

Abstract
The bZIP transcription factor C/EBPβ is a target of Ras signaling that has been implicated in Ras-induced transformation and oncogene-induced senescence (OIS). To gain insights into Ras-C/EBPβ signaling, we investigated C/EBPβ activation by oncogenic Ras. We show that C/EBPβ DNA binding is autorepressed and becomes activated by the Ras-Raf-MEK-ERK-p90RSK cascade. Inducible phosphorylation by RSK on Ser273 in the leucine zipper was required for DNA binding. In addition, three other modifications (phosphorylation on Tyr109 [p-Tyr109], p-Ser111, and monomethylation of Arg114 [me-Arg114]) within an N-terminal autoinhibitory domain were important for Ras-induced C/EBPβ activation and cytostatic activity. Apart from its role in DNA binding, Ser273 phosphorylation also creates an interhelical g↔e′ salt bridge with Lys268 that increases attractive electrostatic interactions between paired leucine zippers and promotes homodimerization. Mutating Ser273 to Ala or Lys268 to Glu decreased C/EBPβ homodimer formation, whereas heterodimerization with C/EBPγ was relatively unaffected. The S273A substitution also reduced the antiproliferative activity of C/EBPβ in RasV12-expressing fibroblasts and decreased binding to target cell cycle genes, while a phosphomimetic substitution (S273D) maintained growth arrest function. Our findings identify four novel C/EBPβ-activating modifications, including RSK-mediated phosphorylation of a bifunctional residue in the leucine zipper that regulates DNA binding and homodimerization and thereby promotes cell cycle arrest.
Oncogenic Ras stimulates the proliferation of many tumors and immortalized cells. However, in primary cells, sustained signaling by Ras or other oncogenes can induce a stable form of cell cycle arrest known as oncogene-induced senescence (OIS) (36). The bZIP transcription factor C/EBPβ (CCAAT/enhancer binding protein β) mediates cellular responses to growth factors and cytokines and is also regulated by oncogenic signals emanating from activated Ras or its downstream effectors (29). Interestingly, C/EBPβ participates in both proliferative and antiproliferative responses to oncogenic Ras (34). C/EBPβ-deficient mice are resistant to development of chemically induced skin tumors that carry Ras mutations, suggesting that C/EBPβ is essential for Ras-dependent tumorigenesis in skin (50). In addition, overexpression of C/EBPβ in human mammary epithelial cells induces features of transformation, including increased proliferation and anchorage-independent growth (6). Conversely, forced C/EBPβ expression blocks cell cycle progression of hepatoma cells and epidermal keratinocytes (5, 49). C/EBPβ-deficient mouse embryo fibroblasts (MEFs) are defective for RasV12-induced proliferation arrest and senescence (35), demonstrating an essential role for C/EBPβ in OIS in primary fibroblasts. The senescence-inducing activity of C/EBPβ requires RB-E2F and involves repression of E2F-regulated cell cycle genes, such as c-MYC (35). C/EBPβ is also required for BRAF-induced senescence of human fibroblasts, in part through upregulation of proinflammatory cytokines, their receptors, and p15INK4b (20). In addition, C/EBPβ mediates cytostatic responses to transforming growth factor beta (TGF-β) through repression of c-MYC gene transcription and activation of p15INK4b (14). C/EBPβ therefore has both pro- and antiproliferative functions, although the mechanistic basis for these opposing activities is not well understood (34).
C/EBPβ is an intrinsically repressed protein whose transcriptional activity can be stimulated by extracellular signals or expression of activated Ras, Raf, or other oncogenic kinases (18, 24, 47, 50) (S. Lee, M. Miller, J. D. Shuman, and P. F. Johnson, submitted for publication). Consistent with these findings, C/EBPβ was recently implicated as a critical target of ERK1/2 signaling in luteinizing hormone-mediated maturation of ovarian granulosa cells (13). Oncogenic Ras-induced activation of C/EBPβ involves modification of several residues, including phosphorylation on Thr189 (rat C/EBPβ; equivalent to human Thr235) by ERK1/2 or cyclin-dependent kinases (CDKs) (24, 37). Thr189 phosphorylation controls C/EBPβ activity in part by causing release of a repressive mediator complex and replacement by an activating mediator assembly (23). Ras also stimulates phosphorylation on Ser64 in the transactivation domain, catalyzed by CDK2/cdc2 (37). In hepatocytes, TGF-α induces phosphorylation on Ser105 (or a functionally analogous site in the murine protein, Thr217) by the ERK1/2-activated kinase p90RSK (4, 21). This modification regulates the proliferative and prosurvival functions of C/EBPβ in hepatic cells (3, 4). Other known C/EBPβ phosphoacceptors include Ser240 (protein kinase A [PKA] (8), Ser277 (calcium/calmodulin-dependent kinase) (46), and Ser181/185 (GSK3) (42).
Several studies have found that C/EBPβ is regulated partly at the level of DNA binding (27, 39, 40, 45), and in vitro experiments using recombinant proteins suggest that N-terminal sequences inhibit the C-terminal bZIP domain (12, 47). Here, we show that C/EBPβ DNA-binding activity in mammalian cells is intrinsically repressed (Lee et al., submitted) and can be activated by oncogenic RasV12 or growth factors via the Raf/MEK/ERK/RSK pathway. RSK-mediated phosphorylation on Ser273 in the C/EBPβ leucine zipper is necessary to overcome autorepression by the N-terminal region; C/EBPβ activation also involves three Ras-induced modifications within an N-terminal autoinhibitory element. In addition to stimulating DNA binding, a negative charge on phospho-Ser273 regulates dimerization specificity, favoring the formation of C/EBPβ homodimers by increasing electrostatic attractions between paired leucine zipper α-helices. Our results suggest that phosphorylation of Ser273 and the resulting increase in DNA binding and homodimerization are critical for Ras-induced senescence and cell cycle arrest in fibroblastic cells.
MATERIALS AND METHODS
Cells and reagents.
L929 cells (L cells), HEK293T cells, and C/EBPβ−/− MEFs (35) were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum; NIH 3T3 cells were grown in DMEM with 10% calf serum. U0126, LY294002, SB203580, and SP600125 were obtained from Calbiochem; fmk was synthesized as described previously (9); and BI-D1870 was purchased from the University of Dundee. C/EBPβ antibody (C-19) was from Santa Cruz, the C/EBPγ C-terminal antibody has been described previously (26), and RSK2 and RSK3 antisera were kindly provided by M. Ernst and N. Rice. Phospho-RSK (S380) antibody was from Cell Signaling Technology.
Plasmid and retroviral constructs.
The 2× C/EBP-luc reporter was a gift from P. Rorth (Carnegie Institution of Washington) and contains two repeats of the consensus C/EBP binding site, TGCAGATTGCGCAATCTGCA. This plasmid carries the minimal thymidine kinase promoter (22) and a BamHI site for proximal insertion of transcription factor-binding sites. pcDNA3.1 expression constructs for rat C/EBPβ, S64A, and T189A have been described previously (37). The C/EBPβ constructs are designed to express the p34 (LAP) isoform, although some LIP is occasionally produced (12). C/EBPβ phosphorylation site mutants were generated by site-directed mutagenesis using the QuikChange mutagenesis kit (Stratagene). Wild-type (WT) and mutant C/EBPβ genes were transferred from pcDNA3.1 to pBABE-puro, and the resulting retroviral vectors were used to infect C/EBPβ−/− MEFs (35) or NIH 3T3 cells. Other expression plasmids were obtained from the following sources: RSK3, T. Sturgill; H-RasV12, C. Der; constitutively active Raf1 (Raf BXB), M. White; constitutively active (CA) MEK1, D. Kalvakolanu; CA-RSK2 (Y707A), J. Blenis; dominant-negative MEK1 (dn-MEK1), D. Kalvakolanu; dn-ERK1 and dn-ERK2, L. Sealy; and dn-RSK1, J. Blenis.
Transfection and preparation of cell lysates.
L cells (8 × 104) were cultured in 6-well plates for 24 h and transfected with 1 μg pcDNA3.1-C/EBPβ with or without 0.3 μg H-RasV12 vector using 2 μl Fugene (Roche) per μg of DNA. 293T cells were transfected with 500 ng C/EBPβ vector and 100 ng RasV12 in 60-mm dishes. Where indicated, 1 μg expression plasmid for constitutively active or dominant-negative kinase was included. After transfection, the cells were cultured in complete medium for 24 h and serum starved overnight prior to harvest. Chemical inhibitors were included upon serum withdrawal and were maintained in the medium overnight. Whole-cell lysates (RIPA) or nuclear extracts were prepared for electrophoretic mobility shift assay (EMSA). For transactivation assays, L cells were transfected with 100 ng 2× C/EBP-luc reporter plasmid and 5 ng WT or mutant C/EBPβ vector with 10 ng RasV12. After overnight starvation, the cells were lysed in 1× passive lysis buffer (Promega).
EMSA.
The EMSA probe was a double-stranded oligonucleotide containing a consensus C/EBP binding site (underlined) (5′-GATCCATATCCCTGATTGCGCAATAGGCTCAAAA-3′) and labeled with [γ-32P]ATP and T4 polynucleotide kinase. The probe was incubated with whole-cell lysate (5 μl RIPA extract) or nuclear extract in 25 μl 20 mM HEPES, pH 7.5, 200 mM NaCl, 5% Ficoll, 5 mM dithiothreitol (DTT), 5 mM EDTA, 1 μg poly(dI-dC), 1 μg bovine serum albumin (BSA) at room temperature for 30 min. RIPA buffer (5 μl) was added to the nuclear extracts to maintain consistent binding conditions. For supershift assays, extracts were preincubated with 200 ng C/EBPβ or C/EBPγ antibody for 30 min on ice before binding reactions were set up. DNA-protein complexes were separated on 6% polyacrylamide-1× Tris-borate-EDTA (TBE) gels. For mixing experiments, individual proteins (WT C/EBPβ, S273A, S273D, and C/EBPγ) were overexpressed in L cells with or without RasV12, and RIPA lysates were prepared. C/EBPβ extracts were mixed with 5 μg C/EBPγ extract, incubated for 15 min at room temperature, and analyzed by EMSA. Dimer ratios were determined by quantitating the appropriate bands using a PhosphorImager and ImageQuant software (Molecular Dynamics).
Recombinant proteins.
His-tagged C/EBPβ proteins were expressed in Escherichia coli using the pET20b+ vector. After IPTG (isopropyl-β-d-thiogalactopyranoside) induction, cells were collected and resuspended in lysis buffer (20 mM Tris-HCl, pH 6.8, 150 mM NaCl, 1 mM EDTA, 1 mM benzamidine, 0.2 mM phenylmethylsulfonyl fluoride [PMSF], 1 μg/ml antipain, 0.1% β-mercaptoethanol, 0.5% NP-40) containing 50 μg/ml lysozyme. After mild sonication and centrifugation, the pellet containing insoluble C/EBPβ was resuspended in solubilization buffer (5 M urea, 50 mM Tris-HCl, pH 8.0, 0.1% β-mercaptoethanol, 1 mM benzamidine, 0.2 mM PMSF, 1 μg/ml antipain) and rotated overnight. The solubilized protein was clarified and bound to an Ni-nitrilotriacetic acid (NTA) column (Promega). The column was washed serially with solubilization buffer and washing buffer (5 M urea, 0.1 M KPO4, pH 6.5, 0.1% β-mercaptoethanol, 1 mM imidazole). Protein was eluted with 0.1 M KPO4, pH 6.5, 0.5 M NaCl, 0.1% β-mercaptoethanol, 500 mM imidazole and dialyzed against 10 mM Tris (pH 6.8), 100 mM NaCl, 0.1% β-mercaptoethanol, 5% glycerol for 1 h at room temperature.
RSK immunokinase assays.
L cells were transfected with 5 μg RSK2 vector for 24 h, serum starved for 8 h, stimulated with 15% serum and 1 μg/ml fibroblast growth factor (FGF) for 20 min, and lysed in 50 mM Tris, pH 7.5, 100 mM NaCl, 1% Triton X-100, 5 mM EDTA, 40 mM β-glycerophosphate, 40 mM p-nitrophenylphosphate, 1 mM sodium vanadate, 10 mM NaF supplemented with protease inhibitors. After centrifugation, the supernatant was mixed with 10 μl antiserum against RSK2, RSK3, or normal rabbit serum and incubated overnight at 4°C. Protein A beads were added and mixed for 1 h. The beads were washed twice each with lysis buffer and kinase buffer (40 mM HEPES, pH 7.4, 20 mM p-nitrophenylphosphate, 1 mM EGTA, 1 mM sodium vanadate, 12.5 mM MgCl2) and incubated with recombinant C/EBPβ protein in the presence of 20 μCi [γ-32P]ATP (3,000 Ci/mmol) and 65 μM cold ATP for 25 min at room temperature. SDS sample buffer was added to stop the reaction, and the samples were loaded onto 12% SDS-polyacrylamide gels, transferred to Immobilon, and subjected to autoradiography.
Metabolic 32P labeling.
L cells were transfected with 6 μg C/EBPβ vector, with or without 2 μg pcDNA3-H-RasV12. After 24 h, the cells were washed with phosphate-buffered saline (PBS), serum starved for 4 h, and then washed with serum-free/phosphate-free medium and incubated in that medium for 20 min. The cells were placed in fresh serum- and phosphate-free medium containing 0.4 mCi/ml [32P]orthophosphoric acid (Amersham-Pharmacia) and incubated for 16 h. Lysates were prepared using RIPA buffer and immunoprecipitated with C/EBPβ antibody (Santa Cruz C-19) and protein A beads. Washed immunoprecipitates were eluted, subjected to SDS-PAGE, and transferred to membranes. After autoradiography, labeled C/EBPβ bands were excised for phosphopeptide analysis.
Phosphopeptide analysis.
Membrane slices containing 32P-labeled proteins were minced, washed twice with distilled H2O, and blocked for 1 h in incubation buffer (50 mM Tris-HCl, 10 mM CaCl2, 5 mM EDTA, pH 8.0) containing 1% Triton X-100. Activation solution (1/10 volume; 50 mM DTT, 5 mM EDTA) was added with 5 μl sequencing grade endoprotease ArgC (Roche); 0.5 M urea and 20 mM methylamine were included to increase protein solubility. The reaction was incubated overnight at 37°C, and the supernatant was applied to a Waters 3.9-mm by 300-mm C18 high-performance liquid chromatography (HPLC) column. The column was developed using a gradient of 0 to 30% acetonitrile in 0.05% trifluoroacetic acid over 90 min at a flow rate of 1 ml/min, and the 32P in each fraction was measured using a Beckman scintillation counter. 32P-containing peptides were coupled to Sequalon disks and subjected to solid-phase Edman degradation using a Model 492 Applied Biosystems sequencer. Cycle fractions were spotted onto Whatman number 1 paper, and radioactivity was determined by phosphorimaging.
Identification of posttranslational modifications (PTMs) in CEBPβ by mass spectrometry.
A hemagglutinin (HA) tag was placed at the N terminus of C/EBPβ (LAP), and the protein was expressed with or without H-RasV12 in 293T cells. Whole-cell lysates were prepared in RIPA buffer and immunoprecipitated using HA antibody-coupled agarose beads (AffinityMatrix monoHA.11; Covance). The beads were washed 6 times with RIPA buffer, and the bound proteins were eluted in sample buffer, separated by SDS-PAGE, and stained with Coomassie blue. The CEBPβ band was excised, destained, and digested with trypsin (Promega). The tryptic peptides were analyzed by nanoflow reversed-phase liquid chromatography (nanoRPLC) mass spectrometry (MS) using a 7 T hybrid linear ion trap-Fourier transform ion cyclotron resonance (FTICR) mass spectrometer operated in a data-dependent mode (LTQ-FT; ThermoElectron). The MS scans were performed in the FTICR portion, and the multistage tandem-MS (MS2 and MS3) scans were acquired in the LTQ portion. The acquisition of neutral-loss MS3 spectra was triggered when a neutral loss of phosphoric acid (H3PO4) was detected among fragment ions in the MS2 scans. Tandem mass spectra were searched against the rat CEBPβ protein sequence using SEQUEST software (ThermoElectron). Phosphorylation of serine, threonine, and tyrosine (+79.9663 Da), methylation of lysine and arginine residues (+14.01565 Da), and oxidation of methionine (+15.9949 Da) were included as dynamic modifications when searching the MS2 spectra, whereas loss of water (−18.0106 Da) at serine and threonine was added in searching the MS3 spectra. The peptides were identified by filtering the SEQUEST results and further examined by manually inspecting the MS2 and MS3 spectra to confirm the reported modifications.
Serum stimulation time course.
L cells (5 × 105) in 60-mm dishes were transfected with 2.5 μg C/EBPβ plasmid; 24 h later, the cells were switched to serum-free medium overnight. The cells were then stimulated with fresh medium containing 15% fetal bovine serum (FBS) and harvested in RIPA buffer or kinase assay lysis buffer at the appropriate time points. C/EBPβ DNA-binding and RSK autophosphorylation assays were performed as described above.
Cell proliferation and transformation assays.
C/EBPβ−/− MEFs or NIH 3T3 cells were infected with an H-RasV12 retrovirus and subsequently with vectors for WT or mutant rat C/EBPβ. After drug selection, the cells were analyzed for proliferation (colony assays and/or growth curves) or plated in soft agar to assess anchorage-independent growth, as described previously (33, 35).
ChIP.
Chromatin immunoprecipitation (ChIP) assays were used to analyze C/EBPβ binding to cell cycle genes in MEFs or to the interleukin 6 (IL-6) gene in 293T cells. ChIP was performed as described previously (35) using C/EBPβ−/− MEFs infected with pWZL(hygro)-RasV12 and pBabe(puro) vectors expressing WT C/EBPβ or S273A. The 293T cells were transfected with C/EBPβ in the absence or presence of RasV12 and dn-RSK vectors for 24 h and then serum starved overnight. Where appropriate, U0126 (see below) was applied during starvation. The PCR primers spanning the C/EBP site in the human IL-6 gene were as follows: 5′-CCATGATCTTGTTCTAACAACTGCC-3′ and 5′-CTTTGTTGGAGGGTGAGGGTGG-3′.
RESULTS
C/EBPβ DNA binding is stimulated by oncogenic Ras via MEK/ERK/RSK signaling.
To assess whether C/EBPβ DNA-binding activity is regulated by Ras signaling in cells undergoing OIS, we performed EMSA on nuclear extracts from low-passage MEFs infected with H-RasV12-expressing or control retroviruses. A canonical C/EBP site was used as the probe. Oncogenic Ras caused a significant increase in two C/EBP complexes (Fig. 1A, lanes 1 and 2). Antibody supershifts showed that these complexes primarily contained C/EBPβ; the upper band was a C/EBPβ homodimer, while the major, faster-migrating band was composed mostly of C/EBPβ-C/EBPγ heterodimers (lanes 3 and 4), consistent with previous observations (26). Thus, Ras signaling stimulates C/EBPβ DNA binding in senescing primary fibroblasts.
FIG. 1.

(A) C/EBPβ DNA binding in MEFs is activated by H-RasV12. Low-passage cells were infected with control or Ras-expressing retroviruses, and nuclear extracts were prepared and analyzed by EMSA using a C/EBP site probe. Lanes 3 and 4 show supershift assays with C/EBPβ or C/EBPγ antibody (Ab), respectively. (B) L cells were transfected with C/EBPβ with or without RasV12 (lanes 2 and 3). After overnight serum starvation, whole-cell lysates were prepared and assayed by EMSA. Mock-transfected cells are shown in lane 1. The arrow indicates a C/EBPβ antibody supershift complex (SS; lane 4). Western blotting (bottom) shows equal amounts of C/EBPβ protein used in the EMSA experiment. The slow-migrating species induced by RasV12 indicates phosphorylation on Ser64 (43). (C) Effects of C/EBPβ phosphorylation site mutants. L cells were transfected with vectors for the indicated mutants, with or without RasV12, and analyzed as described for panel B.
We also tested activation of exogenous C/EBPβ in transiently transfected L929 mouse fibroblasts (L cells). The p34 (LAP) isoform of rat C/EBPβ (12) was expressed in the absence or presence of H-RasV12 (LAP is the predominant activating C/EBPβ isoform expressed in most cells). The cells were serum starved for 16 h to suppress physiological Ras signaling, and extracts were prepared and normalized for C/EBPβ levels by Western blotting (Fig. 1B). The blot revealed a slower-migrating LAP protein in Ras-expressing cells that corresponded to phosphorylation on Ser64 by CDKs (37). EMSA showed that C/EBPβ DNA binding was weak in the absence of RasV12 (Fig. 1B, lane 2), while Ras stimulated the formation of three EMSA complexes (lane 3) that were supershifted by a C/EBPβ antibody (lane 4). The upper band comigrated with a complex formed by purified C/EBPβ (data not shown), identifying this species as a homodimer (26). These data show that DNA binding of endogenous and overexpressed C/EBPβ is activated by Ras signaling. Low basal activity suggests that the protein is autoinhibited in the absence of an activating signal (18, 47). Accordingly, a truncated protein containing only the C-terminal bZIP domain was almost fully derepressed in the absence of Ras (see Fig. 5D) (Lee et al., submitted).
FIG. 5.

Functional analysis of Ser273 mutants. (A) L cells were transfected with WT C/EBPβ (lanes 1 and 2), S273A (lanes 3 and 4), or S273D (lanes 5 and 6) with or without RasV12 and analyzed by EMSA. (B) Transactivation assays were performed with 2× C/EBP-luc reporter and WT or mutant C/EBPβ with or without RasV12. Luciferase activity was determined and normalized to the total protein concentration. Data are expressed as fold increase over the reporter alone and represent the average (± standard deviation [SD]) of three independent experiments. Transfection with oncogenic Ras alone showed that efficient transactivation of the reporter requires both C/EBPβ and Ras. (C) The same experiment as in panel B was performed with CA-RSK2 instead of Ras. (D) Mutation of Ser273 does not affect DNA binding by the minimal C/EBPβ bZIP domain (DBD). L cells were transfected with or without RasV12 with WT C/EBPβ DBD (lanes 1 and 2), S273A DBD (lanes 3 and 4), or S273D DBD (lanes 5 and 6) and analyzed by EMSA. (E) Cells expressing WT C/EBPβ DBD and RasV12 were treated with U0126 (lanes 3 to 4) and analyzed as in panel D.
C/EBPβ contains several Ser/Thr residues that are phosphorylated by Ras-activated kinases and thus could mediate C/EBPβ derepression by RasV12. These sites include Ser64 (Cdk1/2) (37), Ser105 (p90RSK) (4), and Thr189 (ERK1/2 and Cdk1/2) (24, 37). Mutants carrying Ala substitutions at these residues, as well as the protein kinase A site (Ser240) (8) and GSK3 sites at Ser181/185 (39), were tested for activation by RasV12. None of the mutations diminished basal or RasV12-induced binding activity (Fig. 1C and data not shown), indicating that these phosphoacceptors do not regulate DNA binding.
Ras activates several downstream signaling cascades, including the Raf/MEK/ERK and phosphatidylinositol 3-kinase (PI3K)/Akt pathways and stress-induced mitogen-activated protein kinases (MAPKs), such as JNK and p38 (7). To identify the RasV12 effector pathway(s) that controls C/EBPβ activation, we tested pharmacological inhibitors of MEK1/2, PI3K, p38MAPK, and JNK (Fig. 2A). Only the MEK1/2 inhibitor U0126 significantly inhibited Ras-induced DNA binding, indicating a critical role for the MEK/ERK pathway. Coexpression of dominant-negative forms of MEK1, ERK1, and ERK2 also blocked C/EBPβ activation (Fig. 2B). Since the C/EBPβ ERK site (Thr189) is dispensable for DNA binding, we investigated the involvement of p90RSK, which is a downstream target of ERK1/2. Coexpression of dn-RSK1 blocked Ras-induced DNA binding (Fig. 2B). In addition, constitutively active forms of Raf1, MEK1, or RSK2 stimulated C/EBPβ binding activity, albeit not as efficiently as RasV12 (Fig. 2C). Expression of CA-Akt did not induce C/EBPβ, nor did it enhance activation by CA-Raf or CA-MEK1 (data not shown). We conclude that signaling through the Raf-MEK-ERK-RSK cascade, but not the PI3K/Akt pathway, is required to derepress C/EBPβ DNA binding.
FIG. 2.

RasV12-induced C/EBPβ DNA binding requires Raf/MEK/ERK/RSK signaling. (A) Cells were cotransfected with C/EBPβ and RasV12 and serum deprived overnight in the absence (lane 2) or presence of kinase inhibitors: 5 μM U0126 (MEK1/2) (UO), 20 μM LY294002 (PI3K) (LY), 10 μM SB203580 (p38 MAPK) (SB), or 10 μM SP600125 (JNK) (SP). Lane 1, C/EBPβ alone. (B) C/EBPβ and RasV12 were overexpressed with dn-MEK1 (lane 3), dn-ERK1 (lane 4), dn-ERK2 (lane 5), dn-RSK1 (lane 6), or no interfering protein (lane 2). (C) A similar experiment was performed using CA kinases: Raf1 BXB (lane 3), CA-MEK1 (lane 4), and CA-RSK2 (lane 5). Lanes 1 and 2, C/EBPβ expressed alone or with RasV12, respectively. (D) ChIP assay of C/EBPβ binding to the chromosomal IL-6 gene. 293T cells were transfected with the indicated combinations of C/EBPβ, RasV12, and dn-RSK; 24 h later, the cells were serum starved overnight and then harvested for ChIP. After ChIP with the indicated antibodies, DNA was amplified using primers spanning the C/EBP site in the human IL-6 promoter (1). Cells were also treated with U0126 or cotransfected with dn-RSK1 to disrupt MEK-ERK-RSK signaling (lanes 4 and 5).
Further evidence for regulation of C/EBPβ DNA binding was obtained by analyzing its association with an endogenous target gene, the IL-6 gene (1). ChIP assays showed that ectopically expressed C/EBPβ bound to the IL-6 gene proximal promoter, and binding was modestly but reproducibly increased by coexpression of activated Ras (Fig. 2D, lanes 2 to 3). Moreover, treatment of the cells with U0126 or coexpression of dn-RSK significantly diminished C/EBPβ binding to the IL-6 gene (lanes 4 to 5). These data demonstrate the critical importance of the MEK-ERK-RSK cascade in Ras-induced binding to a chromosomal target.
Phosphorylation on C/EBPβ Ser273 by p90RSK.
Since C/EBPβ activation involves ERK/RSK signaling, we performed immunokinase assays to examine C/EBPβ phosphorylation by RSK kinases. Immune complexes containing overexpressed RSK2 or endogenous RSK2/RSK3 efficiently phosphorylated recombinant C/EBPβ in vitro (Fig. 3A, lanes 2, 3, and 5). To identify modified residues in 32P-C/EBPβ, the labeled sample was digested with ArgC protease, and peptides were resolved by reversed-phase HPLC. Three radiolabeled peptides were detected (Fig. 3B), which were then subjected to Edman sequencing. Radioactivity from peak 1 was released at cycles 5 and 9 (Fig. 3C), while no assignments were obtained for the other two peaks. Two possible scenarios could account for 32P-labeled residues at positions 5 and 9 of a C/EBPβ ArgC peptide, both of which involve phosphorylation on Ser273 in the leucine zipper (Fig. 3C). In support of this idea, the Ser273 sequence displays similarity to a consensus RSK site (Fig. 3C). Ser277 also may be weakly phosphorylated and shows partial homology to a RSK site.
FIG. 3.

C/EBPβ is phosphorylated on Ser273 by p90RSK. (A) Phosphorylation of C/EBPβ by immunoprecipitated RSK. Endogenous RSK2 or RSK3 (lanes 1 to 3) or overexpressed RSK2 (lanes 4 and 5) was immunoprecipitated from L cells using antisera against RSK2 (lanes 2, 4, and 5) or RSK3 (lane 3) or normal rabbit serum (NRS) (lane 1). The immunoprecipitates were incubated with recombinant C/EBPβ and [γ-32P]ATP, and the reaction products were analyzed by SDS-PAGE. Lane 4 is a mock reaction without substrate. The upper band is autophosphorylated RSK. (B) 32P-labeled C/EBPβ was digested with ArgC protease, and the peptides were separated by reversed-phase HPLC. (C) Edman sequencing. Peaks 1 to 3 were collected and subjected to 30 cycles of Edman sequencing, and the radiolabel released at each cycle was determined. Assignments were obtained only from peak 1. Below are shown two possible scenarios for assignments at positions 5 and 9. The asterisks denote labeled residues, and the arrowheads indicate possible cleavage sites for ArgC protease. In the second scheme, Ser273 alone is phosphorylated but the HPLC peak is a mixture of two peptides with different N termini. The solid and open boxes depict potential RSK recognition sites at Ser273 and Ser277, each containing one mismatch to the consensus sequence. (D) Recombinant S273A protein was analyzed as described for panels A and B. (E) L cells transfected with WT C/EBPβ or S273A, with or without RasV12, were labeled with [32P]orthophosphate, and the immunoprecipitated C/EBPβ proteins were subjected to ArgC phosphopeptide mapping. Two independent experiments are shown.
To confirm that Ser273 is a RSK site, we mutated this residue to alanine (S273A) and subjected the protein to in vitro phosphorylation and phosphopeptide analysis. Peak 1 was absent in S273A, and peak 2 was greatly diminished (Fig. 3D), indicating that Ser273 is a RSK phosphoacceptor in vitro. Peak 2 is most likely a partial proteolytic product that spans Ser273, consistent with our observation that the leucine zipper resists proteolytic digestion. We also performed metabolic 32P labeling studies to assess Ser273 phosphorylation in transfected cells. ArgC phosphopeptide analysis of WT C/EBPβ expressed without Ras revealed a major radioactive peak centered at fraction 28 (Fig. 3E). Ras increased this peak and induced another peak at fraction 34, which contained exclusively phosphoserine (data not shown). Although we were unable to obtain an Edman-sequencing assignment for phosphoserine in the fraction 34 peptide, this peak was strongly reduced in the S273A mutant. Decreased radioactivity in the mutant peptide was observed in two independent experiments using L cells (Fig. 3E) and in 293T cells (data not shown). In addition, this phosphopeptide fractionates identically to peak 2 from C/EBPβ phosphorylated by RSK in vitro (Fig. 3B). Preliminary tandem mass spectrometry data also supported modification on Ser273 (data not shown). Thus, several lines of evidence demonstrated that Ser273 is inducibly phosphorylated in Ras-expressing cells.
To further examine the role of RSK, we tested two chemical inhibitors: fmk, which specifically inhibits the C-terminal autoregulatory kinase domains of RSK1, -2, and -4 (9), and BI-D1870, which inhibits the N-terminal catalytic kinase domains of all four RSK isoforms (31). Treatment with 5 μM fmk suppressed Ras-induced DNA binding in L cells and 293T cells (Fig. 4A). Analysis of phosphorylation on RSK Ser380 (11) confirmed that RSK was activated in Ras-expressing cells and that this was blocked by fmk (Fig. 4A, lower two gels). BI-D1870 (10 μM) also diminished Ras-activated DNA binding, although its inhibitory effect was less pronounced (Fig. 4B). Treatment of MEFs with fmk or U0126 also blocked the Ras-induced increase in C/EBPβ DNA binding (Fig. 4C), showing that oncogenic Ras activates endogenous C/EBPβ through a MEK/RSK-dependent pathway.
FIG. 4.

RSK inhibitors suppress C/EBPβ DNA binding. (A) L cells and 293T cells were transfected as indicated and treated with 5 μM fmk overnight in the absence of serum. Nuclear extracts from the L cells were analyzed for C/EBPβ expression and DNA-binding activity (left). Whole-cell lysates prepared from transfected 293T cells were analyzed similarly (right). RSK activation was monitored using an antibody against phospho-Ser380 (p-Ser380), and total RSK levels were analyzed using a mixture of RSK2 and RSK3 antibodies (lower two gels). (B) The experiment described for panel A was performed using 10 μM BI-D1870 to inhibit RSK. (C) Activation of endogenous C/EBPβ by oncogenic Ras requires MEK-ERK-RSK signaling. MEFs infected with RasV12 or control retroviruses were serum starved overnight and analyzed by EMSA. In lanes 3 and 4, RasV12-expressing cells were treated with fmk or U0126, respectively, for 16 h prior to harvest.
Mutating Ser273 to Ala disrupts DNA binding induced by Ras or growth factors.
We next examined the effects of Ser273 mutations on DNA binding. The S273A mutation strongly decreased C/EBPβ activation by RasV12 (Fig. 5A, lanes 3 and 4), whereas a phosphomimetic mutant (S273D) displayed increased basal activity compared to the WT, and this binding was further stimulated by Ras (lanes 5 and 6). These results support the notion that Ser273 phosphorylation promotes DNA binding. The fact that S273D remains responsive to RasV12 implies that additional Ras-induced modifications are involved in C/EBPβ derepression (see below). fmk did not noticeably diminish activation of S273D by RasV12 (data not shown), indicating that Ser273 is the principal target of RSK for activation of DNA binding. The Ser273 mutations also affected C/EBPβ activity in transcriptional assays (Fig. 5B). RasV12 and C/EBPβ cooperatively activated transcription of a C/EBP reporter gene, inducing its expression by nearly 100-fold, as reported previously (24, 35, 50). The S273A mutation diminished Ras-induced transcription by ∼60%, while the activity of S273D was increased by ∼2-fold. CA-RSK2 also enhanced C/EBPβ transactivation, albeit much less than Ras, and the S273A mutation reduced this by more than 50% (Fig. 5C). The activity of S273D was not stimulated by CA-RSK2 significantly more than WT C/EBPβ (data not shown), again consistent with Ser273 being a major target of p90RSK. Thus, the effects of Ser273 substitutions on transcriptional activation are qualitatively similar to those observed in DNA-binding assays.
To determine whether Ser273 affects intrinsic DNA binding by the bZIP domain, we introduced Ser273 mutations into a truncated version of C/EBPβ containing only the DNA-binding domain (DBD). The DBD displayed constitutively high DNA-binding activity that was largely unaffected by Ras or the S273A and -D substitutions (Fig. 5D). The altered mobility of the S273A complex may signify differences in charge or protein conformation. DBD binding was also not blocked by U0126 (Fig. 5E). These data show that DNA binding by the isolated bZIP domain occurs independently of Ras signaling and indicate that Ser273 phosphorylation is specifically required to overcome autoinhibition by the N-terminal region.
We next asked whether C/EBPβ is regulated by physiological Ras signaling elicited by serum growth factors. C/EBPβ-transfected L cells were starved, stimulated with serum, and analyzed for binding activity over a time course. DNA binding was transiently induced, peaking at 4 h and declining at 6 h (Fig. 6A). However, the S273A mutant was resistant to serum stimulation with no induction observed at any time point, demonstrating that C/EBPβ activation by growth factors is highly dependent on Ser273. Analysis of RSK activation (autophosphorylation) showed that RSK2 activity is low in starved cells and increases after serum treatment, exhibiting kinetics similar to those of C/EBPβ induction (Fig. 6B). Furthermore, coexpression of dn-RSK blocked C/EBPβ activation by serum (Fig. 6C). The coincident activation of RSK2 and C/EBPβ, together with the inhibitory effect of dn-RSK, indicates that p90RSK is required for transient induction of C/EBPβ DNA binding by growth factor signaling.
FIG. 6.
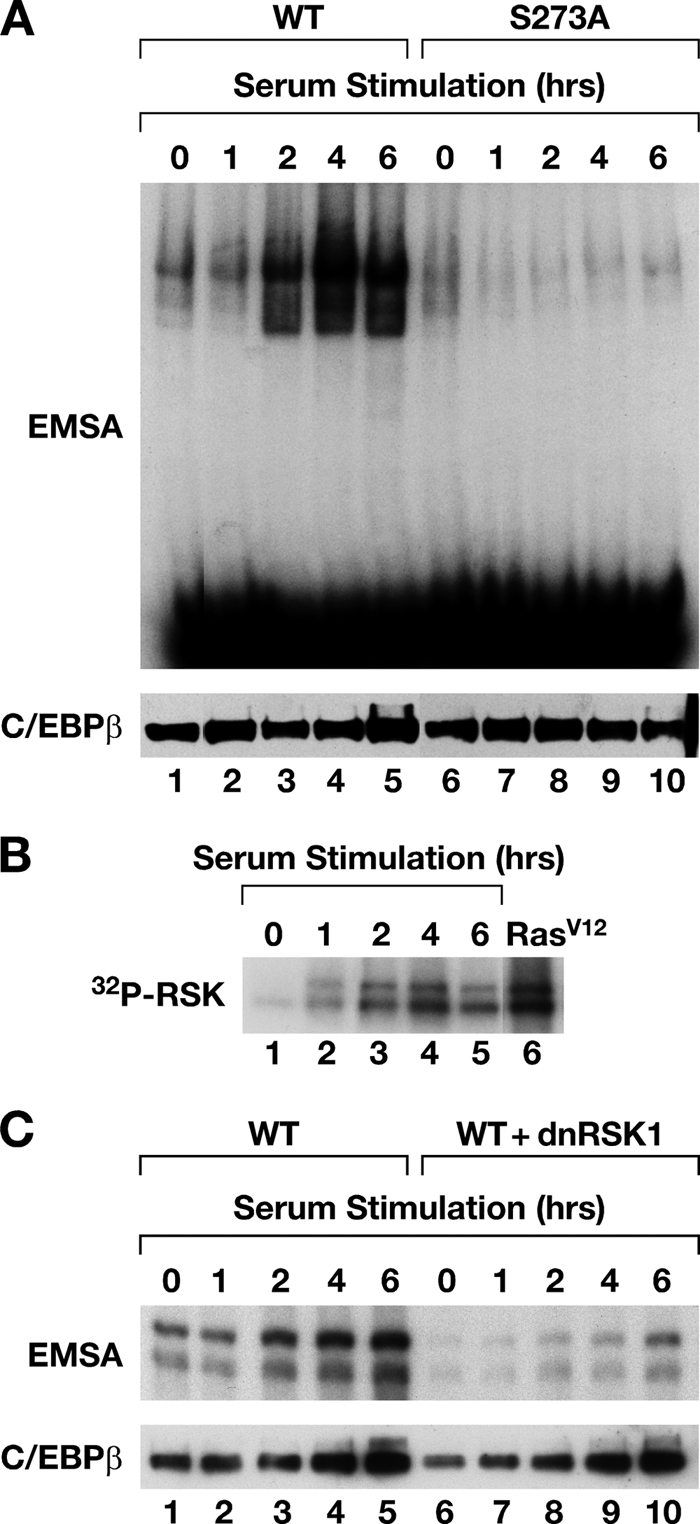
C/EBPβ activation by serum growth factors. (A) L cells were transfected with WT C/EBPβ (lanes 1 to 5) or S273A (lanes 6 to 10), serum starved overnight, and restimulated with 15% FBS for the indicated times. Equivalent C/EBPβ expression at each time point was verified by immunoblotting (bottom). (B) Kinetics of RSK2 activation in serum-stimulated cells. The extracts described in panel A were immunoprecipitated with RSK2 antibody and assayed for autophosphorylation by incubation with [γ-32P]ATP. RSK2 activity in RasV12-transfected cells is shown in lane 6. (C) L cells were transfected with C/EBPβ vector with or without dn-RSK1. The cells were serum starved for 16 h and switched to complete growth medium containing 15% FBS, and extracts were prepared at the indicated time points.
Posttranslational modifications in the CR5/AID autoinhibitory domain also contribute to derepression.
The fact that phosphorylation on Ser273 is necessary but not sufficient for C/EBPβ derepression suggested the involvement of additional Ras-induced PTMs. To identify such candidates, we performed tryptic digestion and multistage tandem mass spectrometry (MS2 and MS3) of C/EBPβ expressed in 293T cells. Several PTMs were detected (Fig. 7 and data not shown). Three of the most prevalent were phosphorylation on Tyr109 (Fig. 7A) and Ser111 (Fig. 7B) and monomethylation of Arg114 (Fig. 7C). All three PTMs were detected when C/EBPβ was expressed with RasV12 but not in its absence (three independent experiments). These sites are clustered within an autoinhibitory region of C/EBPβ termed AID (Lee et al., submitted) or CR5 (18) that suppresses transactivation and DNA binding, suggesting that they contribute to C/EBPβ derepression.
FIG. 7.

Multistage tandem mass spectrometry (MS2 and MS3) spectra of CEBPβ peptides modified by phosphorylation at Tyr109 (A) or Ser111 (B) and methylation at Arg114 (C). Phosphorylation on Ser111 was identified in an MS2 spectrum (B, bottom) and confirmed by a subsequent MS3 scan (top). Y* and S*, phosphorylated Tyr109 and Ser111; S#, Ser111 with loss of water; R∼, monomethylated Arg114. The modifications were detected only in C/EBPβ expressed with RasV12 and were confirmed by at least three independent experiments.
We converted each site to a nonmodifiable amino acid and assessed DNA binding (Fig. 8A). The Y109F and S111A mutations decreased Ras-induced DNA binding relative to WT C/EBPβ, and a double mutant carrying both substitutions displayed even less binding. Converting Arg114 to Lys also decreased DNA binding. Moreover, each mutation reduced C/EBPβ-mediated transcription to various degrees (Fig. 8B). Conversely, a double acidic (phosphomimetic) substitution, Y109E/S111D, increased basal DNA binding in a manner similar to S273D and also potentiated Ras-induced transcription by more than 2-fold (Fig. 8C and D). Thus, three inducible modifications within the CR5/AID region, together with p-Ser273, contribute to C/EBPβ activation by Ras.
FIG. 8.

Functional analysis of Tyr109, Ser111, and Arg114 mutants. (A) Effects of nonphosphorylatable (Ala) or Arg→Lys substitutions on Ras-induced DNA binding in 293T cells. The diagram shows the positions of modified residues (P, phosphorylation; Me, methylation) within the AID autoinhibitory domain (black shading). WB, Western blot. (B) Transactivation assays of the same mutants using the 2× C/EBP-luc reporter. The error bars indicate standard deviations. (C and D) The T109E, S111D, and S273D phosphomimetic substitution mutants were analyzed for DNA binding and transactivation, respectively. (E) WT C/EBPβ and the indicated mutants were stably expressed in 3T3Ras cells, and the cells were analyzed for colony formation (left) or anchorage-independent growth in soft agar (right). Western blotting (F) confirmed equivalent expression of the C/EBPβ proteins. (F) Nuclear extracts from the cells shown in panel E were analyzed by Western blotting and EMSA. Homo- and heterodimeric C/EBPβ EMSA complexes are indicated.
C/EBPβ suppresses proliferation and the transformed phenotype of RasV12-expressing NIH 3T3 cells (3T3Ras) but does not inhibit growth of control cells (33), demonstrating that Ras signaling is required to unmask the cytostatic activity of C/EBPβ. Therefore, we examined the effects of mutating Y109/S111 or R114 on the antiproliferative and tumor suppressor activity of C/EBPβ in 3T3Ras cells. The cells were infected with C/EBPβ retroviruses and plated at low density to assess clonogenic growth or analyzed for anchorage-independent growth in soft agar (Fig. 8E). C/EBPβ strongly inhibited clonogenic growth and also suppressed colony formation in soft agar. The modification-defective Y109F/S111A and R114K mutants were much less effective in inhibiting colony growth than WT C/EBPβ and only weakly suppressed anchorage-independent growth. EMSA of cell extracts showed efficient DNA binding by WT C/EBPβ (Fig. 8F) and revealed homodimers, as well as faster-migrating heterodimeric complexes. Binding by the Y109F/S111A and R114K mutants was significantly impaired, confirming the important roles of these PTMs in derepressing C/EBPβ DNA binding. Thus, the biological phenotypes of mutating these three sites support the effects observed in biochemical and transcriptional assays.
A negative charge on Ser273 alters C/EBPβ dimerization stability and specificity.
Leucine zipper α-helices consist of several heptad repeats, with residues in each repeat designated a to g and leucines occupying the d positions (Fig. 9A). The a and d residues create a hydrophobic interface between dimer subunits, while e and g are usually occupied by polar amino acids that can make electrostatic interactions between paired helices (Fig. 9B). g residues interact with e amino acids located 5 residues C terminally on the opposite subunit [the g(i)↔e(i′ + 5) rule]. These g↔e′ interactions can be attractive, repulsive, or neutral and thus influence dimerization stability and specificity (19, 44). Ser273 occupies the e position of the third heptad repeat in C/EBPβ, while the corresponding g amino acid (Lys268) is positively charged, suggesting that negative charge on phospho-Ser273 could create an attractive g↔e′interaction with Lys268 and increase the stability of C/EBPβ dimers. A potential phosphoacceptor occupies the equivalent position in other C/EBP family members (Fig. 9A). In addition, C/EBPα, -β, and -δ contain Ser/Thr residues at the e position of the first heptad repeat (Thr259 in rat C/EBPβ), with a corresponding Lys at the g position. These configurations suggest that C/EBPβ and other family members could undergo regulated dimerization through phosphorylation of leucine zipper residues.
FIG. 9.

Ser273 phosphorylation is predicted to affect C/EBPβ dimerization. (A) Sequence alignment of bZIP regions of C/EBP proteins. α-helical positions (a to g) within the leucine zipper heptad repeats are shown. The brackets denote g↔e′ interactions, and conserved e-position Ser/Thr residues are shaded in red. Corresponding g-position basic residues are shown in green. Black shading indicate identical residues. BR, basic region; LZ, leucine zipper. (B) Cross-sectional diagram of the leucine zipper coiled coil showing helical positions in the heptad repeat and g↔e′ interactions. (C) Predicted charge-charge interactions for C/EBPβ and C/EBPγ homodimers and C/EBPβ-C/EBPγ heterodimers in basal (unphosphorylated) or phosphorylated (+Ras) states. +, attractive; −, repulsive.
Since C/EBPγ is the major heterodimeric partner of C/EBPβ in most cells analyzed (26), we calculated the predicted g↔e′ electrostatic interactions for homo- and heterodimers of C/EBPβ and C/EBPγ when C/EBPβ Ser273 is either unmodified or phosphorylated (Fig. 9C). This analysis suggested that Ser273 phosphorylation favors formation of β-β homodimers relative to β-γ heterodimers, since homodimers acquire two phosphorylation-induced g↔e′salt bridges while heterodimers gain only one. The formation of C/EBPγ homodimers should be prevented by net repulsive interactions within its LZ interface (Fig. 9C), a prediction that has been confirmed experimentally (16, 25).
We used EMSA to assess the role of Ser273 in C/EBPβ dimerization specificity. C/EBPβ proteins were expressed with or without RasV12, mixed with C/EBPγ extract, and assayed by EMSA (Fig. 10A). A mixing protocol was used to maintain uniform C/EBPγ levels, since C/EBPγ expression differed in the absence and presence of RasV12 (data not shown). C/EBPγ was coexpressed with RasV12, although the results were the same without Ras. The EMSA complexes were quantified to evaluate the levels of each dimeric binding species. Coexpression of RasV12 increased binding of both β-β and β-γ dimers, although enhancement of homodimerization was greater than the increase seen for heterodimers (Fig. 10, compare lanes 8 and 9). The S273A mutation greatly reduced Ras-induced homodimer formation, while β-γ heterodimers were decreased by a lesser amount (Fig. 10, lanes 10 and 11). In contrast, S273D increased the ratio of Ras-activated homodimers to heterodimers by 2-fold relative to WT C/EBPβ. These results indicate that a negative charge on phospho-Ser273 preferentially increases homodimer formation while having a more modest effect on heterodimerization with C/EBPγ, consistent with predictions.
FIG. 10.

Ser273 controls C/EBPβ dimerization. (A) DNA binding of C/EBPβ and C/EBPγ proteins expressed in L cells with or without RasV12 (lanes 1 to 7). In lanes 8 to 13, C/EBPβ extracts were combined with the C/EBPγ extract and incubated at room temperature to allow reassortment of subunits prior to EMSA; lanes 14 and 15 show supershifts with C/EBPβ and C/EBPγ antibodies. EMSA complexes were quantified by phosphorimaging, and the homodimer/heterodimer (β-β/β-γ) ratios were calculated. ND, not determined because the β-β level was undetectable. (B) Ser273 mutations preferentially affect homodimerization relative to β-γ heterodimer formation. The indicated C/EBPβ proteins were coexpressed with RasV12, and increasing amounts of C/EBPγ in 293T cells and nuclear extracts were analyzed by EMSA (top); C/EBPβ and C/EBPγ Western blots are shown below. (C) Effects of Ser273 mutations on transcriptional inhibition by C/EBPγ. L cells were transfected with 2× C/EBP-luc, C/EBPβ vector, and increasing amounts of C/EBPγ plasmid. RasV12 was coexpressed (left) or cells were stimulated with serum (right) to activate C/EBPβ. Normalized luciferase activity in the absence of C/EBPγ (control) was set to 100% for each C/EBPβ protein, and the other values are expressed as percentages of the control. The data are the averages (±SD) of three experiments. (D) Effect of mutating Lys268 to an acidic residue (Glu). The K268E mutant was analyzed for dimerization with C/EBPγ as described for panel B. (E) The K268E mutant shows increased susceptibility to C/EBPγ inhibition in RasV12 transactivation assays. The experiment was performed as described for panel C.
The effect of Ser273 was further analyzed by examining C/EBPβ dimerization in 293T cells. WT C/EBPβ and Ser273 mutants were expressed with RasV12 and increasing amounts of C/EBPγ, and dimers were assessed by EMSA (Fig. 10B). More heterodimers were observed for each C/EBPβ protein as C/EBPγ levels were increased. However, S273A formed fewer homodimers than WT C/EBPβ (Fig. 10B, left), whereas the S273D mutant strongly favored homodimer formation (Fig. 10B, right). S273A β-γ heterodimers predominated over β-β homodimers at the highest level of C/EBPγ (Fig. 10B, left, lane 6), while the reverse was true for S273D (Fig. 10B, right, lane 6). WT C/EBPβ formed equivalent amounts of homo- and heterodimers (lane 3).
Since heterodimerization with C/EBPγ can inhibit transactivation by C/EBPβ (10, 26), we asked whether Ser273 mutations affect C/EBPγ-mediated transcriptional repression in reporter assays. C/EBPβ was transfected with increasing amounts of C/EBPγ in RasV12-expressing or serum-treated cells (Fig. 10C). Transcriptional activation by WT C/EBPβ and S273D was unaffected by C/EBPγ up to the maximum dose used. However, C/EBPγ efficiently inhibited S273A-mediated transcription in a dose-dependent manner. Similar results were obtained when serum stimulation was used to activate C/EBPβ. To examine the importance of positively charged Lys268, which can potentially interact with phosphorylated Ser273, we tested an acidic substitution mutant, K268E. K268E displayed decreased DNA-binding activity relative to WT C/EBPβ in Ras-expressing cells (Fig. 10D, compare lanes 1 and 4). K268E also decreased the ratio of β-β homodimers to heterodimers in cells expressing C/EBPγ (compare lanes 3 and 6). Furthermore, like S273A, the transcriptional activity of this mutant was much more susceptible to inhibition by C/EBPγ than was WT C/EBPβ (Fig. 10E). These data support a model in which phosphorylation on Ser273 creates electrostatic interactions with Lys268, enhancing the stability of C/EBPβ homodimers and increasing their resistance to displacement by C/EBPγ.
Phenotypic consequences of mutating Ser273.
To determine whether Ser273 is important for the Ras-induced cytostatic activity of C/EBPβ, we examined the ability of Ser273 mutants to induce growth arrest in RasV12-expressing C/ΕBPβ−/− MEFs. As observed previously (35), overexpression of RasV12 alone in C/EBPβ−/− cells did not cause proliferative arrest or senescence, whereas coexpression of Ras and C/EBPβ severely inhibited growth (Fig. 11A). The antiproliferative activity of S273A was reduced compared to that of C/EBPβ; conversely, S273D inhibited cell growth more efficiently than WT C/EBPβ. C/EBPβ and S273D displayed strong DNA-binding activity and abundant homodimer formation in MEFs, while S273A DNA binding was weak and few homodimers were evident (Fig. 11A, right). A similar experiment in RasV12-expressing NIH 3T3 fibroblasts showed efficient growth arrest by WT C/EBPβ and S273D, while the S273A mutant had impaired antiproliferative activity (Fig. 11B). Again, C/EBPβ cytostatic activity corresponded to strong DNA binding and high levels of homodimers (Fig. 11B, right). Thus, Ser273 phosphorylation is important for C/EBPβ-mediated cell cycle arrest in RasV12-expressing cells and is associated with an increase in homodimers.
FIG. 11.

Ser273 is required for C/EBPβ-induced cell cycle arrest. (A) Effects of Ser273 mutations on C/EBPβ-induced cell cycle arrest in MEFs. (Left) RasV12-expressing C/EBPβ−/− MEFs were infected with the indicated C/EBPβ retroviruses and analyzed in cell proliferation or colony assays. Proliferation data were normalized to day zero and are plotted as the fold increase in cell number (average of triplicate assays). The brackets and asterisks denote significant differences in cell numbers at day 6 (*, P ≤ 0.05). C/EBPβ expression levels and DNA binding are shown on the right; C/EBPβ homodimers are indicated. (B) The same experiment was performed in NIH 3T3 fibroblasts. (C) ChIP assays of C/EBPβ binding to target genes. C/EBPβ−/− MEFs expressing RasV12 were infected with retroviruses encoding WT C/EBPβ or S273A. Chromatin was immunoprecipitated with C/EBPβ or control (IgG) antibodies, and bound DNA was analyzed by PCR using primers spanning E2F binding sites in the indicated cell cycle genes (35). The β2-microglobulin promoter was used as a negative control.
C/EBPβ induces cell cycle arrest in part by associating with the promoters of E2F-regulated cell cycle genes and repressing their transcription (35). To determine if C/EBPβ binding to these genes is affected by the S273A mutation, we performed ChIP assays using Ras-expressing C/EBPβ−/− MEFs infected with C/EBPβ or S273A virus (Fig. 11C). C/EBPβ interacted with the DHFR and PCNA promoters, as previously reported (35). No signals were obtained with a nonspecific antibody, and binding to the β2-microglobulin gene was not detected. S273A binding to the DHFR and PCNA promoters was significantly reduced compared to WT C/EBPβ. Similar data were obtained by analyzing binding to the IL-6 promoter, another C/EBPβ target that was recently implicated in senescence induction (20). These data show that C/EBPβ binding to senescence-associated genes requires Ser273.
DISCUSSION
We identified C/EBPβ Ser273 as a novel RSK target and demonstrated that Ser273 phosphorylation regulates RasV12-induced DNA binding and dimerization. Our findings also indicate that increased DNA binding and homodimerization of C/EBPβ are important for cell cycle arrest in primary fibroblasts expressing oncogenic Ras. Previous work showed that RSK-C/EBPβ signaling is required for proliferation of hepatocytes in response to TGF-α and involves phosphorylation on Ser105 (rat) or Thr217 (mouse) (4). In the present study, we found that Ser105 is dispensable for activation of DNA binding (Fig. 1C), whereas Ser273 is a critical RSK target in this response. It is possible that Ser105 and Ser273 are both modified by RSK but control different C/EBPβ functions.
C/EBPβ DNA binding is inhibited in unstimulated cells by three hydrophobic domains in the N-terminal region: a transactivation domain element and AID/CR5, as well as a short sequence juxtaposed to Ser273 termed the C-terminal regulatory domain (CRD) (Lee et al., submitted). Gel filtration and glutaraldehyde cross-linking experiments indicated that purified recombinant C/EBPβ is predominantly dimeric in vitro (data not shown), suggesting that autoinhibition suppresses DNA binding and not dimerization. We have proposed that the N-terminal inhibitory region makes intramolecular interactions with the CRD, forming a hydrophobic core that binds the basic region and represses DNA binding. Ser273 is critical for Ras-induced derepression of full-length C/EBPβ, but not for constitutive DNA binding by the bZIP domain alone. Therefore, Ser273 phosphorylation may contribute to C/EBPβ activation by relieving interactions between the N-terminal region and the CRD. The mechanism probably involves charge repulsion between phospho-Ser273 and negatively charged N-terminal sequences in the TAD region and/or phospho-Tyr109/Ser111. In support of this model, the S273D phosphomimetic mutation decreased binding of the bZIP domain to the N-terminal region in pull-down assays, and mutating several acidic residues in the TAD disrupted Ras-induced DNA binding (Lee et al., submitted).
Besides Ser273, we identified two phosphomodifications and a methylated arginine within the N-terminal AID/CR5 element that are necessary for efficient derepression by Ras. Mutation of these residues decreased C/EBPβ DNA binding and impaired Ras-dependent growth arrest activity in NIH 3T3 cells. These PTMs presumably disrupt intraprotein interactions involving AID/CR5 and the CRD that inhibit DNA binding. They may also derepress C/EBPβ transactivation function, since CR5 was originally characterized as inhibiting the TAD (18). While Tyr109 and Ser111 phosphorylation could activate C/EBPβ through a charge repulsion mechanism, the role of Arg114 methylation is less clear. Since methylation of Lys and Arg residues often enhances interactions with other proteins (2, 17), these modifications might not directly disrupt autoinhibition. It is possible that Arg114 methylation creates a recognition site for another modifying enzyme and mutating this residue prevents the induction of other PTMs. In summary, C/EBPβ activation involves multiple modification events whose net effect is to interrupt autoinhibitory folding interactions.
Piwien-Pilipuk et al. reported that C/EBPβ DNA binding is induced by growth hormone (GH) through a dephosphorylation mechanism (27, 28). Although the identity of the inhibitory phosphorylated residues was not determined, they suggested that GSK3 may be the relevant kinase. Subsequent work identified serines 177, 181, and 185 in the serine-rich region adjacent to the DNA-binding domain as GSK3 substrates whose phosphorylation inhibits DNA binding (48). In contradiction to this model, Tang et al. found that GSK3-mediated phosphorylation of 2 residues in this region, primed by ERK1/2 phosphorylation on Thr189, activates C/EBPβ DNA binding in differentiating adipocytes (39). We observed that Thr189 and Ser181/Ser185 are dispensable for Ras-induced DNA binding (Fig. 1B and data not shown). Therefore, the C/EBPβ activation mechanism may depend on the cell type or signaling pathway engaged.
In addition to its role in derepressing C/EBPβ DNA binding, phosphorylation on Ser273 creates two additional attractive g↔e′ interactions between paired leucine zippers that promote homodimerization. This is the first evidence that a physiological phosphorylation event regulates bZIP dimerization. Previous work demonstrated that phosphorylation can induce dimerization of the vitellogenin-binding protein bZIP domain (38). In this study, an artificial PKA site (Ser or Thr) was introduced into an e position of the VBP leucine zipper, with Arg occupying the corresponding g position. PKA-induced phosphorylation on Ser stabilized homodimer formation via interhelical g↔e′ interactions, as well as intrahelical salt bridges (although phosphorylation of Thr had a destabilizing effect). These results support our observation that Ser273 phosphorylation can enhance C/EBPβ homodimerization.
While phospho-Ser273 shifts the equilibrium toward homodimers, the distribution of C/EBPβ dimers in vivo may be influenced by additional factors, such as the relative levels of C/EBPβ and C/EBPγ or other C/EBPβ partners. Furthermore, C/EBPγ contains a potential e site phosphoacceptor equivalent to Ser273 that could also be targeted by kinases and thereby facilitate heterodimer formation (Fig. 7A). However, since RasV12 signaling did not affect the dimerization properties of C/EBPγ in our experiments, phosphorylation of this residue (if it occurs) probably involves a different signaling pathway.
We propose that C/EBPβ dimerization has important consequences for cellular proliferation and transformation. Several observations indicate that C/EBPβ homodimers are growth inhibitory while C/EBPβ-C/EBPγ heterodimers stimulate, or are permissive for, cell proliferation. For example, C/EBPβ-null MEFs are hyperproliferative and fail to undergo Ras-induced cell cycle arrest and senescence (35), while C/EBPβ overexpression arrests proliferation of MEFs, hepatoma cells, and keratinocytes (5, 49). In contrast, C/EBPγ−/− MEFs grow very poorly in culture whereas overexpression of C/EBPγ stimulates growth (R. Malik and P. F. Johnson, unpublished data). These results suggest a model in which C/EBPβ homodimers and β-γ heterodimers exert anti- and proproliferative effects, respectively. In this view, C/EBPγ can neutralize the growth-inhibitory activity of C/EBPβ through its ability to form heterodimers. Loss of C/EBPγ or overexpression of C/EBPβ would allow C/EBPβ homodimers to accumulate to levels that inhibit cell cycle progression. Several studies have demonstrated that C/EBPβ has a proliferative/oncogenic role in some cell types (4, 6, 15, 30, 32, 41), and it is possible that C/EBPβ stimulates proliferation of these cells through the action of β-γ heterodimers.
Constitutive high-level signaling by oncogenic Ras is predicted to cause sustained phosphorylation on Ser273, inducing C/EBPβ homodimerization and proliferative arrest. Accordingly, WT C/EBPβ and S273D induced cell cycle arrest in Ras-expressing cells while S273A showed diminished antiproliferative activity (Fig. 11). In WT MEFs, endogenous levels of C/EBPβ are sufficient to mediate Ras-induced growth arrest (35). In NIH 3T3 cells, small amounts of exogenous C/EBPβ stimulate Ras transformation (50) while higher levels suppress proliferation and inhibit transformation, perhaps because a threshold set by C/EBPγ and/or other dimeric partners is exceeded and C/EBPβ homodimers accumulate. H-RasV12 decreased endogenous C/EBPβ levels in NIH 3T3 cells (33) but increased its expression in primary MEFs (50). Thus, the opposing proliferative responses to oncogenic Ras in primary MEFs (senescence) and immortalized NIH 3T3 fibroblasts (transformation) correlate with C/EBPβ levels, which in turn may affect the ratio of homo- to heterodimers formed by activated C/EBPβ.
Lastly, our findings raise the question of whether phosphorylation-mediated dimerization occurs in other bZIP proteins. Inspection of leucine zippers from human bZIP proteins (35) indicates that such control of dimerization is relatively rare. As mentioned above, all members of the C/EBP family could theoretically undergo regulated dimerization due to potential phosphoacceptors in the first and third heptads that could make attractive g↔e′ interactions (Fig. 9A). Members of the Oasis and ATF2 families and JDP1 and BATF of the Fos family contain Ser/Thr residues at e positions, while Jun family members contain Thr (g)↔Arg (e′) pairs in the third heptad. Although other human bZIP proteins do not contain similar configurations of phosphoacceptors, the possibility that dimerization of C/EBP, ATF, Oasis, Fos, and Jun proteins is modulated by phosphorylation in their leucine zippers warrants further investigation.
Acknowledgments
We thank the individuals mentioned in the text for generously providing plasmids and antibodies, Suzanne Specht for assistance with phosphopeptide mapping, Barb Shankle and Nancy Martin for genotyping and preparation of MEFs, Angie Hackley for animal handling, and Jiro Wada and Allan Kane for preparing figures.
We dedicate this paper to the memory of Barb Shankle.
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and contract N01-CO-12400.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Footnotes
Published ahead of print on 29 March 2010.
REFERENCES
- 1.Akira, S., H. Isshiki, T. Sugita, O. Tanabe, S. Kinoshita, Y. Nishio, T. Nakajima, T. Hirano, and T. Kishimoto. 1990. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 9:1897-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bedford, M. T., and S. G. Clarke. 2009. Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buck, M., V. Poli, T. Hunter, and M. Chojkier. 2001. C/EBPbeta phosphorylation by RSK creates a functional XEXD caspase inhibitory box critical for cell survival. Mol. Cell 8:807-816. [DOI] [PubMed] [Google Scholar]
- 4.Buck, M., V. Poli, P. van der Geer, M. Chojkier, and T. Hunter. 1999. Phosphorylation of rat serine 105 or mouse threonine 217 in C/EBP beta is required for hepatocyte proliferation induced by TGF alpha. Mol. Cell 4:1087-1092. [DOI] [PubMed] [Google Scholar]
- 5.Buck, M., H. Turler, and M. Chojkier. 1994. LAP (NF-IL-6), a tissue-specific transcriptional activator, is an inhibitor of hepatoma cell proliferation. EMBO J. 13:851-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bundy, L. M., and L. Sealy. 2003. CCAAT/enhancer binding protein beta (C/EBPbeta)-2 transforms normal mammary epithelial cells and induces epithelial to mesenchymal transition in culture. Oncogene 22:869-883. [DOI] [PubMed] [Google Scholar]
- 7.Campbell, S. L., R. Khosravi-Far, K. L. Rossman, G. J. Clark, and C. J. Der. 1998. Increasing complexity of Ras signaling. Oncogene 17:1395-1413. [DOI] [PubMed] [Google Scholar]
- 8.Chinery, R., J. A. Brockman, D. T. Dransfield, and R. J. Coffey. 1997. Antioxidant-induced nuclear translocation of CCAAT/enhancer-binding protein beta. A critical role for protein kinase A-mediated phosphorylation of Ser299. J. Biol. Chem. 272:30356-30361. (Erratum, J. Biol. Chem. 273:15308, 1998.) [DOI] [PubMed] [Google Scholar]
- 9.Cohen, M. S., C. Zhang, K. M. Shokat, and J. Taunton. 2005. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 308:1318-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooper, C., A. Henderson, S. Artandi, N. Avitahl, and K. Calame. 1995. Ig/EBP (C/EBP gamma) is a transdominant negative inhibitor of C/EBP family transcriptional activators. Nucleic Acids Res. 23:4371-4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dalby, K. N., N. Morrice, F. B. Caudwell, J. Avruch, and P. Cohen. 1998. Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J. Biol. Chem. 273:1496-1505. [DOI] [PubMed] [Google Scholar]
- 12.Descombes, P., and U. Schibler. 1991. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 67:569-579. [DOI] [PubMed] [Google Scholar]
- 13.Fan, H. Y., Z. Liu, M. Shimada, E. Sterneck, P. F. Johnson, S. M. Hedrick, and J. S. Richards. 2009. MAPK3/1 (ERK1/2) in ovarian granulosa cells are essential for female fertility. Science 324:938-941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomis, R. R., C. Alarcon, C. Nadal, C. Van Poznak, and J. Massague. 2006. C/EBPbeta at the core of the TGFbeta cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell 10:203-214. [DOI] [PubMed] [Google Scholar]
- 15.Greenbaum, L. E., W. Li, D. E. Cressman, Y. Peng, G. Ciliberto, V. Poli, and R. Taub. 1998. CCAAT enhancer-binding protein beta is required for normal hepatocyte proliferation in mice after partial hepatectomy. J. Clin. Invest. 102:996-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hattori, T., N. Ohoka, Y. Inoue, H. Hayashi, and K. Onozaki. 2003. C/EBP family transcription factors are degraded by the proteasome but stabilized by forming dimer. Oncogene 22:1273-1280. [DOI] [PubMed] [Google Scholar]
- 17.Huang, J., and S. L. Berger. 2008. The emerging field of dynamic lysine methylation of non-histone proteins. Curr. Opin. Genet. Dev. 18:152-158. [DOI] [PubMed] [Google Scholar]
- 18.Kowenz-Leutz, E., G. Twamley, S. Ansieau, and A. Leutz. 1994. Novel mechanism of C/EBP β (NF-M) transcriptional control: activation through derepression. Genes Dev. 8:2781-2791. [DOI] [PubMed] [Google Scholar]
- 19.Krylov, D., I. Mikhailenko, and C. Vinson. 1994. A thermodynamic scale for leucine zipper stability and dimerization specificity: e and g interhelical interactions. EMBO J. 13:2849-2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuilman, T., C. Michaloglou, L. C. Vredeveld, S. Douma, R. van Doorn, C. J. Desmet, L. A. Aarden, W. J. Mooi, and D. S. Peeper. 2008. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133:1019-1031. [DOI] [PubMed] [Google Scholar]
- 21.Lee, S. J., and S. G. Kim. 2006. Role of p90 ribosomal S6-kinase-1 in oltipraz-induced specific phosphorylation of CCAAT/enhancer binding protein-beta for GSTA2 gene transactivation. Mol. Pharmacol. 69:385-396. [DOI] [PubMed] [Google Scholar]
- 22.McKnight, S. L., and R. Kingsbury. 1982. Transcriptional control signals of a eukaryotic protein-coding gene. Science 217:316-324. [DOI] [PubMed] [Google Scholar]
- 23.Mo, X., E. Kowenz-Leutz, H. Xu, and A. Leutz. 2004. Ras induces mediator complex exchange on C/EBP beta. Mol. Cell 13:241-250. [DOI] [PubMed] [Google Scholar]
- 24.Nakajima, T., S. Kinoshita, T. Sasagawa, K. Sasaki, M. Naruto, T. Kishimoto, and S. Akira. 1993. Phosphorylation at threonine-235 by a ras-dependent mitogen-activated protein kinase cascade is essential for transcription factor NF-IL6. Proc. Natl. Acad. Sci. U. S. A. 90:2207-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newman, J. R., and A. E. Keating. 2003. Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science 300:2097-2101. [DOI] [PubMed] [Google Scholar]
- 26.Parkin, S. E., M. Baer, T. D. Copeland, R. C. Schwartz, and P. F. Johnson. 2002. Regulation of CCAAT/enhancer-binding protein (C/EBP) activator proteins by heterodimerization with C/EBPgamma (Ig/EBP). J. Biol. Chem. 277:23563-23572. [DOI] [PubMed] [Google Scholar]
- 27.Piwien-Pilipuk, G., O. MacDougald, and J. Schwartz. 2002. Dual regulation of phosphorylation and dephosphorylation of C/EBPbeta modulate its transcriptional activation and DNA binding in response to growth hormone. J. Biol. Chem. 277:44557-44565. [DOI] [PubMed] [Google Scholar]
- 28.Piwien-Pilipuk, G., D. Van Mater, S. E. Ross, O. A. MacDougald, and J. Schwartz. 2001. Growth hormone regulates phosphorylation and function of CCAAT/enhancer-binding protein beta by modulating Akt and glycogen synthase kinase-3. J. Biol. Chem. 276:19664-19671. [DOI] [PubMed] [Google Scholar]
- 29.Ramji, D. P., and P. Foka. 2002. CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem. J. 365:561-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson, G. W., P. F. Johnson, L. Hennighausen, and E. Sterneck. 1998. The C/EBPbeta transcription factor regulates epithelial cell proliferation and differentiation in the mammary gland. Genes Dev. 12:1907-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sapkota, G. P., L. Cummings, F. S. Newell, C. Armstrong, J. Bain, M. Frodin, M. Grauert, M. Hoffmann, G. Schnapp, M. Steegmaier, P. Cohen, and D. R. Alessi. 2007. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem. J. 401:29-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seagroves, T. N., S. Krnacik, B. Raught, J. Gay, B. Burgess-Beusse, G. J. Darlington, and J. M. Rosen. 1998. C/EBPbeta, but not C/EBPalpha, is essential for ductal morphogenesis, lobuloalveolar proliferation, and functional differentiation in the mouse mammary gland. Genes Dev. 12:1917-1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sebastian, T., and P. F. Johnson. 2009. RasV12-mediated down-regulation of CCAAT/enhancer binding protein {beta} in immortalized fibroblasts requires loss of p19Arf and facilitates bypass of oncogene-induced senescence. Cancer Res. 69:2588-2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sebastian, T., and P. F. Johnson. 2006. Stop and go: anti-proliferative and mitogenic functions of the transcription factor C/EBPbeta. Cell Cycle 5:953-957. [DOI] [PubMed] [Google Scholar]
- 35.Sebastian, T., R. Malik, S. Thomas, J. Sage, and P. F. Johnson. 2005. C/EBPbeta cooperates with RB:E2F to implement Ras(V12)-induced cellular senescence. EMBO J. 24:3301-3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Serrano, M., A. W. Lin, M. E. McCurrach, D. Beach, and S. W. Lowe. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88:593-602. [DOI] [PubMed] [Google Scholar]
- 37.Shuman, J. D., T. Sebastian, P. Kaldis, T. D. Copeland, S. Zhu, R. C. Smart, and P. F. Johnson. 2004. Cell cycle-dependent phosphorylation of C/EBPbeta mediates oncogenic cooperativity between C/EBPbeta and H-RasV12. Mol. Cell. Biol. 24:7380-7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szilák, L., J. Moitra, and C. Vinson. 1997. Design of a leucine zipper coiled coil stabilized 1.4 kcal mol-1 by phosphorylation of a serine in the e position. Protein Sci. 6:1273-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang, Q. Q., M. Gronborg, H. Huang, J. W. Kim, T. C. Otto, A. Pandey, and M. D. Lane. 2005. Sequential phosphorylation of CCAAT enhancer-binding protein beta by MAPK and glycogen synthase kinase 3beta is required for adipogenesis. Proc. Natl. Acad. Sci. U. S. A. 102:9766-9771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang, Q. Q., and M. D. Lane. 1999. Activation and centromeric localization of CCAAT/enhancer-binding proteins during the mitotic clonal expansion of adipocyte differentiation. Genes Dev. 13:2231-2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang, Q. Q., T. C. Otto, and M. D. Lane. 2003. CCAAT/enhancer-binding protein beta is required for mitotic clonal expansion during adipogenesis. Proc. Natl. Acad. Sci. U. S. A. 100:850-855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang, Q. Q., T. C. Otto, and M. D. Lane. 2003. Mitotic clonal expansion: a synchronous process required for adipogenesis. Proc. Natl. Acad. Sci. U. S. A. 100:44-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vinson, C., M. Myakishev, A. Acharya, A. A. Mir, J. R. Moll, and M. Bonovich. 2002. Classification of human B-ZIP proteins based on dimerization properties. Mol. Cell. Biol. 22:6321-6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vinson, C. R., T. W. Hai, and S. M. Boyd. 1993. Dimerization specificity of the leucine zipper-containing bZIP motif on DNA binding: prediction and rational design. Genes Dev. 7:1047-1058. [DOI] [PubMed] [Google Scholar]
- 45.Wada, N., M. Matsumura, Y. Ohba, N. Kobayashi, T. Takizawa, and Y. Nakanishi. 1995. Transcription stimulation of the Fas-encoding gene by nuclear factor for interleukin-6 expression upon influenza virus infection. J. Biol. Chem. 270:18007-18012. [DOI] [PubMed] [Google Scholar]
- 46.Wegner, M., Z. Cao, and M. G. Rosenfeld. 1992. Calcium-regulated phosphorylation within the leucine zipper of C/EBP beta. Science 256:370-373. [DOI] [PubMed] [Google Scholar]
- 47.Williams, S. C., M. Baer, A. J. Dillner, and P. F. Johnson. 1995. CRP2 (C/EBPβ) contains a bipartite regulatory domain that controls transcriptional activation, DNA binding and cell specificity. EMBO J. 14:3170-3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao, X., S. Zhuang, Y. Chen, G. R. Boss, and R. B. Pilz. 2005. Cyclic GMP-dependent protein kinase regulates CCAAT enhancer-binding protein beta functions through inhibition of glycogen synthase kinase-3. J. Biol. Chem. 280:32683-32692. [DOI] [PubMed] [Google Scholar]
- 49.Zhu, S., H. S. Oh, M. Shim, E. Sterneck, P. F. Johnson, and R. C. Smart. 1999. C/EBPbeta modulates the early events of keratinocyte differentiation involving growth arrest and keratin 1 and keratin 10 expression. Mol. Cell. Biol. 19:7181-7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu, S., K. Yoon, E. Sterneck, P. F. Johnson, and R. C. Smart. 2002. CCAAT/enhancer binding protein-beta is a mediator of keratinocyte survival and skin tumorigenesis involving oncogenic Ras signaling. Proc. Natl. Acad. Sci. U. S. A. 99:207-212. [DOI] [PMC free article] [PubMed] [Google Scholar]