

Abstract
Proteolytic cleavage of the influenza virus surface glycoprotein hemagglutinin (HA) by host cell proteases is crucial for infectivity and virus spread. The proteases HAT (human airway trypsin-like protease) and TMPRSS2 (transmembrane protease serine S1 member 2) known to be present in the human airways were previously identified as proteases that cleave HA. We studied subcellular localization of HA cleavage and cleavage inhibition of seasonal influenza virus A/Memphis/14/96 (H1N1) and pandemic virus A/Hamburg/5/2009 (H1N1) in MDCK cells that express HAT and TMPRSS2 under doxycycline-induced transcriptional activation. We made the following observations: (i) HA is cleaved by membrane-bound TMPRSS2 and HAT and not by soluble forms released into the supernatant; (ii) HAT cleaves newly synthesized HA before or during the release of progeny virions and HA of incoming viruses prior to endocytosis at the cell surface, whereas TMPRSS2 cleaves newly synthesized HA within the cell and is not able to support the proteolytic activation of HA of incoming virions; and (iii) cleavage activation of HA and virus spread in TMPRSS2- and HAT-expressing cells can be suppressed by peptide mimetic protease inhibitors. The further development of these inhibitors could lead to new drugs for influenza treatment.
Human influenza viruses cause acute infection of the respiratory tract that affects millions of people during seasonal outbreaks every year. Furthermore, the emergence of a new influenza virus for which there is little or no immunity in the human population may provoke an influenza pandemic, as is the case with the currently circulating swine origin H1N1 influenza A virus.
Influenza virus replication is initiated by the surface glycoprotein hemagglutinin (HA) that mediates binding to sialic acid-containing cell surface receptors and fusion of the viral envelope with the endosomal membrane. HA is synthesized as a precursor protein HA0 and needs to be cleaved by a host cell protease into the subunits HA1 and HA2 to gain its fusion capacity (10, 20, 37). Proteolytic cleavage of HA0 enables HA to undergo conformational changes at low pH that expose the N-terminal hydrophobic fusion peptide of HA2 and trigger membrane fusion (36). HA0 of most avian and mammalian influenza viruses contains a single arginine, rarely a single lysine, at the cleavage site. In general, activation of HA0 with a monobasic cleavage site was assumed to occur extracellularly when virions are already released from the cells, and trypsin (21, 22), as well as several trypsin-like proteases such as plasmin (12, 23, 24), tryptase Clara from rat bronchiolar epithelial Clara cells, mast cell tryptase from porcine lung (19), and a protease similar to blood clotting factor Xa from chicken allantoic fluid (13), have been identified as HA-activating enzymes in vitro. Furthermore, some bacterial proteases were shown to support proteolytic activation of HA, too (32, 41). However, the proteases responsible for HA cleavage in the human airways were only poorly defined until recently, when we could demonstrate that the serine proteases HAT (human airway trypsin-like protease, also known as TMPRSS11D) and another airway protease, TMPRSS2 (transmembrane protease serine S1 member 2, also known as epitheliasin), cleave HA containing a single arginine at the cleavage site (4). In full agreement with these observations, it was shown in subsequent studies that TMPRSS2 and the related protease TMPRSS4 cleave HA of the 1918 H1N1 influenza virus (6) and that TMPRSS2 activates the fusion protein F of human metapneumovirus (35).
The proteases HAT and TMPRSS2 belong to the family of type II transmembrane serine proteases (16, 39). They contain a short N-terminal cytoplasmic domain followed by a transmembrane sequence, a variable stem region, and a C-terminal catalytic serine protease domain (Fig. 1). HAT and TMPRSS2 are synthesized as zymogens and require proteolytic cleavage at a highly conserved arginine residue to become enzymatically active. Cleavage activation of TMPRSS2 was shown to occur autocatalytically (1), and studies using an enzymatically inactive HAT mutant [HAT (S368A)] demonstrated that HAT undergoes autocatalytic activation, too (unpublished data). The catalytic domain is linked to the membrane-bound N-terminal chain by a disulfide bridge. However, soluble forms of HAT and TMPRSS2 were described, suggesting that the catalytic domains may be shed from the cell surface (1, 46). The physiological role of TMPRSS2 and HAT in the human airways remains to be determined. TMPRSS2 was suggested to be involved in the regulation of epithelial sodium channels (9). HAT was originally isolated from patients with chronic airway diseases and among other functions was shown to increase mucin gene expression and to stimulate bronchial fibroblast proliferation in airway epithelial cells in vitro (8, 30, 31, 46).
FIG. 1.

Schematic domain structures of HAT and TMPRSS2. HAT and TMPRSS2 are synthesized as single-chain zymogens that consist of an N-terminal transmembrane domain (TM), a stem region containing, e.g., sea urchin sperm protein, enterokinase, and agrin domain (SEA) for HAT or low-density lipoprotein receptor class A domain (LDLRA), and scavenger receptor cysteine-rich domain (SRCR) for TMPRSS2, and a C-terminal trypsin-like serine protease domain that contains the catalytic triad consisting of histidine (H), aspartic acid (D), and serine (S). The zymogens are activated by proteolytic cleavage at arginine residues R186 for HAT and R255 for TMPRSS2 (indicated by arrows). For immunochemical detection of the recombinant proteases HAT and TMPRSS2 are expressed with a C-terminally fused FLAG tag peptide (DYKDDDDK).
We recently established MDCK cells with doxycycline-induced expression of HAT and TMPRSS2 to study cleavage of HA by either protease in more detail (3). Here, we used these cell lines to address the questions of when and where cleavage of HA by HAT and TMPRSS2 takes place. We demonstrate that HA is cleaved by membrane-bound forms of either protease. TMPRSS2 cleaves HA within the cell, while HAT cleaves HA at the cell surface and, thereby, supports cleavage of newly synthesized HA, as well as HA of incoming virions. We further demonstrate that proteolytic activation and spread of influenza viruses in HAT- and TMPRSS2-expressing cells can be blocked by appropriate peptide mimetic protease inhibitors.
MATERIALS AND METHODS
Cells and viruses.
Madin-Darby canine kidney (MDCK) cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum, penicillin, streptomycin and glutamine. Generation of MDCK-HAT and MDCK-TMPRSS2 cells that express HAT and TMPRSS2, respectively, under doxycycline-dependent transcriptional activation were described elsewhere (3). MDCK-HAT and MDCK-TMPRSS2 cells were maintained in growth medium described above supplemented with 0.3 mg of Geneticin (Gibco-BRL)/ml and 2 μg of puromycin (InvivoGen)/ml. Expression of either protease was induced by the addition of 0.2 μg of doxycycline (Clontech)/ml to the growth medium.
Human influenza virus H1N1 A/Memphis/14/96 (A/Memphis/96) was provided by Robert Webster (St. Jude Children's Research Hospital, Memphis, TN) and new pandemic H1N1 virus A/Hamburg/5/2009 (A/Hamburg/09) was provided by Mikhail Matrosovich (Institute of Virology, Philipps University Marburg). Viruses were propagated in MDCK cells in infection medium (DMEM supplemented with 0.1% bovine serum albumin, glutamine, and antibiotics) containing 1 μg of tolylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin (Sigma)/ml, and cell supernatants were cleared by low-speed centrifugation (4,000 rpm, 10 min) and stored at −80°C. To generate stocks of influenza virus A/Memphis/96 (H1N1) containing uncleaved HA0, wild-type MDCK cells were infected at a multiplicity of infection (MOI) of 1, washed carefully with phosphate buffer-saline (PBS) to remove inoculated virus, and incubated in infection medium in the absence of trypsin for 24 h. Cell culture supernatants were cleared by low-speed centrifugation and virus stocks were stored at −80°C. Vesicular stomatitis virus (VSV; Indiana serotype) was propagated in Vero E6 cells in infection medium.
Antibodies.
A monoclonal antibody against the influenza A virus nucleoprotein (NP) was provided by Alexander Klimov (Centers for Disease Control, Atlanta, GA), rabbit sera against H1 was provided by Mikhail Matrosovich (Institute of Virology, Philipps University Marburg, Marburg, Germany), and rabbit sera against H3 was derived from rabbits immunized with A/Aichi/2/68 (H3N2). Polyclonal rabbit anti-FLAG antiserum was purchased from Sigma, and species-specific horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Dako. The rabbit anti-prostasin serum was derived from rabbits immunized with the peptide H2N-DIIPHPSYLQEGSQDC-COOH (amino acids [aa] 119 to 134 of human prostasin). The rabbit serum against VSV was kindly provided by Georg Herrler (University of Veterinary Medicine Hannover, Hannover, Germany).
Plasmids.
The mammalian expression plasmids pCAGGS-HA encoding the cDNA for HA of A/HongKong/1/68 (H3N2) and pCAGGS-HAT encoding HAT have been described previously (4). The cDNA of human prostasin (accession number NM_002773) was amplified from the total RNA of LNCaP cells by using prostasin-specific primers (Pro-SacI-for, 5′-C GTC GAG CTC GGC ATG GCC CAG AAG G-3′; Pro-NotI-rev, 5′-TAT AGC GGC CGC TCA GTG CTC GCT GAG CC-3′) and cloned into pCAGGS expression plasmid using SacI and NotI restriction sites.
Multicycle virus replication in MDCK-HAT and MDCK-TMPRSS2 cells.
To analyze multicycle replication and spread of influenza viruses in MDCK-HAT and MDCK-TMPRSS2 cells, the cells were seeded in 24-well plates and grown in the absence or presence of doxycycline (0.2 μg/ml) for 24 h. Cells were infected at a multiplicity of infection (MOI) of 0.01 in infection medium with or without the addition of doxycycline and then incubated for 24 h at 37°C and 5% CO2. Infected cells were immunostained against NP as described previously (4). Briefly, at 24 h postinfection (p.i.) the cells were fixed with 4% paraformaldehyde in MEM (minimum essential medium) and permeabilized with 0.3% Triton X-100 in PBS. Cells were incubated with mouse anti-NP and HRP-conjugated secondary antibodies and stained using the peroxidase substrate TrueBlue (KPL). To analyze spread of VSV in MDCK-HAT and MDCK-TMPRSS2 cells, the cells were infected at an MOI of 0.001, incubated for 24 h at 37°C, and immunostained by using rabbit sera against VSV, HRP-conjugated secondary antibodies and TrueBlue peroxidase substrate.
Expression of prostasin and coexpression of prostasin and HAT with HA.
For coexpression of HA with prostasin and HAT, respectively, MDCK cells were seeded in 12-well plates and cotransfected with pCAGGS-HA and either pCAGGS-prostasin or pCAGGS-HAT or pCAGGS by using the transfection reagent Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Cells were incubated under serum-free conditions for 24 h at 37°C, and cell lysates were subjected to SDS-PAGE and Western blot analysis as described below.
To analyze expression and shedding of prostasin MDCK cells were grown in 6-cm dishes, transfected with pCAGGS-prostasin or pCAGGS and incubated under serum-free conditions for 24 h at 37°C. Supernatants (5 ml) were centrifuged at 3,000 rpm and 4°C for 10 min and concentrated about 100-fold using Centricon YM-10 filter devices (Millipore). Concentrated supernatants were subjected to SDS-PAGE under reducing conditions and Western blot analysis.
Biotinylation of cell surface proteins.
MDCK-HAT and MDCK-TMPRSS2 cells were grown in 6-well plates with or without the addition of doxycycline for 24 h. The cell monolayers were then put on ice, washed two times with cold PBS containing calcium and magnesium ions (PBS-Ca/Mg), and incubated twice with 1 mg of sulfo-NHS-biotin (Calbiochem)/ml solubilized in PBS-Ca/Mg for 20 min on ice. Biotinylation was quenched by incubation with 0.1 M glycine in PBS-Ca/Mg for 1 min, and the cells were washed several times with PBS-Ca/Mg to remove residual biotin reagent. The cells were lysed in radioimmunoprecipitation assay buffer (0.05 M Tris [pH 7.4], 1% Triton X-100, 1% sodium deoxycholate, 1% SDS, 150 mM NaCl, 25 mM EDTA, 10 μM aprotinin [Trasylol; Bayer HealthCare]), and cell debris was removed by centrifugation at 13,000 rpm and 4°C for 40 min. Biotinylated proteins were precipitated by using Streptavidine Sepharose (GE Healthcare). Precipitated proteins were dissolved in SDS-PAGE sample buffer, heated for 10 min at 95°C, and subjected to SDS-PAGE under reducing conditions and expression of HAT and TMPRSS2 was analyzed by Western blotting with FLAG-specific antibodies.
SDS-PAGE and Western blot analysis of cellular and viral proteins.
Cells were washed with PBS, resuspended in reducing SDS sample buffer, and lysed by sonication and heating to 95°C for 5 min. Proteins were subjected to SDS-PAGE (12% polyacrylamide gel) and subsequently transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare). Proteins were detected by incubation with primary antibodies and species-specific peroxidase-conjugated secondary antibodies and by subsequent incubation with ECL peroxidase substrate (Pierce) and exposure of the PVDF membrane to autoradiography films (CEA). To analyze virus-containing supernatants, the supernatants were cleared from cell debris by low-speed centrifugation (4,000 rpm, 5 min) and then pelleted by ultracentrifugation (28,000 rpm, 2 h, 4°C). Pellets were resuspended in reducing SDS sample buffer, heated for 5 min at 95°C, and subjected to SDS-PAGE and Western blot analysis with HA-specific antibodies as described above.
Protease activity at the cell surface and in cell supernatants.
To determine the enzymatic activity of HAT and TMPRSS2 expressed at the cell surface of MDCK-HAT and MDCK-TMPRSS2 cells, the cells were grown in 96-well plates in the absence or presence of doxycycline for 24 h. Cells were washed with PBS, and the protease activity was assayed by incubation of cells with fluorogenic synthetic peptide Boc-Leu-Gly-Arg-AMC (Bachem, Bubendorf, Switzerland) at a final concentration of 25 μM in assay buffer (50 mM Tris-HCl [pH 7.4]) for 30 min at 37°C. Hydrolysis of the peptide was monitored by the fluorescence intensity of free 7-amino-4-methylcoumarin (AMC) using a Perkin-Elmer LS55 luminescence spectrometer with wavelengths set at 350 nm for excitation and 460 nm for emission.
To examine the enzymatic activity of soluble HAT and TMPRSS2 released into the cell supernatants, MDCK-HAT and MDCK-TMPRSS2 cells were grown in 75-cm2 flasks under serum-free conditions in the absence or presence of doxycycline for 24 h. Cells grown in the absence of doxycycline were used as controls. Supernatants (15 ml) were cleared from cell debris by low-speed centrifugation (3,000 rpm, 10 min, 4°C) and then concentrated ∼150-fold using Centricon YM-10 filter devices (Millipore). To measure the enzymatic activity, supernatants were diluted 1:5 in assay buffer. Samples of 30 μl were incubated 30 to 240 min at 37°C with 30 μl of 50 μM Boc-Leu-Gly-Arg-AMC in assay buffer. The fluorescence intensity of AMC was measured as described above. To examine the enzymatic activity of prostasin-containing supernatants, the concentrated supernatants were diluted 1:10 in prostasin assay buffer (20 mM Tris-HCl [pH 9]). Samples of 30 μl were incubated for 30 to 60 min at 37°C with 30 μl of 50 μM Boc-Leu-Gly-Arg-AMC in prostasin assay buffer, and the fluorescence intensity of AMC was measured.
Cocultivation of protease- and HA-expressing MDCK cells.
MDCK-HAT or MDCK-TMPRSS2 cells were grown on coverslips (18 by 24 mm) in the presence or absence of 0.2 μg of doxycycline/ml for 24 h. Further, wild-type MDCK cells seeded on coverslips of the same size were transfected with pCAGGS-HA. At 24 h after transfection and treatment with doxycycline, respectively, cells were washed with PBS, and one coverslip containing HA-expressing MDCK cells and one coverslip containing MDCK-HAT or MDCK-TMPRSS2 cells were placed next to each other in a culture dish (6 cm in diameter) containing serum-free medium supplemented with or without doxycycline. Cells were cocultivated for 30 h at 37°C and 5% CO2. Growth on separate coverslips prevents that cells overgrow each other, and thus HA cleavage can only be mediated by soluble HAT or TMPRSS2 but not by enzymatic active membrane-bound proteases on the surface of adjacent cells. As a positive control, HA-expressing cells and MDCK-TMPRSS2 cells on separate coverslips were cocultivated in the presence of TPCK-trypsin (1 μg/ml). Finally, cells of each coverslip were harvested separately using cell scrapers and dissolved in SDS sample buffer. Cell lysates were subjected to SDS-PAGE and Western blot analysis with HA- and FLAG-specific antibodies for detection of HA, HAT, and TMPRSS2.
Infection assay of protease expressing cells with virions containing HA0 at 37°C.
MDCK-HAT or MDCK-TMPRSS2 cells were seeded in 96-well plates and cultivated to confluence in growth medium with or without doxycycline for 24 h. Cells were washed two times with PBS and incubated with noninfectious virus A/Memphis/96 (H1N1) with uncleaved HA0 at a ratio of virions to cells of 0.1 in infection medium. The cells were incubated for 30 min on ice to allow adsorption of virions and subsequently shifted to 37°C and 5% CO2, followed by incubation for 10 h to allow single cycle replication. As controls, doxycycline-treated MDCK-HAT and MDCK-TMPRSS2 cells were infected in the presence of 25 μM aprotinin (Trasylol; Bayer HealthCare). At 10 h after inoculation the cells were fixed with 4% paraformaldehyde and immunostained against NP as described above.
Assay for HA0 cleavage by exposure of virions to protease-expressing cells at 4°C.
MDCK-HAT and MDCK-TMPRSS2 cells were grown in 12-well plates in the absence or presence of doxycycline for 24 h. Cells were washed with PBS and incubated with 500 μl of a virus suspension containing virus with uncleaved HA (∼104 virions per ml) in infection medium on ice for 6 h. Preincubated virus was then removed from cell surfaces by extensive shaking of the plate (5 min on a rotation shaker; 200 rpm). The recovered virus suspensions were used to inoculate confluent wild-type MDCK cells grown in 96-well plates by using 100 μl recovered virus suspension per well of wild-type MDCK cells. At 10 h p.i. MDCK cells were immunochemically stained against NP.
Peptide mimetic inhibitors and multicycle viral replication analyses.
Stock solutions of benzamidine-derived peptide mimetic inhibitors synthesized according to previous methods (15, 33, 34) were prepared in double-distilled water (I-1, I-2) or sterile 10% dimethyl sulfoxide (I-3) and stored at −20°C. For analysis of multicycle virus replication in the presence of the inhibitors MDCK-HAT and MDCK-TMPRSS2, the cells were seeded in 24-well plates and grown in the absence or presence of doxycycline for 24 h. Cells were infected with influenza viruses at an MOI of 0.02 or with VSV at an MOI of 0.001 in infection medium for 1 h at 37°C. The inoculum was removed, and the cells were incubated in infection medium supplemented with the respective inhibitor (30 μM) and with or without doxycycline for 24 h at 37°C. Infected cells were immunostained against influenza virus NP and against viral proteins of VSV, respectively.
To analyze viral growth and inhibition of HA cleavage in the presence of protease inhibitors cells grown in 6-well plates in the absence or presence of doxycycline for 24 h were infected at an MOI of 0.01 for 1 h, washed with PBS, and incubated in infection medium with or without the respective inhibitor (30 μM) at 37°C. At 24 h p.i. virus-containing supernatants were subjected to SDS-PAGE and Western blot analysis, and virus titers in PFU were determined by Avicel plaque assay in MDCK cells as described previously (29). Briefly, MDCK cells were inoculated with 10-fold serial dilutions of each sample for 1 h at 37°C. The inoculum was removed and replaced by Avicel overlay containing 1 μg of trypsin/ml. Cells were incubated for 48 h at 37°C and subsequently immunostained against the viral nucleoprotein.
RESULTS
Multicycle viral replication in MDCK-HAT and MDCK-TMPRSS2 cells.
We previously demonstrated that the proteases HAT and TMPRSS2 cleave the HA of human and avian influenza viruses of subtypes H1, H2, and H3 at a monobasic cleavage site (4). Therefore, it was reasonable to assume that both proteases support proteolytic activation of the HA of currently circulating H1N1 swine origin influenza viruses containing a monobasic HA cleavage motif, too. To test this, recently established MDCK cells that express HAT and TMPRSS2, respectively, under doxycycline-dependent transcriptional activation (3) were infected with the seasonal H1N1 isolate A/Memphis/96 and the pandemic isolate A/Hamburg/09 and incubated at 37°C in the absence or presence of doxycycline. At 24 h p.i. virus from cell supernatants was pelleted by ultracentrifugation and subsequently analyzed by SDS-PAGE and Western blotting. Progeny virus of A/Memphis/96 and A/Hamburg/09 released from doxycycline treated cells with induced expression of either HAT or TMPRSS2 contained HA1 and HA2, whereas virions released from cells grown in the absence of doxycycline contained only HA0 (Fig. 2A). In addition, MDCK-TMPRSS2 and MDCK-HAT cells were infected with either H1N1 isolate and incubated with or without doxycycline for 24 h to allow multiple cycles of viral replication. Immunostaining of infected cells against the viral nucleoprotein revealed spread of A/Memphis/96 and A/Hamburg/09 in the presence of doxycycline, providing evidence for proteolytic activation of HA in these cells. In contrast, no viral spread was observed in the absence of doxycycline due to missing cleavage activation of HA in these cells (Fig. 2B). These data demonstrate that HAT and TMPRSS2 activate the HA of currently circulating pandemic H1N1 viruses.
FIG. 2.

Proteolytic processing of HA by the proteases HAT, TMPRSS2, and prostasin. (A) MDCK-TMPRSS2 and MDCK-HAT cells were infected with H1N1 influenza viruses A/Hamburg/09 or A/Memphis/96 at an MOI of 0.01 and incubated in the absence (−) or presence (+) of doxycycline (Dox) for 24 h. Virus-containing cell supernatants were concentrated by ultracentrifugation, subjected to SDS-PAGE under reducing conditions, and subsequently transferred to a PVDF membrane and analyzed by using HA-specific antibodies and HRP-conjugated secondary antibodies. (B) MDCK-HAT and MDCK-TMPRSS2 cells were infected with A/Memphis/96 or A/Hamburg/09 at an MOI of 0.01 and incubated in the absence or presence of Dox for 24 h to allow multiple cycles of viral replication. Infected cells were immunostained against the viral nucleoprotein. (C) MDCK cells were cotransfected with pCAGGS-HA and either pCAGGS, pCAGGS-prostasin, or pCAGGS-HAT. Mock transfections were done with empty pCAGGS. At 24 h posttransfection, cell lysates were analyzed by SDS-PAGE and Western blotting with antibodies against H3 (left panel). Supernatants of pCAGGS-prostasin or pCAGGS (Mock) transfected MDCK cells were concentrated ∼100-fold and subjected to SDS-PAGE and Western blot analysis with prostasin-specific antibodies (middle panel). Enzymatic activity of the supernatants was examined by incubation with the fluorogenic substrate peptide Boc-Leu-Gly-Arg-AMC and release of AMC due to hydrolysis of the substrate was measured. The results are presented as mean enzymatic activities for three independent experiments.
In order to identify HA activating proteases in the human airway epithelium, we considered the protease prostasin as a further candidate. Prostasin is a glycosylphosphatidylinositol (GPI)-anchored trypsin-like serine protease that is expressed widespread in the human respiratory epithelium (7, 27) and has been found to activate the epithelial sodium channel (9, 44). Prostasin can be released from the cell surface as a soluble protease, and both GPI-anchored and soluble forms were shown to be enzymatically active (7). In order to investigate whether prostasin is able to cleave HA, both proteins were coexpressed in MDCK cells, and cell lysates were analyzed by SDS-PAGE and Western blotting with HA-specific antibodies. As shown in Fig. 2C only HA0 was detected in cells expressing HA and prostasin, whereas cleavage of HA0 into HA1 and HA2 was observed in control cells expressing HAT and HA. Prostasin was found to be released from transient prostasin-expressing MDCK cells as a soluble protease with a molecular mass of 40 kDa. Incubation of prostasin-containing supernatants with a fluorogenic peptide substrate revealed that prostasin is released as an enzymatically active protease (Fig. 2C). Thus, lack of HA cleavage during coexpression with prostasin was not due to missing enzymatic activity of prostasin but shows that prostasin is not capable of cleaving HA. These results demonstrate that not all trypsin-like proteases expressed in the human airway epithelium support proteolytic activation of the influenza virus HA.
Expression of TMPRSS2 and HAT at the cell surface and shedding of the catalytic domains.
Type II transmembrane serine proteases (TTSPs) are a family of cell surface-associated proteases. To examine whether HAT and TMPRSS2 are expressed at the cell surface and possess proteolytic activity at the surface, we performed a surface biotinylation analysis of MDCK-HAT and MDCK-TMPRSS2 cells. Therefore, the cells were grown in the absence or presence of doxycycline, and subsequently cell surface proteins were biotin labeled, precipitated, and analyzed by SDS-PAGE and immunoblotting with antibodies against the C-terminal FLAG epitope. Both TMPRSS2 and HAT were expressed on the cell surface as full-length protein (70 and 47 kDa) and in a processed form with an average molecular mass of 30 kDa (Fig. 3A), representing the zymogens and presumably the catalytic domains of the mature forms, respectively. Both forms could be detected under reducing conditions using FLAG-specific antibodies (see Fig. 1). As expected, no protease expression was observed in the absence of doxycycline. To assay the enzymatic activity of HAT and TMPRSS2 at the cell surface, MDCK-HAT and MDCK-TMPRSS2 cells were maintained in the absence or presence of doxycycline for 24 h and subsequently incubated with a fluorogenic peptide substrate. Thereby, high levels of enzymatic activity were measured on the surface of HAT-expressing cells, whereas only low if any protease activity was measured on the surface of TMPRSS2-expressing cells (Fig. 3B). Similar results were obtained by using different fluorogenic peptides containing arginine and various amino acids (data not shown). Taken together, these results demonstrate that HAT is expressed as an enzymatically active protease at the cell surface, whereas TMPRSS2 seems to possess only marginal enzymatic activity at the cell surface, although it is expressed as a processed protease at the plasma membrane as well.
FIG. 3.

Cell surface expression of TMPRSS2 and HAT, shedding from the cell surface, and determination of the enzymatic activity of membrane-bound and soluble forms. (A) MDCK-TMPRSS2 and MDCK-HAT cells were grown in 6-well plates in the absence or presence of doxycycline (Dox) for 24 h. Cell surface proteins were biotinylated; the cells were then lysed, and biotin-labeled proteins were precipitated and subjected to SDS-PAGE under reducing conditions and Western blot analysis with FLAG-specific antibodies. The zymogen and the mature form of either protease are indicated by open and filled arrowheads, respectively. A nonspecific band recognized by the FLAG-specific antiserum is indicated by an asterisk. (B) MDCK-TMPRSS2 and MDCK-HAT cells were grown in 96-well plates with or without Dox for 24 h. Protease activity at the cell surface was determined by incubation with the fluorogenic peptide substrate Boc-Leu-Gly-Arg-AMC, and the fluorescence intensity of the AMC was measured. The results are the mean enzymatic activities for three independent experiments, with values for control cells (−Dox) subtracted from values of Dox-treated cells within each experiment. (C) The soluble forms of HAT and TMPRSS2 released from MDCK-HAT and MDCK-TMPRSS2 cells grown in the absence (−) or presence (+) of Dox were concentrated and analyzed by Western blotting with FLAG-specific antibodies. (D) Enzymatic activity of concentrated TMPRSS2- or HAT-containing supernatants was measured by using the fluorogenic peptide Boc-Leu-Gly-Arg-AMC as described above.
We next investigated whether HAT and TMPRSS2 are released from the plasma membrane. For this purpose, cell supernatants from MDCK-HAT and MDCK-TMPRSS2 cells were concentrated ∼150-fold and subjected to SDS-PAGE and Western blot analysis. Soluble HAT and small amounts of soluble TMPRSS2 were present in the cell supernatants demonstrating that both proteases can be shed from the cell surface (Fig. 3C). Soluble HAT showed a higher molecular mass than expected for the catalytic domain, indicating that HAT undergoes additional proteolytic processing, possibly within the SEA domain, to become released from the cell surface (26). However, incubation of the concentrated supernatants with a fluorogenic peptide to determine the enzymatic activity revealed only very low levels of enzymatic activity for soluble TMPRSS2 and soluble HAT, respectively (Fig. 3D).
Cleavage of influenza virus hemagglutinin by membrane-bound HAT and TMPRSS2.
To investigate whether the low enzymatic activity of soluble TMPRSS2 or HAT is still sufficient to cleave HA in MDCK-TMPRSS2 and MDCK-HAT cells, we performed a cocultivation experiment of the protease-expressing cell lines with transient HA-expressing MDCK cells. Therefore, MDCK-HAT or MDCK-TMPRSS2 cells were seeded on coverslips and cocultivated with transient HA-expressing MDCK cells seeded on a separate coverslip next to each other in the absence or presence of doxycycline for 30 h. In such an experimental approach, HA cleavage can occur by soluble proteases released from protease-expressing cells but not by membrane-bound proteases expressed at the cell surface. Cells of each coverslip were harvested separately and analyzed by SDS-PAGE and Western blotting. No cleavage of HA0 was observed by cocultivation of HA-expressing cells with either TMPRSS2- or HAT-expressing cells (Fig. 4, left panel). Expression of HAT and TMPRSS2 during cocultivation in the presence of doxycycline was confirmed by Western blot analysis with FLAG-specific antibodies (Fig. 4, right panel) demonstrating that the lack of HA cleavage was not due to missing protease expression. Cocultivation of MDCK-TMPRSS2 cells and HA-expressing MDCK cells in the presence of exogenous trypsin was used as a positive control and supported cleavage of HA0. These findings demonstrate that cleavage of HA in MDCK-HAT and MDCK-TMPRSS2 cells is mediated by cell-associated TMPRSS2 and HAT and not by the soluble forms released from the cells.
FIG. 4.

Examination of HA cleavage by soluble HAT and TMPRSS2. MDCK-TMPRSS2 or MDCK-HAT cells grown on coverslips with or without addition of doxycycline (Dox) to the growth medium were cocultivated with transiently HA-expressing MDCK cells grown on separate coverslips next to each other in one cell culture dish for 30 h as described in Materials and Methods. As a positive control, 1 μg of trypsin/ml was added during cocultivation. Cells of each coverslip were harvested and subjected to SDS-PAGE and Western blot analysis with H3-specific antibodies (left panel) or FLAG-specific antibodies (right panel). Zymogens (open arrowheads) and the catalytic domains (filled arrowhead) of the mature forms of TMPRSS2 and HAT are indicated. A nonspecific cross-reacting band is indicated by an asterisk.
Proteolytic activation of virions at the cell surface.
The data described above suggested that HAT, since it is expressed as an enzymatically active protease on the plasma membrane, may be able to support HA cleavage at the cell surface. To prove this idea, we analyzed proteolytic activation of virions containing HA0 at the stage of entry into MDCK-HAT cells. Virions containing noncleaved HA0 are not able to infect cells unless HA0 becomes cleaved at the stage of entry prior to fusion. For this purpose MDCK-HAT cells and as a control MDCK-TMPRSS2 cells were inoculated with A/Memphis/96 virions containing only HA0 and incubated for 10 h at 37°C to allow single cycle replication. As shown in Fig. 5A, MDCK-HAT cells could be infected with virions containing noncleaved HA0, and infection was completely inhibited in the presence of the protease inhibitor aprotinin. The data demonstrate that HAT-expressing cells support proteolytic activation of HA0 of virions at the stage of entry. In contrast, control cells without induction of HAT expression, as well as TMPRSS2-expressing cells, did not support the proteolytic activation of incoming virions.
FIG. 5.
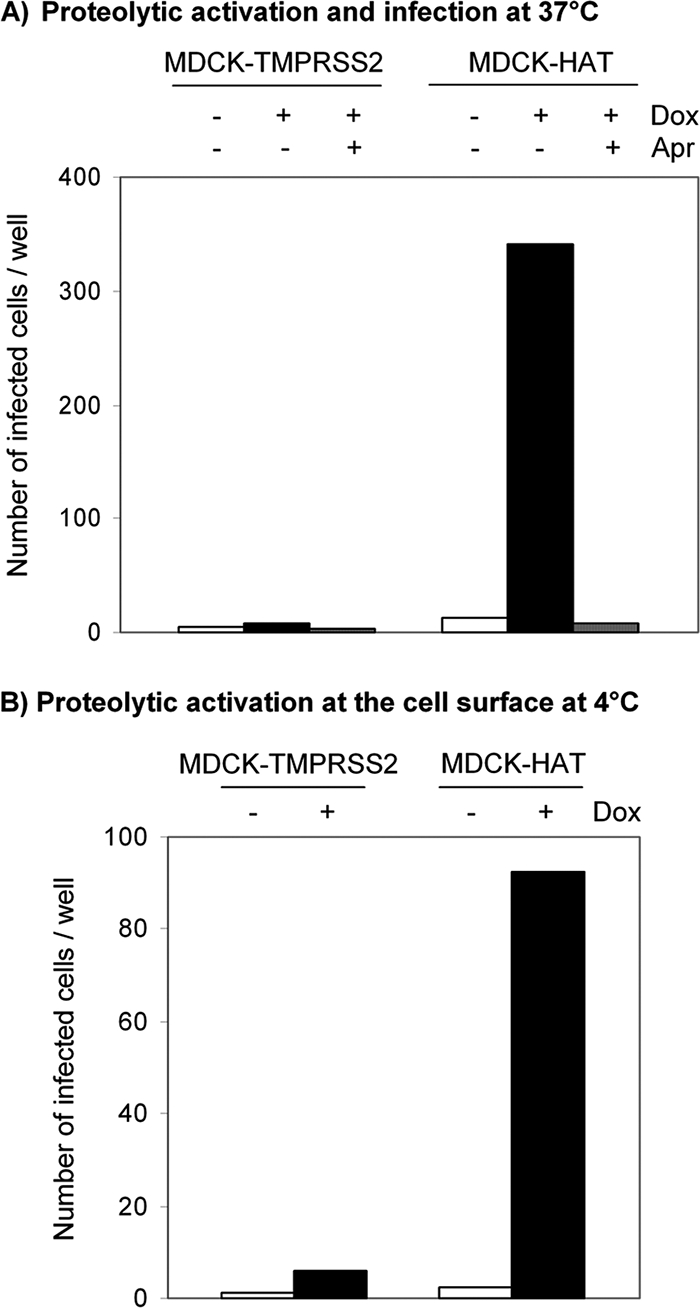
Proteolytic activation of influenza virions at the stage of entry. (A) Proteolytic activation at 37°C. MDCK-TMPRSS2 and MDCK-HAT cells grown in 96-well plates with or without doxycycline (Dox) were inoculated with human influenza virus A/Memphis/96 (H1N1) containing uncleaved HA0 (∼103 virions per well) and incubated for 10 h at 37°C to allow single cycle replication. As a control inoculation with virions was performed in the presence of 25 μM aprotinin (Apr). Cells were immunostained against NP, and the number of infected cells per well was determined. The results are the mean values of a representative experiment. (B) Proteolytic activation at 4°C prior to endocytosis. Influenza virus A/Memphis/96 containing HA0 was adsorbed to MDCK-TMPRSS2 and MDCK-HAT cells for 6 h at 4°C. Virions were removed from the cells and used for inoculation of wild-type MDCK cells seeded in 96-well plates. At 10 h p.i. MDCK cells were immunostained against NP, and infected cells per well were counted. The results are the mean values of a representative experiment.
In order to demonstrate that activation of virions containing HA0 occurs at the cell surface by membrane-bound HAT prior to endocytosis, we analyzed proteolytic activation of HA0 on the surface of MDCK-HAT cells at 4°C. Virions bind to cell surface receptors at 4°C but are not endocytosed. Therefore, MDCK-HAT cells and, as a negative control MDCK-TMPRSS2 cells, were grown with or without doxycycline for 24 h, put on ice, and incubated with influenza virus A/Memphis/96 containing HA0 for several hours to compensate for the reduced enzymatic activity of HAT at 4°C. The preincubated virions were then removed from the cells by extensive shaking and were used for inoculation of wild-type MDCK cells that do not express any HA-activating protease. In doing so, only virions that remained at the cell surface during the extended incubation time at 4°C were verified in this assay. The MDCK cells were incubated at 37°C for 10 h to allow a single cycle of viral replication and subsequently immunostained against NP, and the number of infected cells was determined. The results show that virions that were preincubated at the cell surface of HAT-expressing cells were able to infect MDCK cells, indicating that HA0 was proteolytically activated during adsorption to HAT-expressing cells (Fig. 5B). In contrast, virions exposed to MDCK-TMPRSS2 cells became not activated and therefore were not able to infect wild-type MDCK cells. These data provide strong evidence that proteolytic activation of incoming virions by membrane-bound HAT takes place on the cell surface, whereas TMPRSS2 does not support cleavage activation of HA at the stage of entry.
Suppression of influenza virus spread by peptide mimetic protease inhibitors.
We recently demonstrated that some naturally occurring protease inhibitors as well as synthetic serine protease inhibitors suppress cleavage of HA0 in HAT- and TMPRSS2-expressing MDCK cells (3). We also observed that viral spread in MDCK-HAT cells was more susceptible to exogenous protease inhibitors than in MDCK-TMPRSS2 cells. These data suggested that differences in subcellular localization of either protease may account for the differences in sensitivity to protease inhibitors. To prove this hypothesis, we investigated the effect of three different protease inhibitors on HAT and TMPRSS2 (Fig. 6A). Inhibitors I-1 and I-2 were expected to have negligible cell permeability due to their strongly basic benzamidine moiety. Furthermore, an I-2 homologous and much more hydrophobic inhibitor modified by a long-chain fatty acid designated as I-3, with an expected improvement in cell permeability as shown previously (11), was tested. To examine the efficacy of I-1, I-2, and I-3 to inhibit processing of HA by HAT and TMPRSS2, MDCK-HAT and MDCK-TMPRSS2 cells were infected with A/Hamburg/09 and incubated in the absence or presence of the respective inhibitor for 24 h. To analyze multicycle viral replication and spread of infection cells were immunostained against the viral nucleoprotein (Fig. 6B). In addition, virus titers in the supernatants were determined by plaque titration (Fig. 6C), and virus-containing supernatants were analyzed for HA cleavage by SDS-PAGE and Western blotting with HA-specific antibodies (Fig. 6D). As a control, doxycycline-treated and nontreated cells were infected for 24 h in the absence of protease inhibitors. As expected, viral multicycle replication and spread due to HA0 cleavage was visible in doxycycline treated MDCK-HAT and MDCK-TMPRSS2 cells infected in the absence of any inhibitors (lane 2), while due to missing HA activation in infected cells grown without doxycycline only single cycle replication and no spread of infection was observed (lane 1). Cleavage of HA0 and the spread of A/Hamburg/09 in TMPRSS2-expressing cells were not affected by I-1 and I-2 but strongly inhibited by decanoylated inhibitor I-3 (lane 5). In addition, only low amounts of progeny virions were detected in supernatants of I-3-treated cells at 24 h p.i. due to the suppression of proteolytic activation of HA and thus the suppression of virus propagation (Fig. 6C and D). These data indicate that inhibition of TMPRSS2 requires cellular uptake of the protease inhibitor, confirming the observation that TMPRSS2 cleaves HA intracellularly. In contrast, proteolytic activation of HA and viral spread in HAT-expressing cells were markedly suppressed by all applied inhibitors with strong inhibition of multicycle viral replication by I-1 (Fig. 6B to D). Thus, HA cleavage by HAT can be easily blocked by exogenous inhibitors, supporting the results for the cleavage of HA by membrane-bound HAT at the cell surface. To confirm that suppression of viral growth in I-1-, I-2-, and I-3-treated cells was specifically due to inhibition of proteolytic activation of HA and not due to reduced cell viability, we analyzed viral growth of VSV in inhibitor-treated cells. VSV does not require proteolytic activation by host cell proteases. As shown in Fig. 6B, the growth of VSV was not affected by I-1, I-2, and I-3, as well as by doxycycline treatment, demonstrating that these inhibitors do not affect cell viability at a concentration of 30 μM.
FIG. 6.

Suppression of HA cleavage and influenza virus spread by protease inhibitors. (A) Structural formulas of peptide mimetic inhibitors I-1, I-2, and I-3. (B) MDCK-TMPRSS2 and MDCK-HAT cells were infected with A/Hamburg/09 (H1N1) at an MOI of 0.02 and VSV at an MOI of 0.001, respectively, and incubated with or without doxycycline (Dox) in the absence (Ø) or presence of inhibitor I-1, I-2, or I-3 at a final concentration of 30 μM at 37°C. At 24 h p.i. the cells were immunostained with influenza virus NP- and VSV-specific antibodies, respectively. (C) MDCK-TMPRSS2 and MDCK-HAT cells were infected with A/Hamburg/09 at an MOI of 0.01 and incubated in the absence or presence of doxycycline for 24 h (lanes 1 and 2, see panel B) in the absence or presence of inhibitor I-1 (lane 3), I-2 (lane 4), or I-3 (lane 5). Virus titers were determined by plaque assay at 24 h p.i. Note that the bars representing virus titers in cells without induction of TMPRSS2 and HAT expression (−Dox), respectively, cannot be seen in the figure since the titers were below the limit of determination (102 PFU/ml). The results are the mean values of two independent experiments. (D) Virus-containing cell supernatants shown in Fig. 6C were concentrated by ultracentrifugation and then subjected to SDS-PAGE under reducing conditions and Western blot analysis with HA-specific antibodies.
Taken together, these results confirm the observations for subcellular localization of HA cleavage by TMPRSS2 and HAT. Moreover, the data demonstrate that HAT and TMPRSS2 provide promising drug targets and show that peptide mimetic inhibitors exhibit potential as novel therapeutic compounds for an influenza treatment.
DISCUSSION
The currently available measures to control influenza are vaccination and antiviral medications using the neuraminidase (NA) inhibitors oseltamivir and zanamivir. Considering the high impact of influenza viruses on public health, more extensive profiling of new drug targets is of great interest. Due to the essential role of HA cleavage for influenza virus infectivity and spread, targeting of relevant host cell proteases is a promising therapeutic strategy.
We previously demonstrated that the human airway proteases HAT and TMPRSS2 cleave the HA of human and avian influenza viruses containing a monobasic cleavage site (4) and, consistent with this, we show here that both enzymes also activate the currently circulating pandemic swine origin H1N1 virus. In addition, we demonstrate that the trypsin-like protease prostasin known to be present in the human airway epithelium, too, is not able to cleave HA, emphasizing that not every trypsin-like protease expressed in the human airways is capable of supporting proteolytic activation and spread of influenza viruses.
In the present study we investigated the subcellular localization of HA cleavage by TMPRSS2 and HAT using MDCK cell lines with doxycycline-dependent expression of either protease. We demonstrate that proteolytic activation of HA occurs by membrane-bound TMPRSS2 and HAT and not by soluble forms released from the cell surface. HAT is expressed as an enzymatically active protease at the plasma membrane that is able to cleave HA at the cell surface. To our knowledge, this is the first time that membrane-bound HAT was shown to be an enzymatically active protease at the cell surface. In contrast, TMPRSS2, which was found to be expressed at the plasma membrane as a mature protease, too, shows poor if any enzymatic activity at the cell surface. Furthermore, as described previously (1, 46), we observed shedding of HAT and to some extent of TMPRSS2 into the supernatant. However, the soluble forms showed only marginal enzymatic activity and were not sufficient to support the cleavage of HA in these cells. The reason for the missing enzymatic activity of soluble HAT and TMPRSS2, as well as membrane-bound TMPRSS2 at the plasma membrane, remains unknown thus far but might be related to additional proteolytic processing, conformational changes, or the presence of extracellular protease inhibitors. Regulation of protease activities is important, since uncontrolled proteolysis provides a basis for multiple pathological conditions and diseases. For example, matriptase, another TTSP, is inhibited by the hepatocyte growth factor activator inhibitor 1 (HAI-1) almost immediately after zymogen activation and is released from the cells only as a matriptase-HAI-1 complex (25). Matriptase is associated with human epithelial cancers and even slightly increased expression of the protease relative to HAI-1 was shown to exhibit a strong oncogenic potential (28). Little knowledge exists about expression levels, as well as mechanisms underlying the regulation of protease activity and shedding of TMPRSS2 and HAT in the human airway epithelium, but overexpression of TMPRSS2 and HAT is assumed to be involved in prostate cancer and respiratory diseases, respectively (1, 8, 30, 43). TMPRSS2 was shown to be released from prostate and prostate cancer cells (1). HAT-related enzymatic activity can be detected in the sputum of patients with chronic airway diseases (45, 46). Thus, increased shedding of either protease might play a role in pathogenesis. In this respect, it would be interesting to investigate whether increased amounts of soluble HAT in the sputum are sufficient to support HA cleavage and therefore might play a role in influenza progression in patients with asthma or chronic bronchitis.
By using influenza virions containing HA0, we demonstrate that membrane-bound HAT is able to activate HA of incoming virions at the cell surface. Thus, HAT provides an additional mechanism for viruses to be activated late in infection prior to virus entry into new cells. Proteolytic activation of some influenza viruses containing a monobasic HA cleavage site at the stage of entry was observed before in MDBK cells (5) and in human respiratory adenoid epithelial cells (HAEC) (47). The relevant proteases and the subcellular site of HA activation were not identified. Our data strongly suggest that activation of incoming virions in HAEC occurs by a membrane-bound protease at the cell surface, most likely HAT. Immunohistochemical studies revealed that HAT is expressed exclusively in ciliated cells of the trachea and bronchi, while TMPRSS2 seems to be expressed more widespread in the human airways (18, 40, 42). Considering that different cell types of the airway epithelium express a partially different protease repertoire, HAT may enable proteolytic activation of progeny viruses released from cells that do not express relevant HA activating proteases. We propose that HAT cleaves HA of virions adsorbed to the cell surface receptors. We also found that virions containing cleaved HA are released from HAT-expressing MDCK cells. Thus, HAT does not only activate HA0 at the stage of entry but also newly synthesized HA0, most likely at the plasma membrane during the assembly and budding of progeny virions.
In contrast, membrane-bound TMPRSS2 at the cell surface and soluble TMPRSS2 released from the cells did not show enzymatic activity. Therefore, HA cleavage by TMPRSS2 seems to take place intracellularly, most probably during its transport from the endoplasmic reticulum to the plasma membrane, where virus assembly and budding take place. Consistent with this, we found that virions containing cleaved HA are released from TMPRSS2-expressing cells. In this respect, cleavage activation by TMPRSS2 seems to be similar to cleavage of HA of highly pathogenic avian influenza viruses (HPAIV) containing a multibasic HA cleavage site, which is cleaved in the trans-Golgi network by furin and furin-related proteases (17, 38). Thus, cleavage activation of HA with monobasic and multibasic cleavage site, respectively, in the human airway epithelium might take place in the same intracellular compartment and yet be performed by different proteases. Interestingly, cleavage of HA with monobasic cleavage site in human large intestine epithelial Caco-2 cells was shown to take place intracellularly (51) and by using reverse transcription-PCR analyses we found that TMPRSS2 is expressed in Caco-2 cells (unpublished data). Further studies on expression and subcellular compartmentalization of TMPRSS2 in different cell types and tissues are under investigation in our laboratory.
Targeting of HA-activating proteases presents a therapeutic approach that should exclude the development of drug resistant viruses. Studies by Zhirnov et al. demonstrated that influenza virus replication in chicken embryos, mice, and human airway epithelial cell cultures can be suppressed by inhibition of HA cleavage using the serine protease inhibitor aprotinin (47, 49, 50). Furthermore, aerosolized aprotinin was shown to be effective against respiratory virus diseases (48). We recently demonstrated that specific low-molecular-weight peptide mimetic inhibitors efficiently suppress influenza virus propagation in HAT- or TMPRSS2-expressing MDCK cells by inhibition of HA cleavage (3). In the present study we used these inhibitors to investigate the susceptibility of HAT and TMPRSS2 to exogenous protease inhibitors in more detail. We found that the cell-impermeable inhibitors I-1 and I-2 blocked HA activation in HAT-expressing cells but not in TMPRSS2-expressing cells, whereas the much more hydrophobic decanoylated inhibitor I-3 efficiently blocked proteolytic cleavage of HA in both HAT- and TMPRSS2-expressing cells. These results further support the concept that TMPRSS2 and HAT cleave HA in different cellular compartments. Acylation of peptide derivatives has been shown before to increase their efficacy as HPAIV and HIV inhibitors (2, 11, 14, 38). In the present study, inhibitors with or without a fatty acid modification proved also to be helpful tools to discriminate between extracellular and intracellular cleavage activation of HA.
To date, protease inhibitors are not applied as preventative and therapeutic approaches for influenza treatment. This is largely due to limited knowledge about relevant host cell proteases and their characteristics, complicating the development of specific and selective protease inhibitors. HAT and TMPRSS2 are HA-activating enzymes in the human airways that represent promising drug targets. Further characterization of HAT and TMPRSS2 should help to develop potent protease inhibitors as new drugs for influenza treatment.
Acknowledgments
We thank Mikhail Matrosovich for providing antibodies and viruses and Petra Neubauer-Rädel and Andrea Schultz for technical assistance. We are grateful to Georg Herrler for providing the VSV antiserum.
This study was supported by grants from the Deutsche Forschungsgemeinschaft SFB-593-TPB2.
Footnotes
Published ahead of print on 17 March 2010.
REFERENCES
- 1.Afar, D. E., I. Vivanco, R. S. Hubert, J. Kuo, E. Chen, D. C. Saffran, A. B. Raitano, and A. Jakobovits. 2001. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res. 61:1686-1692. [PubMed] [Google Scholar]
- 2.Becker, G. L., F. Sielaff, M. E. Than, I. Lindberg, S. Routhier, R. Day, Y. Lu, W. Garten, and T. Steinmetzer. 2010. Potent inhibitors of furin and furin-like proprotein convertases containing decarboxylated P1 arginine mimetics. J. Med. Chem. 53:1067-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Böttcher, E., C. Freuer, T. Steinmetzer, H. D. Klenk, and W. Garten. 2009. MDCK cells that express proteases TMPRSS2 and HAT provide a cell system to propagate influenza viruses in the absence of trypsin and to study cleavage of HA and its inhibition. Vaccine 27:6324-6329. [DOI] [PubMed] [Google Scholar]
- 4.Böttcher, E., T. Matrosovich, M. Beyerle, H. D. Klenk, W. Garten, and M. Matrosovich. 2006. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 80:9896-9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boycott, R., H. D. Klenk, and M. Ohuchi. 1994. Cell tropism of influenza virus mediated by hemagglutinin activation at the stage of virus entry. Virology 203:313-319. [DOI] [PubMed] [Google Scholar]
- 6.Chaipan, C., D. Kobasa, S. Bertram, I. Glowacka, I. Steffen, T. S. Tsegaye, M. Takeda, T. H. Bugge, S. Kim, Y. Park, A. Marzi, and S. Pöhlmann. 2009. Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 83:3200-3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, L. M., M. L. Skinner, S. W. Kauffman, J. Chao, L. Chao, C. D. Thaler, and K. X. Chai. 2001. Prostasin is a glycosylphosphatidylinositol-anchored active serine protease. J. Biol. Chem. 276:21434-21442. [DOI] [PubMed] [Google Scholar]
- 8.Chokki, M., S. Yamamura, H. Eguchi, T. Masegi, H. Horiuchi, H. Tanabe, T. Kamimura, and S. Yasuoka. 2004. Human airway trypsin-like protease increases mucin gene expression in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 30:470-478. [DOI] [PubMed] [Google Scholar]
- 9.Donaldson, S. H., A. Hirsh, D. C. Li, G. Holloway, J. Chao, R. C. Boucher, and S. E. Gabriel. 2002. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 277:8338-8345. [DOI] [PubMed] [Google Scholar]
- 10.Garten, W., and H. D. Klenk. 2008. Cleavage activation of the influenza virus hemagglutinin and its role in pathogenesis. Avian influenza: monographs in virology 27. Karger, Basel, Switzerland.
- 11.Garten, W., A. Stieneke, E. Shaw, P. Wikstrom, and H. D. Klenk. 1989. Inhibition of proteolytic activation of influenza virus hemagglutinin by specific peptidyl chloroalkyl ketones. Virology 172:25-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goto, H., and Y. Kawaoka. 1998. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 95:10224-10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gotoh, B., F. Yamauchi, T. Ogasawara, and Y. Nagai. 1992. Isolation of factor Xa from chick embryo as the amniotic endoprotease responsible for paramyxovirus activation. FEBS Lett. 296:274-278. [DOI] [PubMed] [Google Scholar]
- 14.Hallenberger, S., V. Bosch, H. Angliker, E. Shaw, H. D. Klenk, and W. Garten. 1992. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 360:358-361. [DOI] [PubMed] [Google Scholar]
- 15.Hellstern, P., U. Stürzebecher, B. Wuchold, H. Haubelt, U. T. Seyfert, M. Bauer, A. Vogt, and J. Stürzebecher. 2007. Preservation of in vitro function of platelets stored in the presence of a synthetic dual inhibitor of factor Xa and thrombin. J. Thromb. Haemost. 5:2119-2126. [DOI] [PubMed] [Google Scholar]
- 16.Hooper, J. D., J. A. Clements, J. P. Quigley, and T. M. Antalis. 2001. Type II transmembrane serine proteases. Insights into an emerging class of cell surface proteolytic enzymes. J. Biol. Chem. 276:857-860. [DOI] [PubMed] [Google Scholar]
- 17.Horimoto, T., K. Nakayama, S. P. Smeekens, and Y. Kawaoka. 1994. Proprotein-processing endoproteases PC6 and furin both activate hemagglutinin of virulent avian influenza viruses. J. Virol. 68:6074-6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacquinet, E., N. V. Rao, G. V. Rao, W. Zhengming, K. H. Albertine, and J. R. Hoidal. 2001. Cloning and characterization of the cDNA and gene for human epitheliasin. Eur. J. Biochem. 268:2687-2699. [DOI] [PubMed] [Google Scholar]
- 19.Kido, H., Y. Okumura, H. Yamada, T. Q. Le, and M. Yano. 2007. Proteases essential for human influenza virus entry into cells and their inhibitors as potential therapeutic agents. Curr. Pharm. Des. 13:405-414. [DOI] [PubMed] [Google Scholar]
- 20.Klenk, H. D., and W. Garten. 1994. Host cell proteases controlling virus pathogenicity. Trends Microbiol. 2:39-43. [DOI] [PubMed] [Google Scholar]
- 21.Klenk, H. D., R. Rott, M. Orlich, and J. Blödorn. 1975. Activation of influenza A viruses by trypsin treatment. Virology 68:426-439. [DOI] [PubMed] [Google Scholar]
- 22.Lazarowitz, S. G., and P. W. Choppin. 1975. Enhancement of the infectivity of influenza A and B viruses by proteolytic cleavage of the hemagglutinin polypeptide. Virology 68:440-454. [DOI] [PubMed] [Google Scholar]
- 23.Lazarowitz, S. G., A. R. Goldberg, and P. W. Choppin. 1973. Proteolytic cleavage by plasmin of the HA polypeptide of influenza virus: host cell activation of serum plasminogen. Virology 56:172-180. [DOI] [PubMed] [Google Scholar]
- 24.LeBouder, F., E. Morello, G. F. Rimmelzwaan, F. Bosse, C. Péchoux, B. Delmas, and B. Riteau. 2008. Annexin II incorporated into influenza virus particles supports virus replication by converting plasminogen into plasmin. J. Virol. 82:6820-6828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee, M. S., K. Kiyomiya, C. Benaud, R. B. Dickson, and C. Y. Lin. 2005. Simultaneous activation and HAI-1-mediated inhibition of matriptase induced at activation foci in immortal human mammary epithelial cells. Am. J. Physiol. Cell Physiol. 288:C932-C941. [DOI] [PubMed] [Google Scholar]
- 26.Levitin, F., O. Stern, M. Weiss, C. Gil-Henn, R. Ziv, Z. Prokocimer, N. I. Smorodinsky, D. B. Rubinstein, and D. H. Wreschner. 2005. The MUC1 SEA module is a self-cleaving domain. J. Biol. Chem. 280:33374-33386. [DOI] [PubMed] [Google Scholar]
- 27.List, K., J. P. Hobson, A. Molinolo, and T. H. Bugge. 2007. Co-localization of the channel activating protease prostasin/(CAP1/PRSS8) with its candidate activator, matriptase. J. Cell Physiol. 213:237-245. [DOI] [PubMed] [Google Scholar]
- 28.List, K., R. Szabo, A. Molinolo, V. Sriuranpong, V. Redeye, T. Murdock, B. Burke, B. S. Nielsen, J. S. Gutkind, and T. H. Bugge. 2005. Deregulated matriptase causes ras-independent multistage carcinogenesis and promotes ras-mediated malignant transformation. Genes Dev. 19:1934-1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matrosovich, M., T. Matrosovich, W. Garten, and H. D. Klenk. 2006. New low-viscosity overlay medium for viral plaque assays. Virol. J. 3:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsushima, R., A. Takahashi, Y. Nakaya, H. Maezawa, M. Miki, Y. Nakamura, F. Ohgushi, and S. Yasuoka. 2006. Human airway trypsin-like protease stimulates human bronchial fibroblast proliferation in a protease-activated receptor-2-dependent pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 290:L385-L395. [DOI] [PubMed] [Google Scholar]
- 31.Miki, M., Y. Nakamura, A. Takahashi, Y. Nakaya, H. Eguchi, T. Masegi, K. Yoneda, S. Yasuoka, and S. Sone. 2003. Effect of human airway trypsin-like protease on intracellular free Ca2+ concentration in human bronchial epithelial cells. J. Med. Invest. 50:95-107. [PubMed] [Google Scholar]
- 32.Scheiblauer, H., M. Reinacher, M. Tashiro, and R. Rott. 1992. Interactions between bacteria and influenza A virus in the development of influenza pneumonia. J. Infect. Dis. 166:783-791. [DOI] [PubMed] [Google Scholar]
- 33.Schweinitz, A., T. Steinmetzer, I. J. Banke, M. J. Arlt, A. Stürzebecher, O. Schuster, A. Geissler, H. Giersiefen, E. Zeslawska, U. Jacob, A. Krüger, and J. Stürzebecher. 2004. Design of novel and selective inhibitors of urokinase-type plasminogen activator with improved pharmacokinetic properties for use as antimetastatic agents. J. Biol. Chem. 279:33613-33622. [DOI] [PubMed] [Google Scholar]
- 34.Schweinitz, A., A. Stürzebecher, U. Stürzebecher, O. Schuster, J. Stürzebecher, and T. Steinmetzer. 2006. New substrate analogue inhibitors of factor Xa containing 4-amidinobenzylamide as P1 residue, part 1. Med. Chem. 2:349-361. [DOI] [PubMed] [Google Scholar]
- 35.Shirogane, Y., M. Takeda, M. Iwasaki, N. Ishiguro, H. Takeuchi, Y. Nakatsu, M. Tahara, H. Kikuta, and Y. Yanagi. 2008. Efficient multiplication of human metapneumovirus in Vero cells expressing the transmembrane serine protease TMPRSS2. J. Virol. 82:8942-8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531-569. [DOI] [PubMed] [Google Scholar]
- 37.Steinhauer, D. A. 1999. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 258:1-20. [DOI] [PubMed] [Google Scholar]
- 38.Stieneke-Gröber, A., M. Vey, H. Angliker, E. Shaw, G. Thomas, C. Roberts, H. D. Klenk, and W. Garten. 1992. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 11:2407-2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szabo, R., and T. H. Bugge. 2008. Type II transmembrane serine proteases in development and disease. Int. J. Biochem. Cell Biol. 40:1297-1316. [DOI] [PubMed] [Google Scholar]
- 40.Takahashi, M., T. Sano, K. Yamaoka, T. Kamimura, N. Umemoto, H. Nishitani, and S. Yasuoka. 2001. Localization of human airway trypsin-like protease in the airway: an immunohistochemical study. Histochem. Cell Biol. 115:181-187. [DOI] [PubMed] [Google Scholar]
- 41.Tashiro, M., P. Ciborowski, H. D. Klenk, G. Pulverer, and R. Rott. 1987. Role of Staphylococcus protease in the development of influenza pneumonia. Nature 325:536-537. [DOI] [PubMed] [Google Scholar]
- 42.Vaarala, M. H., K. S. Porvari, S. Kellokumpu, A. P. Kyllönen, and P. T. Vihko. 2001. Expression of transmembrane serine protease TMPRSS2 in mouse and human tissues. J. Pathol. 193:134-140. [DOI] [PubMed] [Google Scholar]
- 43.Vaarala, M. H., K. Porvari, A. Kyllönen, O. Lukkarinen, and P. Vihko. 2001. The TMPRSS2 gene encoding transmembrane serine protease is overexpressed in a majority of prostate cancer patients: detection of mutated TMPRSS2 form in a case of aggressive disease. Int. J. Cancer 94:705-710. [DOI] [PubMed] [Google Scholar]
- 44.Vallet, V., A. Chraibi, H. P. Gaeggeler, J. D. Horisberger, and B. C. Rossier. 1997. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature 389:607-610. [DOI] [PubMed] [Google Scholar]
- 45.Yamaoka, K., K. Masuda, H. Ogawa, K. Takagi, N. Umemoto, and S. Yasuoka. 1998. Cloning and characterization of the cDNA for human airway trypsin-like protease. J. Biol. Chem. 273:11895-11901. [DOI] [PubMed] [Google Scholar]
- 46.Yasuoka, S., T. Ohnishi, S. Kawano, S. Tsuchihashi, M. Ogawara, K. Masuda, K. Yamaoka, M. Takahashi, and T. Sano. 1997. Purification, characterization, and localization of a novel trypsin-like protease found in the human airway. Am. J. Respir. Cell Mol. Biol. 16:300-308. [DOI] [PubMed] [Google Scholar]
- 47.Zhirnov, O. P., M. R. Ikizler, and P. F. Wright. 2002. Cleavage of influenza a virus hemagglutinin in human respiratory epithelium is cell associated and sensitive to exogenous antiproteases. J. Virol. 76:8682-8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhirnov, O. P., L. S. Kirzhner, A. V. Ovcharenko, and N. A. Malyshev. 1996. Aerosolized aprotinin is an effective drug against viral respiratory illness. Anti-Infective Drugs Chemother. 14:209-216. [Google Scholar]
- 49.Zhirnov, O. P., A. V. Ovcharenko, and A. G. Bukrinskaya. 1984. Suppression of influenza virus replication in infected mice by protease inhibitors. J. Gen. Virol. 65:191-196. [DOI] [PubMed] [Google Scholar]
- 50.Zhirnov, O. P., A. V. Ovcharenko, and A. G. Bukrinskaya. 1985. Myxovirus replication in chicken embryos can be suppressed by aprotinin due to the blockage of viral glycoprotein cleavage. J. Gen. Virol. 66:1633-1638. [DOI] [PubMed] [Google Scholar]
- 51.Zhirnov, O. P., I. V. Vorobjeva, A. V. Ovcharenko, and H. D. Klenk. 2003. Intracellular cleavage of human influenza a virus hemagglutinin and its inhibition. Biochemistry 68:1020-1026. [DOI] [PubMed] [Google Scholar]