

Abstract
Hepatitis C virus (HCV) is an important human pathogen affecting 170 million chronically infected individuals. In search for cellular proteins involved in HCV replication, we have developed a purification strategy for viral replication complexes and identified annexin A2 (ANXA2) as an associated host factor. ANXA2 colocalized with viral nonstructural proteins in cells harboring genotype 1 or 2 replicons as well as in infected cells. In contrast, we found no obvious colocalization of ANXA2 with replication sites of other positive-strand RNA viruses. The silencing of ANXA2 expression showed no effect on viral RNA replication but resulted in a significant reduction of extra- and intracellular virus titers. Therefore, it seems likely that ANXA2 plays a role in HCV assembly rather than in genome replication or virion release. Colocalization studies with individually expressed HCV nonstructural proteins indicated that NS5A specifically recruits ANXA2, probably by an indirect mechanism. By the deletion of individual NS5A subdomains, we identified domain III (DIII) as being responsible for ANXA2 recruitment. These data identify ANXA2 as a novel host factor contributing, with NS5A, to the formation of infectious HCV particles.
Hepatitis C virus (HCV) infections are characterized by a mostly unapparent acute phase leading to persistence in ca. 70% of all infected individuals. Currently, 170 million people suffer from chronic hepatitis C, and they have a high risk to develop severe liver disease. It has been estimated that HCV accounts for 27% of cirrhosis and 25% of hepatocellular carcinoma cases worldwide (2).
HCV is an enveloped positive-strand RNA virus belonging to the genus Hepacivirus in the family Flaviviridae. The genome of HCV encompasses a single ∼9,600-nucleotide (nt)-long RNA molecule containing one large open reading frame (ORF) that is flanked by nontranslated regions (NTRs), which are important for viral translation and replication. HCV proteins generated from the polyprotein precursor are cleaved by cellular and viral proteases into at least 10 different products (for a review of polyprotein cleavage and the function of the individual proteins, see reference 4). The structural proteins Core, E1, and E2 are located in the amino-terminal portion of the polyprotein, followed by p7, a hydrophobic peptide that is supposed to be a viroporin, and the nonstructural proteins (NS) NS2, NS3, NS4A, NS4B, NS5A, and NS5B. Only the nonstructural proteins NS3 to NS5B are involved in viral RNA replication. NS3 is a multifunctional protein, consisting of an amino-terminal protease domain required for the processing of the NS3 to NS5B region and a carboxyterminal helicase/nucleoside triphosphatase domain. NS4A is a cofactor that activates the NS3 protease function by forming a heterodimer. The hydrophobic protein NS4B induces vesicular membrane alterations involved in RNA replication. NS5A is a phosphoprotein that seems to play an important role in viral replication and assembly (3, 35, 58). NS5B is the RNA-dependent RNA polymerase of HCV.
Positive-strand RNA viruses replicate their RNA in vesicular structures originating from different cellular organelles (36). In the case of HCV, particular membrane alterations have been identified by electron microscopy, designated the membranous web, consisting of accumulations of vesicles primarily derived from the endoplasmic reticulum (17). Important insights into the organization of HCV replication complexes were obtained by the in vitro analysis of viral RNA synthesis in membrane preparations of cells harboring subgenomic HCV replicons, so-called crude replication complexes (CRCs) (1, 20). A current model based on a stoichiometric analysis of CRCs suggests that each vesicular structure contains multiple copies of viral nonstructural proteins and has a connection to the cytoplasm, allowing the constant supply of nucleotides for RNA synthesis (45), presumably analogously to the replication complex of the closely related dengue virus (DV) (64). Viral RNA synthesis in CRCs is highly resistant to proteinases and nucleases (39), and the membranes are detergent resistant at 4°C, resembling features of lipid rafts (54).
Several purification techniques have been established to identify relevant HCV host factors by proteomics, based on either the extraction of detergent-resistant membranes (19, 34) or the immunoprecipitation of vesicles (24), revealing different sets of cellular proteins potentially involved in viral replication. In most of these studies, cell lines harboring persistent subgenomic replicons were utilized (33); however, with the availability of a fully permissive cell culture system supporting the complete HCV replication cycle (31, 63, 66), it became evident that viral RNA replication and assembly are closely linked. Recent work revealed an intimate connection of viral replication complexes and assembly sites in close proximity to cytoplasmic lipid droplets (38), with Core and especially NS5A functioning as central regulators by a poorly defined mechanism. NS5A is phosphorylated at multiple serine and threonine residues, binds RNA, and is composed of three domains, which are separated by trypsin-sensitive low-complexity regions (LCS I and II) (59). An N-terminal amphipathic alpha helix tightly associates NS5A with intracellular membranes. Domain I and LCS1 most likely are involved in viral RNA replication, since replication-enhancing mutations primarily mapped to this region (8, 32). The role of domain II is unknown, while domain III recently has been shown to be dispensable for RNA replication but essential for viral particle assembly (3, 35, 58). One of the proposed mechanisms points to a critical interaction with the Core protein, for which phosphorylation in the C-terminal part of domain III of NS5A appears to be required (35). The interaction of Core and NS5A has been proposed to be important for the recruitment of the replication complexes to lipid droplets (3), thereby allowing a coordinated packaging of the newly synthesized RNA.
In this study, we identified annexin A2 (also called annexin II, calpactin 1, and ANXA2) as an HCV host factor by a proteomic analysis. ANXA2 belongs to a family of proteins characterized by their Ca2+-dependent binding to negatively charged phospholipids. The annexin proteins consist of two principle domains, a variable N-terminal and a conserved C-terminal domain, which harbors the Ca2+ and membrane binding sites (for a review, see references 14 and 15). All annexins show cytosolic and membrane localizations. Membrane recruitment probably is regulated by intracellular Ca2+ fluctuations, and target membrane selection differs for different annexins.
In addition to showing a cytosolic distribution, ANXA2 can associate with the plasma membrane and the membrane of early endosomes. Plasma membrane-associated ANXA2 typically is found in a tight heterotetrameric complex with the S100 protein S100A10 (p11). ANXA2 specifically interacts with phosphatidylinositol(4,5)bisphosphate (PIP2) (22, 48) and binds to membranes enriched in cholesterol, supporting a role in the organization of lipid raft-like membrane microdomains. Due to the direct binding of ANXA2 to F-actin, the protein has been proposed to provide a direct link between cytoskeletal elements and PIP2/cholesterol-rich membrane domains (47).
ANXA2 has been implicated in several cellular transport processes, including the internalization and transport of cholesteryl esters, the biogenesis of multivesicular bodies, the recycling of plasma membrane receptors, and the Ca2+-induced exocytosis of certain secretory granules (14). Here, we show that ANXA2 is present at HCV replication sites within the membranous web. The recruitment of ANXA2 is mediated by domain III of NS5A and probably is required for efficient virus assembly.
MATERIALS AND METHODS
Antibodies.
For the detection of viral antigens, the following antibodies were used: mouse monoclonal antibodies detecting NS5A domain III (9E10; generous gift of Charles M. Rice), NS5A domain I of JFH1 (2F6; generated in cooperation with Hengli Tang), NS3-helicase of JFH1 (2E3; generated in cooperation with Hengli Tang), and NS5B of Con1 (3B1; Darius Moradpour). An NS3-specific rabbit polyclonal antiserum was raised against a fragment of the helicase domain (amino acids [aa] 1230 to 1526 of the polyprotein of Con1; EMBL nucleotide sequence database accession no. AJ238799) as well as NS4B (Con1), NS5A (raised against domain I of JFH1), and NS5B (JFH1). The NS3 and NS4B polyclonal antisera recognize both Con1 and JFH1 proteins. Dengue virus-infected cells were stained with a polyclonal rabbit antiserum directed against aa 127 to 170 of NS4B (37). Semliki Forest virus (SFV) was detected by an affinity-purified polyclonal rabbit antiserum recognizing nsp3, which was kindly provided by Tero Ahola.
Antibodies directed against cellular antigens included polyclonal rabbit antisera specific for annexin A2 (H-50; Santa Cruz), annexin A4 (K411; V. Gerke), calreticulin (SPA-600; Stressgen), and calnexin (N-terminal part; SPA-865; Stressgen), as well as monoclonal mouse antibodies recognizing double-stranded RNA (dsRNA) (J2; English & Scientific Consulting), p11 (42), annexin A5 (A8640; Sigma), annexin A2 (clone 5; BD Bioscience), HH7 (60), protein disulfide isomerase (PDI; Iowa hybridoma bank), and β-actin (clone AC-15; Sigma). Note that HH7, a well-characterized monoclonal antibody specifically recognizing ANXA2, cross-reacts with NS5A of Con1 and JFH1 in immunofluorescence analyses. Unless otherwise stated, all experiments shown in this study were performed with a polyclonal rabbit antibody against ANXA2 (H-50; Santa Cruz), which proved not to be cross-reactive (data not shown).
Cell cultures.
Cell monolayers of the human hepatoma cell line Huh-7 (40) were grown in Dulbecco's modified Eagle medium (DMEM; Invitrogen, Karlsruhe, Germany) supplemented with 2 mM l-glutamine, nonessential amino acids, 100 U per ml penicillin, 100 μg per ml streptomycin, and 10% fetal calf serum. G418 (geneticin; Invitrogen) was added at a final concentration of 1 mg/ml in the case of replicon cell clone 9-13 carrying an HCV replicon expressing neomycin phosphotransferase (33) or a subgenomic genotype 2a replicon (25) in Huh7-Lunet cells. Cured replicon cells were generated by a 2-week treatment with 100 U/ml of each interferon alpha and interferon gamma. The absence of replicons was confirmed by selection with G418, which did not result in the formation of resistant colonies. Before proteomic analysis, cured cells were cultured for more than 10 passages in the absence of interferons. Huh-7/Lunet cells refer to a Huh-7 cell clone that was generated with a selectable replicon and cured from HCV by treatment with a specific inhibitor (7). Huh7-Lunet T7-BLR cells are a cell pool stably expressing T7 RNA polymerase and blasticidin S-deaminase, and they were cultured in the presence of 10 μg/ml blasticidin. Lentiviral particles for the transduction of the gene encoding T7-RNA polymerase were prepared in 293T cells exactly as described previously (27) using pWPI-T7-BLR and packaging constructs pCMVR8.91 and pMD.G (provided by Didier Trono). This cell population is referred to as Lunet-T7 cells in the following. Huh7-Lunet/CD81 cells are highly permissive for HCV infection due to the ectopic expression of CD81 and have been described recently (26).
Core ELISA.
Core protein amounts in cell lysates and supernatants were determined by using the Trak-C Core enzyme-linked immunosorbent assay (ELISA) (Ortho Clinical Diagnostics) as recently described (43).
Electroporation of subgenomic replicons and viral genomes.
For electroporation, single-cell suspensions of Huh7-Lunet cells were prepared by the trypsinization of monolayers, detaching the cells from the culture dish by rinsing with complete DMEM, washing once with PBS, counting, and resuspending at 107 cells per ml in Cytomix (62) containing 2 mM ATP and 5 mM glutathione. Five to 10 μg of in vitro-transcribed RNA was mixed with 400 μl of the cell suspension by pipetting, electroporated, and immediately transferred to 12 ml for replication assays or 20 ml complete DMEM for infectivity assays. In knockdown experiments, 1 nmol small interfering RNA (siRNA) was added to the in vitro transcripts. Electroporation conditions were 975 μF and 270 V using a Gene pulser system (Bio-Rad, Munich, Germany) and a cuvette with a gap width of 0.4 cm (Bio-Rad). Cells were seeded in aliquots and harvested at different time points to determine luciferase activity or perform immunofluorescence analysis. Supernatants of cells transfected with JC1 harvested at 24 to 72 h after electroporation were used to generate virus stocks or to determine 50% tissue culture infectivity dose (TCID50) titers at the indicated time points.
Expression of proteins in Lunet-T7 cells.
pTM plasmids were transfected into Lunet-T7 cells via transfection with Effectene as described by the manufacturer (Qiagen). Transcription was performed by the T7 RNA polymerase constitutively expressed in this cell line. Twenty-four hours posttransfection, cells were fixed for immunofluorescence.
Immunoblot analysis and silver staining of proteins.
For immunoblot analysis, cells or CRCs were directly dissolved in Laemmli sample buffer, and proteins were separated by sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis (SDS-PAGE) using standard techniques (50). After electrophoresis, proteins were either stained with silver or transferred to a nitrocellulose membrane and detected with various primary and secondary antibodies conjugated with horseradish peroxidase (Sigma) and developed with the ECL plus Western blotting detection system by following the instructions of the manufacturer (Amersham Biosciences, Freiburg, Germany). Quantity One (Bio-Rad) was used to quantify relative band intensities.
Immunofluorescence.
Cells were seeded on coverslips in 24-well plates at 4.5 × 104 per well 24 h prior to transfection or infection. For fixation, cells were washed three times with PBS and fixed in 4% paraformaldehyde solution for about 20 min at room temperature. Thereafter, cells were again washed three times with PBS and then stored for 2 to 3 days at 4°C or directly used for further preparation. For permeabilization, cells were incubated for 15 min with 0.5% Triton X-100 in PBS and washed three times with PBS prior to incubation with the first antibody. The primary antibody was diluted to the desired concentration, typically 1:100, with a 1× PBS buffer containing 3% bovine serum albumin (BSA) to prevent the unspecific binding of the antibody. After 60 min of incubation at room temperature, cells were washed three times with PBS and incubated with the secondary antibodies conjugated with Alexa 488 or Alexa 546 (goat anti-rabbit or goat anti-mouse, respectively; Invitrogen) diluted 1:1,000 in PBS with 3% BSA. After 45 min of incubation in the dark, cells were washed once with PBS and counterstained with DAPI (4′, 6′-diamidino-2-phenylindole; Molecular Probes, Karlsruhe, Germany). After three additional washes with PBS, cells were washed once with water and mounted on glass slides with Fluoromount G. Pictures of prepared samples were taken either by the epifluorescence microscope CTR-MIC from Leica or the confocal microscope Nikon C1Si confocal. The pictures were edited and merged in Adobe Photoshop 6.0.
Infectivity assays.
The infectivity of chimeric JC1 virus (43) was determined by using a limiting dilution assay on Huh-7.5 cells (31). Infected cells were detected by using a mouse monoclonal NS5A-specific antibody (9E10) and a peroxidase-conjugated goat anti-mouse polyclonal antibody (Sigma). The TCID50 was calculated as described elsewhere (43). Intracellular infectivity assays were performed according to a published protocol (13) with freeze-thaw lysates of transfected cells. Forty-eight hours posttransfection, Huh7-Lunet cells were washed (PBS), scraped off, and centrifuged for 5 min at 700 × g. Cell pellets were resuspended in 1 ml cell culture medium and subjected to three freeze-thaw cycles using liquid nitrogen and a thermo block at 37°C. Cell debris was removed by centrifugation at 10,000 × g for 10 min at 4°C, and cell-associated infectivity was determined by a TCID50 assay. Culture supernatants from transfected cells were treated alike, and infectivity was determined in parallel.
In vitro replicase assay.
HCV in vitro replicase activity was determined in a reaction mixture containing 20 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 5 mM dithiothreitol (DTT), 5 mM KCl, 40 μg/ml actinomycin D, 20 μCi [α-32P]CTP, 10 μM CTP, 1 mM (each) ATP and UTP, 5 mM GTP, 2.5 mM phosphoenol pyruvate, 1 U pyruvate kinase (Sigma, Taufkirchen, Germany), 1 U of RNasin, and 4 μl sample fraction in a total volume of 10 μl at 35°C for 60 min. Reaction products were purified by phenol-chloroform extraction and isopropanol precipitation and analyzed by denaturing glyoxal agarose gel electrophoresis followed by autoradiography. A radioactively labeled in vitro transcript corresponding to the length of a replicon RNA was used to determine the size of the reaction products.
In vitro transcription.
In vitro transcripts of HCV were generated using the protocol described recently (32). All plasmid constructs used for in vitro transcription in this study contained a genomic hepatitis delta virus ribozyme followed by a T7-terminator sequence to generate authentic 3′ ends of the viral genomes. In vitro transcription reaction mixtures contained 80 mM HEPES, pH 7.5, 12 mM MgCl2, 2 mM spermidine, 40 mM DTT, 3.125 mM each nucleoside triphosphate, 1 U/μl RNasin (Promega, Mannheim, Germany), 0.05 μg/μl restricted plasmid DNA, and 0.6 U/μl T7 RNA polymerase (Promega). After 2 h at 37°C, an additional 0.3 U/μl T7 was added, and the reaction mixture was incubated for another 2 h. Transcription was terminated by the addition of 1 U RNase-free DNase (Promega) per μg plasmid DNA and 30 min of incubation at 37°C. After extraction with acidic phenol and chloroform, DNA was precipitated with isopropanol and dissolved in RNase-free water. The concentration was determined by the measurement of the optical density at 260 nm, and RNA integrity was checked by agarose gel electrophoresis. Capped transcripts of the dengue virus genome and SFV replicons were generated as described recently (30).
Plasmid constructs.
Subgenomic replicons pFK_i389LucNS3-3′JFH_δg, pFK_i389LucNS3-3′ET_δg, and the chimeric full-length construct pFK_JC1 have been described previously (32, 43, 52).
pTM1-2 is a multifunctional vector expressing an ampicillin resistance gene and encoding a T7 promoter that is used for the transient expression of foreign proteins. The following plasmids were generated by inserting the gene of interest into the multiple cloning site (MCS) via NcoI-SpeI restriction sites: pTM_NS3-5B_JFH, pTM_NS3/4A_JFH, pTM_NS4B_JFH, and pTM_NS5B_JFH. The cloning of pTM_NS5A_JFH was done by overlap PCR (overlap primer, TTGAAAAACACGATAATACCATGTCCGGATCCTGGCTCCGC), fusing the encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES) of pTM1-2 with the NS5A sequence and adding the start codon.
pTM NS3-5B JFH served as a vector for the cloning of pTM NS3-5B/5AΔDII (delta2222-2314aa) and ΔDIII (delta2328-2435aa). Deletions were transferred by the NsiI-BsrGI digestion of pFK_i389Luc_EI_JFH1/J6/C-846_delta2222-2314 and pFK_JC1_delta2328-2435 [3]). pTM NS3-5B/5AΔDI (delta2005-2190aa) was established by an overlap PCR product (overlap primer, CAAAAATTGGCTGACCTCTAAATTGGCGGCGCGGCGCTTGGCACGGGG) containing MfeI-RsrII restriction sites and NsiI-MfeI, as well as the NsiI-RsrII digestion of pTM NS3-5B JFH.
pWPI-T7-BLR was used to generate cell lines constitutively expressing T7 RNA polymerase. The gene encoding T7-RNA polymerase was cloned from plasmid pSC6 T7 neo (a generous gift from Martin Billeter) into pWPI-BLR, a derivative of the bicistronic lentiviral vector pWPI (a gift from Didier Trono), generated by the modification of the MCS and replacing the green fluorescent protein (GFP) in the reporter cistron with a blasticidin resistance gene (BLR), using AscI and BamHI restriction sites.
A plasmid encoding the dengue virus type 2 NGC clone was kindly provided by Andrew Davidson and was used to generate infectious dengue virus genomes by in vitro transcription. Semliki Forest virus replicon pSFV3-lacZ was used to generate Semliki Forest virus replicons (30).
Preparation of CRCs and 2D gel electrophoresis.
The protocol for the CRC preparation has been described previously (45). In brief, 2 × 108 Huh-7 cells (replicon 9-13 or cured cells as a negative control) were harvested in PBS and pelleted. Cells were suspended to a density of 2.5 × 107 cells/ml in hypotonic lysis buffer (10 mM Tris-HCl, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM phenylmethylsulfonyl fluoride [PMSF], 2 μg/ml aprotinin) and lysed by 75 strokes with a dounce homogenizer. Nuclei and intact cells were removed by centrifugation. The intracellular membranes in the resulting supernatant were sedimented on 300 μl of 60% (wt/wt) sucrose and treated with proteinase K (final concentration, 8 mg/ml) for 60 min at 25°C. After the addition of 2 mM PMSF, proteinase K-treated CRCs were loaded under a continuous 15 to 60% (wt/wt) sucrose gradient and centrifuged for 16 h at 71,000 × g and 4°C. Ten fractions were collected from the top and analyzed for density, protein concentration, and in vitro replicase activity. The peak fraction of in vitro replicase activity (typically at a density of 1.17 g/ml) and the neighboring fractions were combined, diluted in lysis buffer to 10 ml, and pelleted for 1 h (4°C and 50,500 × g). The pellet was resuspended in 500 μl two-dimensional (2D) protein solubilization buffer {2 M thiourea, 20 mg/ml 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 20 mg/ml SB3-10, 5 M deionized urea solution, 1× protease inhibitor mix (Sigma), 25 μl/ml tributylphosphine (Sigma), 10 μl/ml immobilized pH gradient (IPG) buffer pH 3-11NL (Amersham), 75 U/ml benzonase (Roche)} and incubated for 60 min at room temperature. After the addition of iodoacetamide to a final concentration of 15 mM, samples were incubated for an additional 90 min, cleared by centrifugation (5 min at maximum speed in a tabletop centrifuge), and subjected to isoelectric focusing. Therefore, IPG strips (3-11NL; Amersham) were loaded passively by rehydration in 350 μl 2D protein solubilization buffer containing 1 mg total protein for 10 h. Isoelectric focusing was performed in an IPGphor II unit (Amersham) with the following voltage profile: a 3-h linear gradient from 300 to 3,500 V, 3 h constant at 3,500 V, and then 12 h constant at 5,000 V.
After isoelectric focusing, strips were incubated for 10 min with 2D equilibration buffer (6 M urea, 2% SDS, 50 mM Tris-HCl, pH 8.8, 30% glycerol, 2% DTT) and for a further 10 min with the same buffer containing 2.5% iodoacetamide. The equilibrated strip was placed on top of a 10% acrylamide resolving gel without SDS, overlaid with 0.5% high-purity agarose, and subjected to electrophoresis (50).
Gels were fixed and silver stained using standard protocols (51). Spot patterns were compared by visual examination. Interesting spots were picked and proteins identified as previously described by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) (18) or high-performance liquid chromatography electrospray tandem mass spectrometry (HPLC ESI MSMS) (57) in the Core Facility for Mass Spectrometry and Proteomics at the Centre for Molecular Biology, Heidelberg, Germany.
Silencing of Huh7-Lunet cells.
Cells were seeded on 15-cm-diameter culture dishes and transfected with siRNAs using HiPerfect (Qiagen, Hilden, Germany) 72 h prior to electroporation according to the instructions of the manufacturer. A second dose of siRNA was applied by coelectroporating 1 nmol siRNA with HCV subgenomic or genomic RNA, corresponding to a final concentration of 2.5 μM siRNA. Mock-silenced cells were treated with HiPerfect in the absence of siRNA. Note that the efficient silencing of ANXA2 always required two successive applications of siRNAs, probably due to the high abundance and stability of the protein. Silencing using this protocol had no visible impact on cell growth or cell viability. Cytotoxic and cytostatic effects furthermore were ruled out by CytoTox96 assays (Promega) in lysates and supernatants of cells.
siRNAs.
The siRNA referred to as siControl (AGA AGU CAG GCC AUU ACA ATT) is directed against the 3′ NTR of dengue virus and was used as the negative control throughout this study, with the exception of experiments involving dengue virus. HCV siRNA (AGG UCU CGU AGA CCG UGC ATT) binds to the 5′ NTR of the HCV genome and efficiently inhibits HCV genotype 1b and 2a replication; this siRNA was used as the positive control in experiments involving HCV and as the negative control in the experiments dealing with dengue virus. siRNAs directed against annexins were selected by checking the entire coding sequence of the respective annexin using commercial siRNA design tools (Invitrogen and Eurofins MWG Operon) and by selecting the candidate with the highest score: siANXA2a (UUA ACA GAG UCU ACA AGG ATT), siANXA2b (CCU UAU GAC AUG UUG GAA ATT), siANXA4 (CCA UAU ACA UCU GGU UAC CTT), or siANXA5 (UUA GAA GCU UAG CCU UAG ATT).
Transient replication assay.
For the quantification of the replication efficiency of subgenomic replicons harboring a firefly luciferase gene, luciferase activity was determined in cell lysates at different time points after electroporation. Cells were washed twice with phosphate-buffered saline and scraped off the plate into 500 μl ice-cold lysis buffer (1% Triton X-100, 25 mM glycylglycine, 15 mM MgSO4, 4 mM EGTA, 1 mM DTT). The lysate was cleared by centrifugation (5 min, 14,000 rpm, 4°C). One hundred microliters of cleared lysate was mixed with 360 μl assay buffer (25 mM glycylglycine, 15 mM MgSO4, 4 mM EGTA, 1 mM DTT, 2 mM ATP, 15 mM K2PO4, pH 7.8) and, after the addition of 200 μl of a 200 μM luciferin solution, measured in a luminometer (Lumat LB9507; Berthold, Freiburg, Germany) for 20 s. All luciferase assays were done in duplicate measurements. Luciferase activity was expressed as the ratio of relative light units (RLU) (n-fold) at a given time point relative to the luciferase activity measured 4 h after transfection.
RESULTS
Purification of membrane fractions containing HCV replicase complexes.
The aim of our study was the identification of host cell factors that are associated with the HCV replication complex and play a functional role in the viral life cycle. Therefore, we established a purification protocol for the proteomic analysis of viral replication complexes from Huh-7 cells harboring a subgenomic replicon (isolate Con1, clone 9-13 [33, 44]). Cells from the same clone were cured from the replicon by interferon alpha and interferon gamma treatment in the absence of selective pressure and served as a negative control. The starting point of the purification procedure was the preparation of so-called crude replicase complexes (CRCs) (45) (Fig. 1A). These CRC preparations are capable of viral RNA synthesis in vitro (Fig. 1C, lanes 1 and 2), and their enzymatic activity was used as a quality control throughout the purification procedure. To enrich further potential host factors of viral replication, we treated CRCs excessively with proteinase K to remove all proteins that are not part of viral replication complexes and then purified the membranous structures by floatation in a sucrose gradient (Fig. 1A). This approach is based on our previously published model of the viral replication complex, according to which the viral proteins and RNA, along with host factors engaged in replication, are situated within vesicular structures and thereby are resistant to nuclease and protease treatment (45). Proteinase K treatment had no major effect on replicase activity in vitro (Fig. 1C, lanes 3 and 4); the apparently lower replicase activity in these samples and the gradient fractions (lanes 5 to 17) compared to that of the concentrated CRC preparation (lane 2) was due to the dilution of the samples.
FIG. 1.

Establishment of a purification scheme for the proteomic analysis of HCV replication complexes. (A) Schematic representation of a sample preparation for the proteomic analysis of cell extracts from HCV replicon and cured replicon cells. (B) Analysis of cell extracts from replicon and cured replicon cells. Identical portions of the pellet collected from the peak gradient fractions were subjected to SDS-PAGE. Proteins included in this fraction were either stained with Coomassie brilliant blue (top) or analyzed by immunoblotting using a polyclonal antiserum specific for HCV proteins (given in the right of each panel). (C) Replicase activity of sucrose gradient fractions of CRCs treated with proteinase K (PrK). Gradient fractions were collected from the top, and 4 μl of each gradient fraction as well as nontreated (−PrK) and PrK-treated (+PrK) CRCs were analyzed for in vitro replicase activity. A concentrated CRC fraction from replicon cells served as the positive control, and a corresponding fraction from cured cells served as the negative control.
Peak gradient fractions of replicase activity (Fig. 1C, lanes 9 to 11) were pooled and pelleted by ultracentrifugation. Lysates of cured cells were prepared in parallel, and fractions of the same density, typically 1.13 to 1.17 g/cm3, were collected. Conventional SDS-PAGE of the pellets did not reveal a difference in the overall protein composition between samples (Fig. 1B, top). As expected, the viral proteins NS3, NS5A, and NS5B were present only in the pellet obtained from replicon cells (Fig. 1B, middle and bottom).
Identification of ANXA2 by proteomic analysis.
Since the overall protein composition of the purified fractions from replicon and cured cells still was very complex, we subjected purified extracts of ca. 2 × 108 cells to two-dimensional gel electrophoresis, with isoelectric focusing in the first dimension and SDS-PAGE in the second dimension. The protein pattern was very similar for samples from replicon and cured cells (Fig. 2A) and also among eight independent experiments. Spot patterns were compared by visual examination, and proteins present exclusively or significantly enriched in the CRC fraction of replicon cells were identified by MALDI-TOF MS or HPLC ESI MSMS. The overall number of prominent differences was surprisingly low, and most of them showed up only in individual experiments (Table 1). We furthermore could not identify the HCV nonstructural proteins by 2D SDS-PAGE, although they were clearly detectable by conventional 1D SDS-PAGE and Western blot analysis in the same preparations (Fig. 1B), arguing for a relatively low sensitivity of the technique. The only reproducible difference was a string of three spots with different isoelectric points but identical molecular weights, which were unambiguously identified as ANXA2 (Fig. 2A, indicated by asterisks). They were found in larger amounts in CRC fractions of replicon cells in seven out of eight experiments (data not shown). Interestingly, two other members of the annexin family, annexin A4 and A5, also were identified in this analysis (Table 1), albeit less reproducibly, arguing that the annexins play an important role in the formation or activity of the HCV replication complex. To corroborate this assumption and to determine whether ANXA2 also is enriched in CRCs induced by another HCV isolate of a different genotype, we treated CRC preparations of Huh-7 cells harboring a JFH1 subgenomic replicon (genotype 2a) with proteinase K. As shown in Fig. 2B, ANXA2 was similarly abundant in total lysates and the CRC preparation of JFH1 replicon and control cells. However, upon proteinase K treatment, a larger fraction of ANXA2 was resistant to proteolysis in the presence of HCV. The quantification of the Western blot revealed a 3.4 times larger amount of ANXA2 in HCV-positive CRCs compared to that of cured control cells. An antiserum directed against the N-terminal part of calnexin was used as a control for the proteinase K digest, since this part of the protein is located in the endoplasmic reticulum (ER) lumen and therefore should be protected from proteases in an intact ER structure, whereas the carboxy terminus is protease sensitive in a cell lysate. Almost no full-length calnexin was detectable after protease K treatment, indicating that the proteinase K digestion was complete. In case of the C-terminally truncated fragment, a slight 1.4-fold increase was observed in the HCV-positive CRCs. However, in contrast to the HCV-induced increase in ANXA2 amounts, the change in calnexin was not reproducible, corroborating the sporadic detection of calnexin in the comparative 2D gels (Table 1). Similar results were obtained with genotype 1 (data not shown).
FIG. 2.
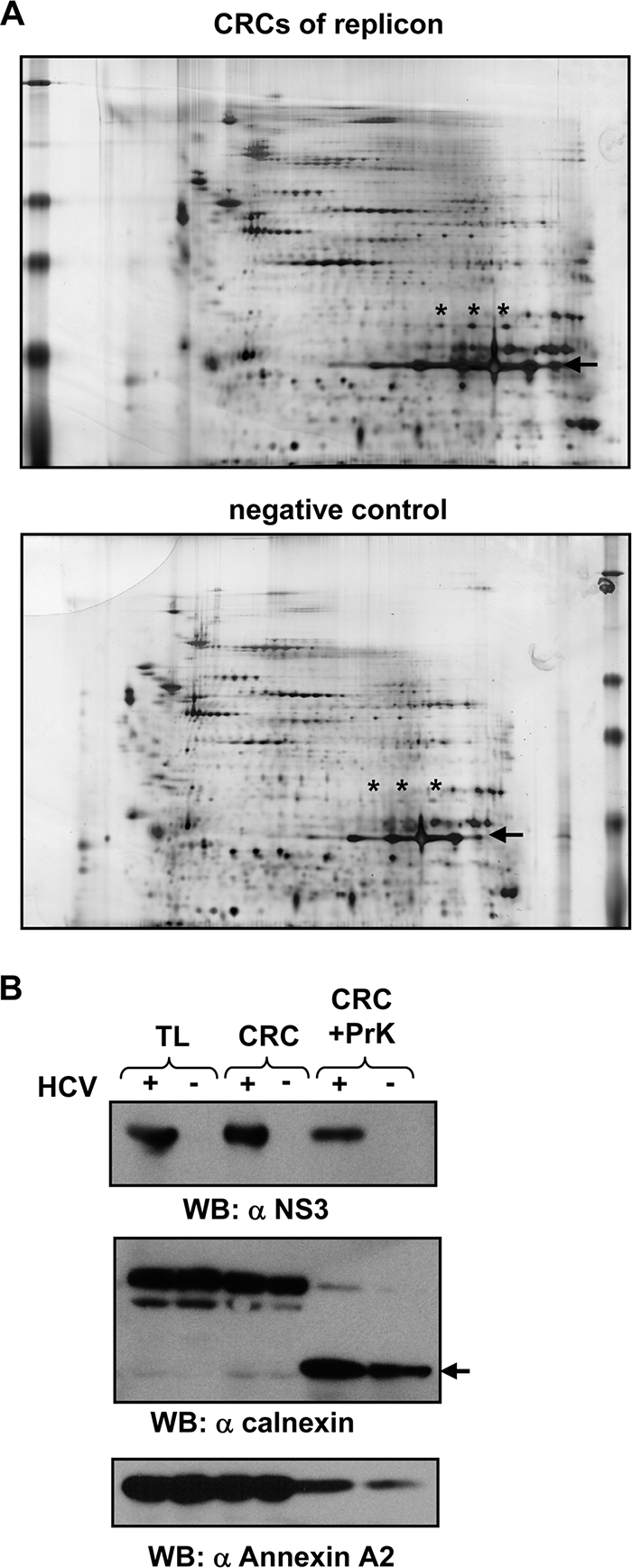
Identification of annexin A2 as a potential component of HCV genotype 1b replication complexes and evaluation by the Western blot analysis (WB) of cell extracts from genotype 2a replicon cells. (A) 2D-PAGE of membrane fractions from cells harboring subgenomic replicons or their cured counterparts treated according to the scheme in Fig. 1A. Proteinase K is marked by the arrow, and ANXA2 spots are marked with asterisks. (B) Detection of viral and cellular proteins in CRCs before and after proteinase K digestion. Viral and cellular antigens were detected in total-lysate (TL), CRC, and digested CRC fractions (CRC+PrK) of cells harboring a subgenomic genotype 2a replicon (isolate JFH1) and their cured counterparts. The viral protein was detected with a polyclonal antiserum directed against the helicase domain of NS3 (α-NS3). The band corresponding to C-terminally truncated calnexin is marked by an arrow. ANXA2 was stained with a monoclonal mouse antibody. Note that 5-fold-larger amounts of the proteinase K-treated CRC fractions were loaded compared to those of untreated samples.
TABLE 1.
Potential host factors in HCV replication enriched in CRCsd
| Identified protein | Gene name | Protein accession no. | Identification methoda | Differential spot intensityb |
|---|---|---|---|---|
| Annexin A2 | ANXA2 | AAH23990 | PMF (12, P < 0.0001) | 7 |
| PMF (11, P < 0,0001) | ||||
| MSMS (4, P < 0.0001) | ||||
| MSMS (10, P < 0.0001) | ||||
| Annexin A4 | ANXA4 | AAH63672 | PMF (10, P < 0.0001) | 2 |
| Annexin A5 | ANXA5 | AAH01429 | PMF (7, P < 0.01) | 2 |
| MSMS (9, P < 0.0001) | ||||
| ATP synthase, beta subunit | ATP5B | AAH16512 | PMF (8, P < 0.01) | 4c |
| PMF (17, P < 0.0001) | ||||
| PMF (7, P < 0.01) | ||||
| MSMS (4, P < 0.0001) | ||||
| ATP synthase, O subunit | ATP5O | AAV38639 | PMF (8, P < 0.001) | 1 |
| Calnexin | CANX | AAH42843 | MSMS (1, P < 0.05) | 1 |
| BiPe | HSPA5 | CAA61201 | PMF (13, P < 0.0001) | 1 |
| Peroxiredoxin 3 | PRDX3 | NP_054817 | PMF (5, P < 0.01) | 1 |
PMF, peptide mass fingerprint using MALDI-TOF MS data; MSMS, peptide sequencing using HPLC ESI MSMS data. The numbers in parentheses refer to the number of matching peptides and the P value for identity or extensive homology.
Number of experiments in which enrichment of the protein in CRCs of replicon cells was found, based on the comparison of the once identified spots on behalf of their location in the 2D gel; eight repetitions were performed.
Various molecular sizes of 27 and 56 kDa; all spots were identified by PMF or MSMS analysis.
Protein spots identified with scores indicating identity (P < 0.05) were included.
BiP, binding immunoglobulin protein.
In summary, we identified ANXA2 as a candidate host cell protein enriched in the HCV replication complex.
Annexin A2 colocalizes with HCV replication sites.
To complement the proteomic analysis of HCV replication complexes with cell-based and phenotypic studies, we next analyzed whether ANXA2 colocalized with these viral structures. In immunofluorescence studies, HCV replication complexes, also referred to as membranous webs, appear in a dot-like pattern with the colocalization of all nonstructural proteins and newly synthesized viral RNA (17) as well as double-stranded replication intermediates (56). The cellular distribution of ANXA2 in naïve Huh-7 cells (Fig. 3, right) and Huh7.5 cells (not shown) appeared to be mainly cytosolic, with minor nuclear and plasma membrane staining. In contrast, ANXA2 was clearly redistributed to dot-like structures induced by HCV in infected cells (Fig. 3A, left) as well as replicon cell lines harboring genotype 1 (Con1) or genotype 2 (JFH1) sequences (Fig. 3A, middle). This colocalization to the putative replication complexes was detectable not only for NS5A but also with antibodies detecting NS3, NS4B, and NS5B (data not shown). In addition, we analyzed JFH1 replicon cells for the colocalization of ANXA2 and double-stranded RNA (Fig. 3B). As expected, ANXA2 and dsRNA clearly colocalized in the dot-like structures exclusively present in the replicon cells, thereby confirming the association of ANXA2 with active HCV replication sites.
FIG. 3.

Subcellular localization of ANXA2. (A) Colocalization of NS5A with annexin A2 in infected Huh7.5 or cell lines harboring persistent genotype 1b (Con1, clone 9-13) or genotype 2a (JFH1) replicons. Cells containing subgenomic replicons and naïve Huh-7 cells were subjected to immunofluorescence analysis 48 h after seeding. Huh7.5 cells were analyzed 48 h after infection. ANXA2 was detected by a polyclonal rabbit antiserum and a secondary goat anti-rabbit antibody labeled with Alexa 488 (middle row). NS5A was detected with monoclonal antibody 9E10, recognizing genotype 1b and 2a subtypes, and a secondary goat anti-mouse antibody labeled with Alexa 546 (upper row). Naïve Huh-7 cells served as the negative control (Huh-7) and were processed in parallel. (B) Colocalization of ANXA2 and double-stranded RNA in HCV JFH1 replicon cells. Cells containing subgenomic replicons and naïve Huh-7 cells were subjected to immunofluorescence analysis 48 h after seeding. ANXA2 was detected by a polyclonal rabbit antiserum and a secondary goat anti-rabbit antibody labeled with Alexa 488. dsRNA was detected with monoclonal mouse antibody J2 and a secondary goat anti-mouse antibody labeled with Alexa 546. (C) Localization of annexin A2 and the replication complexes of Semliki Forest virus (SFV) in BHK-21 cells or dengue virus (DV) in Huh-7 cells. Cells were fixed 24 h posttransfection or infection and stained with monoclonal mouse ANXA2 antibody HH7 and a secondary goat anti-mouse antibody labeled with Alexa 546. Viral antigens were detected with polyclonal antisera directed against SFV nsp3 or DV NS4B, respectively, and a goat anti-rabbit antibody labeled with Alexa 488.
To exclude that the redistribution of ANXA2 was simply a consequence of the reorganization of the ER induced by HCV, we also analyzed the distribution of the ER marker PDI in the presence and absence of HCV replicons (Fig. 4A). PDI partially colocalized with NS5A, another component of the HCV replication complex, but showed no obvious redistribution in replicon cells compared to the distribution in naïve cells. This was in line with the assumption that the membranous web is derived from ER membranes but that PDI was not specifically recruited by HCV. We also were interested in a possible colocalization of S100A10 (p11) with the HCV replication sites (Fig. 4B), since this protein can form heterotetrameric complexes with ANXA2, which are crucial for many of its functions (14). The distribution of S100A10 was similar to that of ANXA2 in naïve Huh-7 cells, but in contrast to the annexin, no specific colocalization to the membranous web was found, indicating that the functions of ANXA2 in the HCV life cycle are independent of S100A10.
FIG. 4.

Localization of HCV nonstructural proteins and different cellular proteins in Huh7-Lunet/CD81 cells infected with HCV JC1. Infected cells (left rows) or naive Huh7-Lunet/CD81 cells (right rows) were subjected to immunofluorescence analysis 48 h after seeding. Subcellular localization of PDI (middle row) and NS3 (top row) (A), p11 (S100/A10, middle row) and NS3 (top row) (B), annexin A4 (middle row) and NS5A (top row) (C), and annexin A5 (middle row) and NS3 (top row) (D). Note that NS3 and NS5A almost perfectly colocalize in HCV infected cells.
We next analyzed a potential colocalization of other annexins identified in our proteomic analysis with HCV proteins (Table 1). Annexin A4 showed a clear colocalization with HCV replication sites in HCV-infected cells (Fig. 4C), whereas the distribution of annexin A5 was unaltered by HCV and no specific colocalization to dot-like structures was observed (Fig. 4D). The same colocalization patterns were found in genotype 1 replicon cells (data not shown).
Finally, we wanted to know whether the recruitment of ANXA2 was specific for HCV or a general feature of replication sites of other positive-strand RNA viruses. We could not observe a specific colocalization of ANXA2 with the respective replication sites of dengue virus (DV) or Semliki Forest virus (SFV), indicating that the recruitment of ANXA2 is a specific feature of HCV (Fig. 3C).
Taking these findings together, annexin A2 and A4 are recruited to and colocalize with the replication sites of HCV. The colocalization of ANXA2 was independent of S100A10 and was not found for SFV and dengue virus replication sites.
Knockdown of annexin A2 does not impair viral RNA replication but significantly reduces infectious HCV titers.
Given the association of annexin A2 with the HCV replicase complex in subgenomic replicon cells, it seemed plausible that the protein is required for viral RNA replication. To address this question, we silenced ANXA2 mRNA in Huh7-Lunet cells using two independent siRNAs. An siRNA directed to the HCV genome served as the positive control and an unrelated siRNA as the negative control (Fig. 5). Seventy-two hours after siRNA transfection, silenced cells were electroporated with a subgenomic luciferase replicon of genotype 2a (JFH1) (Fig. 5B) or genotype 1b (Con1) (Fig. 5C) and a second dose of the same siRNA. Cells were harvested at different time points after transfection and analyzed for ANXA2 knockdown efficiency by Western blotting and luciferase activity as a correlate of HCV replication. siANXA2a proved to be more efficient than siANXA2b and substantially reduced intracellular ANXA2 levels (Fig. 5A, lower Western blot panel) without any cytostatic or cytopathic effects (data not shown). Despite the efficient silencing of ANXA2, no reduction in viral RNA replication was observed for JFH1 (Fig. 5B) or for Con1 (Fig. 5C). In contrast, siRNA directed against the HCV genome substantially reduced viral replication, indicating that the silencing approach could affect RNA replication. Similar results were obtained with cell lines harboring persistent replicons of different genotypes. In this case, we also determined RNA replication directly by reverse transcription-quantitative PCR (RT-qPCR) on HCV positive-strand genomes after ANXA2 knockdown (data not shown). However, we again found no indication for a direct effect of reduced ANXA2 levels on HCV RNA replication.
FIG. 5.
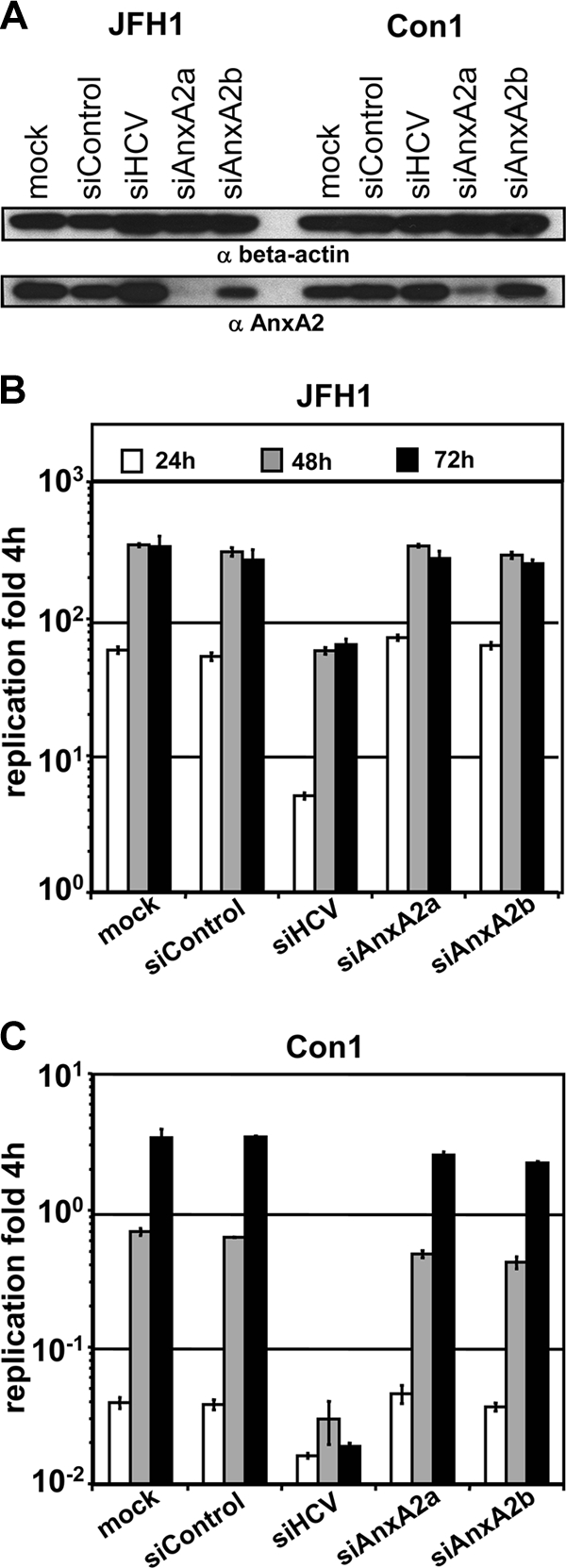
Effect of annexin A2 knockdown on replication of subgenomic reporter replicons. Huh7-Lunet cells were transfected with the indicated siRNAs, and 3 days later they were electroporated with a second dose of the same siRNA together with JFH1 or Con1 subgenomic replicons encoding firefly luciferase and analyzed for ANXA2 knockdown efficiency (A) and HCV replication (B and C). (A) Knockdown efficiency of ANXA2 siRNAs analyzed by Western blotting. Total cell lysates were harvested 48 h after electroporation with the indicated siRNAs and HCV replicon RNAs and subjected to Western blot analysis. β-Actin (upper) as well as ANXA2 (lower) were stained with monoclonal mouse antibodies. (B and C) The replication efficiency of HCV genotype 1b (B) or 2a (C) replicons was determined by luciferase reporter assay. Luciferase activity is expressed as the ratio of relative light units (RLU) obtained at 24 (white bars), 48 (gray bars), and 72 h (black bars) relative to the luciferase activity 4 h after electroporation to normalize for transfection efficiency. Mean values and standard deviations from a representative experiment out of five independent experiments are shown.
To investigate whether ANXA2 plays a role in other steps of the viral life cycle, we used the same experimental setup as that described above but transfected a full-length HCV genome instead of subgenomic replicons and measured viral infectivity titers in the supernatant of transfected cells. We chose a chimeric genome termed JC1, which was composed of the structural genes and a part of NS2 of isolate J6 and the remaining parts of JFH1, because this chimera was shown to yield high titers (43). Western blot analysis revealed that ANXA2 knockdown with siANXA2a was very efficient (Fig. 6A, bottom), whereas NS5A levels were comparable in mock-treated and ANXA2-silenced cells (Fig. 6A, top), confirming that the silencing of ANXA2 had no impact on RNA replication. Titers of infectious virus were determined in the supernatant 24 and 48 h postelectroporation by TCID50 measurement. Interestingly, viral titers were reduced 5- to 7-fold on average upon ANXA2 knockdown compared to titers from siControl or mock-treated cells (Fig. 6B), suggesting a role of ANXA2 in either the assembly or release of HCV particles. In contrast, the knockdown of annexin A4 or A5 had just a minor effect on viral titers (Fig. 6C), indicating that only ANXA2 facilitates the production of infectious HCV particles. To exclude that ANXA2 silencing has a general effect on virus exocytosis, we also analyzed the impact of ANXA2 silencing on infectious titers of the related dengue virus. However, no significant effect of ANXA2 silencing on dengue virus titers was observed (Fig. 6D).
FIG. 6.

Impact of the silencing of ANXA2 and other annexins on HCV or dengue virus titers. Huh7-Lunet cells were transfected with the indicated siRNAs, electroporated with a second dose of the same siRNA together with viral genomes, and analyzed for ANXA2 knockdown efficiency (A) and titers of HCV (B and C) or dengue virus (D). (A) Reduction of ANXA2 levels after knockdown. Shown is a Western blot of cell lysates harvested 48 h postelectroporation of Huh7-Lunet cells with HCV JC1 in vitro transcripts and the indicated siRNAs. Antigens were detected by monoclonal mouse antibodies directed against NS5A, β-actin, and ANXA2 as indicated on the right. (B) ANXA2 knockdown impairs the production of infectious HCV. The relative infectivity in supernatants at 24 and 48 h after electroporation with JC1 and the indicated siRNA is shown. Mean values and standard deviations of viral titers from seven independent experiments are given in percentages normalized to TCID50 titers in mock-silenced cells. (C) No impact of ANXA4 or ANXA5 knockdown on viral titers. HCV infectivity titers in supernatants harvested 48 h after the electroporation of cells with HCV JC1 full-length RNA and the indicated siRNAs are shown. The TCID50/ml of a representative experiment are shown. Knockdown efficiency was checked by Western blotting (not shown). (D) No reduction of dengue virus titers upon ANXA2 silencing. Titers of dengue virus genotype 2 in supernatants harvested 48 h after the electroporation of presilenced cells with DV full-length RNA and siRNAs shown as the TCID50/ml of a representative experiment. An siRNA directed against HCV was used as a negative control in this experiment.
In summary, the silencing of annexin A2 but not of A4 or A5 clearly reduced HCV but not dengue virus titers, suggesting an important role of this protein in the production of infectious HCV virions.
Annexin A2 knockdown reduces the secretion of viral Core and most likely affects HCV assembly.
To gain more mechanistic insight into the function of ANXA2 in HCV particle production, we first analyzed whether the reduction in infectious virus titer was due to an inhibition of the secretion of assembled virus particles by determining the intra- and extracellular levels of HCV Core protein. At 4 and 48 h postelectroporation, intracellular Core levels were almost identical in mock-, control, and ANXA2-silenced cells (Fig. 7A), arguing for comparable transfection, translation, and RNA replication efficiencies in all three experimental settings. However, we observed a 5-fold drop of extracellular Core levels upon ANXA2 knockdown 48 h after the transfection of the viral RNA (Fig. 7B), roughly reflecting the extent of the reduction in infectious virus titers. Therefore, the silencing of ANXA2 expression seemed to impair the production of viral particles rather than the specific infectivity of virions.
FIG. 7.
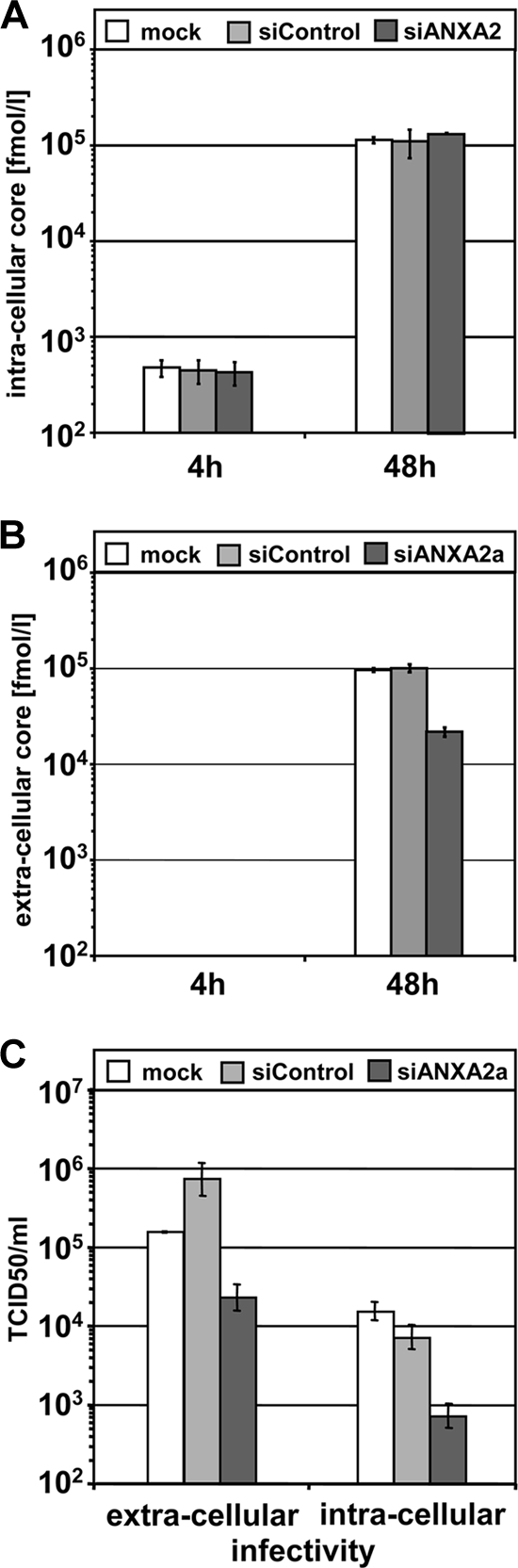
Extra- and intracellular amounts of viral Core protein and infectivity titers after ANXA2 knockdown. Huh7-Lunet cells were transfected with the indicated siRNAs, electroporated with a second dose of the same siRNA together with HCV JC1 full-length viral genome, and analyzed for intra- and extracellular Core protein and TCID50. (A) Intracellular Core levels (fmol/liter) in freeze-thaw lysates of silenced cells harvested 4 and 48 h after electroporation with the indicated siRNAs and HCV full-length RNA. (B) Extracellular Core levels (fmol/liter) in supernatants of cells 4 and 48 h after electroporation. (C) Extra- and intracellular infectivity titers in freeze-thaw lysates and supernatants of cells harvested 48 h after electroporation. Mean values and standard deviations from one out of three (A and B) or five (C) representative experiments are shown.
To discern whether this reduction in the amount of extracellular HCV particles was due to a block in assembly or release, we compared titers of infectious virus in the cell culture supernatant to the titers in cell lysates obtained by repeated freezing and thawing (Fig. 7C). A block in release should result in an intracellular accumulation of infectious virus. Conversely, a block in assembly was expected to impair intra- as well as extracellular viral titers. In fact, a comparable reduction of intra- and extracellular infectivity titers was observed after ANXA2 silencing (Fig. 7C). Therefore, we conclude that ANXA2 might play a role in assembly rather than the release of infectious HCV virions.
Annexin A2 recruitment is mediated by domain III of nonstructural protein 5A.
ANXA2 was identified in a proteomic analysis of HCV replication complexes and colocalized extensively with these structures in replicon cells. Therefore, we assumed that this cellular protein is recruited to replication sites by direct interaction with one of the HCV nonstructural proteins. However, numerous experimental strategies, including the immunoprecipitation of [35S]methionine-labeled cell lysates, immunoprecipitation and Western blotting, glutathione S-transferase pulldown assays, and yeast-two-hybrid assays, did not reveal an unambiguous interaction of ANXA2 with any of the nonstructural proteins of HCV (not shown). Due to the potential involvement of ANXA2 in HCV assembly, we extended this analysis by immunoprecipitation studies with the HCV structural proteins and NS2, again with negative results (data not shown). Therefore, it seemed plausible that ANXA2 was recruited by an indirect mechanism, either by an adaptor protein or by one of those lipids specifically binding ANXA2, which might be enriched at the HCV replication sites. Since the clear colocalization and redistribution of endogenous ANXA2 provided a robust readout, we used an immunofluorescence-based approach to further explore the indirect involvement of HCV nonstructural proteins in the recruitment of ANXA2 to HCV replication sites. We expressed the NS3 to NS5B coding region of isolate JFH1 under the control of a T7 promoter in Huh7-Lunet cells constitutively expressing T7 RNA polymerase (Fig. 8A and B). The expression of NS3 to NS5B led to a typical dot-like distribution of the nonstructural proteins reminiscent of the pattern observed in replicon cells and in HCV-infected cells (compare Fig. 8B, upper, to Fig. 3 and 4). Endogenous ANXA2 clearly colocalized with the NS protein signal in these dot-like structures, indicating that the recruitment of ANXA2 was independent of HCV RNA replication. We next expressed the nonstructural proteins NS3/4A, NS4B, NS5A, and NS5B individually (Fig. 8A and B). In this setting, each NS protein exhibited a different subcellular distribution, which was mainly ER-like with more or less distinct substructures. As previously described, the expression of NS4B alone induced distinct dot-like membrane alterations. However, ANXA2 was not specifically recruited to these structures. NS3/4A and NS5B showed no characteristic substructures and only minor, nonspecific colocalization with ANXA2. Moreover, the ANXA2 pattern was not significantly changed compared to that of naïve cells. In contrast, individually expressed NS5A showed a distinct punctate staining pattern and ANXA2 was clearly recruited to these structures. These results suggested that NS5A was responsible for the recruitment of ANXA2 to viral replication complexes.
FIG. 8.
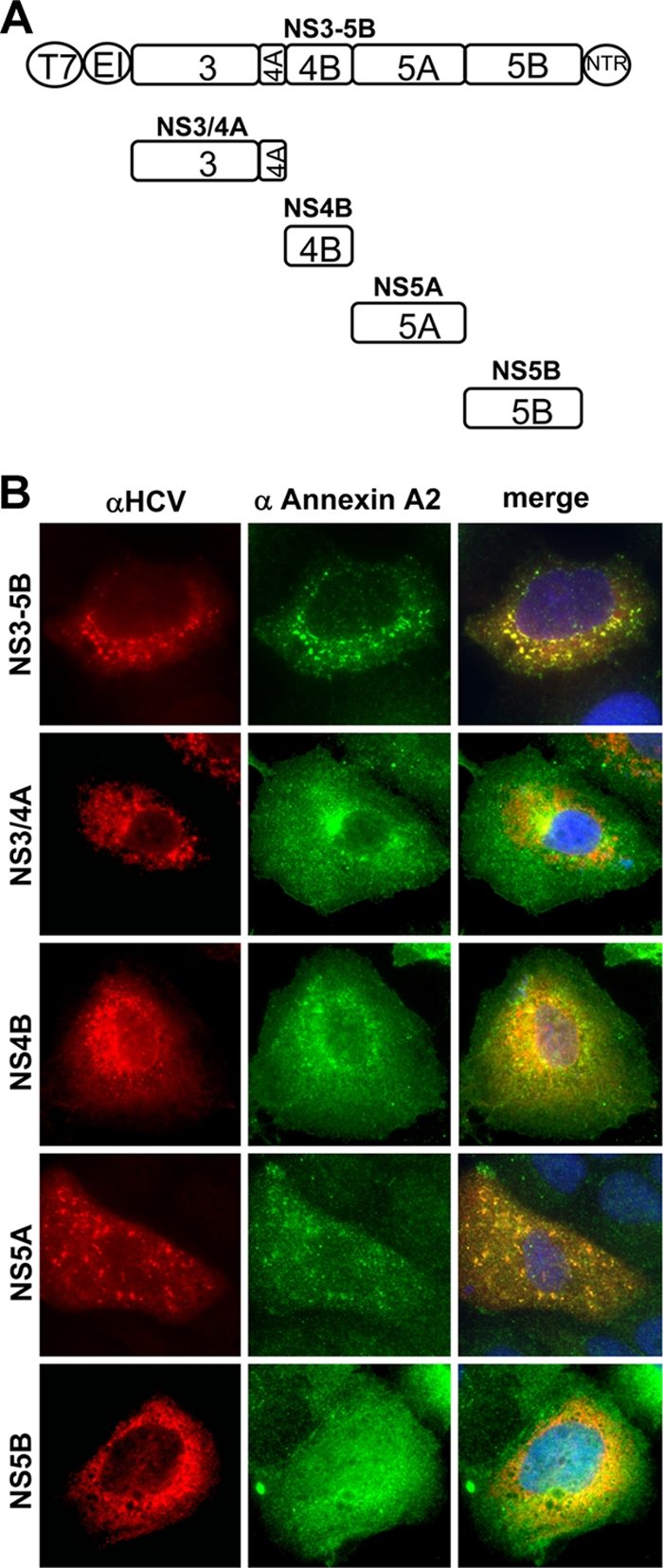
Localization of annexin A2 and HCV antigens in cells expressing individual nonstructural proteins (genotype 2a). (A) Schematic representation of expression constructs used in this experiment. Viral nonstructural proteins are represented as squares; the T7 promoter (T7), EMCV IRES (EI), and 3′ nontranslated regions (NTR) are depicted as circles. The individual NS proteins are shown in relation to the NS3-5B polyprotein. (B) Lunet T7 cells were transfected 24 h after seeding with pTM constructs encoding viral proteins as indicated on the left and subjected to immunofluorescence analysis 24 h after transfection either using primary antibodies detecting HCV antigens and secondary antibodies labeled with Alexa 546 (left) or primary antibodies detecting ANXA2 and secondary antibodies labeled with Alexa 488 (middle). Cells expressing NS3/4A, 4B, and 5B were stained with polyclonal antisera against the individual viral proteins and monoclonal anti-ANXA2 antibody HH7. In cells expressing NS3-5B or NS5A, the viral antigen was detected by mouse monoclonal anti-NS5A antibody 9E10 and ANXA2 with a polyclonal antiserum.
NS5A is a multifunctional protein with three distinct subdomains linked by so-called low-complexity sequences. To map the domain of NS5A responsible for inducing the recruitment of ANXA2, we deleted individual domains (Fig. 9A, ΔDI, ΔDII, and ΔDIII) within the context of the NS3-5B polyprotein. In this experimental setting, the subcellular distribution pattern of the polyprotein was maintained, allowing for a better judgment of colocalization events compared to those of individually expressed NS5A. Although the localization of the nonstructural proteins NS3 to NS5B was slightly altered upon the deletion of the individual NS5A subdomains (Fig. 9B), ANXA2 still redistributed and colocalized extensively after the deletion of NS5A domain I (ΔDI; delta2005-2190aa) and II (ΔDII; delta2222-2314aa). In contrast, colocalization was lost upon the deletion of domain III (ΔDIII; delta2328-2435aa), and the overall ANXA2 distribution appeared similar to that of naïve Huh7-Lunet cells. The same result was obtained with cells harboring a persistent subgenomic replicon with an identical deletion in domain III of NS5A (data not shown), which only slightly affects viral RNA replication (3). Deletions of smaller parts of domain III of NS5A (e.g., aa 2354 to 2435, 2354 to 2404, and 2405 to 2435 [3]) all resulted in a loss of the colocalization of NS5A and ANXA2, indicating that the entire domain III is required to recruit ANXA2 (data not shown).
FIG. 9.
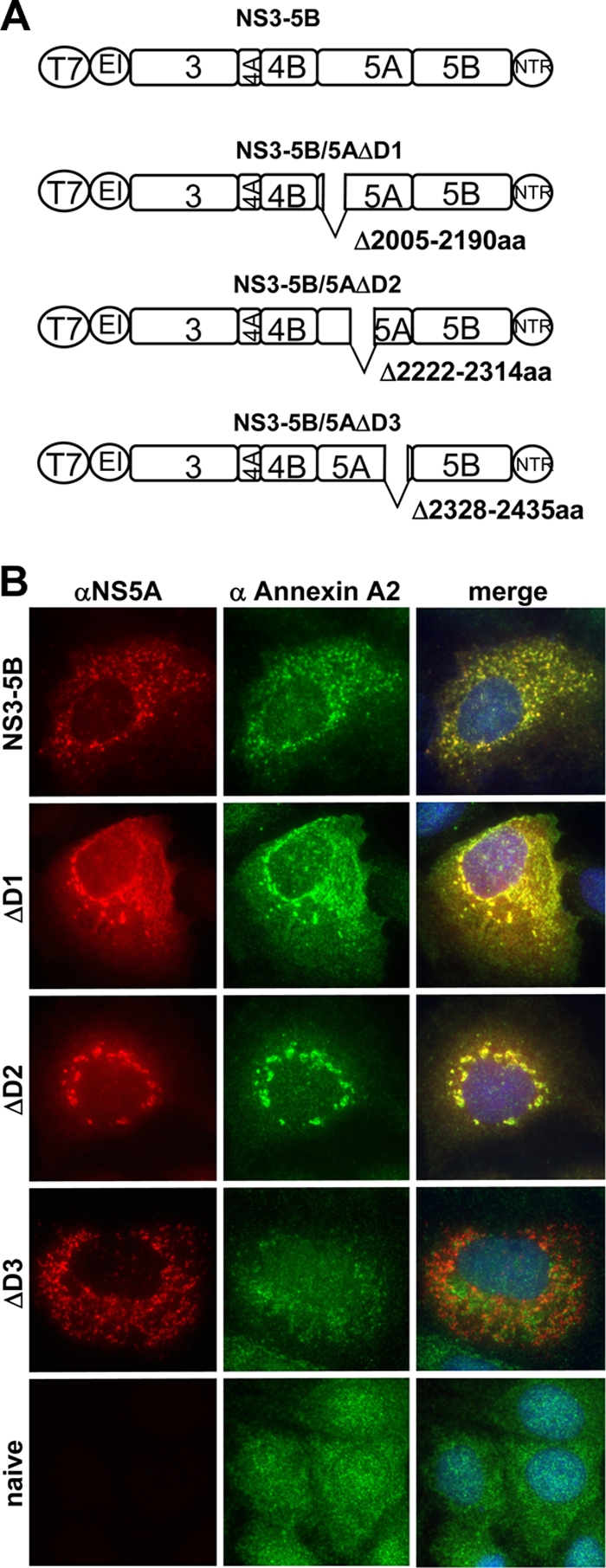
Subcellular localization of annexin A2 and NS5A after expression of NS3 to NS5B coding sequences (genotype 2a) harboring deletions of individual NS5A subdomains. (A) Schematic representation of expression constructs used in this experiment. Internal deletions of NS5A are marked by gaps at the relevant positions; numbers next to the gap refer to the amino acid position of the deletion. Note that the size of the deletions is not drawn to scale. For further explanation, see the legend to Fig. 8A. (B) Lunet T7 cells were transfected 24 h after seeding with pTM constructs encoding viral proteins as indicated on the left and subjected to immunofluorescence analysis 24 h after transfection either using primary antibodies detecting HCV NS5A and secondary antibodies labeled with Alexa 546 (left) or a polyclonal antiserum detecting ANXA2 and secondary antibodies labeled with Alexa 488 (middle). NS5A was stained by monoclonal mouse antibody 9E10 or 2F6 in the case of ΔDIII, since DIII contains the epitope detected by 9E10.
We conclude from these results that NS5A domain III was responsible for inducing the recruitment of ANXA2 to the HCV replication sites. Since this part of NS5A recently has been shown to play an important role in viral assembly and since ANXA2 knockdown impairs assembly, it is tempting to speculate that ANXA2 at least in part mediates this function of NS5A.
DISCUSSION
Several techniques have been established to identify potential HCV host factors by the proteomic analysis of purified replication complexes making use of their particular properties. We focused on the proteinase K resistance of active replication complexes, since we have shown recently that this treatment removes the majority of viral and cellular proteins but does not interfere with viral RNA synthesis in vitro (45). The major disadvantage was the low sensitivity of the technique, which was reflected by the low number of candidates we were able to identify and by the fact that the HCV nonstructural proteins could not be detected by the silver staining of the 2D-PAGE but only by Western blotting. Therefore, our analysis does not provide a comprehensive view of the host factor composition of HCV replication sites but rather identifies only the most abundant and/or most protected proteins. In line with our results, Huang et al. (24) identified the same set of annexins (ANXA2, ANXA4, and ANXA5) using a completely different purification technique based on the affinity capture of FLAG-tagged NS5A in a functional replicon. In contrast, studies relying on the extraction of detergent-resistant membranes provided other sets of candidates with little overlap and did not identify any annexin (19, 34). Purification procedures in the absence of detergents might preserve the complex membrane accumulations without perturbing their integrity, whereas the isolation of detergent-resistant membranes might destroy the three-dimensional architecture of the membranous web and thereby release factors that were bound in these higher-order structures.
Results from our colocalization studies indicated that domain III of NS5A was responsible for the recruitment of annexin A2 to the HCV replication sites. Three recently published reports clearly show that this part of NS5A is essential for viral assembly but not for viral RNA replication (3, 35, 58). The phenotype obtained by ANXA2 silencing therefore resembled the deletion of domain III of NS5A and indicates that ANXA2 mediates the contribution of NS5A to virus assembly. However, a deletion of the entire domain III (2328 to 2435) almost completely abrogated viral assembly (3), whereas the silencing of ANXA2 reduced viral titers at most by one order of magnitude. Although the reduction of intracellular ANXA2 protein levels by silencing was not complete, this difference in effects indicates that annexin A2 is not the only determinant required for mediating the function of domain III in virus assembly.
Previous studies already pointed to an important role of the phosphorylation of particular serine residues at the C terminus of NS5A DIII (35, 58) that are involved in an essential interaction with the Core protein (35). This interaction seems to be important for a recruitment of Core and NS5A on the same lipid droplet, thereby probably linking replication and packaging (3). The replacement of serines 2428/2430/2433 by alanines completely abrogated the interaction with Core (35) and a deletion in domain III (2405 to 2435) resulted in a loss of the colocalization of Core and NS5A (3). In contrast, the same triple-serine mutant had no impact on ANXA2-NS5A colocalization (data not shown), indicating that the function of ANXA2 is independent of the Core-NS5A interaction. The recruitment of a host factor involved in assembly but not in RNA replication into the HCV replication complex, even in the absence of structural proteins, is not unique to ANXA2. ApoB and ApoE also are recruited to the HCV replication sites (24), and these proteins appear to be essential for the envelopment and infectivity of progeny virus. In this respect, NS5A appears to prepare the environment for assembly before and during viral RNA replication, further highlighting the close link between these two processes in the HCV life cycle.
In general, the assembly of HCV is poorly understood, particularly in terms of host factor requirements and their mechanism of action. The situation is further complicated by the intimate connection of replication and assembly sites to lipid droplets (38) and the association of virions with lipoproteins, resulting in a very heterogeneous density of infectious HCV, even in cell culture (11). Therefore, the definition of HCV assembly probably has to be broadened to involve the association of lipoproteins or other yet-to-be-defined host cell components to the viral particle. The heterogeneity of HCV virions also might reflect different assembly pathways with various host factor requirements and might explain the relatively moderate reduction in viral titers that we can achieve upon the silencing of ANXA2 or other assembly cofactors like ApoE, which is, in our hands, also in the range of only about 10-fold (data not shown).
Our colocalization analysis clearly revealed that NS5A was involved in the recruitment of ANXA2; still, we found no evidence for a direct interaction of the proteins. However, we cannot completely rule out a weak interaction between ANXA2 and NS5A. More likely, ANXA2 might be recruited by means of a particular lipid composition of the HCV replication sites. Annexin A2 binds to negatively charged phospholipids, showing a preference for PIP2 (48). This is particularly interesting because HCV has been linked to phosphatidylinositol biosynthesis. Recent siRNA screens revealed an important role of phosphatidylinositol-4-kinase III alpha (PI4KCA) in HCV replication (6, 9, 21, 55, 61). This enzyme generates phosphatidylinositol-4-phosphate, the precursor lipid of PIP2. In addition, we found a strong colocalization of PIP2 with HCV replication sites (S. Reiss, P. Backes, V. Lohmann, I. Woerz, and R. Bartenschlager, unpublished data). Although we do not yet have experimental proof, we speculate that ANXA2 is recruited by PIP2 to the HCV replication sites and that domain III of NS5A plays a critical role in generating or stabilizing the correct lipid composition of the membranes attracting ANXA2. Interestingly, PIP2 also has been proposed to be involved in the interaction of HIV-1 gag and ANXA2 (21), which stimulates HIV production in 293T cells.
The exact mechanistic role of ANXA2 in HCV assembly remains speculative. In most cells, ANXA2 is present as a cytosolic monomer and a heterotetrameric complex with S100A10 (p11), which is found in the cytosol and at the plasma membrane-cytoskeleton interface. Currently, we favor a role of monomeric ANXA2, since we did not detect the colocalization of HCV NS proteins with S100/A10. ANXA2 is involved in a number of cellular processes (for a review, see references 14 and 15) that include different endocytic events, the Ca2+-induced exocytosis of secretory granules, actin dynamics, and the organization of membrane microdomains, probably in conjunction with the actin cytoskeleton. It has further been shown that ANXA2 can bind RNA (12), leaving several possibilities for a functional role of this protein in HCV assembly. However, a general effect on exocytosis is unlikely, since we observed a clear reduction in intracellular HCV titers upon ANXA2 silencing and no significant impairment in the secretion of dengue virus particles. Therefore, we favor the idea that monomeric ANXA2 is recruited by the particular lipid composition of the HCV replication sites, which itself is controlled by NS5A. Once present, ANXA2 participates in the formation of higher-order structures of the membranous web via its membrane organization functions (67), possibly stabilizing membrane microdomains involved in virion morphogenesis. Alternatively, it also could provide contacts to the actin cytoskeleton (23, 48), which has been shown to be linked to HCV replication complexes (10, 28). Interestingly, ANXA2 has been identified in a pulldown of NS3 from HEK 293T cells expressing NS3/4A together with components of the cytoskeleton, providing some evidence for this concept (28). Owing to the high endogenous ANXA2 expression levels in Huh-7 cells, we have not yet succeeded in generating a cell line with efficient stable ANXA2 knockdown, which would allow for the complementation of the functions of ANXA2 in trans. This approach would allow the analysis of mutants of the protein to get a deeper insight into its mechanism of action.
Interestingly, ANXA2 and other annexins have been identified as important host factors of several unrelated viruses and at different stages of their life cycle. ANXA2 is incorporated into cytomegalovirus (65) and influenza virus (29) particles, promotes the entry of a calicivirus (16) and of human cytomegalovirus (46), and plays a role in HIV-1 assembly (21, 49) and bluetongue virus release (5). In the case of HIV-1, gag is targeted to lipid rafts by PIP2 (41) and interacts with ANXA2 in macrophages to facilitate viral assembly (21, 49). Although life cycles of lentiviruses and HCV seem to have little in common, this example provides several parallels to our results, particularly the involvement of PIP2 in ANXA2 recruitment, which we also speculate to be involved in the case of HCV and might point to related mechanisms of action. This is reminiscent of a study pointing out fundamental similarities between retroviral capsids and replication complexes of positive-strand RNA viruses, suggesting common evolutionary origins (53).
In summary, we have identified the PIP2 binding protein annexin A2 as a host factor recruited to HCV replication sites and being involved in viral assembly at an as-yet unknown step. Therefore, annexin A2 represents the first host cell factor that could play a role in organizing the unique membrane arrangement orchestrating the intimate connection between HCV replication and virion morphogenesis.
Acknowledgments
We thank Charles M. Rice for Huh7.5 cells and monoclonal antibody 9E10, Stephen Moss and Katharina Hajjar for embryonic fibroblasts of ANXA2 knockout mice, Tero Ahola for the generous gift of affinity-purified SFV nsp3 antiserum, Martin Billeter for plasmid pSC6T7 neo, Didier Trono for the lentiviral transduction system, Takaji Wakita for JFH1 constructs, Darius Moradpour for monoclonal antibody 3B1, Andrew Davidson for the gift of the DENV NGC clone, and the Nikon Imaging Center for access and support to perform confocal microscopy. We are grateful to Ulrike Herian and Rahel Klein for excellent technical assistance and Sven Miller and Wolfgang Fischl for help with experiments involving dengue virus.
This work was funded by the Deutsche Forschungsgemeinschaft (Lo 1506/1-1 to V.L.; SFB 629, TPA1 to V.G.; GRK 1409 to U.R. and V.G.; and SFB 638, TP5 to R.B.).
Footnotes
Published ahead of print on 24 March 2010.
REFERENCES
- 1.Ali, N., K. D. Tardif, and A. Siddiqui. 2002. Cell-free replication of the hepatitis C virus subgenomic replicon. J. Virol. 76:12001-12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alter, M. J. 2007. Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 13:2436-2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Appel, N., M. Zayas, S. Miller, J. Krijnse-Locker, T. Schaller, P. Friebe, S. Kallis, U. Engel, and R. Bartenschlager. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 4:e1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartenschlager, R., M. Frese, and T. Pietschmann. 2004. Novel insights into hepatitis C virus replication and persistence. Adv. Virus Res. 63:71-180. [DOI] [PubMed] [Google Scholar]
- 5.Beaton, A. R., J. Rodriguez, Y. K. Reddy, and P. Roy. 2002. The membrane trafficking protein calpactin forms a complex with bluetongue virus protein NS3 and mediates virus release. Proc. Natl. Acad. Sci. USA 99:13154-13159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger, K. L., J. D. Cooper, N. S. Heaton, R. Yoon, T. E. Oakland, T. X. Jordan, G. Mateu, A. Grakoui, and G. Randall. 2009. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 106:7577-7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Binder, M., G. Kochs, R. Bartenschlager, and V. Lohmann. 2007. Hepatitis C virus escape from the interferon regulatory factor 3 pathway by a passive and active evasion strategy. Hepatology 46:1365-1374. [DOI] [PubMed] [Google Scholar]
- 8.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972-1974. [DOI] [PubMed] [Google Scholar]
- 9.Borawski, J., P. Troke, X. Puyang, V. Gibaja, S. Zhao, C. Mickanin, J. Leighton-Davies, C. J. Wilson, V. Myer, I. Cornellataracido, J. Baryza, J. Tallarico, G. Joberty, M. Bantscheff, M. Schirle, T. Bouwmeester, J. E. Mathy, K. Lin, T. Compton, M. Labow, B. Wiedmann, and L. A. Gaither. 2009. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 83:10058-10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bost, A. G., D. Venable, L. Liu, and B. A. Heinz. 2003. Cytoskeletal requirements for hepatitis C virus (HCV) RNA synthesis in the HCV replicon cell culture system. J. Virol. 77:4401-4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang, K. S., J. Jiang, Z. Cai, and G. Luo. 2007. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81:13783-13793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Filipenko, N. R., T. J. MacLeod, C. S. Yoon, and D. M. Waisman. 2004. Annexin A2 is a novel RNA-binding protein. J. Biol. Chem. 279:8723-8731. [DOI] [PubMed] [Google Scholar]
- 13.Gastaminza, P., S. B. Kapadia, and F. V. Chisari. 2006. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J. Virol. 80:11074-11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerke, V., C. E. Creutz, and S. E. Moss. 2005. Annexins: linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 6:449-461. [DOI] [PubMed] [Google Scholar]
- 15.Gerke, V., and S. E. Moss. 2002. Annexins: from structure to function. Physiol. Rev. 82:331-371. [DOI] [PubMed] [Google Scholar]
- 16.González-Reyes, S., A. Garcia-Manso, G. Del Barrio, K. P. Dalton, L. Gonzalez-Molleda, J. Arrojo-Fernandez, I. Nicieza, and F. Parra. 2009. Role of annexin A2 in cellular entry of rabbit vesivirus. J. Gen. Virol. 90:2724-2730. [DOI] [PubMed] [Google Scholar]
- 17.Gosert, R., D. Egger, V. Lohmann, R. Bartenschlager, H. E. Blum, K. Bienz, and D. Moradpour. 2003. Identification of the hepatitis C virus RNA replication complex in huh-7 cells harboring subgenomic replicons. J. Virol. 77:5487-5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gotthardt, D., V. Blancheteau, A. Bosserhoff, T. Ruppert, M. Delorenzi, and T. Soldati. 2006. Proteomics fingerprinting of phagosome maturation and evidence for the role of a Gα during uptake. Mol. Cell Proteomics 5:2228-2243. [DOI] [PubMed] [Google Scholar]
- 19.Hara, H., H. Aizaki, M. Matsuda, F. Shinkai-Ouchi, Y. Inoue, K. Murakami, I. Shoji, H. Kawakami, Y. Matsuura, M. M. Lai, T. Miyamura, T. Wakita, and T. Suzuki. 2009. Involvement of creatine kinase B in hepatitis C virus genome replication through interaction with the viral NS4A protein. J. Virol. 83:5137-5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hardy, R. W., J. Marcotrigiano, K. J. Blight, J. E. Majors, and C. M. Rice. 2003. Hepatitis C virus RNA synthesis in a cell-free system isolated from replicon-containing hepatoma cells. J. Virol. 77:2029-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harrist, A. V., E. V. Ryzhova, T. Harvey, and F. Gonzalez-Scarano. 2009. Anx2 interacts with HIV-1 Gag at phosphatidylinositol (4,5) bisphosphate-containing lipid rafts and increases viral production in 293T cells. PLoS One 4:e5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayes, M. J., C. J. Merrifield, D. Shao, J. Ayala-Sanmartin, C. D. Schorey, T. P. Levine, J. Proust, J. Curran, M. Bailly, and S. E. Moss. 2004. Annexin 2 binding to phosphatidylinositol 4,5-bisphosphate on endocytic vesicles is regulated by the stress response pathway. J. Biol. Chem. 279:14157-14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayes, M. J., D. M. Shao, A. Grieve, T. Levine, M. Bailly, and S. E. Moss. 2009. Annexin A2 at the interface between F-actin and membranes enriched in phosphatidylinositol 4,5,-bisphosphate. Biochim. Biophys. Acta 1 793:1086-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang, H., F. Sun, D. M. Owen, W. Li, Y. Chen, M. Gale, Jr., and J. Ye. 2007. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. USA 104:5848-5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato, T., T. Date, M. Miyamoto, A. Furusaka, K. Tokushige, M. Mizokami, and T. Wakita. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808-1817. [DOI] [PubMed] [Google Scholar]
- 26.Koutsoudakis, G., E. Herrmann, S. Kallis, R. Bartenschlager, and T. Pietschmann. 2007. The level of CD81 cell surface expression is a key determinant for productive entry of hepatitis C virus into host cells. J. Virol. 81:588-598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koutsoudakis, G., A. Kaul, E. Steinmann, S. Kallis, V. Lohmann, T. Pietschmann, and R. Bartenschlager. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 80:5308-5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai, C. K., K. S. Jeng, K. Machida, and M. M. Lai. 2008. Association of hepatitis C virus replication complexes with microtubules and actin filaments is dependent on the interaction of NS3 and NS5A. J. Virol. 82:8838-8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LeBouder, F., E. Morello, G. F. Rimmelzwaan, F. Bosse, C. Pechoux, B. Delmas, and B. Riteau. 2008. Annexin II incorporated into influenza virus particles supports virus replication by converting plasminogen into plasmin. J. Virol. 82:6820-6828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liljestrom, P., and H. Garoff. 2001. Expression of proteins using Semliki Forest virus vectors. Curr. Protoc. Mol. Biol. 16:20. [DOI] [PubMed] [Google Scholar]
- 31.Lindenbach, B. D., M. J. Evans, A. J. Syder, B. Wolk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, R. O. Hynes, D. R. Burton, J. A. McKeating, and C. M. Rice. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623-626. [DOI] [PubMed] [Google Scholar]
- 32.Lohmann, V., S. Hoffmann, U. Herian, F. Penin, and R. Bartenschlager. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77:3007-3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lohmann, V., F. Körner, J. O. Koch, U. Herian, L. Theilmann, and R. Bartenschlager. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110-113. [DOI] [PubMed] [Google Scholar]
- 34.Mannova, P., R. Fang, H. Wang, B. Deng, M. W. McIntosh, S. M. Hanash, and L. Beretta. 2006. Modification of host lipid raft proteome upon hepatitis C virus replication. Mol. Cell Proteomics 5:2319-2325. [DOI] [PubMed] [Google Scholar]
- 35.Masaki, T., R. Suzuki, K. Murakami, H. Aizaki, K. Ishii, A. Murayama, T. Date, Y. Matsuura, T. Miyamura, T. Wakita, and T. Suzuki. 2008. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 82:7964-7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller, S., and J. Krijnse-Locker. 2008. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 6:363-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller, S., S. Sparacio, and R. Bartenschlager. 2006. Subcellular localization and membrane topology of the Dengue virus type 2 non-structural protein 4B. J. Biol. Chem. 281:8854-8863. [DOI] [PubMed] [Google Scholar]
- 38.Miyanari, Y., K. Atsuzawa, N. Usuda, K. Watashi, T. Hishiki, M. Zayas, R. Bartenschlager, T. Wakita, M. Hijikata, and K. Shimotohno. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089-1097. [DOI] [PubMed] [Google Scholar]
- 39.Miyanari, Y., M. Hijikata, M. Yamaji, M. Hosaka, H. Takahashi, and K. Shimotohno. 2003. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 278:50301-50308. [DOI] [PubMed] [Google Scholar]
- 40.Nakabayashi, H., K. Taketa, K. Miyano, T. Yamane, and J. Sato. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858-3863. [PubMed] [Google Scholar]
- 41.Ono, A., S. D. Ablan, S. J. Lockett, K. Nagashima, and E. O. Freed. 2004. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 101:14889-14894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osborn, M., N. Johnsson, J. Wehland, and K. Weber. 1988. The submembranous location of p11 and its interaction with the p36 substrate of pp60 src kinase in situ. Exp. Cell Res. 175:81-96. [DOI] [PubMed] [Google Scholar]
- 43.Pietschmann, T., A. Kaul, G. Koutsoudakis, A. Shavinskaya, S. Kallis, E. Steinmann, K. Abid, F. Negro, M. Dreux, F. L. Cosset, and R. Bartenschlager. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 103:7408-7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pietschmann, T., V. Lohmann, G. Rutter, K. Kurpanek, and R. Bartenschlager. 2001. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 75:1252-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinkert, D., R. Bartenschlager, and V. Lohmann. 2005. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 79:13594-13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raynor, C. M., J. F. Wright, D. M. Waisman, and E. L. Pryzdial. 1999. Annexin II enhances cytomegalovirus binding and fusion to phospholipid membranes. Biochemistry 38:5089-5095. [DOI] [PubMed] [Google Scholar]
- 47.Rescher, U., and V. Gerke. 2004. Annexins—unique membrane binding proteins with diverse functions. J. Cell Sci. 117:2631-2639. [DOI] [PubMed] [Google Scholar]
- 48.Rescher, U., D. Ruhe, C. Ludwig, N. Zobiack, and V. Gerke. 2004. Annexin 2 is a phosphatidylinositol (4,5)-bisphosphate binding protein recruited to actin assembly sites at cellular membranes. J. Cell Sci. 117:3473-3480. [DOI] [PubMed] [Google Scholar]
- 49.Ryzhova, E. V., R. M. Vos, A. V. Albright, A. V. Harrist, T. Harvey, and F. Gonzalez-Scarano. 2006. Annexin 2: a novel human immunodeficiency virus type 1 Gag binding protein involved in replication in monocyte-derived macrophages. J. Virol. 80:2694-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 51.Sasse, J., and S. R. Gallagher. 2003. Staining proteins in gels. Curr. Protoc. Immunol. 8:9.. [DOI] [PubMed] [Google Scholar]
- 52.Schaller, T., N. Appel, G. Koutsoudakis, S. Kallis, V. Lohmann, T. Pietschmann, and R. Bartenschlager. 2007. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J. Virol. 81:4591-4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwartz, M., J. Chen, M. Janda, M. Sullivan, J. den Boon, and P. Ahlquist. 2002. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell 9:505-514. [DOI] [PubMed] [Google Scholar]
- 54.Shi, S. T., K. J. Lee, H. Aizaki, S. B. Hwang, and M. M. Lai. 2003. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J. Virol. 77:4160-4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tai, A. W., Y. Benita, L. F. Peng, S. S. Kim, N. Sakamoto, R. J. Xavier, and R. T. Chung. 2009. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5:298-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Targett-Adams, P., S. Boulant, and J. McLauchlan. 2008. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J. Virol. 82:2182-2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tegha-Dunghu, J., B. Neumann, S. Reber, R. Krause, H. Erfle, T. Walter, M. Held, P. Rogers, K. Hupfeld, T. Ruppert, J. Ellenberg, and O. J. Gruss. 2008. EML3 is a nuclear microtubule-binding protein required for the correct alignment of chromosomes in metaphase. J. Cell Sci. 121:1718-1726. [DOI] [PubMed] [Google Scholar]
- 58.Tellinghuisen, T. L., K. L. Foss, and J. Treadaway. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 4:e1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tellinghuisen, T. L., J. Marcotrigiano, A. E. Gorbalenya, and C. M. Rice. 2004. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J. Biol. Chem. 279:48576-48587. [DOI] [PubMed] [Google Scholar]
- 60.Thiel, C., M. Osborn, and V. Gerke. 1992. The tight association of the tyrosine kinase substrate annexin II with the submembranous cytoskeleton depends on intact p11- and Ca(2+)-binding sites. J. Cell Sci. 103 (Pt 3):733-742. [DOI] [PubMed] [Google Scholar]
- 61.Vaillancourt, F. H., L. Pilote, M. Cartier, J. Lippens, M. Liuzzi, R. C. Bethell, M. G. Cordingley, and G. Kukolj. 2009. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 387:5-10. [DOI] [PubMed] [Google Scholar]
- 62.van den Hoff, M. J., A. F. Moorman, and W. H. Lamers. 1992. Electroporation in ‘intracellular’ buffer increases cell survival. Nucleic Acids Res. 20:2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Welsch, S., S. Miller, I. Romero-Brey, A. Merz, C. K. Bleck, P. Walther, S. D. Fuller, C. Antony, J. Krijnse-Locker, and R. Bartenschlager. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wright, J. F., A. Kurosky, E. L. Pryzdial, and S. Wasi. 1995. Host cellular annexin II is associated with cytomegalovirus particles isolated from cultured human fibroblasts. J. Virol. 69:4784-4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102:9294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zibouche, M., M. Vincent, F. Illien, J. Gallay, and J. Ayala-Sanmartin. 2008. The N-terminal domain of annexin 2 serves as a secondary binding site during membrane bridging. J. Biol. Chem. 283:22121-22127. [DOI] [PubMed] [Google Scholar]