 plasmid, and the PCR product was digested with the NdeI and XhoI enzymes and subsequently cloned into the pET16b vector (Novagen). The resulting clones were sequenced to confirm XylS-NTD sequence.
Gel filtration chromatography.
XylS-CTD molecular weight was estimated by gel filtration chromatography on a Superdex 75 HR 10/30 column (Amersham Biosciences) equilibrated with a mixture of 30 mM Tris-HCl (pH 8), 200 mM KCl, 2 mM DTT, 10% (vol/vol) glycerol at room temperature. A 300-μl solution containing 60 to 100 μM protein was loaded per run. Bovine serum albumin (66 kDa), ovalbumin (45 kDa), carbonic anhydrase (29 kDa), trypsin inhibitor (20 kDa), and cytochrome c (12.4 kDa) were used as molecular mass standards. Proteins were detected at 280 nm. Eluted fractions of XylS-CTD were pooled and analyzed by SDS-PAGE (Fig. 2).
FIG. 2.

Size exclusion chromatography of the XylS C-terminal domain. The elution profile of XylS-CTD is shown (continuous line). The following molecular mass standards were used: bovine serum albumin (66 kDa), ovalbumin (45 kDa), carbonic anhydrase (29 kDa), trypsin inhibitor (20 kDa), and cytochrome c (12.4 kDa). R2 = 0.9979. mAU, milli-absorbance units. (Insert) SDS-PAGE of 10 μl of the four 0.5-ml fractions forming the protein peak (numbered as 1, 2, 3, and 4 in the elution profile).
Gel retardation assays (EMSA).
Complementary oligonucleotides containing one (AB) or two (ABAB) intact XylS binding sites (Fig. 1) were annealed and blunt-end ligated into HincII-linearized pUC19. DNA probes (130- or 150-bp DNA fragments, respectively) were obtained by PCR amplification of these constructs with M13 universal and reverse primers. The PCR products were isolated from agarose gels using the QIAquick gel extraction kit (Qiagen) and end labeled with [γ-32P]ATP using T4 polynucleotide kinase (59). Labeled probes (1 nM) were incubated with increasing amounts of purified XylS-CTD domain at 30°C for 15 min in 10 μl of binding buffer [5 mM Tris, 24 mM HEPES, pH 8, 50 mM potassium glutamate, 20 mM NaCl, 1.4 mM EDTA, 0.4 mg/ml bovine serum albumin (BSA), 9% [vol/vol] glycerol, 0.5 μg of poly(dI-dC), and 1 mM DTT]. Samples were loaded onto 8% (wt/vol) nondenaturing polyacrylamide gels in Tris-glycine buffer (0.2 M glycine, 0.025 M Tris-HCl [pH 8.6]). The gels were run for 3 h at 7 V/cm and 4°C, vacuum dried, and exposed to PhosphorImager plates. The results were analyzed using Molecular Imager FX equipment and QuantityOne software (Bio-Rad, Madrid, Spain).
Determination of XylS-CTD DNA stoichiometry by native PAGE.
The molecular weight of XylS-CTD-DNA complexes was determined electrophoretically using the procedure of Ferguson (14), as adapted by Orchard and May for protein-DNA complexes (42). Briefly, binding reaction mixtures containing 500 nM to 5 μM XylS-CTD and 50 bp of target DNA or molecular weight markers were analyzed on a series of polyacrylamide gels (7 to 10% [wt/vol] polyacrylamide at 37.5:1) electrophoresed in Tris-glycine buffer (described above) until the bromophenol blue front in flanking sample lanes reached the bottom of the gels. The gels were silver stained. The distance from the loading well to each complex was measured and standardized with the bromophenol blue migration distance to determine the relative mobility (Rf). The logarithm of the Rf was plotted against the gel concentration for each complex and protein standards, and best-fit lines were obtained. The negative slopes of these lines were then plotted against the molecular weights of the protein standards on a double-logarithmic scale, and best-line fit was obtained. The slopes for free DNA and protein-DNA complexes (the CI and CII fast- and slow-migrating complexes, respectively) were depicted, and their molecular weight was extrapolated according to the equation y = 0.1039x + 2.1505. The marker proteins used were carbonic anhydrase, α-lactalbumin, bovine serum albumin dimer, bovine serum albumin monomer, and ovalbumin.
DNase I footprints.
A 158-bp DNA fragment covering XylS binding sites (positions from −113 to +35 of the Pm promoter) was specifically radiolabeled at either strand 5′ end using [γ-32P]ATP and T4 polynucleotide kinase. End-labeled DNA probes (10 nM) were preincubated for 15 min at 30°C in binding buffer with different amounts of purified XylS-CTD. DNase I was added to a final concentration of 0.5 mU/μl and incubated for 5 min at 30°C, and the reaction was stopped by addition of EDTA. Footprint patterns were analyzed on polyacrylamide sequencing gels calibrated with Maxam-Gilbert sequence ladders.
Association constants and cooperativity analysis for XylS-CTD binding.
Gel retardation assays of a DNA fragment containing the two XylS binding sites were carried out as described above. The results were quantified using Molecular Imager FX equipment and QuantityOne software (Bio-Rad). The fraction of free DNA (E0), complex I (E1), or complex II (E2) is given by equations 1 to 3. To estimate KI and KII, data from gel mobility shift assays were fit to equations 1 to 3 described by Chen et al. (9) using a nonlinear curve-fitting algorithm (Dynamic Curve Fitting from Sigma Plot; Systat Software, Inc.), where P is the total protein concentration and KI and KII are the macroscopic association constants for XylS-CTD binding to one or two sites, respectively, of the A1B1A2B2 DNA fragment.
For complex CI, DNA + mP ⇔ DNA-Pm, where KI = [DNA-Pm]/[DNA][P]m.
For complex CII, DNA-Pm + nP ⇔ DNA-Pm + n, where KII = [DNA-Pm + n]/KI [DNA][P]m + n.
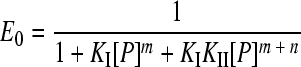 |
(1) |
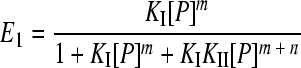 |
(2) |
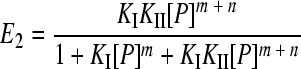 |
(3) |
where m and n represent the number of protein monomers per site in each case, and equal to 1 in both cases, and DNA-Pm and DNA-Pm + n represent CI and CII, respectively.
DNA-bending assays.
The pBend2 plasmid derivative pBend2-Pm80 (18) was digested with different enzymes to yield a series of DNA fragments with permuted positions of XylS binding sites. These fragments were end labeled, and gel retardation assays were performed as described above. To estimate the apparent bend angle (α) and to locate the center of the bend, changes in the mobility of the protein-bound DNA fragments (relative to the well position) were fitted to a cosine function (E) (60), (E) cos α/2 = μM/μE, where μM represents the relative migration of the complex with the lowest mobility and μE represents the relative migration of the complex with the highest mobility.
β-Galactosidase assays.
E. coli CC118λPm::lacZ cells bearing pGP1-2 plasmid were transformed with pET-XylS-C, pET-XylS, pET-XylS-N, or the control plasmid, pET16b (Table 1). Transformants were grown overnight on LB medium containing the appropriate antibiotics. Three independent clones of each strain were used. Quadruplicate cultures were prepared by diluting cells from overnight cultures to 1/100. When cultures had reached an optical density at 600 nm (OD600) of 0.4, one of them was maintained as a noninduced control, while the other three were supplemented with 3MB (1 mM), IPTG (0.5 mM), or both, respectively. Samples for β-galactosidase activity determination were taken 1 h after induction. β-Galactosidase activity was determined in permeabilized whole cells according to Miller (38).
RESULTS
XylS-N-terminal domain is responsible of XylS insolubility.
As for many members of the AraC family, previous attempts to purify the complete XylS protein were unsuccessful and produced protein aggregates (3, 7, 49, 61). To investigate the basis for this insolubility, we analyzed the two protein domains independently. We found that a His10 fusion protein of the C-terminal domain could be overproduced and directly purified in a soluble form. In contrast, XylS N-terminal domain with a His10 fusion (XylSN) aggregated and formed inclusion bodies under every expression condition tested (12), showing that the NTD was the main contributor to the low solubility of full-length XylS. Since the HTH DNA binding elements of XylS are located at the C-terminal end, we tested if both His-tagged XylS wild-type protein and XylS C-terminal domain were able to activate in vivo expression from the Pm promoter (Table 2). E. coli CC118 (Pm::lacZ, pGP1-2) bears a Pm::lacZ fusion in the chromosome as well as the T7 RNAP gene under the control of the phage λ PL promoter. This strain was transformed with either pET-XylS or pET-XylS-C, which carry the wild-type or truncated XylS version, respectively, under the control of T7 promoter or with the control plasmid pET16b (Table 1). The values of β-galactosidase activity determined in the presence and absence of 1 mM 3MB showed that in the absence of any inducer, activity was 1,490 ± 132 Miller units (MU) for XylS-CTD protein and 2,290 ± 234 MU in the case of full-length XylS, while in the control strain bearing pET16b the activity was negligible (Table 2). These high effector-independent levels correspond to high basal T7 polymerase-dependent XylS expression. It is well established that the high level of XylS protein obtained from a high-copy-number vector induces Pm transcription in the absence of effector (45, 49). Activity in the E. coli CC118 (Pm::lacZ, pGP1-2) strain transformed with pET-XylSN was negligible under all conditions (6 MU) and similar to values obtained with the control plasmid, pET16b. In E. coli strain CC118 (Pm::lacZ, pGP1-2), carrying the XylS-CTD fusion protein gene, no significant increase in Pm activity was detected in the presence of either 3MB or IPTG, while in the case of full-length XylS, the addition of 3MB promoted a 2-fold increase in Pm expression level (Table 2). These results confirm that both the DNA-binding domain devoid of the N-terminal domain and the full-length XylS are capable, when overexpressed, to activate transcription even in the absence of effector (19, 28). However, our results demonstrate that the 3MB response relies solely on XylS N terminal.
TABLE 2.
Transcription activation of Pm by different XylS derivatives
| XylS protein | β-Galactosidase activity (MU) with additiona: |
|||
|---|---|---|---|---|
| None | 3MB | IPTG | 3MB + IPTG | |
| Control | 8 ± 4 | 3 ± 2 | 6 ± 3 | 6 ± 4 |
| XylS-CTD | 1,490 ± 132 | 1,780 ± 178 | 2,130 ± 145 | 2,260 ± 163 |
| XylS wild type | 2,290 ± 234 | 4,250 ± 329 | 3,100 ± 289 | 4,150 ± 347 |
| XylS-N | 6 ± 2 | 5 ± 4 | 6 ± 3 | 7 ± 3 |
Values were calculated from four replicas of three independent experiments.
Pm half-sites are necessary and sufficient for XylS binding.
Most AraC family members appear to form dimers in solution, and dimerization determinants have been proposed to reside in the N-terminal domain (6, 8, 43, 44, 49). If this were the case for XylS, a truncated protein devoid of its N-terminal domain would behave as a monomer in solution. We determined the XylS-CTD oligomeric state using gel filtration through a Superdex G-75 column as described in Materials and Methods. A single peak was detected at an elution volume corresponding to a molecular mass of 16 kDa, in good correlation with the molecular mass of XylS-CTD (16.81 kDa), thus implying that the protein was a monomer under our experimental conditions (Fig. 2).
We next examined XylS-CTD binding to its operator, defined as two binding sites organized as direct repeats, one overlapping the RNA polymerase binding site (proximal site) and the second one located 6 nucleotides upstream (distal site). Each binding site is composed of two different boxes, A1/A2 and B1/B2, conserved in both sites and separated by a sequence of six nonconserved nucleotides (13, 19) (Fig. 1). To test if a single binding site was sufficient for XylS-CTD binding, a DNA fragment containing the distal XylS binding site composed of A1 and B1 half-sites was titrated with increasing XylS-CTD concentrations. A single shifted band was observed, corresponding to complex CI (Fig. 3a). As expected (12, 28), when an A1B1A2B2 DNA fragment containing the complete two-site wild-type binding region was titrated, a second, slower-migrating shifted band was formed (complex CII) (Fig. 3b). The figure shows that as the XylS-CTD concentration increased, the CI complex was progressively replaced by the lower-mobility CII complex.
FIG. 3.

XylS-CTD binding to Pm operator. (a) Electrophoretic mobility shift assay of XylS-CTD binding to a Pm template containing only one binding site, symbolized as A1B1 (Fig. 1). The labeled DNA fragment (1 nM) was incubated with 50 nM, 100 nM, 200 nM, 400 nM, 600 nM, 1 μM, 2 μM, 4 μM, and 8 μM purified XylS-CTD (lanes 2 to 10, respectively). Lane 1, free DNA. One XylS-CTD-DNA complex was formed (CI). The scheme depicts the DNA fragment used in the assay, including only one binding site (arrow) composed of A1 (black) and B1 (gray) half-sites. (b) Electrophoretic mobility shift assay of XylS-CTD binding to A1B1A2B2 template. The labeled DNA fragment was incubated with 50 nM, 100 nM, 200 nM, 400 nM, 600 nM, 1 μM, 2 μM, 4 μM, and 8 μM purified XylS-CTD (lanes 2 to 10, respectively). Lane 1, free DNA. XylS-CTD-DNA complexes CI and CII were formed. The scheme depicts DNA fragment used in the assay, including distal (D) and proximal (P) binding sites (arrows), each composed of A (black) and B (gray) boxes. A small arrow indicates the loading wells in the gel.
One XylS-CTD monomer per single binding site.
To determine the DNA/protein stoichiometry of CI and CII complexes, we measured the molecular weight of the two complexes using the electrophoretic method adapted by Orchard and May (42), based on changes in gel mobility at different acrylamide concentrations. Briefly, gel mobility of CI and CII complexes and of a series of molecular weight markers was first plotted against polyacrylamide concentration, and slopes were calculated in each case (Fig. 4a). The linear regression slopes obtained for each molecular weight marker were plotted against the logarithm of the molecular weight (Fig. 4b). When slope values of protein-DNA complexes CI and CII were fitted to the plot, an apparent molecular mass of 46 kDa was estimated for the CI XylS-CTD-DNA complex. The molecular mass of the protein component of the CI complex was extrapolated as 17 kDa after subtracting 29 kDa corresponding to the apparent molecular mass of the 50-bp A1B1A2B2 DNA fragment, consistent with CI resulting from a single XylS-CTD monomer (16.81 kDa) bound to the promoter probe. Likewise, an apparent molecular mass of 57 kDa was obtained for CII complex (28 kDa corresponding to the protein component), showing a slight deviation from the expected mass of 33.62 kDa for two XylS-CTD monomers.
FIG. 4.

Determination of the stoichiometry of XylS-CTD-DNA complexes by native PAGE. Protein markers and XylS bound to DNA sequence were electrophoresed at different polyacrylamide concentrations. (a) The logarithms of the relative mobilities of XylS-CTD-DNA complexes and marker proteins with respect to bromophenol blue were first plotted as a function of polyacrylamide concentration. (b) The slope of the regression equation obtained for each marker is plotted against their molecular weight (MW). The values for free DNA (black squares), CI (light gray triangles), and CII (dark gray diamonds) are depicted, and their molecular weights were extrapolated according to the equation y = 0.1039x + 2.1505. The marker proteins used were carbonic (c.) anhydrase, α-lactalbumin, bovine serum albumin dimer, bovine serum albumin monomer, and ovalbumin.
Binding of XylS-CTD to the Pm promoter induces bending.
Previous work showed that Pm promoter DNA region was intrinsically curved, with an apparent bent angle of 35° ± 5° (16, 18, 19). To explore if binding of XylS-CTD induced further distortion in the DNA, we applied the circular permutation assay developed by Kim and coworkers (30, 64) (see Materials and Methods). A series of 195-bp DNA fragments with permuted positions of the XylS binding sites were incubated with XylS-CTD, and the mobility of the two complexes (CI and CII) was analyzed by native polyacrylamide gel electrophoresis (64) (Fig. 5). The intrinsic bend angles (α) and the location of the center of the bend were estimated according to Thompson and Landy (60). Although this method does not resolve the degree of curvature at each single position in the DNA, we could observe that the Pm intrinsic curvature centered within the A track spanning positions −41 to −46 increased to a global value of 50° ± 5° after binding of one XylS-CTD monomer and to 98° ± 2° after binding of two monomers (Fig. 5). The new center of the bent region was located between positions −49 and −52 in both CI and CII complexes, evidencing a shift from the previously determined free DNA bending center within the XylS binding site toward the DNA region between the two binding sites (Fig. 1 and 5).
FIG. 5.
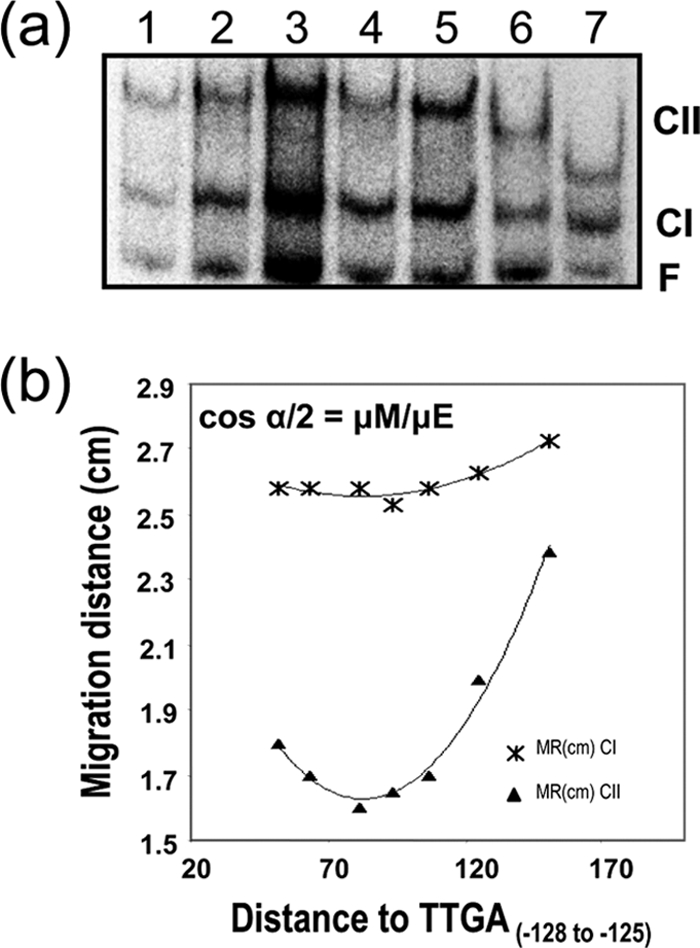
XylS-CTD binding to DNA provokes DNA bending. Gel retardation assays of XylS-CTD on a series of 195 bp DNA fragments, containing XylS binding sites in permutated locations were run in 8% polyacrylamide (wt/vol). The migration distance of each complex (*, CI; ▴, CII) was plotted against the distance between XylS binding site 5′ end and fragment 5′ end in each fragment tested. The bending angle for each complex was estimated according to the equation cos α/2 = μM/μE, where μM is the migration of the complex with lowest mobility and μE is the migration of the complex with highest mobility. Values are the average of six independent determinations.
Stepwise binding of XylS-CTD to Pm binding sites.
In mobility shift assays, we observed that at a low XylS-CTD concentration, CI was preferentially formed, followed by a progressive accumulation of CII concomitant with a decrease of CI at a higher protein concentration (Fig. 3). To analyze if this could result from the first monomer at the proximal site facilitating binding of the second one in a cooperative manner, we performed a series of quantitative analyses of the binding interactions. To this end, five repetitions of titration assays of a DNA fragment containing one or two binding sites with XylS-CTD were performed and the amounts of CI and CII complexes of XylS-CTD bound to an A1B1A2B2 fragment were plotted as a function of XylS-CTD concentration (Fig. 6; see equations 1 to 3 in Materials and Methods) (9). XylS binding sites are not identical in sequence; thus, separate estimates of intrinsic binding affinity for each site could not be derived from two-site gel shift experiments. We used equations 1 to 3 as derived by Chen et al. (9) to estimate the macroscopic association constants, KI and KII. The average data were fit to equations 1 to 3, and KI and KII constants were derived using nonlinear curve fitting (see Materials and Methods). The data obtained (KI = 1.56 × 106 M−1 and KII = 7.3 × 105 M−1) are not supportive of significant cooperative interaction in the sequential binding of two XylS-CTD monomers to their recognition sites.
FIG. 6.
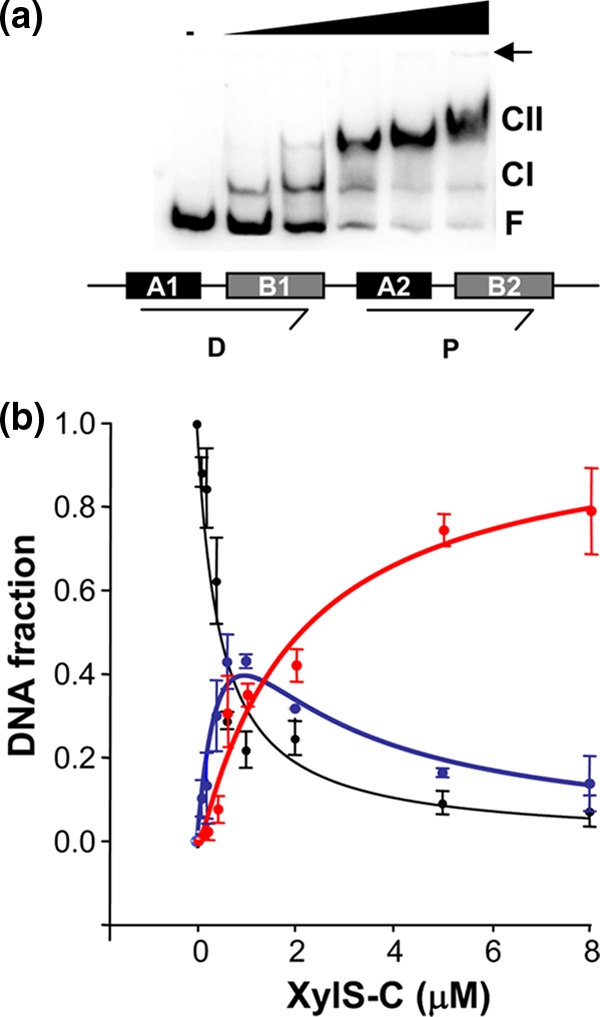
Analysis of XylS-CTD binding to Pm. (a) Representative EMSA of XylS-CTD binding to the DNA containing the XylS binding sites shown under the gel as a scheme. The XylS-CTD concentrations were 0, 80 nM, 400 nM, 1.5 μM, 2.5 μM, and 5 μM. CI, complex 1; CII, complex 2; F, free DNA. A small arrow indicates the loading wells in the gel. (b) Quantitation average of gel shift assay data from five independent experiments. The free DNA (black circles), CI complex (blue), and the CII complex (red) were quantified in a PhosphorImager. Lines represent the best fit of the data to equations 1 to 3 (equation 1, black; equation 2, blue; and equation 3, red [see Materials and Methods]). The regression coefficients for each fit were Rfree = 0.952, RCI = 0.957, and RCII = 0.978. Error bars correspond to standard deviation values.
To analyze if XylS-CTD monomer preferentially bound any of its binding sites, we carried out DNase I footprinting assays of XylS-CTD bound to a Pm fragment. Figure 7 shows that XylS-CTD protected two regions centered at positions −60 and −40, corresponding to the two 15-bp direct repeats (distal and proximal sites). We observed that at a low protein concentration, the proximal site was fully protected, whereas the distal site was only progressively protected by increasing XylS-CTD concentration (see especially the bottom strand footprinting in Fig. 7b). Interestingly, a hyperreactive band corresponding to a T residue at position −42, visible in free DNA, almost disappeared at low XylS-CTD concentrations (Fig. 7a). Concomitant with this, a band corresponding to a T residue at position −51 became hyperreactive, suggesting a shift in the bending center at the Pm promoter (Fig. 7a). These results support sequential and directional binding of the two XylS-CTD monomers at Pm, generating a progressive curvature in the DNA between the two binding sites. Surprisingly, the hyperreactive band had not been previously observed except as a very weak signal in footprint assays using an antigen-tagged XylS C-terminal domain derivative (28).
FIG. 7.

DNase I footprint analysis of XylS-Pm complexes. (a) Upper strand; (b) bottom strand. Lane 1, free DNA. The XylS-CTD concentrations used were as follows: (a) 1 μM (lane 2), 2 μM (lane 3), 5 μM (lane 4), and 10 μM (lane 5); (b) 500 nM (lane 2), 1 μM (lane 3), and 2 μM (lane 4). The position of the region corresponding to the distal (D) and proximal (P) sites is indicated. BC, bending center. The band position in free DNA was determined by comparison with a Maxam and Gilbert sequence ladder (not shown).
DISCUSSION
In this work, we have analyzed XylS DNA binding properties based on two facts: (i) the XylS DNA binding domain was soluble when overproduced, unlike most proteins of the family (4); and (ii) this domain could reproduce all XylS features except for 3MB responsiveness. Gel filtration column chromatography of purified XylS-CTD showed that it behaved as a monomer in solution, and the protein was able to activate transcription in vivo (Table 2) and in vitro (12, 28). This is analogous to the situation in RhaS and ExsA, two members of the AraC family in which the C-terminal domain contains all of the determinants essential for activation (4, 63). The situation in XylS, ExsA, and RhaS contrasts with that of AraC and MelR. In AraC, the DNA binding domain strictly required a leucine zipper or an artificial covalent linkage-mediated dimerization to activate transcription from the PBAD promoter (61), and the MelR C-terminal domain did not activate transcription despite the fact it bound DNA and provoked a similar bend to full-length MelR (2, 25). Thus, XylS-CTD resembles those AraC family proteins related to stress response such as MarA, Sox, and Rob, which activate transcription as monomers critically and exclusively depending upon their intracellular concentrations.
XylS-CTD was able to bind a DNA probe containing only one binding site, rendering a single shift product (Fig. 3a). When the entire binding site including the two direct repeats was probed, XylS-CTD binding gave rise to two distinct complexes that were formed sequentially (Fig. 3b). Gel shift analysis at different acrylamide concentrations showed that CI represents one molecule of XylS-CTD bound to the promoter (Fig. 4), which is consistent with our observation that the protein is a monomer in solution (Fig. 2). The data also supported that CII represents two XylS-CTD molecules bound to the promoter. The DNase footprint protection pattern showed that XylS-CTD bound the two direct repeats at the Pm promoter in two consecutive steps, where the first XylS monomer occupied the proximal binding site, facilitating the binding of the second monomer to the distal site (Fig. 7). Previous results indicated that interactions between XylS HTH α6 and base pairs at B boxes were the most important contacts for XylS binding to its target sequence and subsequent transcription activation (13). However, it is well established that noncontacted bases can also have important effects on protein affinity by affecting DNA flexibility or intrinsic curvature (54). In fact, the Pm 35° ± 5° intrinsic curvature centered in the A track in the proximal binding site (19) increased progressively to 50° ± 5° and 98° ± 2° after binding of the first and second XylS-CTD monomers, respectively (Fig. 5). Concomitantly, the bending center moved toward the region between XylS binding sites (Fig. 1 and 5), in good correlation with the band of enhanced DNase sensitivity located at −51 (Fig. 7). AT base pairs have been referred to as “natural hinges” for protein-induced DNA bending (41). The A track present at XylS proximal binding site between boxes A2 and B2 might act as a flexible motif, increasing XylS-CTD affinity. Analysis of the MarA-DNA cocrystals showed that the two recognition helices are arranged in a parallel manner and separated by 27 Å, whereas the distance between consecutive major grooves of canonical DNA B form is 34 Å. Thus, MarA binding to its target DNA induces a 35° bend in that region. In Pm, the 35° bend is centered in the proximal site and probably explains the observation in DNase footprint assays that at low concentrations XylS-CTD preferentially occupies the proximal binding site. The movement of the bending center toward the distal site, together with the increase of the bending angle to 50°, seems to be required to allow binding of the second monomer, which finally entails an increase of the overall curvature to 98°. The solution structures of AraC-CTD and especially of ToxT containing a buried cis-palmitoleic acid ligand show that the two recognition helices are not parallel but divergently oriented, thus suggesting that not only the DNA but also the protein is distorted in the protein-DNA complex (33, 48). In ToxT, this conformational change is accomplished by the release of the fatty acid from the N-terminal domain, allowing the regulator to bind DNA (33). If protein affinity and complex stability could be enhanced by flexibility in the DNA binding site, it is tempting to speculate that in XylS more extensive protein-DNA contacts and interactions with RNA polymerase would be allowed as the Pm promoter gradually bends, despite the absence of a specific interprotein contact domain involved in dimerization. It should be noted that from our results we cannot distinguish if stability of the complex increased exclusively through the protein-DNA contacts favored by bent DNA or if additional effects caused by unexpected protein-protein interactions in DNA-bound XylS-CTD monomers also play a role (61).
Accumulated evidence suggests XylS native protein functions as a dimer (49). The absence of cooperativity observed in the binding of the two XylS-CTD monomers is probably due to the absence of the N-terminal dimerization domain present in the wild-type protein. This is the case for the Pseudomonas aeruginosa ExsA regulator, where binding of the entire protein to its PexsC target promoter showed a cooperativity that disappeared when only the C-terminal domain of the protein was assayed, suggesting cooperative binding was dependent upon the N-terminal domain (4). In XylS full-length protein, using cell extracts of the wild-type protein we have shown that only CII was formed (12). Complexes with a single monomer are only detectable when the proteins are unable to dimerize in solution, as in the dimerization mutant XylS3L (12) or in XylS-CTD (Fig. 3). Unfortunately, experiments with pure preparations of full-length XylS retaining the dimerization domain cannot be carried out because of the protein's poor solubility.
The Pm promoter can be classified as a class II promoter (5), in which multiple interactions between XylS and RNA polymerase would be established. In fact, XylS establishes interactions with the α-CTD subunit of the RNA polymerase (51, 52). The above-discussed XylS-induced bending process is likely to facilitate and stabilize XylS-RNAP contacts in vivo to form an active transcription initiation complex. In fact, besides protein-protein interactions induced by DNA curvature, some activators stimulate contacts of DNA sequences with the RNA polymerase, favoring the transition from a closed to open complex (24, 57). We have previously shown in chromatin immunoprecipitation assays that XylS recruits RNA polymerase to the Pm promoter in response to 3MB and also promotes open complex formation in permanganate footprinting assays (12). Preliminary results using XylS T283A and E284A mutations in the α-5 helix conserved patch involved in transcription activation (21) rendered XylS derivatives severely defective in their ability to activate transcription from the Pm promoter (P. Domínguez-Cuevas and Marqués, unpublished data). Further studies to define specific amino acid contacts between XylS and the sigma RNA polymerase subunit are required to determine the influence of XylS-induced DNA bending on transcription activation.
Figure 8 illustrates our current model of the molecular events underlying XylS-CTD Pm binding and transcriptional activation. Pm promoter intrinsic 35° bending centered in the proximal XylS binding site overlapping the RNA polymerase −35 hexamer (Fig. 8, step 1) facilitates interaction at the proximal binding site such that a XylS-CTD monomer first recognizes and binds this sequence (step 2). This first event would induce a shift in DNA bending angle toward the intersite region, together with a significant increase in the bending angle (50°), triggering a fast binding of the second monomer and recruitment of the RNA polymerase (step 3) (12). Contacts with RNA polymerase through α-CTD and probably also through the sigma factor would then favor open complex formation and transcription initiation (step 4). The set of results from this and previous works could point to wild-type XylS being a dimer in solution and binding Pm sequentially, with monomer 1 first contacting the proximal site, rapidly followed by DNA bending and setting of the second monomer to obtain a stable dimer-DNA complex.
FIG. 8.

Scheme of the sequential binding model of XylS to Pm. (Step 1) Free DNA. The −10/−35 RNAP binding site and the two XylS binding sites (D, distal; and P, proximal) are depicted. The bending angle is 35°, centered in the XylS proximal binding site. (Step 2) A first XylS monomer binds Pm at the proximal site, shifts the bent center to DNA sequence between the two sites, and increases the bending angle to 50°. (Step 3) This change favors binding of a second monomer at the distal site, further increasing the DNA curvature to an overall value of 98° (here schematized as a right angle). Contacts are established with RNAP through the α-CTD and probably with the σ subunit, which as shown in step 4 favors open complex formation and transcription initiation (54).
Acknowledgments
This work was supported by the Ministerio de Educación y Ciencias grants BCM 2001-0515 to S.M. and BIO2006-05668 and VEM2004-08560 to J.L.R. and grant CIV344 from the Junta de Andalucía. P.D.-C. was financed by a Junta de Andalucía predoctoral fellowship.
We thank Patricia Marín for excellent technical assistance and Chechu Rodríguez-Herva for artwork.
Footnotes
Published ahead of print on 2 April 2010.
REFERENCES
- 1.Bhende, P. M., and S. M. Egan. 1999. Amino acid-DNA contacts by RhaS: an AraC family transcription activator. J. Bacteriol. 181:5185-5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bourgerie, S. J., C. M. Michán, M. S. Thomas, S. J. Busby, and E. I. Hyde. 1997. DNA binding and DNA bending by the MelR transcription activator protein from Escherichia coli. Nucleic Acids Res. 25:1685-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunker, P., M. Hils, J. Altenbuchner, and R. Mattes. 1998. The mannitol utilization genes of Pseudomonas fluorescens are regulated by an activator: cloning, nucleotide sequence and expression of the mtlR gene. Gene 215:19-27. [DOI] [PubMed] [Google Scholar]
- 4.Brutinel, E. D., C. A. Vakulskas, and T. L. Yahr. 2009. Functional domains of ExsA, the transcriptional activator of the Pseudomonas aeruginosa type III secretion system. J. Bacteriol. 191:3811-3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busby, S., and R. H. Ebright. 1997. Transcription activation at class II CAP-dependent promoters. Mol. Microbiol. 23:853-859. [DOI] [PubMed] [Google Scholar]
- 6.Bustos, S. A., and R. F. Schleif. 1993. Functional domains of the AraC protein. Proc. Natl. Acad. Sci. U. S. A. 90:5638-5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caswell, R., C. Webster, and S. Busby. 1992. Studies on the binding of the Escherichia coli MelR transcription activator protein to operator sequences at the MelAB promoter. Biochem. J. 287:501-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caswell, R., J. Williams, A. Lyddiatt, and S. Busby. 1992. Overexpression, purification and characterization of the Escherichia coli MelR transcription activator protein. Biochem. J. 287:493-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, S., M. Iannolo, and J. M. Calvo. 2005. Cooperative binding of the leucine-responsive regulatory protein (Lrp) to DNA. J. Mol. Biol. 345:251-264. [DOI] [PubMed] [Google Scholar]
- 10.Dangi, B., P. Pelupessey, R. G. Martin, J. L. Rosner, J. M. Louis, and A. M. Gronenborn. 2001. Structure and dynamics of MarA-DNA complexes: an NMR investigation. J. Mol. Biol. 314:113-127. [DOI] [PubMed] [Google Scholar]
- 11.Domínguez-Cuevas, P., P. Marin, J. L. Ramos, and S. Marqués. 2005. RNA polymerase holoenzymes can share a single transcription start site for the Pm promoter. Critical nucleotides in the −7 to −18 region are needed to select between RNA polymerase with sigma38 or sigma32. J. Biol. Chem. 280:41315-41323. [DOI] [PubMed] [Google Scholar]
- 12.Domínguez-Cuevas, P., P. Marín, S. J. Busby, J. L. Ramos, and S. Marqués. 2008. Roles of effectors in XylS-dependent transcription activation. Intramolecular domain de-repression and DNA binding. J. Bacteriol. 190:3118-3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Domínguez-Cuevas, P., P. Marín, S. Marqués, and J. L. Ramos. 2008. XylS-Pm promoter interactions through two HTHs: identifying XylS residues important for DNA binding and activation. J. Mol. Biol. 375:59-69. [DOI] [PubMed] [Google Scholar]
- 14.Ferguson, K. A. 1964. Starch-gel electrophoresis—application to the classification of pituitary proteins and polypeptides. Metabolism 13(Suppl.):985-1002. [DOI] [PubMed] [Google Scholar]
- 15.Gallegos, M. T., S. Marqués, and J. L. Ramos. 1996. Expression of the TOL plasmid xylS gene in Pseudomonas putida occurs from a sigma 70-dependent promoter or from sigma 70- and sigma 54-dependent tandem promoters according to the compound used for growth. J. Bacteriol. 178:2356-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallegos, M. T., S. Marqués, and J. L. Ramos. 1996. The TACAN4TGCA motif upstream from the −35 region in the sigma70-sigmaS-dependent Pm promoter of the TOL plasmid is the minimum DNA segment required for transcription stimulation by XylS regulators. J. Bacteriol. 178:6427-6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallegos, M. T., R. Schleif, A. Bairoch, K. Hofmann, and J. L. Ramos. 1997. Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 61:393-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.González-Pérez, M. M., S. Marqués, P. Domínguez-Cuevas, and J. L. Ramos. 2002. XylS activator and RNA polymerase binding sites at the Pm promoter overlap. FEBS Lett. 519:117-122. [DOI] [PubMed] [Google Scholar]
- 19.González-Pérez, M. M., J. L. Ramos, M. T. Gallegos, and S. Marqués. 1999. Critical nucleotides in the upstream region of the XylS-dependent TOL meta-cleavage pathway operon promoter as deduced from analysis of mutants. J. Biol. Chem. 274:2286-2290. [DOI] [PubMed] [Google Scholar]
- 20.González-Pérez, M. M., J. L. Ramos, and S. Marqués. 2004. Cellular XylS levels are a function of transcription of xylS from two independent promoters and the differential efficiency of translation of the two mRNAs. J. Bacteriol. 186:1898-1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grainger, D. C., C. L. Webster, T. A. Belyaeva, E. I. Hyde, and S. J. Busby. 2004. Transcription activation at the Escherichia coli melAB promoter: interactions of MelR with its DNA target site and with domain 4 of the RNA polymerase sigma subunit. Mol. Microbiol. 51:1297-1309. [DOI] [PubMed] [Google Scholar]
- 22.Griffith, K. L., and R. E. Wolf, Jr. 2004. Genetic evidence for pre-recruitment as the mechanism of transcription activation by SoxS of Escherichia coli: the dominance of DNA binding mutations of SoxS. J. Mol. Biol. 344:1-10. [DOI] [PubMed] [Google Scholar]
- 23.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 24.Harrington, R. E. 1992. DNA curving and bending in protein-DNA recognition. Mol. Microbiol. 6:2549-2555. [DOI] [PubMed] [Google Scholar]
- 25.Howard, V. J., T. A. Belyaeva, S. J. Busby, and E. I. Hyde. 2002. DNA binding of the transcription activator protein MelR from Escherichia coli and its C-terminal domain. Nucleic Acids Res. 30:2692-2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jair, K. W., R. G. Martin, J. L. Rosner, N. Fujita, A. Ishihama, and R. E. Wolf, Jr. 1995. Purification and regulatory properties of MarA protein, a transcriptional activator of Escherichia coli multiple antibiotic and superoxide resistance promoters. J. Bacteriol. 177:7100-7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaldalu, N., T. Mandel, and M. Ustav. 1996. TOL plasmid transcription factor XylS binds specifically to the Pm operator sequence. Mol. Microbiol. 20:569-579. [DOI] [PubMed] [Google Scholar]
- 28.Kaldalu, N., U. Toots, V. de Lorenzo, and M. Ustav. 2000. Functional domains of the TOL plasmid transcription factor XylS. J. Bacteriol. 182:1118-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kessler, B., S. Marqués, T. Kohler, J. L. Ramos, K. N. Timmis, and V. de Lorenzo. 1994. Cross talk between catabolic pathways in Pseudomonas putida: XylS-dependent and -independent activation of the TOL meta operon requires the same cis-acting sequences within the Pm promoter. J. Bacteriol. 176:5578-5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim, J., C. Zwieb, C. Wu, and S. Adhya. 1989. Bending of DNA by gene-regulatory proteins: construction and use of a DNA bending vector. Gene 85:15-23. [DOI] [PubMed] [Google Scholar]
- 31.Kwon, H. J., M. H. Bennik, B. Demple, and T. Ellenberger. 2000. Crystal structure of the Escherichia coli Rob transcription factor in complex with DNA. Nat. Struct. Biol. 7:424-430. [DOI] [PubMed] [Google Scholar]
- 32.Lobell, R. B., and R. F. Schleif. 1990. DNA looping and unlooping by AraC protein. Science 250:528-532. [DOI] [PubMed] [Google Scholar]
- 33.Lowden, M. J., K. Skorupski, M. Pellegrini, M. G. Chiorazzo, R. K. Taylor, and F. J. Kull. 2010. Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proc. Natl. Acad. Sci. U. S. A. 107:2860-2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin, R. G., W. K. Gillette, and J. L. Rosner. 2000. Promoter discrimination by the related transcriptional activators MarA and SoxS: differential regulation by differential binding. Mol. Microbiol. 35:623-634. [DOI] [PubMed] [Google Scholar]
- 35.Martin, R. G., K. W. Jair, R. E. Wolf, Jr., and J. L. Rosner. 1996. Autoactivation of the marRAB multiple antibiotic resistance operon by the MarA transcriptional activator in Escherichia coli. J. Bacteriol. 178:2216-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin, R. G., and J. L. Rosner. 2001. The AraC transcriptional activators. Curr. Opin. Microbiol. 4:132-137. [DOI] [PubMed] [Google Scholar]
- 37.Michán, C. M., S. J. Busby, and E. I. Hyde. 1995. The Escherichia coli MelR transcription activator: production of a stable fragment containing the DNA-binding domain. Nucleic Acids Res. 23:1518-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller, J. 1972. Experiments in molecular genetics, p. 352-355. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 39.Munson, G. P., and J. R. Scott. 2000. Rns, a virulence regulator within the AraC family, requires binding sites upstream and downstream of its own promoter to function as an activator. Mol. Microbiol. 36:1391-1402. [DOI] [PubMed] [Google Scholar]
- 40.Niland, P., R. Huhne, and B. Muller-Hill. 1996. How AraC interacts specifically with its target DNAs. J. Mol. Biol. 264:667-674. [DOI] [PubMed] [Google Scholar]
- 41.Olson, W. K., A. A. Gorin, X. J. Lu, L. M. Hock, and V. B. Zhurkin. 1998. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. U. S. A. 95:11163-11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orchard, K., and G. E. May. 1993. An EMSA-based method for determining the molecular weight of a protein-DNA complex. Nucleic Acids Res. 21:3335-3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poore, C. A., C. Coker, J. D. Dattelbaum, and H. L. Mobley. 2001. Identification of the domains of UreR, an AraC-like transcriptional regulator of the urease gene cluster in Proteus mirabilis. J. Bacteriol. 183:4526-4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prouty, M. G., C. R. Osorio, and K. E. Klose. 2005. Characterization of functional domains of the Vibrio cholerae virulence regulator ToxT. Mol. Microbiol. 58:1143-1156. [DOI] [PubMed] [Google Scholar]
- 45.Ramos, J. L., N. Mermod, and K. N. Timmis. 1987. Regulatory circuits controlling transcription of TOL plasmid operon encoding meta-cleavage pathway for degradation of alkylbenzoates by Pseudomonas. Mol. Microbiol. 1:293-300. [DOI] [PubMed] [Google Scholar]
- 46.Ramos, J. L., A. Stolz, W. Reineke, and K. N. Timmis. 1986. Altered effector specificities in regulators of gene expression: TOL plasmid xylS mutants and their use to engineer expansion of the range of aromatics degraded by bacteria. Proc. Natl. Acad. Sci. U. S. A. 83:8467-8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rhee, S., R. G. Martin, J. L. Rosner, and D. R. Davies. 1998. A novel DNA-binding motif in MarA: the first structure for an AraC family transcriptional activator. Proc. Natl. Acad. Sci. U. S. A. 95:10413-10418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodgers, M. E., and R. Schleif. 2009. Solution structure of the DNA binding domain of AraC protein. Proteins 77:202-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruíz, R., S. Marqués, and J. L. Ramos. 2003. Leucines 193 and 194 at the N-terminal domain of the XylS protein, the positive transcriptional regulator of the TOL meta-cleavage pathway, are involved in dimerization. J. Bacteriol. 185:3036-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruíz, R., and J. L. Ramos. 2002. Residues 137 and 153 at the N terminus of the XylS protein influence the effector profile of this transcriptional regulator and the sigma factor used by RNA polymerase to stimulate transcription from its cognate promoter. J. Biol. Chem. 277:7282-7286. [DOI] [PubMed] [Google Scholar]
- 51.Ruíz, R., and J. L. Ramos. 2001. Residues 137 and 153 of XylS influence contacts with the C-terminal domain of the RNA polymerase alpha subunit. Biochem. Biophys. Res. Commun. 287:519-521. [DOI] [PubMed] [Google Scholar]
- 52.Ruíz, R., J. L. Ramos, and S. M. Egan. 2001. Interactions of the XylS regulators with the C-terminal domain of the RNA polymerase alpha subunit influence the expression level from the cognate Pm promoter. FEBS Lett. 491:207-211. [DOI] [PubMed] [Google Scholar]
- 53.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 54.Sarai, A., and H. Kono. 2005. Protein-DNA recognition patterns and predictions. Annu. Rev. Biophys. Biomol. Struct. 34:379-398. [DOI] [PubMed] [Google Scholar]
- 55.Soisson, S. M., B. MacDougall-Shackleton, R. Schleif, and C. Wolberger. 1997. The 1.6 A crystal structure of the AraC sugar-binding and dimerization domain complexed with D-fucose. J. Mol. Biol. 273:226-237. [DOI] [PubMed] [Google Scholar]
- 56.Soisson, S. M., B. MacDougall-Shackleton, R. Schleif, and C. Wolberger. 1997. Structural basis for ligand-regulated oligomerization of AraC. Science 276:421-425. [DOI] [PubMed] [Google Scholar]
- 57.Straney, D. C., S. B. Straney, and D. M. Crothers. 1989. Synergy between Escherichia coli CAP protein and RNA polymerase in the lac promoter open complex. J. Mol. Biol. 206:41-57. [DOI] [PubMed] [Google Scholar]
- 58.Tabor, S., and C. C. Richardson. 1985. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. U. S. A. 82:1074-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Terán, W., A. Felipe, A. Segura, A. Rojas, J. L. Ramos, and M. T. Gallegos. 2003. Antibiotic-dependent induction of Pseudomonas putida DOT-T1E TtgABC efflux pump is mediated by the drug binding repressor TtgR. Antimicrob. Agents Chemother. 47:3067-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson, J. F., and A. Landy. 1988. Empirical estimation of protein-induced DNA bending angles: applications to lambda site-specific recombination complexes. Nucleic Acids Res. 16:9687-9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Timmes, A., M. Rodgers, and R. Schleif. 2004. Biochemical and physiological properties of the DNA binding domain of AraC protein. J. Mol. Biol. 340:731-738. [DOI] [PubMed] [Google Scholar]
- 62.Tobes, R., and J. L. Ramos. 2002. AraC-XylS database: a family of positive transcriptional regulators in bacteria. Nucleic Acids Res. 30:318-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wickstrum, J. R., J. M. Skredenske, A. Kolin, D. J. Jin, J. Fang, and S. M. Egan. 2007. Transcription activation by the DNA-binding domain of the AraC family protein RhaS in the absence of its effector-binding domain. J. Bacteriol. 189:4984-4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu, H. M., and D. M. Crothers. 1984. The locus of sequence-directed and protein-induced DNA bending. Nature 308:509-513. [DOI] [PubMed] [Google Scholar]