

Abstract
Virus-infected cells secrete a broad range of interferons (IFN) which confer resistance to yet uninfected cells by triggering the synthesis of antiviral factors. The relative contributions of the various IFN subtypes to innate immunity against virus infections remain elusive. IFN-α, IFN-β, and other type I IFN molecules signal through a common, universally expressed cell surface receptor, whereas type III IFN (IFN-λ) uses a distinct cell-type-specific receptor complex for signaling. Using mice lacking functional receptors for type I IFN, type III IFN, or both, we found that IFN-λ plays an important role in the defense against several human pathogens that infect the respiratory tract, such as influenza A virus, influenza B virus, respiratory syncytial virus, human metapneumovirus, and severe acute respiratory syndrome (SARS) coronavirus. These viruses were more pathogenic and replicated to higher titers in the lungs of mice lacking both IFN receptors than in mice with single IFN receptor defects. In contrast, Lassa fever virus, which infects via the respiratory tract but primarily replicates in the liver, was not influenced by the IFN-λ receptor defect. Careful analysis revealed that expression of functional IFN-λ receptor complexes in the lung and intestinal tract is restricted to epithelial cells and a few other, undefined cell types. Interestingly, we found that SARS coronavirus was present in feces from infected mice lacking receptors for both type I and type III IFN but not in those from mice lacking single receptors, supporting the view that IFN-λ contributes to the control of viral infections in epithelial cells of both respiratory and gastrointestinal tracts.
The interferon (IFN) system represents a major element of the innate immune response against viral infections (10, 13, 14). Virus-induced IFN is a complex mixture of biologically active molecules, which includes type I and type III IFN. Type I IFN consists of 14 different IFN-α subtypes in the mouse as well as IFN-β, IFN-κ, IFN-ɛ, and limitin, which all signal through the same universally expressed cell surface receptor complex (IFNAR) (30). Type III IFN includes IFN-λ1, IFN-λ2, and IFN-λ3 (21, 28), of which only the latter two are encoded by genes that are expressed in the mouse (22). Type III IFN uses a distinct receptor complex (IL28R) for signaling (21, 28), which appears to be expressed on only a few cell types, including epithelial cells (29). Binding of type I IFN and type III IFN to their cognate receptor complexes triggers signaling cascades that result in the activation of a large number of genes, many of which encode antiviral proteins (10, 32). Type I IFN and type III IFN trigger highly similar gene expression profiles in responsive cells, suggesting that both IFN types might serve similar functions. However, it has to date been largely unclear to which extent IFN-λ might contribute to innate immunity.
Using knockout mouse strains that lack receptors for type I IFN (IFNAR10/0), type III IFN (IL28Rα0/0), or both (IFNAR10/0IL28Rα0/0), we have recently shown that IFN-λ contributes to resistance against influenza A virus (FLUAV) (26). Here, we used the same mouse strains to investigate the relative contribution of IFN-λ in resistance against additional viral pathogens that infect the respiratory and gastrointestinal tract and to visualize IFN-λ-responsive cells. We found that the double-knockout mice showed enhanced susceptibility to various viruses that primarily replicate in lung epithelial cells. Our analysis further revealed that epithelial cells of both lung and gastrointestinal tracts can strongly respond to IFN-λ and that IFN-λ inhibited the replication of severe acute respiratory syndrome coronavirus (SARS-CoV) in both lung and gastrointestinal tracts.
MATERIALS AND METHODS
Ethics statement.
All animal experiments were performed in compliance with the German animal protection law (TierSchG). The mice were housed and handled in accordance with good animal practice as defined by FELASA (www.felasa.eu/guidelines.php) and the national animal welfare body GV-SOLAS (www.gv-solas.de/index.html). The animal welfare committees of the universities of Freiburg, Bonn, and Hamburg as well as the local authorities (Regierungspräsidium Freiburg; Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen; and Behörde für Soziales, Familie, Gesundheit und Verbraucherschutz, Hamburg) approved all animal experiments.
Mice.
If not otherwise indicated, all animals used were of C57BL/6 genetic background. B6.A2G-Mx1 mice (16) carrying intact Mx1 alleles (designated wild type) and B6.A2G-Mx1-IFNAR10/0 mice (20) lacking functional type I IFN receptors (designated IFNAR10/0) were bred locally. B6.A2G-Mx1-IL28Rα0/0 mice lacking functional type III IFN receptors (designated IL28Rα0/0) and B6.A2G-Mx1-IL28Rα0/0IFNAR10/0 mice lacking both type I and type III IFN receptors (designated IFNAR10/0IL28Rα0/0) were previously described (26). C57BL/6 mice lacking functional STAT10/0 (11) were a kind gift from Thomas Decker, Vienna, Austria. BALB/c mice were obtained from Charles River, Sulzfeld, Germany. Six- to eight-week-old animals were used for all infection experiments, which were performed in accordance with the guidelines of the local animal care committee. Animals were euthanized if severe symptoms developed or body weight loss approached 30% of the initial value.
Viruses.
Influenza A viruses included the pandemic swine origin H1N1 isolate A/HH/05/2009 (a kind gift from Markus Eickmann, Marburg, Germany) (7) and mutant laboratory strain SC35M-ΔNS1 (H7N7), which lacks the IFN-antagonistic factor NS1 (19). We further used influenza B virus (FLUBV) strain B/Lee/40 (a kind gift from Thorsten Wolff), human respiratory syncytial virus (RSV) strain A2 (originally obtained from Peter Openshaw, Imperial College, London, United Kingdom), human metapneumovirus (HMPV) strain DO3-574 (17), SARS-CoV strain Frankfurt-1 (9), and Lassa fever virus strain AV (12). For systemic induction of IFN, we made use of the attenuated “clone 13” strain of Rift Valley fever virus (RVFV), which lacks functional IFN-antagonistic factor NSs (4). Experiments with SARS-CoV and Lassa fever virus were performed under BSL3 and BSL4 conditions, respectively. All other viruses used in this study are classified as BSL2 pathogens in Germany. Stocks of the various influenza A virus strains were prepared in MDCK cells, stocks of RSV in Hep-2 cells, stocks of HMPV in LLC-MK2 cells, stocks of SARS-CoV and RVFV in Vero cells, and stocks of Lassa fever virus in BHK-21 cells. Influenza B virus was grown in embryonated chicken eggs.
Virus infections.
Animals were anesthetized by intraperitoneal injection of a mixture of ketamine (100 μg per gram body weight) and xylazine (5 μg per gram body weight) before intranasal infection with the indicated doses of the respiratory viruses in 50 μl of phosphate-buffered saline (PBS) containing 0.3% bovine serum albumin (BSA). For RVFV infections, 100-μl samples of diluted virus stocks were applied intraperitoneally without anesthesia. The units of infection were numbers of PFU per ml for HMPV, Lassa fever virus, RVFV, and SARS-CoV and numbers of focus-forming units (FFU) per ml for FLUAV, FLUBV, and RSV.
Plasmid electrotransfer-mediated expression of IFN.
The experiments were performed as previously described (29). Briefly, 10 or 25 μg of plasmid DNA was electrotransfered in each tibialis muscle. Mx1 responsiveness was examined by immunohistochemistry on day 7 postelectrotransfer, as described previously (29).
Immunohistochemistry.
Perfused tissue was immersed in 4% buffered formaldehyde for 12 h at 4°C before being embedded in paraffin. Tissue sections of approximately 5 μm were placed on SuperFrost Plus slides, dried at room temperature overnight, and processed by standard methods for immunohistochemistry. Briefly, sections were deparaffinized, permeabilized for 5 min in PBS containing 0.5% Triton X-100, and washed in PBS. Blocking was performed by incubating sections for 90 min with 10% normal goat serum (Sigma) diluted in PBS. Sections were then treated for 10 min at 121°C in 0.01 M sodium citrate buffer at pH 6.0 to unmask antigens. Then, immunolabeling was done in blocking solution containing the antibodies. Mx1 protein was detected with rabbit polyclonal antibody AP5, which recognizes the C-terminal 16 amino acids of Mx1 (25). AP5 was used at a dilution of 1/500. For immunofluorescence labeling, the secondary antibody (at 1/800) was a goat anti-rabbit antibody coupled to Alexa 488 (Invitrogen) or a donkey anti-rabbit antibody coupled to Alexa 555 (Invitrogen).
Titration of virus in lungs.
Lung homogenates of animals infected with RSV, HMPV, A/HH/05/2009, or FLUBV were prepared using the FastPrep24 system (MP Biomedicals). After addition of 800 μl of PBS containing 0.3% BSA, organs were subjected to two rounds of mechanical treatment for 15 s each at 6.5 m/s, with 5 min of incubation on ice between the cycles. Tissue debris was removed by low-speed centrifugation. Virus titers in supernatants were determined by immunofluorescence assays with MDCK II cells (FLUBV and A/HH/05/2009), Hep2 cells (RSV), or Vero cells (SARS-CoV) by serial 10-fold dilutions in PBS containing 0.3% BSA. We prepared Lassa virus stocks and measured viral infectivity in blood samples as previously described (2).
RT-PCR.
SARS-CoV RNA concentrations were determined by reverse transcription-PCR (RT-PCR) as previously described in reference 9, using Qiagen RNeasy columns for extraction of RNA from organ homogenates according to provided protocols. HMPV was detected by RT-PCR based on a method described previously (8). Briefly, a Qiagen one-step RT-PCR kit (Qiagen, Hilden, Germany) was used according to the manufacturer's instructions. This 25-μl reaction mixture consisted of 1× OneStep buffer, 400 μM deoxynucleoside triphosphate (dNTP) mix, 400 nM forward primer, 400 nM reverse primer, Qiagen OneStep RT-PCR enzyme mix, 200 nM probe, template RNA, and sterile water. Amplification was performed using a LightCycler instrument (Roche Applied Science) with the following cycling conditions: an RT step at 50°C for 30 min and a Taq activation step at 95°C for 15 min, followed by 45 cycles of PCR at 95°C for 10 s and 60°C for 30 s.
Statistical analysis.
Statistical significance for comparison of two groups was calculated using Student's t tests, using Microsoft Excel software.
RESULTS
IFN-λ contributes to resistance of mice against various pneumotropic viruses.
We previously showed that IFNAR10/0IL28Rα0/0 mice, which lack receptors for both type I and type III IFN, exhibit higher susceptibility toward certain laboratory strains of influenza A virus than IFNAR10/0 mice, which lack only a functional type I IFN receptor (26). To determine whether IFN-λ might also contribute to resistance against other pneumotropic viruses, we compared IFNAR10/0IL28Rα0/0 and IFNAR10/0 mice with respect to virus-induced disease and viral titers in the lung tissue. In the first experiment, we used the pandemic swine origin influenza virus strain A/HH/05/2009 (H1N1) for challenge. IFNAR10/0 mice survived this infection even if a very high virus dose (106 PFU per animal) was used (Fig. 1A). In contrast, all IFNAR10/0IL28Rα0/0 mice succumbed within 6 days if the challenge virus dose was 105 PFU per animal or higher. Even at 104 PFU of virus per animal, roughly 50% of the challenged double-knockout mice developed severe disease and had to be killed (Fig. 1A). To corroborate these findings, 48 h after infection with 103 PFU of virus per animal, the viral lung titers of both mouse lines were determined. The average titers were 3 × 105 PFU per lung in IFNAR10/0 mice and 107 PFU per lung in IFNAR10/0IL28Rα0/0 mice (Fig. 1B). This difference was statistically significant, clearly demonstrating that IFN-λ contributes to resistance against swine origin influenza A virus.
FIG. 1.
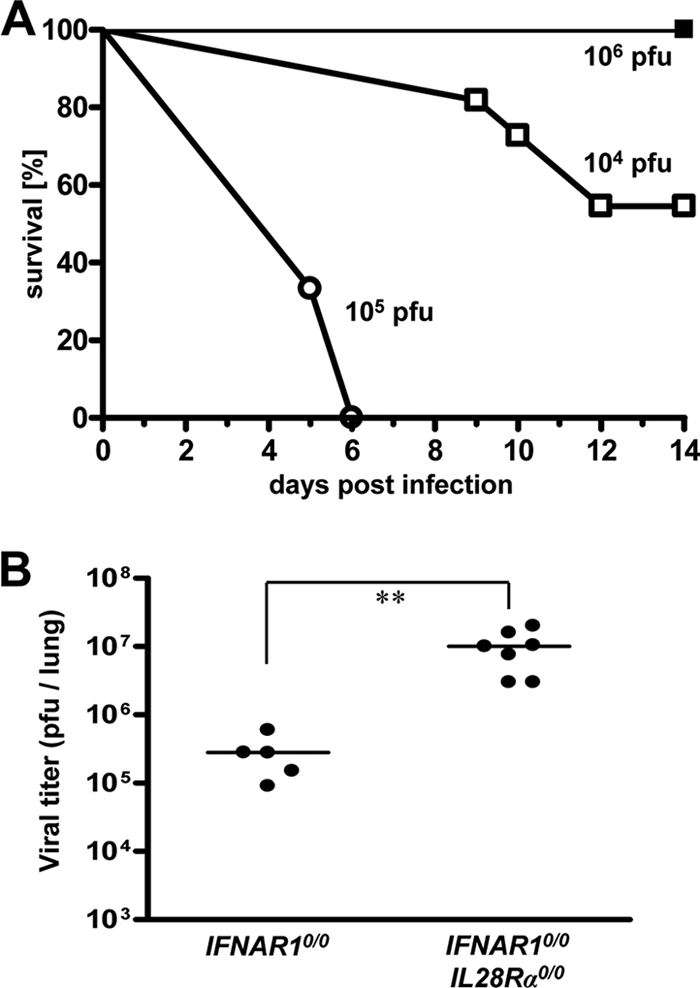
Mice lacking functional receptors for both IFN-α/β and IFN-λ exhibit high susceptibility toward influenza virus strain A/HH/05/2009 (H1N1). (A) Survival of IFNAR10/0 mice (filled squares; n = 4) and IFNAR10/0IL28Rα0/0 double-knockout mice (open symbols; 104 PFU [n = 11] and 105 PFU [n = 3]) after intranasal infection with the indicated doses of virus. (B) Virus titers in lungs at 48 h following intranasal infection with 103 PFU of virus. Combined data for two independent experiments are shown. Each dot represents the data for one animal. **, P < 0.01.
We next used influenza virus strain B/Lee/40 for challenge. If infected with 5 × 102 FFU of B/Lee/40 per animal, all IFNAR10/0IL28Rα0/0 double-knockout mice developed severe disease within 5 to 10 days, whereas all IFNAR10/0 mice survived (Fig. 2A). If challenged with 100 times more virus (5 × 104 FFU), about 80% of the single-knockout IFNAR10/0 mice still survived. Wild-type and IL28Rα0/0 mice were even more resistant and survived the highest virus dose (5 × 105 FFU) that we tested (data not shown). In contrast, conventional C57BL/6 mice, which lack functional alleles of the Mx1 gene, were highly susceptible toward B/Lee/40 (50% lethal dose [LD50] = 2 × 103 FFU) (data not shown), demonstrating that Mx1 is a major factor contributing to resistance against influenza B virus in vivo. Replication of B/Lee/40 in the lungs of the various mouse lines was assessed at 72 h postinfection with 5 × 104 FFU of virus. Titers in lungs of wild-type and IL28Rα0/0 mice were near or below the detection limit (Fig. 2B). In contrast, B/Lee/40 replicated to very high titers in lungs of IFNAR10/0IL28Rα0/0 double-knockout mice. Viral titers in lungs of IFNAR10/0 mice were nonuniform but, on average, were about 20-fold reduced compared to the level for double-knockout mice (Fig. 2B). Thus, IFN-λ clearly plays a substantial role in host resistance toward influenza B virus.
FIG. 2.
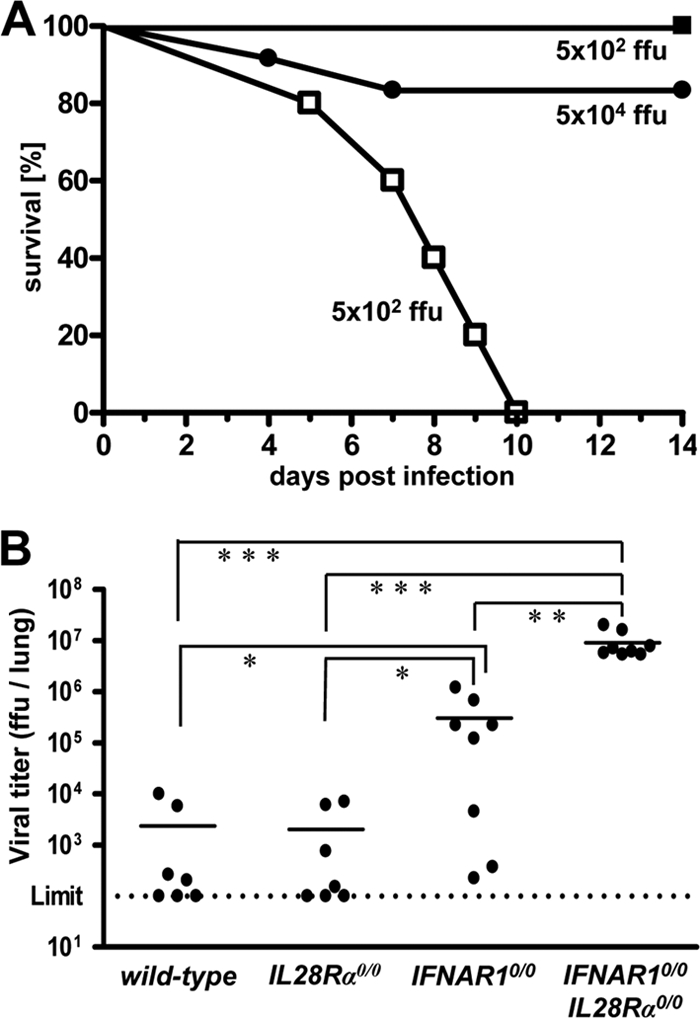
Mice lacking functional receptors for both IFN-α/β and IFN-λ exhibit high susceptibility toward influenza virus strain B/Lee/40. (A) Survival of IFNAR10/0 mice (filled symbols; 5 × 102 PFU [n = 7] and 5 × 104 PFU [n = 10]) and IFNAR10/0IL28Rα0/0 double-knockout mice (open squares; n = 4) after intranasal infection with the indicated doses of virus. Combined data for two independent experiments are shown. (B) Virus titers in lungs at 72 h following intranasal infection with 5 × 104 FFU of virus. Combined data for several independent experiments are shown. Each dot represents the data for one animal. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We also assessed the contribution of IFN-λ to resistance against respiratory syncytial virus (RSV). Replication of RSV is strain dependent. Four days after intranasal inoculation of 8 × 106 FFU of virus, RSV titers reached about 105 FFU per lung in BALB/c mice, which are frequently used for RSV research (27), and about 5 × 103 FFU per lung in wild-type C57BL/6 mice, irrespective of the presence (wild type) or absence (C57BL/6) of functional Mx1 alleles (Fig. 3). As was previously reported (18), IFNAR1 deficiency does not increase susceptibility to RSV replication in C57BL/6 mice, whereas STAT1 deficiency does. In agreement with these findings, we detected only low titers of RSV in the lungs of both wild-type and IFNAR10/0 mice, while RSV titers in lungs of STAT10/0 mice were at least 50-fold higher than those in wild-type mice (Fig. 3). Virus control was not impaired by the lack of functional receptors for IFN-λ (Fig. 3). Surprisingly, however, IFNAR10/0IL28Rα0/0 double-knockout mice showed RSV titers of approximately 106 FFU per lung (Fig. 3). These results demonstrate that either IFN-λ- or IFN-α/β-mediated signals are sufficient for RSV control. The absence of both, however, leads to a drastic increase in viral replication. Interestingly, there was a significant difference in viral titers between STAT1-deficient and IFNAR10/0IL28Rα0/0 double-deficient mice, implicating possible STAT1-independent effects of IFN-λ or IFN-α/β on RSV replication.
FIG. 3.

Mice lacking functional receptors for both IFN-α/β and IFN-λ exhibit high susceptibility toward RSV. Virus titers in lungs at day 4 following intranasal infection with 8 × 106 FFU of RSV. Wild-type mice and C57BL/6 mice differ only by the presence or absence of functional Mx1 alleles. Combined data for several independent experiments are shown. Each dot represents the data for one animal. ***, P < 0.001.
Human metapneumovirus (HMPV) does not replicate to high titers in lungs of standard C57BL/6 mice and does not induce substantial signs of illness in these animals (unpublished data). After intranasal infection with 105 PFU of HMPV, our single-knockout IFNAR10/0 mice also remained healthy and showed no weight loss during the 9-day observation period (Fig. 4A). In contrast, IFNAR10/0IL28Rα0/0 mice experienced substantial respiratory disease and showed weight losses of up to 23% around day 6 postinfection, and in infrequent cases, they had to be killed (Fig. 4A). Analysis of viral RNA in lungs revealed significantly elevated HMPV-RNA load in IFNAR10/0IL28Rα0/0 double-knockout mice compared to the level for IFNAR10/0 single-knockout mice (Fig. 4B), demonstrating that IFN-λ contributes to resistance against HMPV.
FIG. 4.

Mice lacking functional receptors for both IFN-α/β and IFN-λ exhibit high susceptibility toward HMPV. (A) Body weight changes of IFNAR10/0 (n = 4) and IFNAR10/0IL28Rα0/0 (n = 5) mice intranasally infected with 105 PFU of HMPV. Crosses indicate that animals had to be killed due to severe symptoms. (B) HMPV-RNA load in lungs at day 4 following intranasal infection with 105 PFU of HMPV was determined by qRT-PCR. Combined data for two independent experiments are shown. Each dot represents the data for one animal. ***, P < 0.001.
To determine whether IFN-λ is also active against pathogens, which can enter the body through the respiratory tract but eventually replicate in cells of other organs, we performed infection experiments with Lassa fever virus. Groups of IFNAR10/0 single-knockout and IFNAR10/0IL28Rα0/0 double-knockout mice were infected by the intranasal route with either 3 × 105 PFU (high dose) or 3 × 103 PFU (low dose) of Lassa fever virus strain AV. At various times postinfection, blood samples were examined for infectious virus. In addition, serum transaminase levels were monitored to assess liver damage. Peak titers of Lassa fever virus (>105 PFU per ml) were measured around day 8 postinfection for both mouse lines. Importantly, we did not observe any significant differences in viral growth kinetics (Fig. 5) or transaminase levels (data not shown) between IFNAR10/0 and IFNAR10/0IL28Rα0/0 mice. Thus, Lassa fever virus replication was not influenced by IFN-λ, although in our experimental setting the virus was forced to enter the host via the respiratory tract.
FIG. 5.

Lassa fever virus replication is not affected by IFN-λ. IFNAR10/0 and IFNAR10/0IL28Rα0/0 mice were infected by the intranasal route with either 3 × 105 PFU (high dose) or 3 × 103 PFU (low dose) of Lassa fever virus. Blood samples were taken at the indicated times postinfection and tested for presence of virus. Each group consisted of three mice, except the IFNAR10/0IL28Rα0/0 low-dose group, which consisted of two mice.
Lung epithelial cells preferentially express functional IFN-λ receptors.
To identify the cell types in the lung that respond to IFN-λ, we used an indirect assay which takes advantage of the fact that expression of the mouse Mx1 gene is under tight transcriptional control by type I and type III IFN (29). Since IFNAR10/0 mice cannot respond to type I IFN, Mx1 expression in such mice is solely dependent on IFN-λ (26). Thus, cells of IFNAR10/0 mice containing Mx1 protein are likely to express functional IFN-λ receptor complexes. This assay is highly reliable, as Mx1 is a nuclear protein that forms aggregates with a characteristic appearance (29).
To make sure that IFN-λ is abundantly present in the lung, IFNAR10/0 mice were infected with the influenza A virus mutant strain SC35M-ΔNS1, which is a known inducer of type I and type III IFN in mouse lungs (26). At 20 h postinfection, the animals were sacrificed and lung sections were prepared for immunohistochemical analysis. Mx1 staining was readily detected in lungs of IFNAR10/0 mice (Fig. 6A and C). Mx1-positive cells were mainly clustered around bronchioles. From their locations and shapes, it seemed that most Mx1-positive cells are epithelial cells (Fig. 6C). In contrast, Mx1 staining in lungs of SC35M-ΔNS1-infected IFNAR10/0IL28Rα0/0 mice was very weak (Fig. 6B and D). In such animals, only a few cells, which were of unknown identity, expressed Mx1, but strikingly, lung epithelial cells showed no substantial Mx1 staining (Fig. 6D).
FIG. 6.

Lung epithelial cells express functional IFN-λ receptor complexes. IFNAR10/0 (A, C) and IFNAR10/0IL28Rα0/0 (B, D) mice were intranasally infected with 5 × 104 PFU of SC35M-ΔNS1, a potent inducer of type I and type III IFN (26). At 20 h postinfection, the lungs were removed and stained for Mx1 by immunohistofluorescence. (A, B) Low magnification overview. Mx1-positive cells (stained nuclei) are mainly clustered around bronchioles in IFNAR10/0 mice but mostly absent in IFNAR10/0IL28Rα0/0 mice. (B, D) High magnification of bronchioles and surrounding tissue. Epithelial cells were prominently stained for Mx1 in IFNAR10/0 but not in IFNAR10/0IL28Rα0/0 mice.
The responsiveness of bronchiolar epithelial cells to IFN-λ was confirmed for mice expressing a plasmid for mouse IFN-λ3 in the tibialis muscle (29). In such mice, which produce systemic IFN-λ, Mx1 was also prominently expressed in bronchiolar epithelial cells. Mx1 expression in these cells depended on functional IFN-λ receptors, as Mx1 was detected in wild-type mice (data not shown) and IFNAR10/0 mice but not in IL28Rα0/0 mice (see Fig. S1 in the supplemental material). The fact that lung epithelial cells abundantly express functional IFN-λ receptor complexes readily explains our results indicating that IFNAR10/0IL28Rα0/0 mice are more susceptible to several pneumotropic viruses than IFNAR10/0 mice. This fact further suggests that Lassa fever virus may not be restricted by IFN-λ, because this virus presumably uses other cells for initial replication in the respiratory tract before reaching the bloodstream.
Epithelial cells in the intestine express functional IFN-λ receptor complexes.
We further determined whether epithelial cells of other organs, such as the intestine (which is prone to infection with important pathogenic viruses), might also carry functional receptors for IFN-λ. Again, we employed the above-described indirect assay in which expression of Mx1 is used to identify IFN-λ-responsive cells in mice expressing systemic IFN-λ (29). Prominently stained cells were abundantly present in various parts of the small intestine of IFNAR10/0 mice (Fig. 7A), whereas Mx1-positive cells were virtually absent from small intestine sections of mock-treated IFNAR10/0 mice (data not shown) or IFN-λ3-expressing IFNAR10/0IL28Rα0/0 mice (Fig. 7C). Careful inspection revealed that the majority of IFN-λ-responsive cells in the intestines of IFNAR10/0 mice lined the lumen. From their morphology, it appeared that these Mx1-positive cells are epithelial cells (Fig. 7B). Analysis of additional tissue samples from other parts of the gastrointestinal tract showed that epithelial cells in the esophagus (Fig. 7D), stomach (Fig. 7E), small intestine (Fig. 7F), and colon (Fig. 7G) of IFN-λ3-expressing IFNAR10/0 mice were strongly positive for Mx1. In the large intestine but not in the esophagus, stomach, or small intestine, a number of cells of unknown identity also expressed Mx1 in the absence of IFN-λ (data not shown).
FIG. 7.

Epithelial cells of the intestine express functional IFN-λ receptor complexes. The response to in vivo electroporation of a plasmid encoding mouse IFN-λ3 was monitored by immunohistofluorescence staining for Mx1. The duodenum (section of the villi) of IFNAR10/0 mice is shown in low (A) and high (B) magnification. Prominent labeling of nuclear Mx1 is detected mainly in epithelial cells of the villi. (C) No such staining was observed in IFNAR10/0IL28Rα0/0 mice, which cannot respond to IFN-λ. Strongly stained epithelial cells were detected in all regions of the gastrointestinal tract of IFN-λ-treated IFNAR10/0 mice, including esophagus (D), stomach (E), small intestine (F), and colon (G).
To corroborate the epithelial cell specificity of the IFN response in infected mice, we performed additional experiments, in which IFNAR10/0 mice were infected with Rift Valley fever virus clone 13, which lacks the IFN-antagonistic NSs protein and therefore induces large amounts of type I and type III IFN in mice (4). IFNAR10/0 mice were killed at 48 h postinfection when they showed signs of virus-induced disease and when serum IFN titers were reported to reach peak values (4). Under these conditions, large numbers of Mx1-positive epithelial cells were readily detected in the small intestine and other parts of the gastrointestinal tract (see Fig. S2 in the supplemental material). Thus, virus-induced and plasmid-mediated expression of IFN-λ in IFNAR10/0 mice yielded similar results. Taken together, these data demonstrate that epithelial cells are the main targets of IFN-λ in all parts of the gastrointestinal tract.
IFN-λ reduces replication of SARS-CoV in lungs and virus excretion in feces.
Patients infected with SARS-CoV during the outbreak in 2003, besides developing severe respiratory symptoms, excreted infectious virus in their feces (6). Studies with SARS-CoV-related viruses of bats further support the view that these viruses can replicate in epithelial cells of both lung and gastrointestinal tract (23). Therefore, we hypothesized that experiments with SARS-CoV might reveal a possible role for IFN-λ during viral infection of the gastrointestinal tract.
Although replication of SARS-CoV in lungs of IFNAR10/0 and STAT10/0 mice is substantially enhanced compared to the level for wild-type mice (5, 15), a possible contribution of type III IFN has not previously been documented. To address this issue, we infected mice with 106 PFU of SARS-CoV by the intranasal route and determined viral titers in lungs at day 4 postinfection. We found that SARS-CoV titers in lungs of IFNAR10/0 and IL28Rα0/0 single-knockout mice were, on average, at least 10-fold higher than those in wild-type mice (Fig. 8A). In agreement with these observations, viral lung titers in IFNAR10/0IL28Rα0/0 double-knockout mice were 10-fold higher than those in single-knockout mice (Fig. 8A).
FIG. 8.

IFN-λ contributes to restriction of SARS-CoV replication in both lungs and intestinal tracts of mice. (A) Virus titers in lungs of various mouse lines at day 4 following intranasal infection with 106 PFU of SARS-CoV per animal. Combined data for two independent experiments are shown. Each dot represents the data for one animal. *, P < 0.05; ***, P < 0.001. (B) At days 4 and 14 postinfection, RNA samples extracted from 20 droppings of cages harboring the different mouse lines were analyzed by qRT-PCR for SARS-CoV. (C) RNA samples from intestines of infected wild-type and IFNAR10/0IL28Rα0/0 double-knockout mice were analyzed by qRT-PCR for SARS-CoV. Viral RNA levels were calculated using appropriate standards.
To determine whether virus excretion was affected by IFN-λ, fecal samples of the various SARS-CoV-infected mice were analyzed for the presence of viral RNA on days 4 and 14 postinfection. Neither wild-type nor IL28Rα0/0 or IFNAR10/0 single-knockout mice contained detectable levels of viral RNA in their feces. However, fecal samples from cages of IFNAR10/0IL28Rα0/0 double-knockout mice tested positive on day 4 and day 14 postinfection (Fig. 8B). When intestine samples of individual infected mice were analyzed for the presence of viral RNA by quantitative RT-PCR (qRT-PCR), we observed that three of five IFNAR10/0IL28Rα0/0 mice were positive on days 4 and 14 postinfection (Fig. 8C). In contrast, none of the intestine samples of infected wild-type mice scored positive in this assay. These results demonstrate that IFN-λ contributes to restriction of SARS-CoV in both respiratory and gastrointestinal tracts.
DISCUSSION
Previous infection studies with knockout mice lacking functional receptors for type I IFN, type III IFN, or both clearly indicated that IFN-λ contributes to resistance against influenza A virus (26). A limitation of the published work was that highly adapted laboratory viruses had been used and that enhanced virus susceptibility of IFN-λ receptor-deficient mice was detected only if infection experiments were performed with attenuated virus mutants that induce a strong innate immune response. Consequently, it remained unclear whether IFN-λ also plays a role in the defense of wild-type influenza A virus. Here, we addressed this issue by employing a nonadapted primary isolate of the new pandemic swine-origin H1N1 influenza virus. This virus was unable to induce disease in Mx1-positive mice lacking either functional type I or functional type III IFN receptors. However, it replicated to high titers and induced disease in mice lacking both IFN receptor systems. These results show that our earlier conclusion that IFN-λ plays a role in host defense against influenza A virus (26) remains valid and also applies for primary virus isolates.
By using a panel of different pneumotropic viruses, we managed to demonstrate that IFN-λ also has a beneficial effect during infections with other important pathogens, such as influenza B virus, RSV, HMPV, and SARS coronavirus. All these viruses did not grow well and were mainly benign in single-knockout mice lacking functional receptors for either type I or type III IFN, but they replicated to high titers and were able to induce clinical symptoms in mice lacking both IFN receptor systems. In agreement with these results, other researchers previously noted that type I IFN receptor-deficient mice exhibited a degree of resistance to RSV similar to that observed for wild-type mice but that STAT1-deficient mice were highly susceptible (18). The molecular basis for this difference remained unexplained at the time. Our new findings suggest that the type I IFN receptor-deficient mice remained resistant to RSV because the functionally redundant IFN-λ system was still active in these mice. In STAT1-deficient mice, in contrast, both the type I and the type III IFN systems are severely compromised, resulting in enhanced virus susceptibility.
Interestingly, experiments in which we performed intranasal infections of mice with Lassa fever virus yielded no evidence for a protective role for IFN-λ against this pathogen. We believe that this result can be explained by our finding that expression of functional IFN-λ receptors in the lung was restricted to epithelial cells. As the strictly pneumotropic viruses discussed above preferentially use lung epithelial cells for productive replication, it is understandable that these viruses are susceptible to IFN-λ. Lassa fever virus, on the other hand, before becoming systemic, presumably infects other cell types in the respiratory tract that do not express or express only very low levels of functional IFN-λ receptors. Consequently, virus-induced IFN-λ cannot efficiently restrict initial replication of Lassa fever virus in the respiratory tract. It should be noted that once the virus has reached the bloodstream, IFN-λ is not expected to afford any further protection, as previously reported for Rift Valley fever virus and Thogoto virus (26), presumably due to restricted expression of the type III IFN receptor.
One of the most exciting findings of our study is that the protective function of IFN-λ was not restricted to the respiratory tract but also included the gastrointestinal tract. We observed that SARS-CoV-infected mice excreted virus in the feces only if these mice lacked both type I and type III IFN receptors. If either IFN system remained functional, no virus replication could be detected in the intestine of the infected mice. This finding can be explained by the results of our type III IFN receptor expression analysis, which showed that epithelial cells in all areas of the gastrointestinal tract readily responded to IFN-λ. Thus, a picture emerges which suggests that IFN-λ mainly serves to strengthen the antiviral defense of mucosal surfaces that are potential entry sites for pathogenic viruses.
Since IFNAR10/0 mice are highly sensitive toward many viruses (9), whereas mice lacking IFN-λ receptors do not show or show only weak phenotypes in respect to viral infection (1, 26), it remains obvious that the type I IFN system certainly is the dominant system in terms of antiviral protection. However, our data clearly demonstrate that the presence of a functional type III IFN system does indeed provide beneficial and protective antiviral responses that can influence survival. Additionally, we showed that IFN-λ also plays a previously unappreciated role in preventing viral spread to and shedding from the gastrointestinal tract. It will be both interesting and imperative to investigate which other mucosal surfaces and/or exposed body tissues are expressing functional type III IFN receptor complexes and thus may contribute to IFN-λ-mediated antiviral responses. For example, previous studies yielded indirect (3) and direct (31) hints that the skin, which represents a body part that is heavily exposed to pathogens if physically injured, may acquire virus resistance from IFN-λ.
The recently identified IFN genes of fish seem to represent components of an evolutionarily well conserved type III rather than type I IFN system (24). Although this view is not generally accepted (32), it would indicate that IFN-λ is a primordial antiviral cytokine that may serve basic functions. Our finding that IFN-λ of the mouse seems to selectively contribute to innate immunity of mucosal surfaces, which are the most frequent entry sites of viruses, is certainly compatible with this hypothesis.
Supplementary Material
Acknowledgments
We thank Thorsten Wolff for providing a seed stock of B/Lee/40, Markus Eickmann for providing a seed stock of A/HH/05/2009, Thomas Decker for providing STAT10/0 mice, and Otto Haller for helpful comments on the manuscript.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 620) to P.S. and S.E., the German Ministry of Education and Research (Project Code “Ökologie und Pathogenese von SARS”) to C.D., FRSM to T.M., and the University of Louvain (FSR) to F.S.
Footnotes
Published ahead of print on 24 March 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Ank, N., M. B. Iversen, C. Bartholdy, P. Staeheli, R. Hartmann, U. B. Jensen, F. Dagnaes-Hansen, A. R. Thomsen, Z. Chen, H. Haugen, K. Klucher, and S. R. Paludan. 2008. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J. Immunol. 180:2474-2485. [DOI] [PubMed] [Google Scholar]
- 2.Asper, M., T. Sternsdorf, M. Hass, C. Drosten, A. Rhode, H. Schmitz, and S. Gunther. 2004. Inhibition of different Lassa virus strains by alpha and gamma interferons and comparison with a less pathogenic arenavirus. J. Virol. 78:3162-3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartlett, N. W., K. Buttigieg, S. V. Kotenko, and G. L. Smith. 2005. Murine interferon lambdas (type III interferons) exhibit potent antiviral activity in vivo in a poxvirus infection model. J. Gen. Virol. 86:1589-1596. [DOI] [PubMed] [Google Scholar]
- 4.Bouloy, M., C. Janzen, P. Vialat, H. Khun, J. Pavlovic, M. Huerre, and O. Haller. 2001. Genetic evidence for an interferon-antagonistic function of rift valley fever virus nonstructural protein NSs. J. Virol. 75:1371-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cervantes-Barragan, L., R. Zust, F. Weber, M. Spiegel, K. S. Lang, S. Akira, V. Thiel, and B. Ludewig. 2007. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 109:1131-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng, P. K., D. A. Wong, L. K. Tong, S. M. Ip, A. C. Lo, C. S. Lau, E. Y. Yeung, and W. W. Lim. 2004. Viral shedding patterns of coronavirus in patients with probable severe acute respiratory syndrome. Lancet 363:1699-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Childs, R. A., A. S. Palma, S. Wharton, T. Matrosovich, Y. Liu, W. Chai, M. A. Campanero-Rhodes, Y. Zhang, M. Eickmann, M. Kiso, A. Hay, M. Matrosovich, and T. Feizi. 2009. Receptor-binding specificity of pandemic influenza A (H1N1) 2009 virus determined by carbohydrate microarray. Nat. Biotechnol. 27:797-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dare, R., S. Sanghavi, A. Bullotta, M. C. Keightley, K. S. George, R. M. Wadowsky, D. L. Paterson, K. R. McCurry, T. A. Reinhart, S. Husain, and C. R. Rinaldo. 2007. Diagnosis of human metapneumovirus infection in immunosuppressed lung transplant recipients and children evaluated for pertussis. J. Clin. Microbiol. 45:548-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drosten, C., S. Gunther, W. Preiser, S. van der Werf, H. R. Brodt, S. Becker, H. Rabenau, M. Panning, L. Kolesnikova, R. A. Fouchier, A. Berger, A. M. Burguiere, J. Cinatl, M. Eickmann, N. Escriou, K. Grywna, S. Kramme, J. C. Manuguerra, S. Muller, V. Rickerts, M. Sturmer, S. Vieth, H. D. Klenk, A. D. Osterhaus, H. Schmitz, and H. W. Doerr. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348:1967-1976. [DOI] [PubMed] [Google Scholar]
- 10.Dumoutier, L., A. Tounsi, T. Michiels, C. Sommereyns, S. V. Kotenko, and J. C. Renauld. 2004. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-lambda 1: similarities with type I interferon signaling. J. Biol. Chem. 279:32269-32274. [DOI] [PubMed] [Google Scholar]
- 11.Durbin, J. E., R. Hackenmiller, M. C. Simon, and D. E. Levy. 1996. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84:443-450. [DOI] [PubMed] [Google Scholar]
- 12.Gunther, S., P. Emmerich, T. Laue, O. Kuhle, M. Asper, A. Jung, T. Grewing, J. ter Meulen, and H. Schmitz. 2000. Imported Lassa fever in Germany: molecular characterization of a new Lassa virus strain. Emerg. Infect. Dis. 6:466-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haller, O., G. Kochs, and F. Weber. 2006. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 344:119-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haller, O., P. Staeheli, and G. Kochs. 2009. Protective role of interferon-induced Mx GTPases against influenza viruses. Rev. Sci. Tech. 28:219-231. [DOI] [PubMed] [Google Scholar]
- 15.Hogan, R. J., G. Gao, T. Rowe, P. Bell, D. Flieder, J. Paragas, G. P. Kobinger, N. A. Wivel, R. G. Crystal, J. Boyer, H. Feldmann, T. G. Voss, and J. M. Wilson. 2004. Resolution of primary severe acute respiratory syndrome-associated coronavirus infection requires Stat1. J. Virol. 78:11416-11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horisberger, M. A., P. Staeheli, and O. Haller. 1983. Interferon induces a unique protein in mouse cells bearing a gene for resistance to influenza virus. Proc. Natl. Acad. Sci. U. S. A. 80:1910-1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huck, B., D. Neumann-Haefelin, A. Schmitt-Graeff, M. Weckmann, J. Mattes, S. Ehl, and V. Falcone. 2007. Human metapneumovirus induces more severe disease and stronger innate immune response in BALB/c mice as compared with respiratory syncytial virus. Respir. Res. 8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson, T. R., S. E. Mertz, N. Gitiban, S. Hammond, R. Legallo, R. K. Durbin, and J. E. Durbin. 2005. Role for innate IFNs in determining respiratory syncytial virus immunopathology. J. Immunol. 174:7234-7241. [DOI] [PubMed] [Google Scholar]
- 19.Kochs, G., I. Koerner, L. Thiel, S. Kothlow, B. Kaspers, N. Ruggli, A. Summerfield, J. Pavlovic, J. Stech, and P. Staeheli. 2007. Properties of H7N7 influenza A virus strain SC35M lacking interferon antagonist NS1 in mice and chickens. J. Gen. Virol. 88:1403-1409. [DOI] [PubMed] [Google Scholar]
- 20.Koerner, I., G. Kochs, U. Kalinke, S. Weiss, and P. Staeheli. 2007. Protective role of beta interferon in host defense against influenza A virus. J. Virol. 81:2025-2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kotenko, S. V., G. Gallagher, V. V. Baurin, A. Lewis-Antes, M. Shen, N. K. Shah, J. A. Langer, F. Sheikh, H. Dickensheets, and R. P. Donnelly. 2003. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 4:69-77. [DOI] [PubMed] [Google Scholar]
- 22.Lasfar, A., A. Lewis-Antes, S. V. Smirnov, S. Anantha, W. Abushahba, B. Tian, K. Reuhl, H. Dickensheets, F. Sheikh, R. P. Donnelly, E. Raveche, and S. V. Kotenko. 2006. Characterization of the mouse IFN-lambda ligand-receptor system: IFN-lambdas exhibit antitumor activity against B16 melanoma. Cancer Res. 66:4468-4477. [DOI] [PubMed] [Google Scholar]
- 23.Lau, S. K., P. C. Woo, K. S. Li, Y. Huang, H. W. Tsoi, B. H. Wong, S. S. Wong, S. Y. Leung, K. H. Chan, and K. Y. Yuen. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U. S. A. 102:14040-14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levraud, J. P., P. Boudinot, I. Colin, A. Benmansour, N. Peyrieras, P. Herbomel, and G. Lutfalla. 2007. Identification of the zebrafish IFN receptor: implications for the origin of the vertebrate IFN system. J. Immunol. 178:4385-4394. [DOI] [PubMed] [Google Scholar]
- 25.Meier, E., J. Fah, M. S. Grob, R. End, P. Staeheli, and O. Haller. 1988. A family of interferon-induced Mx-related mRNAs encodes cytoplasmic and nuclear proteins in rat cells. J. Virol. 62:2386-2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mordstein, M., G. Kochs, L. Dumoutier, J. C. Renauld, S. R. Paludan, K. Klucher, and P. Staeheli. 2008. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 4:e1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peebles, R. S., Jr., and B. S. Graham. 2005. Pathogenesis of respiratory syncytial virus infection in the murine model. Proc. Am. Thorac. Soc. 2:110-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheppard, P., W. Kindsvogel, W. Xu, K. Henderson, S. Schlutsmeyer, T. E. Whitmore, R. Kuestner, U. Garrigues, C. Birks, J. Roraback, C. Ostrander, D. Dong, J. Shin, S. Presnell, B. Fox, B. Haldeman, E. Cooper, D. Taft, T. Gilbert, F. J. Grant, M. Tackett, W. Krivan, G. McKnight, C. Clegg, D. Foster, and K. M. Klucher. 2003. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 4:63-68. [DOI] [PubMed] [Google Scholar]
- 29.Sommereyns, C., S. Paul, P. Staeheli, and T. Michiels. 2008. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 4:e1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Pesch, V., H. Lanaya, J. C. Renauld, and T. Michiels. 2004. Characterization of the murine alpha interferon gene family. J. Virol. 78:8219-8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Witte, K., G. Gruetz, H. D. Volk, A. C. Looman, K. Asadullah, W. Sterry, R. Sabat, and K. Wolk. 2009. Despite IFN-lambda receptor expression, blood immune cells, but not keratinocytes or melanocytes, have an impaired response to type III interferons: implications for therapeutic applications of these cytokines. Genes Immun. 10:702-714. [DOI] [PubMed] [Google Scholar]
- 32.Zhou, Z., O. J. Hamming, N. Ank, S. R. Paludan, A. L. Nielsen, and R. Hartmann. 2007. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J. Virol. 81:7749-7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.