

Abstract
Leishmania alternates between two morphologically different stages, promastigotes and amastigotes. While the majority of reports focused on how the promastigote form can alter macrophage (Mφ) signaling and function, fewer reports investigated signaling alterations mediated by amastigotes, and there is a lack of comparative studies. In this study, we performed a comparison between the ability of both forms of the parasite to alter Mφ signaling and functions. Here, we show that both promastigotes and amastigotes were able to rapidly activate host protein tyrosine phosphatases (PTPs), importantly the Src homology 2 domain-containing PTP (SHP-1). However, we found that PTP-1B is specifically activated by promastigote but not amastigote infection and that lmcpb−/− promastigotes were no longer able to activate PTP-1B. We also show a similarity in the way promastigotes and amastigotes inactivate the transcription factors (TFs) STAT-1α and AP-1, but we show differences in the modulation of NF-κB, with promastigotes cleaving the p65 subunit, generating a smaller p35 subunit, and amastigotes fully degrading the p65 subunit with no p35 production. Importantly, we show that the cysteine proteinase LmCPb plays a key role in the alteration of NF-κB, STAT-1α, and AP-1 by promastigote and amastigote infections, ultimately leading to the inability of these TFs to translocate to the nucleus in response to gamma interferon (IFN-γ) stimulation and thus contributing to the ability of both parasite forms to effectively block IFN-γ-mediated nitric oxide (NO) production in Mφs.
Leishmania parasites are prevalent in more than 80 countries in the world. It is estimated that there are 12 million cases of leishmaniasis worldwide, with 2 million new cases emerging every year (8). The most common manifestations of the disease are as follows: (i) visceral leishmaniasis caused by Leishmania donovani and Leishmania chagasi and responsible for the majority of mortality cases, (ii) cutaneous leishmaniasis caused principally by Leishmania major and Leishmania mexicana and is the most common manifestation of the disease, and (iii) the disfiguring mucocutaneous leishmaniasis caused by Leishmania braziliensis (23).
Leishmania is a dimorphic protozoan alternating between the promastigote and amastigote stages. Promastigotes have an elongated shape with long flagella and live inside the sand fly vector. Once promastigotes are deposited into mammalian skin while the sand fly is having a blood meal, they are phagocytosed by cells of the monocyte/macrophage (Mφ) lineage and rapidly transform into round amastigotes that are smaller than the promastigotes and lack external flagella (23). In addition to being morphologically different, the two life stages of the parasite have different surface molecule compositions. While infectious metacyclic promastigotes have a thick glycocalyx, this cover is almost completely absent in amastigotes (33). The glycocalyx is made of glycoproteins and other glycosylated species anchored to the surface membrane by a glycosylphosphatidylinositol (GPI) linkage (10). The promastigote surface is predominantly covered by lipophosphoglycan (LPG), a GPI-anchored molecule made of repeating units of a disaccharide and a phosphate. Buried in a sea of LPG, promastigotes have another important GPI-anchored molecule, the surface protease gp63. Interestingly, amastigotes have been shown to produce very little LPG compared to promastigotes (21) and have less gp63 (38) that, in the case of L. mexicana, has been shown to lack a GPI anchor and to be confined to the flagellar pocket (22).
Although LPG and, to a lesser extent, gp63 are the most studied virulence factors in Leishmania, other important virulence factors found in several Leishmania species include clan CA, family C1 cysteine proteinases (CPs) (26). L. mexicana has been shown to have cathepsin L-like CP genes (lmcpb) that are present in multiple copies and occur in a tandem array (39) and two single-copy CP genes, the cathepsin L-like gene (lmcpa) and the other cathepsin B-like gene (lmcpc). Unlike LPG and gp63, which are plentiful in promastigotes and are downregulated in amastigotes, LmCPb (L. mexicana cysteine proteinase b) is expressed at low levels in metacyclic promastigotes and is strongly upregulated in amastigotes (39), indicating that the protein may play a crucial role in the intracellular survival of the parasite.
Given that promastigotes have to avoid Mφ microbicidal action in order to establish themselves in the host and that amastigotes have to suppress Mφ killing abilities when they try to invade new Mφs in the course of a persistent Leishmania infection, it is not surprising that both forms of the parasite can alter key Mφ signaling pathways (31). Indeed, several studies have previously shown that promastigotes (1, 12, 35, 36) and amastigotes (24, 36, 41), or molecules derived from them, are able to block nitric oxide (NO) production by host Mφs in response to activating stimuli, such as gamma interferon (IFN-γ) or bacterial lipopolysaccharide (LPS). The promastigote surface contains several glycoconjugates allowing interaction with Mφs and internalization via several types of receptors, such as complement receptors 1, 3, and 4 (18, 40), the mannose fucose receptor (3), the C-reactive protein receptor (9), and the fibronectin receptor (6). On the other hand, amastigotes lack many of those glycoconjugates and seem to interact with Mφs mainly through glycosylinositol phospholipids (GIPLs) and to be phagocytosed via the Fc receptor following opsonization by antibodies or via complement receptors. Although redundancies might exist in the way promastigotes and amastigotes interact with Mφs and modulate their signaling in order to block their killing functions, the differences between both forms, whether at the gene expression, metabolic, or surface molecule level suggest to us that some differences ought to exist in the way those two forms can modulate Mφ signaling to their own favor. This work is an effort to compare the similarities and differences between promastigotes and amastigotes of L. mexicana in terms of their ability to alter key signaling molecules, namely, protein tyrosine phosphatases (PTPs) and the transcription factors (TFs) nuclear factor kappa B (NF-κB), signal transducer and activator of transcription 1alpha (STAT-1α), and activating protein 1 (AP-1), known to play a pivotal role in NO production (19, 42) as well as other Mφ functions detrimental to the survival of the Leishmania parasite (31). In addition, given the established role of LmCPb as a virulence factor and an immunomodulator (2, 27), this work explores the role of this proteinase in the Leishmania-induced alterations of signaling molecules that we observed.
MATERIALS AND METHODS
Cell culture and reagents.
The immortalized B10R bone marrow-derived Mφs (BMDMs) were derived from B10A.Bcgr mice (37). The immortalized me-3 (SHP-1−/−) and LM-1 (wild-type [WT]) BMDMs were generated from motheaten mice (Ptpn6me/me; C3HeBFeJ me/me) and their respective wild-type littermates (C3HeBFeJ me/+) as previously described (14). Recombinant murine IFN-γ was purchased from Cedarlane, NC. Antibodies used to immunoprecipitate SHP-1 and PTP-1B were purchased from Upstate, NY.
In vitro infection.
Promastigotes of L. mexicana (MNYC/BZ/62/M379), and L. mexicana deficient for LmCPb generated by targeted gene deletion as previously described (27) were kept in SDM medium (10% fetal bovine serum [FBS]) (32), and stationary-phase parasites were used to infect cells in a parasite-to-Mφ ratio ranging from 5:1 to 20:1. Axenic amastigotes of WT and lmcpb−/− L. mexicana were transformed from promastigote cultures, kept in MAA medium (medium for axenically grown amastigotes, pH 5.6, 20% FBS) (27), and incubated at a temperature of 32°C. When parasites were used for infections, noninternalized ones were removed by washing the plates with phosphate-buffered saline (PBS), after which Mφs were collected for subsequent experiments.
Western blot analysis.
Western blotting was performed as previously described (32). Proteins were detected using antibodies directed against gp63 (provided by Robert McMaster, University of British Columbia, Canada), LACK (provided by Eric Prina, Institut Pasteur, France), A2 (provided by Greg Matlashewski, McGill University, Canada), SHP-1 (Chemicon, CA), and PTP-1B (Upstate). Proteins were detected using a horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse antibody (Amersham, QC, Canada) and visualized using ECL Western blotting detection system (Amersham).
NO assay.
NO production was evaluated by measuring the accumulation of nitrite in the culture medium by the Griess reaction, as previously described (13).
pNPP phosphatase assay.
Mφs were collected, lysed in PTP lysis buffer (50 mM Tris [pH 7.0], 0.1 mM EDTA, 0.1 mM EGTA, 0.1% β-mercaptoethanol, 1% Igepal, 25 μg/ml aprotinin, and 25 μg/ml leupeptin), and kept on ice for 45 min. Lysates were cleared by centrifugation, and protein content was determined by using the Bradford reagent. For experiments measuring phosphatase activity in total cell lysates, the lysates (30 μg) were incubated in a phosphatase reaction mix (50 mM HEPES [pH 7.5], 0.1% β-mercaptoethanol, 10 mM p-nitrophenylphosphate [pNPP]) for 30 min at 37°C, and the optical density (OD) at 405 nm was read. For experiments measuring phosphatase activity in immunoprecipitates (IPs), cell lysates were subjected to immunoprecipitation using protein A/G agarose beads (Santa Cruz, CA) and 3 μg of the SHP-1, PTP-1B, or the anti-rat (Sigma-Aldrich, ON, Canada) antibody for nonspecific binding. The beads were spun down and washed three times with the PTP lysis buffer and then incubated with the phosphatase reaction mix for 4 to 6 h at 37°C, and the OD at 405 nm was read.
In-gel PTP assay.
Cell lysates (30 μg) were obtained using the PTP lysis buffer previously described. Samples were loaded on a gel containing a γ-32P-labeled poly(Glu4Tyr) peptide (Sigma-Aldrich), and PTP bands were observed by in-gel PTP assay as previously described (20).
EMSA.
Nuclear extracts were prepared by a standard protocol, and electrophoretic mobility shift assays (EMSAs) were performed as previously described (17). Briefly, nuclear extracts were incubated with binding buffer (11) containing 1.0 ng of [γ-32P]dATP radiolabeled double-stranded DNA oligonucleotide for 20 min at room temperature. The DNA binding consensus sequences used for transcription factors are as follows: 5′-AGTTGAGGGGACTTTCCCAGGC-3′ for NF-κB, 5′-AAGTACTTTCAGTTTCATATTACTCTA-3′ for STAT-1 (gamma interferon activation site [GAS]/interferon-stimulated response element [ISRE] consensus sequence), and 5′-AGCTCGCGTGACTCAGCTG-3′ for AP-1. Sp1 consensus oligonucleotide was used as a nonspecific control (5′-ATTCGATCGGGGCGGGGCGAGC-3′) (all DNA oligonucleotides were purchased from Santa Cruz). DNA-protein complexes were resolved by electrophoresis in native 4% (wt/vol) polyacrylamide gels. The gels were then dried and autoradiographed.
RESULTS
Promastigotes and amastigotes of L. mexicana efficiently block IFN-γ-mediated NO production in Mφs.
To investigate whether both promastigotes and amastigotes were able to inhibit IFN-γ-mediated NO production in our experimental system, NO levels were measured in B10R Mφs infected with one of the two forms, followed by IFN-γ stimulation. Results showed that both promastigotes and axenic amastigotes of L. mexicana were able to significantly block IFN-γ-mediated NO production in Mφs (Fig. 1A). Successful differentiation of L. mexicana promastigotes to amastigotes was confirmed by several methods: morphological changes seen by phase-contrast microscopy (data not shown), the remarkably decreased level of production of gp63 in amastigotes compared to promastigotes (Fig. 1B, top panel), and the detection of the amastigote-specific A2 protein in amastigote but not promastigote lysates (Fig. 1B, middle panel). The LACK D protein was used as a loading control (Fig. 1B, bottom panel).
FIG. 1.

Inhibition of IFN-γ-mediated NO production by L. mexicana promastigotes (Pro) and amastigotes (Ama). (A) NO assay of B10R Mφs left untreated (−) (bar 1), stimulated with IFN-γ (100 U/ml) for 24 h (+) (bar 2), infected with Leishmania (overnight [O/N], 20:1 ratio) (+) (bars 3 and 5), or infected with Leishmania (O/N, 20:1 ratio) and then stimulated with IFN-γ (100 U/ml) for 24 h (bars 4 and 6). The values are means plus standard errors of the means (SEMs) (error bars). Values that were significantly different (P < 0.05, analysis of variance [ANOVA] test) are indicated by an asterisk. (B) Western blot analysis of L. mexicana promastigote and amastigote cell lysates (40 μg). The membrane was cut and blotted for gp63 (top panel) and A2 (middle panel). The membrane was then stripped and reblotted for LACK D (bottom panel) to demonstrate equal loading. All results are representative of at least three independent experiments. w.b., Western blotted.
Promastigotes and amastigotes of L. mexicana activate PTPs in Mφs.
We have previously reported that Leishmania donovani promastigotes can rapidly increase total PTP activity in Mφs to alter their signaling pathways (5). Here, we performed pNPP phosphatase assays to compare the ability of promastigotes and amastigotes of L. mexicana to rapidly activate PTPs in Mφs. Results indicated that both promastigotes and amastigotes were able to rapidly increase total PTP activity in infected Mφs, reaching a peak activation value at 6 h postinfection and decreasing thereafter (Fig. 2A). To obtain a better understanding of the specific PTPs involved in this activation, we performed in-gel PTP assays screening for different Mφ PTPs and monitoring their alterations with promastigote or amastigote infections over a 24-h infection period. In-gel PTP assays showed that both promastigotes and amastigotes were able to rapidly activate the Src homology 2 domain-containing protein tyrosine phosphatase (SHP-1) seen by the appearance of a cleavage product associated with the activation of SHP-1 (Fig. 2B, top two arrows). Interestingly, while promastigote infection clearly activated PTP-1B, resulting in a cleavage fragment, the generation of this active cleaved form by amastigotes was minimal (Fig. 2B, bottom two arrows).
FIG. 2.

Activation of host PTPs by L. mexicana promastigotes and amastigotes. (A) B10R Mφs were infected with L. mexicana promastigotes or amastigotes for the specified time course (0 to 24 h, 20:1 ratio), and total PTP activity of Mφ lysates (20 μg) was measured using the pNPP phosphatase assay. (B) Cell lysates (30 μg) of Mφs infected with promastigotes or amastigotes of L. mexicana for the specified time course (0 to 24 h, 20:1 ratio) subjected to an in-gel PTP assay. The rightmost two lanes contain lysates (30 μg) from WT and SHP-1−/− Mφs to confirm the band corresponding to SHP-1. All results are representative of at least three independent experiments.
L. mexicana's cysteine proteinase LmCPb plays a role in PTP-1B but not SHP-1 activation in Mφs.
One important virulence factor expressed in metacyclic promastigotes and in amastigotes of L. mexicana is the cysteine proteinase LmCPb. To evaluate a possible role for LmCPb in the activation of SHP-1 and PTP-1B, we performed in-gel PTP assays, Western blotting, and pNPP phosphatase assays on lysates and immunoprecipitates (IPs) of Mφs infected with WT or lmcpb−/− promastigotes and amastigotes and evaluated the effects of these parasites on SHP-1 and PTP-1B cleavage and activation. Although there was some reduced cleavage of SHP-1's top band in Mφs infected with lmcpb−/− promastigotes and amastigotes (Fig. 3A, top arrow, and Fig. 3B, top panel), its lower cleavage band was still prominent (Fig. 3A, second arrow from the top, and Fig. 3B, top panel), and SHP-1's IP from Mφs infected by lmcpb−/− parasites exhibited elevated phosphatase activity, similar to that caused by WT parasites (Fig. 3C, top graph), suggesting that LmCPb is not required in the Leishmania-induced SHP-1 activation.
FIG. 3.

Role of LmCPb in PTP-1B activation. (A) B10R Mφs were infected with L. mexicana promastigotes and amastigotes of WT and lmcpb−/− parasites for the specified time course (0 to 6 h, 20:1 ratio), and cell lysates (30 μg) were subjected to an in-gel PTP assay. The rightmost two lanes contain lysates (30 μg) from WT and SHP-1−/− Mφs to confirm the band corresponding to SHP-1. (B) Samples in panel A were run on gels by SDS-PAGE and blotted for SHP-1 (top panel) and PTP-1B (bottom panel). w.b., Western blotted. (C) B10R Mφs were infected with L. mexicana promastigotes (P) and amastigotes (A) of WT and lmcpb−/− parasites (3 h, 20:1 ratio) followed by lysis and immunoprecipitation of SHP-1 or PTP-1B. The SHP-1 and PTP-1B IPs were subjected to a pNPP phosphatase assay (top and bottom graphs, respectively). The values are means plus standard errors of the means (SEMs) (error bars). Values that were significantly different (P < 0.05, ANOVA test) are indicated by an asterisk. Rabbit anti-rat IgG was used as a negative control for the IPs. All results are representative of at least three independent experiments.
Conversely, infection with lmcpb−/− parasites did not lead to the cleavage of the top PTP-1B band seen with WT promastigotes (Fig. 3A, third arrow from the top, and Fig. 3B, bottom panel), resulting in a strong reduction in the cleavage band that we observe in WT promastigote infection (Fig. 3A, bottom arrow, and Fig. 3B, bottom panel). This decrease in PTP-1B cleavage was associated with the inability of lmcpb−/− parasites to cause PTP-1B activation compared to WT promastigote infection (Fig. 3C, bottom graph). This set of data suggests that LmCPb seems to play a role in the activation of PTP-1B in L. mexicana promastigotes.
Ability of L. mexicana promastigotes and amastigotes to alter Mφ TFs.
Because TFs NF-κB, STAT-1α, and AP-1 are strong modulators of Mφ signaling, we decided to study the ability of promastigotes and amastigotes of L. mexicana to alter key Mφ TFs, namely, NF-κB, STAT-1α, and AP-1. We also evaluated the lowest parasite-to-cell ratio required to cause these alterations. EMSAs revealed that both promastigotes and amastigotes were able to cause the disappearance of the p65-containing subunit of NF-κB but with different results. Promastigotes cleaved p65 into a p35-containing subunit, while amastigotes caused a total degradation of this subunit with no observable production of the p35 subunit (Fig. 4A). Interestingly, both promastigotes and amastigotes inhibited STAT-1α and AP-1 activity, but generally, a stronger effect was seen with amastigote infection (Fig. 4B and C). Additionally, a parasite-to-cell ratio of 5:1 was sufficient to observe the previously mentioned alterations (Fig. 4A, B, and C) and was thus selected for the rest of the EMSA experiments conducted.
FIG. 4.

Modulation of Mφ TFs by L. mexicana promastigotes and amastigotes. B10R Mφs were infected with L. mexicana promastigotes and amastigotes at the specified parasite-to-cell ratios (3-h infection). (A to C) Nuclear proteins were extracted and subjected to EMSA to evaluate DNA-binding activity of NF-κB (A), STAT-1α (B), and AP-1 (C). S, specific competition (100-fold excess of specific nonradioactive oligonucleotide); NS, nonspecific competition (100-fold excess of nonspecific, nonradioactive Sp-1 oligonucleotide).
LmCPb plays a crucial role in the ability of promastigotes and amastigotes of L. mexicana to modulate NF-κB, STAT-1α, and AP-1.
To study the role of LmCPb in the modulation of the different transcription factors, we performed EMSAs to evaluate the ability of lmcpb−/− parasites to alter NF-κB, STAT-1α, and AP-1. Results revealed that both promastigotes and amastigotes of lmcpb−/− parasites were not able to alter the p65 subunit of NF-κB (Fig. 5A) or to inhibit STAT-1α (Fig. 5B) and AP-1 (Fig. 5C) activity. The previously mentioned TFs of Mφs infected with lmcpb−/− parasites remained intact and similar to that of uninfected Mφs, suggesting a crucial role for LmCPb in the cleavage/degradation of NF-κB and in the blockage of STAT-1α and AP-1 activity in host Mφs.
FIG. 5.

Role of LmCPb in the modulation of Mφ TFs. B10R Mφs infected for the specified time course (0.5 to 3 h) with WT or lmcpb−/− promastigotes and amastigotes of L. mexicana in a 5:1 parasite-to-cell ratio. Nuclear proteins were extracted and subjected to EMSA to evaluate DNA-binding activity of NF-κB (A), STAT-1α (B), and AP-1 (C). S, specific competition (100-fold excess of specific nonradioactive oligonucleotide); NS, nonspecific competition (100-fold excess of nonspecific, nonradioactive Sp-1 oligonucleotide).
lmcpb-rescued parasites alter Mφ signaling in a similar manner to WT parasites.
To rule out the possibility that the protection of transcription factor alterations that we observe when Mφs are infected with lmcpb−/− parasites is due to the attenuation of those parasites as they are multiply passaged in the knocking-out process, we investigated whether lmcpb add-back parasites were also able to inhibit NF-κB the way WT parasites did. Results showed that indeed this was the case (Fig. 6), suggesting that the lack of effect observed in lmcpb−/− parasite-infected Mφs is indeed due to the absence of this protease and not an effect of parasite attenuation.
FIG. 6.
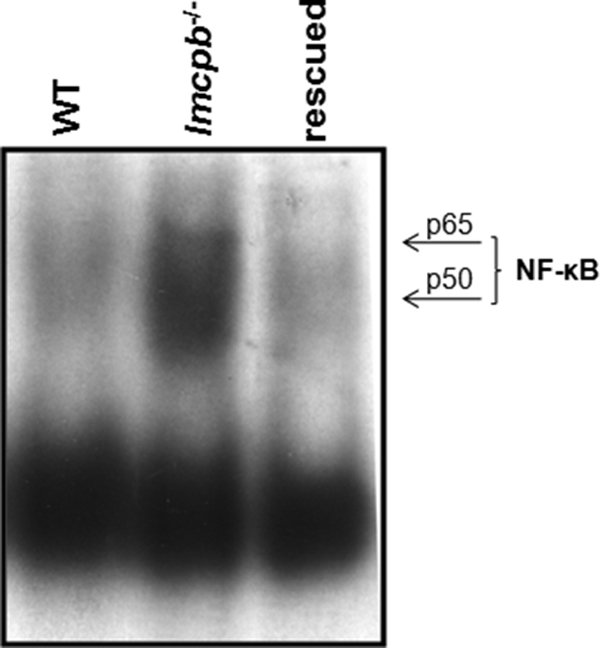
Effect of lmcpb-rescued parasites on Mφ NF-κB. B10R Mφs were infected for 1 h with L. mexicana amastigotes of WT, lmcpb−/−, or lmcpb-rescued parasites. Nuclear proteins were extracted and subjected to EMSA to evaluate DNA-binding activity of NF-κB.
Unlike infection by WT parasites, TFs of Mφs infected with lmcpb−/− parasites retain their ability to be activated by IFN-γ.
To evaluate whether the alteration of NF-κB, STAT-1α, and AP-1 by WT promastigotes and amastigotes interferes with the ability of those TFs to translocate to the nucleus and to evaluate the role of LmCPb in this context, we performed EMSAs. Results indicated that infection with WT promastigotes and amastigotes of L. mexicana rendered p65-containing subunits of NF-κB, STAT-1α, and AP-1 unresponsive to IFN-γ stimulation (Fig. 7A, B, and C, respectively). Interestingly, when Mφs were infected with lmcpb−/− parasites (Fig. 7A, B, and C), these TFs were able to translocate to the nucleus and bind their consensus sequences in a manner similar to that of the IFN-γ-only positive control.
FIG. 7.

Nuclear translocation of TFs in response to IFN-γ in Mφs infected with WT or lmcpb−/− L. mexicana promastigotes (P) and amastigotes (A). B10R Mφs were left uninfected, stimulated with IFN-γ (100 U/ml) for 6 h (+), and infected (overnight [O/N]) with WT or lmcpb−/− promastigotes and amastigotes of L. mexicana in a 5:1 parasite-to-cell ratio, or they were infected (O/N) and then stimulated with IFN-γ (100 U/ml) for 6 h. (A to C) Nuclear proteins were extracted and subjected to EMSA to evaluate the DNA-binding activity of NF-κB (A), STAT-1α (B), and AP-1 (C). S, specific competition (100-fold excess of specific nonradioactive oligonucleotide); NS, nonspecific competition (100-fold excess of nonspecific, nonradioactive Sp-1 oligonucleotide).
Promastigotes and amastigotes of WT L. mexicana but not lmcpb−/− parasites are able to inhibit Mφ NO production in response to IFN-γ stimulation.
To evaluate the impact of LmCPb on Mφ function, we performed NO assays to test whether lmcpb−/− parasites are able to block IFN-γ-mediated NO production by Mφs in a fashion similar to that observed by WT parasites. Results showed that while WT promastigotes and amastigotes successfully inhibited IFN-γ-mediated NO production as illustrated in Fig. 1A, lmcpb−/− parasites were not able to do so. The levels of NO produced in response to lmcpb−/− parasites were comparable to those produced by uninfected cells stimulated with IFN-γ (Fig. 8).
FIG. 8.
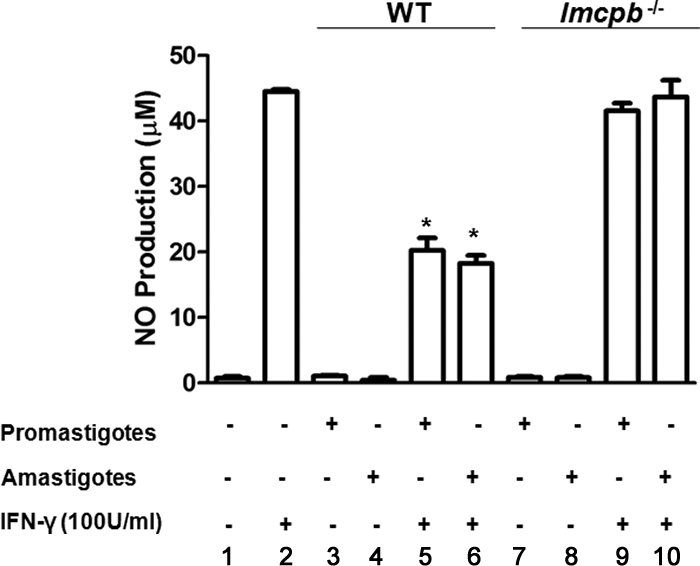
Inability of lmcpb−/− parasites to inhibit IFN-γ-mediated Mφ NO production. NO assay of B10R Mφs left untreated (−) (bar 1), stimulated with IFN-γ (100 U/ml) for 24 h (+) (bar 2), infected with WT (bars 3 and 4) or lmcpb−/− (bars 7 and 8) L. mexicana (O/N, 20:1 ratio) (+), or infected (O/N, 20:1 ratio) and then stimulated with IFN-γ (100 U/ml) for 24 h (bars 5 and 6 and 9 and 10). The values are means plus standard errors of the means (SEMs) (error bars). Values that were significantly different (P < 0.05, ANOVA test) are indicated by an asterisk. The results are representative of at least three independent experiments.
DISCUSSION
NO production by Mφs plays a key role in the resolution of Leishmania infections. Indeed, there are several publications reporting the ability of promastigotes, amastigotes, or various parasite molecules, such as lipophosphoglycan (LPG) (35) and glycosylinositol phospholipids (GIPLs) (36), to inhibit Mφ NO production in response to activating stimuli. Of utmost interest, none of those previous studies compared the ability of both forms of the parasite to inhibit Mφ NO production and the negative regulatory mechanisms they employ to cause this inhibition.
We have previously reported the ability of Leishmania promastigotes to rapidly activate host PTPs (5). Furthermore, we and others have shown that host SHP-1 is one of the key PTPs activated by Leishmania (5, 12-14, 30). However, very little work has been done concerning the ability of amastigotes to activate host PTPs and SHP-1 in particular. The key role that SHP-1 plays in amastigote infection can be deduced from in vivo studies where amastigotes are exclusively found and where the absence of SHP-1 was associated with increased resistance to Leishmania infection (14). Additionally, it has been previously reported that Leishmania donovani amastigotes are able to activate host SHP-1, yet the effect was claimed to be observable after 17 h of infection (29). In this work, we confirm previous observations regarding the ability of promastigotes to activate host PTPs (e.g., SHP-1). Importantly, we demonstrate that amastigotes can rapidly activate host PTP activity (as early as 0.5 h postinfection) (Fig. 2A) and that SHP-1 is a key phosphatase rapidly activated by amastigote infection (Fig. 2B and Fig. 3A, B, and C). This suggests that SHP-1 is a critical phosphatase utilized by both forms of the parasite to inactivate key Mφ signaling pathways.
Interestingly, PTP-1B was activated by promastigote but not amastigote infection (Fig. 2B and Fig. 3A, B, and C). This phosphatase has been reported to bind to and negatively regulate JAK2 (28), while knockdown and overexpression studies suggested its role in the regulation of MyD88-dependent proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), and the inhibition of NF-κB and STAT-1 activation in Toll-like receptor (TLR)-triggered Mφs (43). Given that SHP-1 has also been associated with the ability to negatively regulate JAK2 (5), block LPS-mediated TNF production (1), and control NF-κB and AP-1 activity (4), this finding suggests that PTP-1B activated by promastigotes might have an additive effect to SHP-1 action to help establish the parasite in host cells. Indeed, data from our laboratory demonstrated that Leishmania promastigote gp63 activated PTP-1B in a cleavage-dependent manner and that PTP-1B−/− mice infected with L. major promastigotes showed a significant delay in the onset and progression of footpad inflammation as well as lower parasite burden (15). Therefore, the selective activation of PTP-1B by promastigotes is plausible, given that they are less adapted to the host environment compared to amastigotes that might find SHP-1 action sufficient to ensure their safe entry to new uninfected phagocytes, especially since they seem to affect some signaling molecules more drastically upon initial contact with Mφs, such as the strong inhibition they cause to NF-κB, STAT-1, and AP-1 compared to promastigotes (Fig. 4). It is worth mentioning, however, that the utilization of host PTPs other than SHP-1 by amastigotes is a possibility that cannot be disregarded and deserves further investigation.
Consistent with a previous finding from our group, we have shown that promastigotes cause cleavage of NF-κB's p65 subunit, generating a smaller p35 subunit (16). On the other hand, amastigotes caused complete NF-κB degradation (Fig. 4A) as previously shown (7). Also, consistent with previous findings, promastigotes were able to cause degradation of STAT-1α (11) and inhibition of AP-1 (12) in Mφs, but whether LmCPb degrades STAT-1α on its own or in conjunction with the proteasome, as we previously described for L. donovani (11), remains to be determined. Importantly, we were able to demonstrate for the first time that amastigotes of L. mexicana can also cause rapid STAT-1α and AP-1 inhibition (Fig. 4). Very interestingly, all the alterations caused by promastigotes and amastigotes to NF-κB, STAT-1α, and AP-1 did not take place when Mφs were infected with lmcpb−/− parasites. LmCPb has been previously reported to play a role in the degradation of IκB and NF-κB (7), interleukin 2 receptor (IL-2R), and the immunoglobulin E receptor(IgER) (34). Our data confirm the role of LmCPb in NF-κB degradation (Fig. 5A) and demonstrate for the first time that LmCPb leads to the inhibition of STAT-1α and AP-1 nuclear translocation and DNA-binding activity, possibly by causing their degradation (Fig. 5B and C) or by interfering with key phosphorylation steps (25, 44) required for their translocation to the nucleus. The protection of these transcription factors seen when Mφs were infected with lmcpb−/− parasites is reflected by the ability of these transcription factors to translocate to the nucleus and bind their target sequences in response to IFN-γ stimulation (Fig. 7), which correlated with the ability of Mφs to produce NO when stimulated with IFN-γ following infection with lmcpb−/− parasites (Fig. 8). Importantly, the protection observed with lmcpb−/− parasites cannot be due to their lower ability to enter Mφs, since staining experiments that we performed showed that WT and lmcpb−/− parasites could infect Mφs at similar rates (around 35% of total Mφs become infected in a 1-h period [data not shown]), nor can it be due to their attenuation while they were multiply passaged in the knocking-out process, as signaling alterations could again be seen when the Mφs were infected with lmcpb-rescued parasites (Fig. 6). Collectively, the LmCPb-related findings presented here could help partially explain the previously observed low infectivity of cpb mutants (27), as well as further confirm the role of this cysteine proteinase as a Leishmania virulence factor.
In conclusion, our comparison of promastigotes and amastigotes of L. mexicana in terms of their ability to alter Mφ signaling yielded several interesting similarities as well as differences. While both forms were able to activate total PTP activity and to rapidly activate SHP-1, PTP-1B was activated only by promastigotes. Additionally, while both forms caused full inhibition of STAT-1α and AP-1, NF-κB seemed to be altered differently. Promastigotes cleaved NF-κB's p65 subunit to p35 while amastigotes caused complete p65 inhibition with no significant production of the smaller subunit. Importantly, this study also revealed new roles for LmCPb as a virulence factor by demonstrating its ability to interfere with STAT-1α and AP-1 transcriptional activity, therefore contributing to the parasite's ability to block IFN-γ-induced NO production in Mφs, securing its survival and propagation within its mammalian host.
Acknowledgments
This study is supported by operating grants from the Canadian Institute of Health Research (CIHR) to M.O. and the Wellcome Trust (WT 079356) to J.C.M. I.A.-D. is a recipient of a Fonds de la Recherche en Santé du Québec (FRSQ) Ph.D. Studentship Award.
Editor: J. F. Urban, Jr.
Footnotes
Published ahead of print on 5 April 2010.
REFERENCES
- 1.Abu-Dayyeh, I., M. T. Shio, S. Sato, S. Akira, B. Cousineau, and M. Olivier. 2008. Leishmania-induced IRAK-1 inactivation is mediated by SHP-1 interacting with an evolutionarily conserved KTIM motif. PLoS Negl. Trop. Dis. 2:e305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander, J., G. H. Coombs, and J. C. Mottram. 1998. Leishmania mexicana cysteine proteinase-deficient mutants have attenuated virulence for mice and potentiate a Th1 response. J. Immunol. 161:6794-6801. [PubMed] [Google Scholar]
- 3.Blackwell, J. M., R. A. B. Ezekowitz, M. B. Roberts, J. Y. Channon, R. B. Sim, and S. Gordon. 1985. Macrophage complement and lectin-like receptors bind Leishmania in the absence of serum. J. Exp. Med. 162:324-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blanchette, J., I. Abu-Dayyeh, K. Hassani, L. Whitcombe, and M. Olivier. 2009. Regulation of macrophage nitric oxide production by the protein tyrosine phosphatase Src homology 2 domain phosphotyrosine phosphatase 1 (SHP-1). Immunology 127:123-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanchette, J., N. Racette, R. Faure, K. A. Siminovitch, and M. Olivier. 1999. Leishmania-induced increases in activation of macrophage SHP-1 tyrosine phosphatase are associated with impaired IFN-gamma-triggered JAK2 activation. Eur. J. Immunol. 29:3737-3744. [DOI] [PubMed] [Google Scholar]
- 6.Brittingham, A., G. Chen, B. S. McGwire, K. P. Chang, and D. M. Mosser. 1999. Interaction of Leishmania gp63 with cellular receptors for fibronectin. Infect. Immun. 67:4477-4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cameron, P., A. McGachy, M. Anderson, A. Paul, G. H. Coombs, J. C. Mottram, J. Alexander, and R. Plevin. 2004. Inhibition of lipopolysaccharide-induced macrophage IL-12 production by Leishmania mexicana amastigotes: the role of cysteine peptidases and the NF-κB signaling pathway. J. Immunol. 173:3297-3304. [DOI] [PubMed] [Google Scholar]
- 8.Choi, C. M., and E. A. Lerner. 2001. Leishmaniasis as an emerging infection. J. Investig. Dermatol. Symp. Proc. 6:175-182. [DOI] [PubMed] [Google Scholar]
- 9.Culley, F. J., R. A. Harris, P. M. Kaye, K. McAdam, and J. G. Raynes. 1996. C-reactive protein binds to a novel ligand on Leishmania donovani and increases uptake into human macrophages. J. Immunol. 156:4691-4696. [PubMed] [Google Scholar]
- 10.Ferguson, M. A. 1997. The surface glycoconjugates of trypanosomatid parasites. Philos. Trans. R. Soc. Lond. B Biol. Sci. 352:1295-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forget, G., D. J. Gregory, and M. Olivier. 2005. Proteasome-mediated degradation of STAT1 alpha following infection of macrophages with Leishmania donovani. J. Biol. Chem. 280:30542-30549. [DOI] [PubMed] [Google Scholar]
- 12.Forget, G., D. J. Gregory, L. A. Whitcombe, and M. Olivier. 2006. Role of host protein tyrosine phosphatase SHP-1 in Leishmania donovani-induced inhibition of nitric oxide production. Infect. Immun. 74:6272-6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forget, G., C. Matte, K. A. Siminovitch, S. Rivest, P. Pouliot, and M. Olivier. 2005. Regulation of the Leishmania-induced innate inflammatory response by the protein tyrosine phosphatase SHP-1. Eur. J. Immunol. 35:1906-1917. [DOI] [PubMed] [Google Scholar]
- 14.Forget, G., K. A. Siminovitch, S. Brochu, S. Rivest, D. Radzioch, and M. Olivier. 2001. Role of host phosphotyrosine phosphatase SHP-1 in the development of murine leishmaniasis. Eur. J. Immunol. 31:3185-3196. [DOI] [PubMed] [Google Scholar]
- 15.Gomez, M. A., I. Contreras, M. Halle, M. L. Tremblay, R. W. McMaster, and M. Olivier. 2009. Leishmania GP63 alters host signaling through cleavage-activated protein tyrosine phosphatases. Sci. Signal. 2:ra58. [DOI] [PubMed] [Google Scholar]
- 16.Gregory, D. J., M. Godbout, I. Contreras, G. Forget, and M. Olivier. 2008. A novel form of NF-kappa B is induced by Leishmania infection: involvement in macrophage gene expression. Eur. J. Immunol. 38:1071-1081. [DOI] [PubMed] [Google Scholar]
- 17.Jaramillo, M., and M. Olivier. 2002. Hydrogen peroxide induces murine macrophage chemokine gene transcription via extracellular signal-regulated kinase and cyclic adenosine 5′-monophosphate (cAMP)-dependent pathways: involvement of NF-kappa B, activator protein 1, and cAMP response element binding protein. J. Immunol. 169:7026-7038. [DOI] [PubMed] [Google Scholar]
- 18.Kane, M. M., and D. M. Mosser. 2000. Leishmania parasites and their ploys to disrupt macrophage activation. Curr. Opin. Hematol. 7:26-31. [DOI] [PubMed] [Google Scholar]
- 19.Lowenstein, C. J., E. W. Alley, P. Raval, A. M. Snowman, S. H. Snyder, S. W. Russell, and W. J. Murphy. 1993. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon-gamma and lipopolysaccharide. Proc. Natl. Acad. Sci. U. S. A. 90:9730-9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Markova, B., P. Gulati, P. A. Herlich, and F. D. Bohmer. 2005. Investigation of protein-tyrosine phosphatases by in-gel assays. Methods 35:22-27. [DOI] [PubMed] [Google Scholar]
- 21.McConville, M. J., and J. M. Blackwell. 1991. Developmental changes in the glycosylated phosphatidylinositols of Leishmania donovani: characterization of the promastigote and amastigote glycolipids. J. Biol. Chem. 266:15170-15179. [PubMed] [Google Scholar]
- 22.Medina-Acosta, E., R. E. Karess, H. Schwartz, and D. G. Russell. 1989. The promastigote surface protease (gp63) of Leishmania is expressed but differentially processed and localized in the amastigote stage. Mol. Biochem. Parasitol. 37:263-273. [DOI] [PubMed] [Google Scholar]
- 23.Melby, P. C. 2002. Recent developments in leishmaniasis. Curr. Opin. Infect. Dis. 15:485-490. [DOI] [PubMed] [Google Scholar]
- 24.Melby, P. C., B. Chandrasekar, W. G. Zhao, and J. E. Coe. 2001. The hamster as a model of human visceral leishmaniasis: progressive disease and impaired generation of nitric oxide in the face of a prominent Th1-like cytokine response. J. Immunol. 166:1912-1920. [DOI] [PubMed] [Google Scholar]
- 25.Morton, S., R. J. Davis, A. McLaren, and P. Cohen. 2003. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 22:3876-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mottram, J. C., G. H. Coombs, and J. Alexander. 2004. Cysteine peptidases as virulence factors of Leishmania. Curr. Opin. Microbiol. 7:375-381. [DOI] [PubMed] [Google Scholar]
- 27.Mottram, J. C., A. E. Souza, J. E. Hutchison, R. Carter, M. J. Frame, and G. H. Coombs. 1996. Evidence from disruption of the lmcpb gene array of Leishmania mexicana that cysteine proteinases are virulence factors. Proc. Natl. Acad. Sci. U. S. A. 93:6008-6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myers, M. P., J. N. Andersen, A. Cheng, M. L. Tremblay, C. M. Horvath, J.-P. Parisien, A. Salmeen, D. Barford, and N. K. Tonks. 2001. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J. Biol. Chem. 276:47771-47774. [DOI] [PubMed] [Google Scholar]
- 29.Nandan, D., R. Lo, and N. E. Reiner. 1999. Activation of phosphotyrosine phosphatase activity attenuates mitogen-activated protein kinase signaling and inhibits c-FOS and nitric oxide synthase expression in macrophages infected with Leishmania donovani. Infect. Immun. 67:4055-4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nandan, D., T. L. Yi, M. Lopez, C. Lai, and N. E. Reiner. 2002. Leishmania EF-1 alpha activates the Src homology 2 domain containing tyrosine phosphatase SHP-1 leading to macrophage deactivation. J. Biol. Chem. 277:50190-50197. [DOI] [PubMed] [Google Scholar]
- 31.Olivier, M., D. J. Gregory, and G. Forget. 2005. Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin. Microbiol. Rev. 18:293-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olivier, M., B. J. Romero-Gallo, C. Matte, J. Blanchette, B. I. Posner, M. J. Tremblay, and R. Faure. 1998. Modulation of interferon-gamma-induced macrophage activation by phosphotyrosine phosphatases inhibition: effect on murine leishmaniasis progression. J. Biol. Chem. 273:13944-13949. [DOI] [PubMed] [Google Scholar]
- 33.Pimenta, P. F. P., E. M. B. Saraiva, and D. L. Sacks. 1991. The comparative fine structure and surface glycoconjugate expression of three life stages of Leishmania major. Exp. Parasitol. 72:191-204. [DOI] [PubMed] [Google Scholar]
- 34.Pollock, K. G. J., K. S. McNeil, J. C. Mottram, R. E. Lyons, J. M. Brewer, P. Scott, G. H. Coombs, and J. Alexander. 2003. The Leishmania mexicana cysteine protease, CPB2.8, induces potent Th2 responses. J. Immunol. 170:1746-1753. [DOI] [PubMed] [Google Scholar]
- 35.Proudfoot, L., A. V. Nikolaev, G. J. Feng, X. Q. Wei, M. A. J. Ferguson, J. S. Brimacombe, and F. Y. Liew. 1996. Regulation of the expression of nitric oxide synthase and leishmanicidal activity by glycoconjugates of Leishmania lipophosphoglycan in murine macrophages. Proc. Natl. Acad. Sci. U. S. A. 93:10984-10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Proudfoot, L., C. A. Odonnell, and F. Y. Liew. 1995. Glycoinositolphospholipids of Leishmania major inhibit nitric oxide synthesis and reduce leishmanicidal activity in murine macrophages. Eur. J. Immunol. 25:745-750. [DOI] [PubMed] [Google Scholar]
- 37.Radzioch, D., T. Hudson, M. Boule, L. Barrera, J. W. Urbance, L. Varesio, and E. Skamene. 1991. Genetic resistance/susceptibility to mycobacteria: phenotypic expression in bone marrow derived macrophage lines. J. Leukoc. Biol. 50:263-272. [DOI] [PubMed] [Google Scholar]
- 38.Schneider, P., J. P. Rosat, J. Bouvier, J. Louis, and C. Bordier. 1992. Leishmania major: differential regulation of the surface metalloprotease in amastigote and promastigote stages. Exp. Parasitol. 75:196-206. [DOI] [PubMed] [Google Scholar]
- 39.Souza, A. E., S. Waugh, G. H. Coombs, and J. C. Mottram. 1992. Characterization of a multicopy gene for a major stage-specific cysteine proteinase of Leishmania mexicana. FEBS Lett. 311:124-127. [DOI] [PubMed] [Google Scholar]
- 40.Talamas-Rohana, P., S. D. Wright, M. R. Lennartz, and D. G. Russell. 1990. Lipophosphoglycan from Leishmania mexicana promastigotes binds to members of the CR3, p150,95 and LFA-1 family of leukocyte integrins. J. Immunol. 144:4817-4824. [PubMed] [Google Scholar]
- 41.Wanderley, J. L. M., M. E. C. Moreira, A. Benjamin, A. C. Bonomo, and M. A. Barcinski. 2006. Mimicry of apoptotic cells by exposing phosphatidylserine participates in the establishment of amastigotes of Leishmania (L) amazonensis in mammalian hosts. J. Immunol. 176:1834-1839. [DOI] [PubMed] [Google Scholar]
- 42.Xie, Q. W., R. Whisnant, and C. Nathan. 1993. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon-gamma and bacterial lipopolysaccharide. J. Exp. Med. 177:1779-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu, H. M., H. Z. An, J. Hou, C. F. Han, P. Wang, Y. Z. Yu, and X. T. Cao. 2008. Phosphatase PTP1B negatively regulates MyD88- and TRIF-dependent proinflammatory cytokine and type I interferon production in TLR-triggered macrophages. Mol. Immunol. 45:3545-3552. [DOI] [PubMed] [Google Scholar]
- 44.Zhu, X., Z. Wen, L. Z. Xu, and J. E. Darnell, Jr. 1997. Stat1 serine phosphorylation occurs independently of tyrosine phosphorylation and requires an activated Jak2 kinase. Mol. Cell. Biol. 17:6618-6623. [DOI] [PMC free article] [PubMed] [Google Scholar]