

Abstract
In addition to their classical roles as carbon or nitrogen sources, amino acids can be used for bacterial virulence, colonization, or stress resistance. We found that original deamidase-transport systems impact colonization by Helicobacter pylori, a human pathogen associated with gastric pathologies, including adenocarcinoma. We demonstrated that l-asparaginase (Hp-AnsB) and γ-glutamyltranspeptidase (Hp-γGT) are highly active periplasmic deamidases in H. pylori, producing ammonia and aspartate or glutamate from asparagine and glutamine, respectively. Hp-GltS was identified as a sole and specialized transporter for glutamate, while aspartate was exclusively imported by Hp-DcuA. Uptake of Gln and Asn strictly relies on indirect pathways following prior periplasmic deamidation into Glu and Asp. Hence, in H. pylori, the coupled action of periplasmic deamidases with their respective transporters enables the acquisition of Glu and Asp from Gln and Asn, respectively. These systems were active at neutral rather than acidic pH, suggesting their function near the host epithelial cells. We showed that Hp-DcuA, the fourth component of these novel deamidase-transport systems, was as crucial as Hp-γGT, Hp-AnsB, and Hp-GltS for animal model colonization. In conclusion, the pH-regulated coupled amino acid deamidase-uptake system represents an original optimized system that is essential for in vivo colonization of the stomach environment by H. pylori. We propose a model in which these two nonredundant systems participate in H. pylori virulence by depleting gastric or immune cells from protective amino acids such as Gln and producing toxic ammonia close to the host cells.
In microorganisms, amino acids are generally taken up for protein synthesis or as a source of energy and/or nitrogen. In addition, amino acids can be used for other functions important for bacterial virulence, colonization, or stress resistance. Some examples are amino acid decarboxylases required for acid resistance (18), proline and glycine-betaine uptake required for osmoprotection (59), and arginase required for depletion of arginine from the macrophages to limit the nitric oxide-dependent immune response (20). In the present study, we found two distinct deamidase-transport systems, linking amino acid metabolism and the ability to colonize in Helicobacter pylori. H. pylori is a bacterial pathogen that colonizes the stomach of half of the human population in the world (4). Persistent colonization by H. pylori is associated with the development of gastric pathologies, including peptic ulcer disease and adenocarcinoma (4). H. pylori preferentially uses amino acids as a sole energy source and consumes large amounts of aspartate (Asp), glutamate (Glu), and their respective amides (Asn and Gln) (33, 52). In H. pylori, strong asparaginase and glutaminase activities have been reported that produce ammonia (NH3) from hydrolysis of Asn and Gln, respectively (52). The role of these activities in the nutrition or survival of H. pylori in the human host remained elusive. The major NH3 producer of H. pylori is a potent urease that hydrolyzes urea whose availability is regulated by UreI, a dedicated acid-gated urea channel (50, 57). Production of NH3, a buffering compound, by the cytoplasmic urease is essential for successful colonization of H. pylori, since it allows this bacterium to resist gastric acidity. It is generally admitted that a pH gradient exists in the stomach ranging from median pH 2 in the lumen to pH 4.5 in the gastric mucus layer and to a pH close to neutrality near to the epithelial cells where about 20% of total bacteria adhere (1, 46). The absolute pH value at the epithelial cell surface has been questioned by a couple of groups (5, 14) who, using pH-sensitive dyes, have found it to be close to pH 4. However, this controversial result raises a major concern for how epithelial cells may resist at such a low pH.
Recent work showed that the extracellular Gln hydrolysis activity of H. pylori is provided by the secreted and apoptosis-inducing γ-glutamyltranspeptidase (Hp-γGT; EC 2.3.2.2), HP1118 in strain 26695 (6, 48). Hp-γGT is essential (13) or at least provides some advantages to H. pylori for mouse colonization (32). Purified Hp-γGT disturbed the proliferation of gastric cells by upregulating growth factors (10), inducing mitochondria-mediated apoptosis (29), and inhibiting T-cell proliferation (45). In H. pylori, both Glu and Gln transport were shown to depend on sodium ions, and the latter additionally on Hp-γGT activity (48, 49).
The purified recombinant product of hp0723 (that we designated Hp-AnsB) was shown to hydrolyze in vitro Asn, as well as Gln to a lower extent, and to exert cytotoxic activities on cell lines (11). In addition, a Δhp0723 mutant of H. pylori was shown to be defective for in vivo competition in the gerbil model (34). Although consumption of Asp, Asn, Glu, and Gln in H. pylori has been previously reported, H. pylori transporters for these amino acids remained unidentified. Moreover, little was known about urease-independent NH3 production in H. pylori, the mechanisms associated to glutaminase and asparaginase activities, their localization, regulation, and function in vivo.
In the present study, we have identified two nonredundant systems involved in amino acid uptake coupled with NH3 production. Two sole genes responsible for Asp and Glu uptake activities in H. pylori were identified. We demonstrated that the Asp transporter is essential to colonize the gastric mucosa of mice infected with H. pylori. Asparaginase activity was periplasmic and hydrolyzed Asn in vivo but not Gln. We demonstrated that the coupled deamidase-uptake activities are the exclusive systems responsible for direct (Asp and Glu) and indirect (Asn and Gln) amino acids assimilation by H. pylori. Moreover, these activities are functional at pH values corresponding to those found close to the epithelial cells of the host.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Plasmids employed to transform H. pylori were constructed using Escherichia coli strain MC1061 (12) (Table 1) grown at 37°C on solid or in liquid Luria-Bertani medium (35). Spectinomycin was used at 100 μg·ml−1 for the selection of E. coli transformants. The H. pylori strains used in this study were strains 26695 (56) and SS1 (30). H. pylori strains were grown on blood agar base 2 (Oxoid) plates supplemented with 10% defibrinated horse blood and with an antibiotics-fungicide mix consisting of vancomycin (final concentration, 12.5 μg·ml−1), polymyxin B (0.31 μg·ml−1), amphotericin B (2.5 μg·ml−1), and trimethoprim (6.25 μg·ml−1). H. pylori was incubated at 37°C under microaerobic conditions (6% O2, 10% CO2, and 84% N2). For selection of H. pylori transformants, kanamycin and chloramphenicol were added to the growth medium at concentrations of 20 μg·ml−1 and 4 μg·ml−1, respectively. The different H. pylori mutants were obtained by natural transformation, as described previously (13), with 2 μg of plasmid DNA. Liquid cultures were grown in brain heart infusion (BHI) (Oxoid) supplemented with 4% decomplemented fetal calf serum (Eurobio) or with 0.2% β-cyclodextrin (used for quantitative real-time reverse transcriptase PCR [qRT-PCR]; Sigma) and the antibiotics-fungicide mix.
TABLE 1.
Bacterial strains used in this study
| Strain | Relevant characteristic | Reference |
|---|---|---|
| E. coli | ||
| MC1061 | Used for plasmid constructions | 12 |
| H. pylori | ||
| 26695 | Sequenced reference strain | 56 |
| 26695 ΔansB | HP0723::Cmr | This work |
| 26695 ΔdcuA | HP0724::Kmr | This work |
| 26695 Δggt | HP1118::Kmr | This work |
| 26695 ΔgltS | HP1506::Kmr | This work |
| 26695 ΔansB Δggt | HP0723::Kmr HP1118::Cmr | This work |
| SS1 | Mouse-adapted strain | 30 |
| SS1 ΔdcuA | HP0724::Kmr | This work |
Molecular techniques.
Standard procedures were used for endonuclease digestions, ligation, agarose gel electrophoresis, and elution of DNA fragments from agarose gels (44). Qiagen midi- or maxicolumns and a QIAamp DNA extraction kit (Qiagen) were used for large-scale plasmid and rapid chromosomal DNA preparations, respectively. PCR was carried out according to the manufacturer's recommendations using either the Taq DNA polymerase kit (Amersham) or the Phusion Hot Start high-fidelity DNA polymerase (Finnzymes). The pCR8/GW/TOPO TA cloning kit (Invitrogen) was used for the construction of suicide plasmids.
Construction of H. pylori mutants.
Chromosomal inactivation of hp0723 (encoding Hp-AnsB), hp0724 (encoding Hp-DcuA), hp1118 (encoding Hp-γGT), and hp1506 (encoding Hp-GltS) genes was performed in H. pylori strain 26695 or SS1. Deletions were introduced by allelic exchange using plasmids (derived from the pCR8/GW/TOPO TA vector) in which around 500 bp of the 5′-end and the 3′-end regions flanking the open reading frame of the target gene and an antibiotic resistance cassette (nonpolar kanamycin cassette [50] or nonpolar chloramphenicol cassette) were cloned. These plasmids were constructed and amplified in E. coli and used as suicide plasmids in H. pylori. H. pylori mutants were obtained by natural transformation with these suicide plasmids as previously described (7). The double mutants were constructed in two steps, as follows: a first inactivation was carried out with the nonpolar kanamycin cassette, and the second one with the nonpolar chloramphenicol cassette. Correct chromosomal insertion of the nonpolar kanamycin or chloramphenicol cassettes and correct allelic exchange were verified by PCR.
Measurement of asparaginase and glutaminase activity levels.
The glutaminase and asparaginase activity levels of H. pylori were measured using the ammonia assay kit (Sigma) with culture supernatants standardized as follows. H. pylori strains were amplified on plates for 24 h, grown overnight in liquid medium, inoculated in liquid culture at an optical density of 600 nm (OD600) of 0.2, and left to grow for 5 h until an OD600 of 0.5. These fresh log-phase cells were harvested and washed once in phosphate-buffered saline (PBS; Roche), and 3 × 108 CFU of bacteria (corresponding to 0.135 mg dry mass of bacterial cells) were resuspended in 1 ml of buffer with 5 mM Gln or Asn. Buffers used were either PBS at pH 7 or citrate-phosphate buffer (citric acid, 0.1 M; Na2HPO4, 0.2 M) for pH 7, 6.5, 6, 5.5, 5, or 4.5 in order to avoid changes in pH during the pH range experiments. Aliquots were taken after 15, 30, or 90 min of incubation at 37°C and centrifuged to pellet the bacteria. The NH3 concentration of the supernatant was measured immediately with the ammonia assay kit (Sigma), according to the manufacturer's recommendations. This assay is based on the following reaction: in the presence of NH3, α-ketoglutaric acid, and NADPH, the enzyme glutamate dehydrogenase produces Glu and NADP+. The oxidation of NADPH to NADP+ results in a decrease in the absorbance at 340 nm that is proportional to the concentration of NH3. The samples were diluted when necessary in order to measure NH3 amounts in the region of linearity of the assay, according to the manufacturer's recommendations, and the NH3 production was calculated from a standard curve.
Measurement of glutamine, glutamate, asparagine, and aspartate uptake by H. pylori.
The procedure is adapted from that used by Shibayama et al. (49). H. pylori parental strains and the isogenic knockout single or double mutants were grown under the same standardized conditions as those used for the enzymatic assays described above. Bacterial cells (0.27 mg dry mass of bacterial cells) were collected by centrifugation and washed twice with 5 mM morpholineethanesulfonic acid (MES) buffer, pH 6.6, with 150 mM NaCl. Uptake was initiated in the same buffer by adding 10 μl of 14C-labeled glutamine (l-[U-14C]glutamine; specific activity, 9.47 GBq/mmol) (196 μM; Amersham Bioscience), 10 μl of 14C-labeled glutamate (l-[U-14C]glutamic acid; specific activity, 9.36 GBq/mmol) (198 μM; Amersham Bioscience), 3 μl of 14C-labeled asparagine (l-[U-14C]asparagine; specific activity, 5.55 GBq/mmol) (666 μM; American Radiolabeled Chemicals), or 8.3 μl of 14C-labeled aspartate (l-[U-14C]aspartic acid; specific activity, 7.66 GBq/mmol) (241 μM; Amersham Bioscience) to the bacterial suspension. Final concentrations were 1.96 μM glutamine, 1.98 μM glutamate, 2.00 μM asparagine, or 2.00 μM aspartate. Aliquots of the samples were taken after various incubation times at 37°C and immediately filtrated on Durapore 0.45-μm membrane filters (Millipore) that were abundantly washed with 10 ml of the washing buffer that contained cold glutamine, glutamate, glutamine, or aspartate at 0.2 mM (depending on the 14C-labeled amino acid tested). Washing avoids unspecific binding of the radiolabeled amino acids to the bacteria. Finally, filters were transferred into scintillation vials with scintillation liquid (EcoLite+; MP Biomedicals). Radioactivity of bacteria retained on the filters was quantified by liquid scintillation counting between 5 and 150 keV.
Mouse model of colonization.
Aliquots of 100 μl containing 1.6 × 108 H. pylori SS1 parental strain or of 100 μl containing 1.1 × 108 SS1 ΔdcuA mutant in peptone broth were inoculated orogastrically into eight NMRI-specific pathogen-free mice each (Charles River Laboratories), as previously described (17). One month after inoculation, the mice were killed, and stomachs were removed for assessment of colonization by H. pylori. Viable H. pylori in the stomachs of these mice were enumerated by quantitative culturing of serial dilutions of the homogenized tissues in peptone broth and plating on blood agar plates supplemented with bacitracin (200 μg·ml−1) and nalidixic acid (10 μg·ml−1).
Quantitative real-time PCR.
An overnight culture of H. pylori 26695 was harvested by centrifugation at 4°C for 10 min at 3,000 × g and resuspended in liquid BHI supplemented with 0.2% β-cyclodextrin and the antibiotics-fungicide mix, which was adjusted at pH 7 or pH 5 by HCl addition. Total RNAs were extracted from exponential-phase H. pylori liquid cultures (optical density at 600 nm = 0.5). RNA was extracted using the phenol-chloroform method as previously described (8). DNA was removed from RNA preparations by DNase I digestion with 5 U RNase-free DNase I recombinant (Roche) for 20 min at 37°C followed, by a second phenol-chloroform purification. A second DNase treatment was carried out with the Turbo DNA-free kit (Ambion), according to the manufacturer's instructions. Total RNA was quantified on a NanoDrop spectrophotometer and visualized on an ethidium bromide-stained agarose gel.
Total RNA served as a template for cDNA synthesis using the AMV reverse transcriptase (Promega). Synthesis reactions were carried out by following the manufacturer's protocol, starting with using 1 μg total RNA and 50 ng random hexamers (Roche) per 20-μl reaction mixture. cDNA was diluted to 100 ng/μl. RNA transcripts were quantified on an Applied Biosystems StepOnePlus PCR machine using Power SYBR green PCR master mix (Applied Biosystems) in a 20-μl reaction mixture containing 50 ng of total cDNA. For each experiment, the transcript level was normalized to the level of the ppk gene (polyphosphate kinase; hp1010). Table S1 in the supplemental material lists the primer sequences used for qRT-PCR.
Bioinformatic analysis and signal peptide detection.
Signal peptide detection was performed with the SignalP 3.0 interface (16). For the study of the distribution of the ureA, ureB, ureI, ggt, gltS, ansB, and dcuA genes in the Helicobacter genus presented in Table 2, we used manual tBLASTn analyses with the genomic BLAST tool available on the NCBI website (http://www.ncbi.nlm.nih.gov/sutils/genom_table.cgi) and with the dedicated BLAST server for Helicobacter mustelae analysis (http://www.sanger.ac.uk/cgi-bin/blast/submitblast/h_mustelae). The sequence alignment shown in Fig. S2 in the supplemental material was performed using ClustalW with default settings (55), followed by manual modifications of the asparaginase sequences obtained from the NCBI nucleotide database, with the exception of the sequence of H. mustelae asparaginase, obtained from the Sanger website (http://www.sanger.ac.uk/Projects/H_mustelae/).
TABLE 2.
Distribution of the ureA, ureB, ureI, ggt, gltS, dcuA, and ans genes in Helicobacter species for which complete genome sequences are available
| Helicobacter strain | Host | Presence (+) or absence (−) of indicated gene |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| ureA | ureB | ureI | ggt | gltS | dcuA | ans (SP)a | ans (no SP)a | ||
| Gastric | |||||||||
| H. pylori 26695 | Human | + | + | + | + | + | + | + | − |
| H. pylori 98-10 | Human | + | + | + | + | + | + | + | − |
| H. pylori B128 | Human | + | + | + | + | + | + | + | − |
| H. pylori G27 | Human | + | + | + | + | + | + | + | − |
| H. pylori HPAG1 | Human | + | + | + | + | + | + | + | − |
| H. pylori HPKX_438_AG0C1 | Human | + | + | + | + | + | + | + | − |
| H. pylori HPKX_438_CA4C1 | Human | + | + | + | + | + | + | + | − |
| H. pylori J99 | Human | + | + | + | + | + | + | + | − |
| H. pylori P12 | Human | + | + | + | + | + | + | + | − |
| H. pylori Shi470 | Human | + | + | + | + | + | + | + | − |
| H. acinonychis Sheeba | Wildcat | + | + | + | + | + | + | + | − |
| H. mustelae ATCC 43772 | Ferret | + | + | + | + | + | − | − | + |
| Enterohepatic | |||||||||
| H. canadensis MIT 98-5491 | Human | − | − | − | − | + | + | − | + |
| H. cinaedi CCUG 18818 | Hamster | − | − | − | − | − | + | − | + |
| H. pullorum MIT 98-5489 | Bird | − | − | − | − | + | + | + | − |
| H. winghamensis ATCC BAA-430 | Human | − | − | − | − | + | + | + | − |
| H. bilis ATCC 43879 | Human, mouse, and dog | + | + | + | + | + | + | + | − |
| H. hepaticus ATCC 51449 | Mouse | + | + | + | − | − | + | + | − |
ans, gene encoding asparaginase. No assignment to either ansA or ansB genes encoding closely related asparaginases was done, since our phylogenetic analysis (see tree shown in Fig. S4 in the supplemental material) revealed several inconsistencies with the previous annotations. SP, a signal peptide was detected on the sequence (SignalP 3.0 hidden Markov model [HMM] score ≥ 0.750); no SP, a signal peptide was not detected (SignalP 3.0 HMM score < 0.750).
RESULTS
H. pylori asparaginase activity is provided by Hp-AnsB.
To identify the enzyme responsible for the H. pylori asparaginase activity, we measured Asn deamidation activities in the H. pylori 26695 strain and different mutants. The ansB hp0723 gene is annotated as coding for type II l-asparaginase. We constructed a deletion mutant of this gene in the H. pylori 26695 strain. In addition, a deletion mutant of the gene encoding Hp-γGT (hp1118) and a double mutant ΔansB Δggt strain (Table 1) were constructed. Growth in liquid medium of these mutants was unchanged compared with that of the wild-type strain (data not shown). The asparaginase activity level was measured in whole bacteria by detection of NH3 production in the presence of Asn. No detectable spontaneous hydrolysis (SH) of Asn was detected during the assay. Figure 1A shows the NH3 production resulting from H. pylori asparaginase activity, which corresponds to 1.2 μmol per mg of bacterial cells after 15 min of incubation using 5 mM Asn as a substrate. The same level of asparaginase activity was measured with the Δggt mutant, indicating that Hp-γGT does not display asparaginase activity. An important decrease in asparaginase activity of more than 90% was observed with the ΔansB mutant. The same decreased activity was measured with the ΔansB Δggt double mutant. These data demonstrate that Hp-AnsB is responsible for all the asparaginase activity in H. pylori.
FIG. 1.

Ammonia production by H. pylori. H. pylori parental strain 26695 or the isogenic single or double mutant ΔansB, Δggt, and ΔansB Δggt strains at 3 × 108 bacteria·ml−1 were incubated in phosphate-buffered saline (PBS) with 5 mM Asn or Gln. NH3 production was measured after 15 min of incubation at 37°C. Asn (A) or Gln (B) hydrolysis, measured by NH3 production from the parental and mutant strains. Spontaneous hydrolysis (SH) corresponds to the hydrolysis of Gln or Asn under the assay conditions without bacteria. Error bars represent the standard deviations obtained from at least three measurements. wt, wild type.
H. pylori glutaminase activity is provided by Hp-γGT.
To characterize the enzymes responsible for in vivo glutaminase activity of H. pylori, we measured this activity level in H. pylori strain 26695 and in isogenic single and double mutants carrying deletions of the ansB and ggt genes. The glutaminase activity level was measured by the production of NH3 after incubation of whole bacteria with Gln. The wild-type strain produced 2.8 μmol of NH3 per mg of bacterial cells after 15 min of incubation with 5 mM Gln, an activity level that was about twice that of Asn deamidation under the same test conditions (Fig. 1B). In the Δggt and the ΔansB Δggt mutants, we measured a strong decrease in glutaminase activity of ∼80%. The residual Gln deamidation activity of Δggt mutant was found to increase with time, suggesting that it is carried by another enzyme (see Fig. S1 in the supplemental material). A previous study showed that recombinant Hp-AnsB purified from E. coli exhibited in vitro asparaginase activity as well as low Gln deamidation activity (11), suggesting that the glutaminase activity observed in the Δggt mutant could be Hp-AnsB dependent. This was not the case in our in vivo assay since (i) the glutaminase activities of H. pylori 26695 and ΔansB strains were comparable and (ii) the ΔansB Δggt mutant presented a NH3 production level similar to that of the single mutant Δggt (Fig. 1B). Taken together, our results demonstrated that Hp-AnsB is not implicated in vivo in the production of NH3 from Gln and that Hp-γGT is the major enzyme with glutaminase activity in H. pylori. The residual activity detected in the ΔansB Δggt double mutant suggested the existence of another enzyme with weak glutaminase activity that is still to be identified.
Thus, Hp-γGT is responsible for the major part of Gln deamidation and Hp-AnsB is responsible for the major part of Asn deamidation in H. pylori.
Identification and characterization of the sole H. pylori glutamate transporter Hp-GltS.
During its growth, H. pylori takes up large amounts of amino acids, with Gln and Glu being among the most consumed (33, 52). No specific carriers of these amino acids have been characterized in H. pylori. A previous study showed that the uptake of Gln and Glu is carried out by an unknown sodium-dependent transporter (49). We found that gltS (hp1506), annotated to exhibit homology to Na+/Glu symporters, is a good candidate for glutamate uptake in H. pylori.
In order to characterize amino acid transport in H. pylori, we measured the uptake of l-[U-14C]Glu, l-[U-C14]Gln, and l-[U-14C]Asp by whole cells of the H. pylori 26695 wild-type strain and its isogenic ΔgltS mutant. There was a rapid accumulation of radiolabeled Glu and Gln in the wild-type strain but no detectable intracellular radioactivity in the ΔgltS mutant after 14 min (Fig. 2). These results indicated that Glu and Gln are incorporated in an Hp-GltS-dependent way. The wild-type and the ΔgltS strains rapidly incorporated radiolabeled Asp, indicating that Asp transport does not depend on Hp-GltS (data not shown).
FIG. 2.

Uptake of Glu and Gln by H. pylori. Incorporation of radiolabeled Glu or Gln by the H. pylori 26695 parental strain or by ΔgltS and Δggt mutants at pH 7 or at pH 5.5. Measurements were performed after 14 min of incubation of the bacteria in 5 mM MES buffer and 150 mM NaCl. Uptake was initiated by the addition of 1.98 μM l-[U-14C]Glu (A) or 1.96 μM l-[U-14C]Gln (B). Error bars represent the standard deviations obtained from at least three measurements.
Unlike the H. pylori 26695 wild-type strain, the Δggt mutant is unable to incorporate radiolabeled Gln, while Glu transport in this mutant was unaffected (Fig. 2). As the Gln used is radiolabeled on every carbon, the result also showed that no degradation product of this amino acid was incorporated in the Δggt mutant. These results indicated that Gln transport is exclusively dependent on its deamidation into Glu by the periplasmic Hp-γGT activity.
Our results demonstrated that Hp-GltS is an amino acid transporter, allowing Glu-specific incorporation but not that of Gln or Asp. Thus, we concluded that Hp-GltS, which was previously found to be essential for colonization in the Mongolian gerbil model in a signature-tagged mutagenesis study (28), is the sole Glu transporter in H. pylori.
Identification and characterization of the sole H. pylori aspartate transporter.
In addition to the consumption of large amounts of Glu and Gln, H. pylori preferentially uses other amino acids such as Asn and Asp (52). Again, no specific carrier of these amino acids has been identified in H. pylori. We found that the hp0724 gene (dcuA), adjacent to the asparaginase, has homology to anaerobic C4-dicarboxylate transport proteins. As Asp is a C4-dicarboxylate, a ΔdcuA mutant was constructed in strain 26695. To test the physiological implication of Hp-DcuA in the incorporation of Asp, we measured the uptake of l-[U-14C]Asp by H. pylori strain 26695 and by the isogenic ΔdcuA mutant. Rapid accumulation of radiolabeled Asp was measured in H. pylori 26695 but not in the ΔdcuA mutant (Fig. 3A). These results demonstrate that DcuA is the sole Asp transporter in H. pylori 26695.
FIG. 3.

Uptake of Asp and Asn by H. pylori. Incorporation of radiolabeled Asp or Asn by the H. pylori 26695 parental strain or by the ΔdcuA and ΔansB mutants at pH 7 or at pH 5.5. Measurements were performed after 14 min of incubation of the bacteria in 5 mM MES buffer and 150 mM NaCl. (A) Uptake was initiated by the addition of 2 μM l-[U-14C]Asp. (B) Relative uptake of radiolabeled Asp at pH 7 by H. pylori strains without or with the addition of 0.2 mM cold Asn compared to that of the parental H. pylori 26695 strain without added Asn. (C) Uptake was initiated by the addition of 2 μM l-[U-14C]Asn. Error bars represent the standard deviations obtained from at least three measurements.
The H. pylori asparaginase is active in the periplasm.
As it is annotated, open reading frame (ORF) hp0723 (ansB) did not contain a sequence encoding a signal peptide. Close examination of the H. pylori 26695 genomic sequence revealed an alternative start codon preceded by a Shine-Dalgarno sequence upstream of hp0723, including a characteristic signal peptide conserved in all available Helicobacter species (see Fig. S2 in the supplemental material), suggesting that Hp-AnsB is secreted.
To test whether Hp-AnsB activity is periplasmic, two experiments were performed. First, we measured the uptake of l-[U-14C]Asp by H. pylori 26695 and by its isogenic ΔansB mutant with or without an excess of cold Asn. Without the addition of Asn, transport of radiolabeled Asp was comparable in the wild-type 26695 strain and the ΔansB mutant (Fig. 3B). In contrast, the addition of 0.2 mM Asn inhibited the uptake of 14C-labeled Asp in wild-type H. pylori 26695 but not in the ΔansB mutant. If asparaginase was active inside the bacterium, Asp production by Hp-AnsB-dependent Asn hydrolysis should not affect the incorporation of radiolabeled Asp by H. pylori. In contrast, if asparaginase were active in the periplasm, Hp-AnsB production of Asp resulting from hydrolysis of cold Asn should compete and inhibit the accumulation of 14C-labeled Asp in H. pylori. These results demonstrated that Hp-AnsB is active in the periplasm of H. pylori. Moreover, the results imply that Asn is not a substrate for DcuA, since Asn does not inhibit uptake of Asp in the absence of the periplasmic asparaginase activity.
To strengthen these data, we directly measured the uptake of l-[U-14C]Asn by H. pylori 26695 and its isogenic ΔansB mutant. We observed that unlike the H. pylori 26695 wild-type strain, the ΔansB mutant was unable to incorporate radiolabeled Asn (Fig. 3C).
These results indicated that Asn is exclusively transported by an indirect pathway involving a prior step of deamidation into Asp by Hp-AnsB, whose activity is restricted to the periplasm.
The sole aspartate transporter of H. pylori is essential for in vivo colonization.
To evaluate the importance of Hp-DcuA activity in vivo, we compared the abilities of the SS1 strain, a mouse-adapted H. pylori strain (30), and its isogenic ΔdcuA mutant to colonize orogastrically inoculated NMRI mice. Animals were sacrificed after 1 month. The stomachs were removed from the animals, and colonization by SS1 and the ΔdcuA mutant was assessed by quantitative culturing of the stomachs homogenates (17). Contrary to the SS1 parental strain, the SS1 ΔdcuA mutant was not recovered from any of the infected animals (Fig. 4). These results indicated that the ΔdcuA mutant is unable to colonize the mouse stomach. This demonstrated that Hp-DcuA is essential for the establishment of an infection by H. pylori.
FIG. 4.
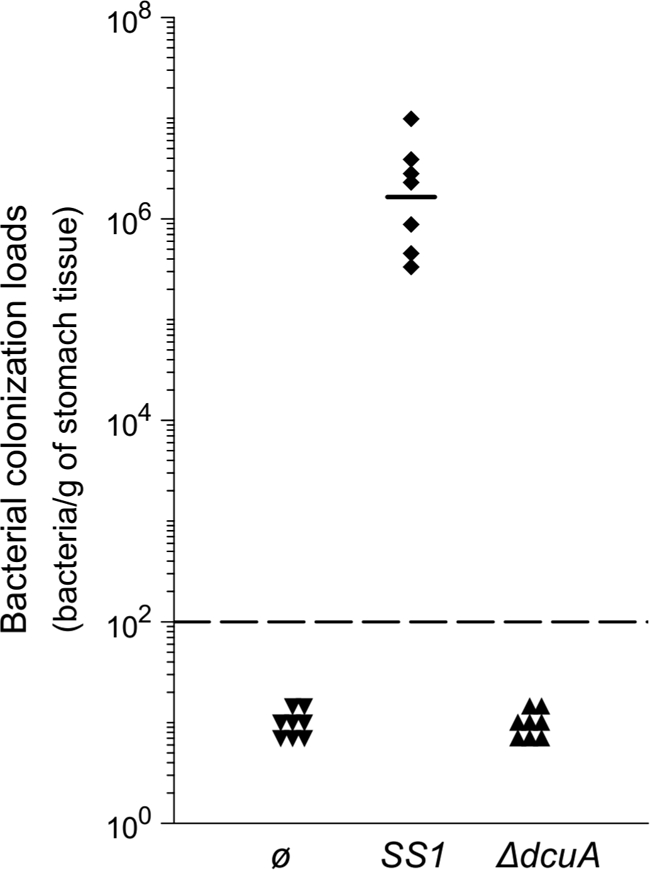
The dcuA gene is required for mouse stomach colonization by the H. pylori SS1 strain. Each point corresponds to the colonization load for one mouse. The solid horizontal bar represents the geometric mean for the mice infected by SS1 wild-type strain. The detection limit is shown by a dashed horizontal line.
pH modulation of the coupled amino acid deamidase-transport systems.
Since H. pylori is facing significant changes in the pH of the stomach, we examined whether the expression of the ansB, ggt, gltS, and dcuA genes responded to pH variation. H. pylori cultures were performed at pH 5 or 7 during one generation time, as previously described (8). Expression of the ansB, ggt, dcuA, and gltS genes was measured by quantitative real-time reverse transcriptase PCR (qRT-PCR). For all genes studied, pH-dependent changes in the amount of the corresponding mRNAs did not exceed 2-fold. At pH 5, the ggt gene is slightly induced (fold change, 1.42) and other genes are slightly repressed (fold changes, 0.62 for gltS, 0.57 for ansB, and 0.70 for dcuA). This showed that there is no major effect of pH on gene regulation at the transcriptional level.
We examined Asp, Asn, Glu, and Gln incorporation at neutral and acidic pHs. Uptake of l-[U-14C]Asp, l-[U-14C]Asn, l-[U-14C]Glu, and l-[U-14C]Gln by H. pylori 26695 was measured at pH 7 and pH 5.5. For each of these four amino acids, we observed an accumulation of radioactivity in the bacteria at neutral pH that was strongly diminished at acidic pH (Fig. 2 and 3). These results indicated that, at pH 5.5 in H. pylori 26695, accumulation of Asp, Asn, Glu, and Gln is strongly diminished. Since Asp and Glu accumulation in H. pylori was pH dependent, we asked whether the periplasmic producers of Glu and Asp (Hp-γGT and Hp-AnsB, respectively) were also pH regulated.
To test this hypothesis, the pH response profiles of the Hp-γGT and the Hp-AnsB activities were measured in whole cells of the H. pylori 26695 wild-type strain or the Δggt and ΔansB isogenic mutants. During the assay, pH variations caused by NH3 production were controlled to be negligible, and viability of the bacteria was maintained (data not shown) (see Fig. S3 in the supplemental material). Under the assay conditions used, the spontaneous hydrolysis (SH) of Gln and Asn is negligible, indicating that NH3 production from these amino acids is exclusively dependent on the presence of H. pylori (Fig. 5). Both deamidase activities present a maximum at neutral pH and decline rapidly below pH 5.5 (Fig. 5). Urease activities measured under the same conditions with 5 mM urea were 17.9 and 64.4 μmol of NH3 per mg bacterial cells at pH 7 and 4, respectively.
FIG. 5.

pH profiles of ammonia production by the H. pylori 26695, ΔansB, and Δggt strains. Catalytic activity of NH3 production was measured with 3 × 108 bacteria incubated for 30 min in citrate-phosphate buffer adjusted at different pH values at 37°C, with 5 mM Gln (A) or 5 mM Asn (B). SH corresponds to the hydrolysis of Gln or Asn under the assay conditions without bacteria. Error bars represent the standard deviations obtained from at least three measurements.
Thus, Hp-γGT and Hp-AnsB activities are pH modulated and form two coherent incorporation pathways together with their respective sole transporters, Hp-GltS and Hp-DcuA.
Distribution of the ansB, ggt, gltS, and dcuA genes among Helicobacter species.
Since we showed that H. pylori possesses two coupled incorporation pathways (AnsB plus DcuA and γGT plus GltS) important for the colonization of the stomach, we examined the distribution of the ansB, ggt, gltS and dcuA genes in 12 gastric (10 H. pylori strains and 2 other gastric Helicobacter sp. strains) and 6 enterohepatic Helicobacter species, for which entire genomic sequences are available (Table 2). For this analysis, we added the ureI, ureA, and ureB genes that encode the pH-regulated urea transporter and the urease structural subunits (7). As reported in Table 2, the ureA, ureB, ureI, ansB, ggt, gltS and dcuA genes are present in all H. pylori strains and in Helicobacter acinonychis, which colonizes the stomachs of large felines. Interestingly, out of these seven genes, the gastric H. mustelae strain lacks both dcuA and ansB genes. The H. mustelae genome possesses an open reading frame that potentially encodes a cytoplasmic asparaginase (no signal peptide detected) whose sequence is distant from the AnsB sequences and from the AnsA type I cytoplasmic asparaginases (39). The absence of an AnsB-type periplasmic asparaginase in H. mustelae is a characteristic shared with the six enterohepatic Helicobacter species examined. A phylogenetic tree of the asparaginases is presented in Fig. S4 in the supplemental material (drawn using TreeView [40]). Indeed, based on a 16S RNA similarity matrix, H. mustelae has been previously shown to be phylogenetically closer to the enterohepatic species than to the gastric species (9, 51). The presence of cytoplasmic asparaginase in H. mustelae correlates with this phylogenetic classification. Finally, none of the ureA, ureB, ureI, ggt, and gltS genes are conserved in every enterohepatic Helicobacter species. We concluded that the coupled transport/incorporation pathways described in this study are strictly conserved in H. pylori and the closely related H. acinonychis and have not been maintained during evolution in the enterohepatic Helicobacter species.
DISCUSSION
Here we deciphered in H. pylori two coupled deamidase-transport systems active in the periplasm and at the inner membrane, respectively. These systems guarantee the acquisition of amino acids Glu and Asp from Gln and Asn, respectively. As a consequence, these systems simultaneously consume Glu/Gln and Asp/Asn from the environment and produce NH3 in the periplasm. We showed that (i) Hp-AnsB and Hp-γGT are responsible for asparaginase and glutaminase periplasmic activities, respectively, (ii) Hp-DcuA and Hp-GltS are the specific and sole transporters of Asp and Glu, and (iii) Gln and Asn are exclusively incorporated by indirect pathways, requiring their prior deamidation. Importantly, each of the four functions provided by Hp-AnsB, Hp-DcuA, Hp-γGT, and Hp-GltS is individually essential or very important for colonization of animal models by H. pylori (34, 13, 28) and conserved in the H. pylori strains.
In contrast to Glu and Asp transport, uptake of the corresponding amides Gln and Asn proceeds through an indirect pathway involving a prior periplasmic deamidation step. Each of these indirect uptake pathways is unique in agreement with the small genome size (1.6 Mb) and the documented low functional redundancy of H. pylori. On the H. pylori 26695 genome, the annotation of HP1169-HP1172 as a putative Gln ABC transporter should be revised, since we demonstrated that H. pylori is unable to import Gln directly. In contrast to H. pylori, another member of Campylobacterales, Campylobacter jejuni, was found to possess different redundant transporters for Asp, Glu, and Gln (22, 31).
In H. pylori whole cells, the Hp-GltS/Hp-DcuA transport and the Hp-γGT/Hp-AnsB hydrolysis activities were optimal at neutral pH and rapidly declined at and below pH 5.5. The impact of the external pH on the periplasmic pH is difficult to evaluate. However, the reported pH dependence on the activity of purified recombinant Hp-AnsB presented a distinct profile, with 70% of the activities preserved at pH 4.5 compared to that preserved at pH 7 (11). The pH profiles of Gln hydrolysis measured with purified Hp-γGT were also different. They showed a dramatic decline of the in vitro activity below pH 7 (49). In addition, the minimal in vitro activity of Hp-AnsB on Gln reported by Cappelletti et al. (11) was not observed by our measurements using whole cells. No major pH-dependent transcriptional regulation was measured for the four genes encoding the two deamidase-uptake systems. Previous transcriptomic studies reported contradictory results for ansB, finding either upregulation after a 30-min acid shock in brain heart infusion (BHI) medium (58) or downregulation at pH 5 in brucella broth supplemented with 10% fetal bovine serum (34). None of the transcriptomic analyses of the response to pH (2, 3, 8, 34, 58) identified dcuA and ggt as acid-responsive genes. In agreement with the present results, we previously found that the expression of the gltS gene is slightly downregulated by acidity (8). Thus, functional regulation of the deamidase-transport proteins provides the cell with a strong and rapid response to pH that would not be achieved by transcriptional regulation of the corresponding genes. Interestingly, the pH responses of the periplasmic NH3 producers Hp-AnsB and Hp-γGT are precisely inverse to that of urea uptake by the acid-activated urea channel UreI, which optimally provides urea to urease below pH 5 (50, 57).
What is the function of these coupled amino acid uptake and periplasmic NH3 production systems in H. pylori? First, Asp, Asn, Glu, and Gln are four of the eight amino acids most consumed by H. pylori (33, 52). In contrast with E. coli, H. pylori possesses only one sugar transporter and preferentially uses amino acids as a carbon source (33, 52). It is thus possible that the dominant role of these four amino acids is to serve as a carbon source in H. pylori. However, none of the H. pylori strains examined (n = 34) presented auxotrophy for these amino acids (37, 42, 54). Previous observations and our present data point to a role of these systems that is not restricted to nutrient acquisition.
Most interesting is the observation that Gln and Asn are not taken up directly by H. pylori (Fig. 6). Analysis of the predicted metabolic pathways of H. pylori (25-27) indicated that periplasmic AnsB is the only enzyme using free Asn as a substrate. In addition, it has been demonstrated that in H. pylori, neither Asn nor Gln are incorporated directly during protein synthesis. In all Helicobacter species, as in several other bacteria, Gln-tRNAGln and Asn-tRNAAsn are synthesized in the following two steps: (i) Glu and Asp are mischarged on tRNAGln and tRNAAsn, respectively, and (ii) an amidotransferase complex (GatABC) transfers a molecule of NH3 from a donor (Gln) to the mischarged tRNA to obtain the correctly loaded aminoacyl-tRNA (47). Thus, it seems that no Asn is required in the cytoplasm, pointing to an original role of the periplasmic asparaginase. Gln is not directly incorporated into proteins; however, it is synthesized from Glu by the cytoplasmic and essential glutamine synthetase (GlnA; EC 6.3.1.2). Intracellular Gln serves directly or indirectly as an NH3 donor for the synthesis of essential molecules such as peptidoglycan precursors and pyrimidine. Paradoxically, instead of directly taking up essential Gln, GlnA cytoplasmically synthesizes Gln from Glu at the cost of ATP and ammonia, again supporting a specific function of Gln periplasmic hydrolysis. In addition, the absence of direct Gln uptake explains why mutations of the glnA gene encoding GlnA were lethal in H. pylori even under conditions of extracellular Gln supplementation (19).
FIG. 6.

Schematic representation of the direct and indirect Glu, Gln, Asp, and Asn incorporation pathways associated with ammonia production in H. pylori. H. pylori (top) is represented in contact with a host epithelial cell (bottom). The proteins described in this work are represented by filled circles. At neutral pH, Glu and Asp are transported directly into the cytoplasm. In contrast, Gln and Asn are exclusively taken up by indirect pathways (illustrated by the two crosses). Gln and Asn hydrolysis in the periplasm generates ammonia plus Glu and Asp, respectively. A model is presented in which these two nonredundant systems participate in H. pylori virulence by depleting gastric or immune cells from protective amino acids and producing toxic ammonia in contact with the host cells. Abbreviations: Glu, l-glutamate; Gln, l-glutamine; Asp, l-aspartate; Asn, l-asparagine; NH3, ammonia; γGT, γ-glutamyltranspeptidase; AnsB, asparaginase.
The fate of periplasmic NH3 generated by Hp-AnsB and Hp-γGT is intriguing. Two roles seem unlikely, as follows: (i) use of this NH3 as a source of nitrogen (indeed, urease activity generates important amounts of intracellular NH3 that are at least in part incorporated directly into Gln by GlnA [GlnA physically interacts with UreA {53}], and H. pylori presents no NH3 uptake system, such as, for instance, the AmtB transporter), and (ii) this production of ammonia in resistance to strong acidity (the enzymatic activities are specifically optimal at neutral pH). However, we cannot exclude a role of the two periplasmic deamidases in resistance to weakly acidic conditions, like pH 6, under which the enzymes are still fully active (Fig. 5). Interestingly, it was been shown previously that the acid resistance of H. pylori in the presence of low concentrations of urea (0.1 mM) is reinforced by the addition of 1 mM Gln (41). In contrast to the acid-activated urease activity, the deamidase-transport pathways are specifically functional at neutral pH, encountered by H. pylori inside the gastric mucus near the host epithelial cells or attached to them where they are potentially exposed to the immune system.
The protective effect of dietary Gln in human gastric pathologies (38) and in animal models (23) has been extensively documented. In addition, purified Hp-AnsB and Hp-γGT proteins present cytotoxic properties (11). Hp-γGT has been shown to be proapoptotic (48) and to inhibit T-cell proliferation (45). NH3 was shown to kill gastric epithelial cells in a dose-dependent manner (36). Finally, Gln and Glu protect cultured gastric cells from NH3-induced cell death (36), and depletion of glutamine limits the host immune response (43, 45). Asparaginase is used in the treatment of lymphoblastic leukemia because of its ability to deplete the tumor cells from essential Asn (15). The coupled mechanisms described here enable H. pylori to acquire nutrients (among which, Glu and Gln) and consequently cause their depletion from the cellular environment of the host with simultaneous ammonia production. Partially in line with Shibayama et al. (49), we propose a model (Fig. 6) that we are presently investigating in which these essential activities have deleterious effects on the immune cells and generate lesions of the epithelial cells by (i) depleting the gastric and immune cells from protective amino acids and/or (ii) delivering NH3 directly in contact with the epithelial cells.
The ansB, ggt, gltS, and dcuA genes are not essential for H. pylori growth on plates but are required for full virulence in animal models (34, 13, 28). A previous study using whole-genome microarrays revealed the presence of the ansB, dcuA, ggt, and gltS genes in the 56 H. pylori strains examined and in the 4 closely related H. acinonychis strains available (21). Our analysis of the distribution of the ansB, dcuA, ggt, and gltS genes showed a strong conservation of the sequences in H. pylori and H. acinonychis. However, genes encoding asparaginases were also detected in gastric H. mustelae and in the six enterohepatic Helicobacter species examined. A closer analysis of the corresponding proteins by construction of a phylogenetic tree, to which other epsilonproteobacteria were added (Campylobacter jejuni and Wolinella succinogenes) (see Fig. S4 in the supplemental material), demonstrated the following: (i) characteristic AnsB proteins with signal peptides in H. pylori and H. acinonychis; (ii) asparaginase without a signal peptide in H. mustelae; and (iii) clustering of asparaginases from enterohepatic Helicobacter species with those of Campylobacter species, associated with a disparity in the prediction of signal peptides (presence or absence) (see Fig. S4 in the supplemental material). Interestingly, a recent study reported that the acquisition of a signal peptide sequence to otherwise cytoplasmic asparaginase enhances the capacity of certain Campylobacter jejuni strains to utilize asparagine for in vitro growth and to more efficiently colonize the liver (24). However, the impact of asparaginase on radioactive amino acid uptake by the redundant C. jejuni transporters was not tested (24). It is tempting to speculate that in the Campylobacterales, including Helicobacter and Campylobacter species, the acquisition of periplasmic localized asparaginase enhances the capacity to colonize specific niches. However, contrary to what we found in H. pylori in which the AnsB signal peptide is strictly conserved, the periplasmic localization of C. jejuni asparaginase is only strain specific. These data suggest a selective pressure on the stomach-colonizing organisms for the conservation of periplasmic asparaginase. Similarly, the presence of γGT in some C. jejuni strains has been associated with enhanced virulence (24). In contrast with C. jejuni, the asparaginase and γGT activities of H. pylori are essential for full colonization, as are the coupled amino acid uptake systems. Such dramatic phenotypes have not frequently been reported in bacterial pathogens.
In conclusion, we showed that H. pylori possesses two essential and exclusive coregulated systems involved in amino acid hydrolysis in the periplasm and uptake that are coupled with periplasmic NH3 production. We propose a model in which these two coupled mechanisms correspond to an original diversion of conventional pathways of amino acids incorporation that might provide H. pylori with additional strategies for its proliferation and persistence in the stomach and may participate in the pathogenesis induced by H. pylori.
Supplementary Material
Acknowledgments
We thank Cécile Muller for her interest in this work and her help with the qRT-PCR experiments. We thank Jost Enninga, Hannu Myllykallio, and Ivo Boneca for discussions and critical reading of the manuscript.
This work was funded by Projet transversal de recherche (PTR) grant 310 from the Institut Pasteur. D.L. was a recipient of the Bourse Roux-Institut Pasteur postdoctoral fellowship.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 5 April 2010.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Algood, H. M. S., and T. L. Cover. 2006. Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin. Microbiol. Rev. 19:597-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allan, E., C. L. Clayton, A. McLaren, D. M. Wallace, and B. W. Wren. 2001. Characterization of the low-pH responses of Helicobacter pylori using genomic DNA arrays. Microbiology 147:2285-2292. [DOI] [PubMed] [Google Scholar]
- 3.Ang, S., C. Z. Lee, K. Peck, M. Sindici, U. Matrubutham, M. A. Gleeson, and J. T. Wang. 2001. Acid-induced gene expression in Helicobacter pylori: study in genomic scale by microarray. Infect. Immun. 69:1679-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atherton, J. C. 2006. The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu. Rev. Pathol. 1:63-96. [DOI] [PubMed] [Google Scholar]
- 5.Baumgartner, H. K., and M. H. Montrose. 2004. Regulated alkali secretion acts in tandem with unstirred layers to regulate mouse gastric surface pH. Gastroenterology 126:774-783. [DOI] [PubMed] [Google Scholar]
- 6.Bumann, D., S. Aksu, M. Wendland, K. Janek, U. Zimny-Arndt, N. Sabarth, T. F. Meyer, and P. R. Jungblut. 2002. Proteome analysis of secreted proteins of the gastric pathogen Helicobacter pylori. Infect. Immun. 70:3396-3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bury-Moné, S., S. Skouloubris, A. Labigne, and H. De Reuse. 2001. The Helicobacter pylori UreI protein: role in adaptation to acidity and identification of residues essential for its activity and for acid activation. Mol. Microbiol. 42:1021-1034. [DOI] [PubMed] [Google Scholar]
- 8.Bury-Moné, S., J. Thiberge, M. Contreras, A. Maitournam, A. Labigne, and H. De Reuse. 2004. Responsiveness to acidity via metal ion regulators mediates virulence in the gastric pathogen Helicobacter pylori. Mol. Microbiol. 53:623-638. [DOI] [PubMed] [Google Scholar]
- 9.Bury-Moné, S., S. Skouloubris, C. Dauga, J. Thiberge, D. Dailidiene, D. E. Berg, A. Labigne, and H. De Reuse. 2003. Presence of active aliphatic amidases in Helicobacter species able to colonize the stomach. Infect. Immun. 71:5613-5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Busiello, I., R. Acquaviva, A. Di Popolo, T. G. Blanchard, V. Ricci, M. Romano, and R. Zarrilli. 2004. Helicobacter pylori gamma-glutamyltranspeptidase upregulates COX-2 and EGF-related peptide expression in human gastric cells. Cell. Microbiol. 6:255-267. [DOI] [PubMed] [Google Scholar]
- 11.Cappelletti, D., L. R. Chiarelli, M. V. Pasquetto, S. Stivala, G. Valentini, and C. Scotti. 2008. Helicobacter pylori L-asparaginase: a promising chemotherapeutic agent. Biochem. Biophys. Res. Commun. 377:1222-1226. [DOI] [PubMed] [Google Scholar]
- 12.Casadaban, M. J., and S. N. Cohen. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J. Mol. Biol. 138:179-207. [DOI] [PubMed] [Google Scholar]
- 13.Chevalier, C., J. M. Thiberge, R. L. Ferrero, and A. Labigne. 1999. Essential role of Helicobacter pylori gamma-glutamyltranspeptidase for the colonization of the gastric mucosa of mice. Mol. Microbiol. 31:1359-1372. [DOI] [PubMed] [Google Scholar]
- 14.Chu, S., S. Tanaka, J. D. Kaunitz, and M. H. Montrose. 1999. Dynamic regulation of gastric surface pH by luminal pH. J. Clin. Invest. 103:605-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Earl, M. 2009. Incidence and management of asparaginase-associated adverse events in patients with acute lymphoblastic leukemia. Clin. Adv. Hematol. Oncol. 7:600-606. [PubMed] [Google Scholar]
- 16.Emanuelsson, O., S. Brunak, G. von Heijne, and H. Nielsen. 2007. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2:953-971. [DOI] [PubMed] [Google Scholar]
- 17.Ferrero, R. L., J. M. Thiberge, M. Huerre, and A. Labigne. 1998. Immune responses of specific-pathogen-free mice to chronic Helicobacter pylori (strain SS1) infection. Infect. Immun. 66:1349-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foster, J. W. 2004. Escherichia coli acid resistance: tales of an amateur acidophile. Nat. Rev. Microbiol. 2:898-907. [DOI] [PubMed] [Google Scholar]
- 19.Garner, R. M., J. Fulkerson, and H. L. Mobley. 1998. Helicobacter pylori glutamine synthetase lacks features associated with transcriptional and posttranslational regulation. Infect. Immun. 66:1839-1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gobert, A. P., D. J. McGee, M. Akhtar, G. L. Mendz, J. C. Newton, Y. Cheng, H. L. Mobley, and K. T. Wilson. 2001. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc. Natl. Acad. Sci. U. S. A. 98:13844-13849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gressmann, H., B. Linz, R. Ghai, K. Pleissner, R. Schlapbach, Y. Yamaoka, C. Kraft, S. Suerbaum, T. F. Meyer, and M. Achtman. 2005. Gain and loss of multiple genes during the evolution of Helicobacter pylori. PLoS Genet. 1:e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guccione, E., M. D. R. Leon-Kempis, B. M. Pearson, E. Hitchin, F. Mulholland, P. M. van Diemen, M. P. Stevens, and D. J. Kelly. 2008. Amino acid-dependent growth of Campylobacter jejuni: key roles for aspartase (AspA) under microaerobic and oxygen-limited conditions and identification of AspB (Cj0762), essential for growth on glutamate. Mol. Microbiol. 69:77-93. [DOI] [PubMed] [Google Scholar]
- 23.Hagen, S. J., M. Ohtani, J. Zhou, N. S. Taylor, B. H. Rickman, G. L. Blackburn, and J. G. Fox. 2009. Inflammation and foveolar hyperplasia are reduced by supplemental dietary glutamine during Helicobacter pylori infection in mice. J. Nutr. 139:912-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hofreuter, D., V. Novik, and J. E. Galán. 2008. Metabolic diversity in Campylobacter jejuni enhances specific tissue colonization. Cell Host Microbe 4:425-433. [DOI] [PubMed] [Google Scholar]
- 25.Kanehisa, M., and S. Goto. 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28:27-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa, M., M. Araki, S. Goto, M. Hattori, M. Hirakawa, M. Itoh, T. Katayama, S. Kawashima, S. Okuda, T. Tokimatsu, and Y. Yamanishi. 2008. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36:D480-D484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanehisa, M., S. Goto, M. Hattori, K. F. Aoki-Kinoshita, M. Itoh, S. Kawashima, T. Katayama, M. Araki, and M. Hirakawa. 2006. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34:D354-D357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kavermann, H., B. P. Burns, K. Angermuller, S. Odenbreit, W. Fischer, K. Melchers, and R. Haas. 2003. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J. Exp. Med. 197:813-822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim, K., S. Lee, M. Park, J. Song, H. Kang, W. Lee, M. Cho, K. Rhee, H. Youn, and S. Baik. 2007. Gamma-glutamyltranspeptidase of Helicobacter pylori induces mitochondria-mediated apoptosis in AGS cells. Biochem. Biophys. Res. Commun. 355:562-567. [DOI] [PubMed] [Google Scholar]
- 30.Lee, A., J. O'Rourke, M. C. De Ungria, B. Robertson, G. Daskalopoulos, and M. F. Dixon. 1997. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112:1386-1397. [DOI] [PubMed] [Google Scholar]
- 31.Lin, A. E., K. Krastel, R. I. Hobb, S. A. Thompson, D. G. Cvitkovitch, and E. C. Gaynor. 2009. Atypical roles for Campylobacter jejuni amino acid ATP binding cassette transporter components PaqP and Pa in bacterial stress tolerance and pathogen-host cell dynamics. Infect. Immun. 77:4912-4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGovern, K. J., T. G. Blanchard, J. A. Gutierrez, S. J. Czinn, S. Krakowka, and P. Youngman. 2001. γ-Glutamyltransferase is a Helicobacter pylori virulence factor but is not essential for colonization. Infect. Immun. 69:4168-4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mendz, G. L., and S. L. Hazell. 1995. Aminoacid utilization by Helicobacter pylori. Int. J. Biochem. Cell Biol. 27:1085-1093. [DOI] [PubMed] [Google Scholar]
- 34.Merrell, D. S., M. L. Goodrich, G. Otto, L. S. Tompkins, and S. Falkow. 2003. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect. Immun. 71:3529-3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller, J. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 36.Nakamura, E., and S. J. Hagen. 2002. Role of glutamine and arginase in protection against ammonia-induced cell death in gastric epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 283:G1264-G1275. [DOI] [PubMed] [Google Scholar]
- 37.Nedenskov, P. 1994. Nutritional requirements for growth of Helicobacter pylori. Appl. Environ. Microbiol. 60:3450-3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okabe, S., K. Takeuchi, Y. Takata, T. Naganuma, and K. Takagi. 1976. Effects of L-glutamine on various gastric lesions in rats and guinea pigs. Digestion 14:325-331. [DOI] [PubMed] [Google Scholar]
- 39.O'Toole, P. W., W. J. Snelling, C. Canchaya, B. M. Forde, K. R. Hardie, C. Josenhans, R. L. Graham, G. McMullan, J. Parkhill, E. Belda, and S. D. Bentley. 2010. Comparative genomics and proteomics of Helicobacter mustelae, an ulcerogenic and carcinogenic gastric pathogen. BMC Genomics 11:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Page, R. D. 1996. TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12:357-358. [DOI] [PubMed] [Google Scholar]
- 41.Rektorschek, M., D. Weeks, G. Sachs, and K. Melchers. 1998. Influence of pH on metabolism and urease activity of Helicobacter pylori. Gastroenterology 115:628-641. [DOI] [PubMed] [Google Scholar]
- 42.Reynolds, D. J., and C. W. Penn. 1994. Characteristics of Helicobacter pylori growth in a defined medium and determination of its amino acid requirements. Microbiology 140:2649-2656. [DOI] [PubMed] [Google Scholar]
- 43.Roth, E. 2008. Nonnutritive effects of glutamine. J. Nutr. 138:2025S-2031S. [DOI] [PubMed] [Google Scholar]
- 44.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 45.Schmees, C., C. Prinz, T. Treptau, R. Rad, L. Hengst, P. Voland, S. Bauer, L. Brenner, R. M. Schmid, and M. Gerhard. 2007. Inhibition of T-cell proliferation by Helicobacter pylori gamma-glutamyl transpeptidase. Gastroenterology 132:1820-1833. [DOI] [PubMed] [Google Scholar]
- 46.Schreiber, S., M. Konradt, C. Groll, P. Scheid, G. Hanauer, H. Werling, C. Josenhans, and S. Suerbaum. 2004. The spatial orientation of Helicobacter pylori in the gastric mucus. Proc. Natl. Acad. Sci. U. S. A. 101:5024-5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sheppard, K., P. Akochy, J. C. Salazar, and D. Söll. 2007. The Helicobacter pylori amidotransferase GatCAB is equally efficient in glutamine-dependent transamidation of Asp-tRNAAsn and Glu-tRNAGln. J. Biol. Chem. 282:11866-11873. [DOI] [PubMed] [Google Scholar]
- 48.Shibayama, K., K. Kamachi, N. Nagata, T. Yagi, T. Nada, Y. Doi, N. Shibata, K. Yokoyama, K. Yamane, H. Kato, Y. Iinuma, and Y. Arakawa. 2003. A novel apoptosis-inducing protein from Helicobacter pylori. Mol. Microbiol. 47:443-451. [DOI] [PubMed] [Google Scholar]
- 49.Shibayama, K., J. Wachino, Y. Arakawa, M. Saidijam, N. G. Rutherford, and P. J. F. Henderson. 2007. Metabolism of glutamine and glutathione via gamma-glutamyltranspeptidase and glutamate transport in Helicobacter pylori: possible significance in the pathophysiology of the organism. Mol. Microbiol. 64:396-406. [DOI] [PubMed] [Google Scholar]
- 50.Skouloubris, S., J. M. Thiberge, A. Labigne, and H. De Reuse. 1998. The Helicobacter pylori UreI protein is not involved in urease activity but is essential for bacterial survival in vivo. Infect. Immun. 66:4517-4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Solnick, J. V., and D. B. Schauer. 2001. Emergence of diverse Helicobacter species in the pathogenesis of gastric and enterohepatic diseases. Clin. Microbiol. Rev. 14:59-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stark, R. M., M. S. Suleiman, I. J. Hassan, J. Greenman, and M. R. Millar. 1997. Amino acid utilisation and deamination of glutamine and asparagine by Helicobacter pylori. J. Med. Microbiol. 46:793-800. [DOI] [PubMed] [Google Scholar]
- 53.Stingl, K., K. Schauer, C. Ecobichon, A. Labigne, P. Lenormand, J. Rousselle, A. Namane, and H. de Reuse. 2008. In vivo interactome of Helicobacter pylori urease revealed by tandem affinity purification. Mol. Cell. Proteomics 7:2429-2441. [DOI] [PubMed] [Google Scholar]
- 54.Testerman, T. L., P. B. Conn, H. L. T. Mobley, and D. J. McGee. 2006. Nutritional requirements and antibiotic resistance patterns of Helicobacter species in chemically defined media. J. Clin. Microbiol. 44:1650-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tomb, J. F., O. White, A. R. Kerlavage, R. A. Clayton, G. G. Sutton, R. D. Fleischmann, K. A. Ketchum, H. P. Klenk, S. Gill, B. A. Dougherty, K. Nelson, J. Quackenbush, L. Zhou, E. F. Kirkness, S. Peterson, B. Loftus, D. Richardson, R. Dodson, H. G. Khalak, A. Glodek, K. McKenney, L. M. Fitzegerald, N. Lee, M. D. Adams, E. K. Hickey, D. E. Berg, J. D. Gocayne, T. R. Utterback, J. D. Peterson, J. M. Kelley, M. D. Cotton, J. M. Weidman, C. Fujii, C. Bowman, L. Watthey, E. Wallin, W. S. Hayes, M. Borodovsky, P. D. Karp, H. O. Smith, C. M. Fraser, and J. C. Venter. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539-547. [DOI] [PubMed] [Google Scholar]
- 57.Weeks, D. L., S. Eskandari, D. R. Scott, and G. Sachs. 2000. A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science 287:482-485. [DOI] [PubMed] [Google Scholar]
- 58.Wen, Y., E. A. Marcus, U. Matrubutham, M. A. Gleeson, D. R. Scott, and G. Sachs. 2003. Acid-adaptive genes of Helicobacter pylori. Infect. Immun. 71:5921-5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wood, J. M. 2007. Bacterial osmosensing transporters. Methods Enzymol. 428:77-107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.