

Abstract
Poliovirus (PV) 2Apro has been considered important for PV replication and is known to be toxic to host cells. A 2Apro-deficient PV would potentially be less toxic and ideal as a vector. To examine whether 2Apro is needed to form progeny virus, a full-length cDNA of dicistronic (dc) PV with (pOME) or without (pOMEΔ2A) 2Apro was constructed in the strain PV1(M)OM. RNAs of both pOME and pOMEΔ2A were capable of forming progeny viruses, called OME and OMEΔ2A, respectively. In their ability to induce a cytopathic effect (CPE), the strains ranked as OMEΔ2A < OME ≒ PV1(M)OM. These results suggest that 2Apro is not essential for full-length dc PV to form progeny virus and that it contributes to the efficient viral replication and/or induction of a CPE. To clarify whether 2Apro is essential for P1-null (lacking the entire coding sequence for capsid proteins) PV, the RNA replication activity of P1-null PV (pOMΔP1) or P1-null PV without 2Apro (pOMΔP1Δ2A) or without both 2Apro and 2B (pOMΔP1Δ2AΔ2B) was examined. The RNAs of pOMΔP1 and pOMΔP1Δ2A could replicate and form progeny viruses under a trans supply of P1 protein, whereas the RNA of pOMΔP1Δ2AΔ2B could not. These results suggest that 2Apro is not needed for the replication of P1-null PV, although it is important for PV RNA replication and inducing a CPE. To know whether a 2Apro-deficient PV can be used as a vector, a P1-null PV containing the enhanced green fluorescent protein (EGFP) coding sequence with or without 2Apro was examined. It expressed fluorescent protein. This result suggests that 2Apro-deficient PV can express foreign genes.
Poliomyelitis is an acute disease of the central nervous system caused by the poliovirus (PV), a human enterovirus that belongs to the Picornaviridae family. Humans are the only natural hosts of PV. In humans, an infection is initiated by oral ingestion of the virus followed by multiplication in the alimentary mucosa (7, 38), from where the virus spreads through the bloodstream. Viremia is considered essential for leading to paralytic poliomyelitis in humans.
PV is a nonenveloped particle that consists of a positive single-stranded RNA genome and 60 copies each of four capsid proteins and occurs in three serologically distinct types, type 1, type 2, and type 3. The genome, composed of approximately 7,500 nucleotides (nt), is polyadenylylated and covalently linked at the 5′ end to a small protein, VPg (31, 40, 44). The RNA alone is infectious; cells transfected with the RNA produce infectious progeny virions. The polyprotein is cotranslationally cleaved by virus-specific proteinases to form viral capsid proteins (VP0, VP1, and VP3) and noncapsid proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D). VP0 is further cleaved into VP2 and VP4 during the formation of virions. 2Apro, 3Cpro, and 3CDpro are viral proteinases involved in processing specific to PV polyproteins (26). The translation of the viral mRNA is controlled by an internal ribosomal entry site (IRES), a 400-nt RNA segment of the viral genome that precedes the open reading frame (ORF) (33, 34). An IRES element with a similar function exists in the genomic RNA of the encephalomyocarditis virus (EMCV) (19, 20). Molla et al. (29) inserted the type 2 EMCV IRES element into the ORF of PV (at the P1*P2 junction), thereby generating a virus carrying a dicistronic (dc) RNA genome. In this dc virus, viral proteins are produced by proteolytic processing of two distinct polyproteins, P1 and P2-P3, specifying the capsid proteins and the nonstructural proteins, respectively.
PV defective interfering particles (DIs) have been isolated from laboratory-propagated viral populations (12, 13, 21, 28) and from manipulated cloned infectious cDNAs (16) and, in all cases, retain translational as well as replication competence (28, 32). It has been reported that foreign gene sequences could be substituted into the PV P1 region without affecting the replication of the RNA (4, 37) as long as the translational reading frame was maintained (16). Since the replicons do not encode capsid proteins, they were encapsidated when transfected into cells previously infected with a recombinant vaccinia virus (VV-P1) which expresses the PV capsid precursor, P1 (36). Serial passage of these replicons in the presence of VV-P1 resulted in increasing titers of encapsidated replicons, allowing the generation of stocks of the recombinant PV vectors (36).
In addition to cleaving viral polyproteins, 2Apro is known to cleave several cellular proteins. Cleavage of the eukaryotic translation initiation factor eIF4G by 2Apro inhibits the cap-dependent translation of cellular mRNA without affecting the translation of viral RNA (15, 24). Independent of the shutoff of host protein synthesis, 2Apro also stimulated the translation of PV RNA (17). The stimulation of translation was later shown to be mediated, at least in part, by the C-terminal cleavage product of eIF4G that is generated by 2Apro (8, 18, 45). Therefore, 2Apro enhances viral protein synthesis in infected cells both by inhibiting host cell protein synthesis and by stimulating the translation of viral RNA. Several genetic studies suggest 2Apro to also have an essential role in PV RNA replication. The replication of a subgenomic RNA replicon which contained a deletion mutation in 2Apro was severely inhibited in transfected cells (14). Interestingly, the replication of this RNA could be rescued in trans when 2Apro was provided by a wild-type helper RNA (14). In addition, studies using a dc PV RNA, where the cis cleavage function of 2Apro was not required to cleave the viral polyprotein, suggested that 2Apro activity was essential for efficient viral RNA replication (30). The C-terminal region of 2Apro has also been implicated in viral RNA replication (27). 2Apro also targets nuclear factors, including several transcription factors (42) and a structural component of small nuclear ribonucleoproteins (snRNPs), gemin-3, which is implicated in the removal of eukaryotic introns mediated by the spliceosome machinery (3). PV 2Apro induces alterations in the nuclear pore complex, which inhibits the nuclear export of U snRNA, rRNA, and mRNA but not tRNA. The inhibition of trafficking of de novo-synthesized mRNAs occurs early after 2Apro expression, suggesting that this protease could prevent host responses to viral infections (11).
PV induces an apoptotic response when its growth is markedly suppressed, for example, in the presence of guanidine hydrochloride. A temperature-sensitive (ts) mutant of PV also had suppressed viral growth and induced an apoptotic response. In contrast, a productive infection in these apoptotic cells was accompanied by a canonical necrotic cytopathic effect (CPE) (1, 2, 41). The viral infection triggers an apoptotic pathway involving the consecutive activation of caspase-9 and caspase-3. The productive viral infection suppresses the implementation of this apoptotic program, at least in part, by aberrant processing and degradation of procaspase-9 (6).
Here, we show that a 2Apro-deficient full-length dc PV is replication competent, although the efficiency of its replication is decreased. Moreover, a 2Apro-deficient P1-null (lacking the entire coding sequence for the capsid proteins) PV with or without enhanced green fluorescent protein (EGFP) can also produce progeny virions under a trans supply of P1 protein. Therefore, 2Apro is not needed for the replication of PV with or without the P1 coding region in the viral genome, although 2Apro plays important roles in PV RNA replication and inducing a CPE.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
Monolayers of HeLa and African green monkey kidney (AGMK) cells were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 5% newborn calf serum (NCS; Mitsubishi Kasei), 0.11% NaHCO3 (Wako Pure Chemical Industries Ltd.), and 0.1 mg/ml kanamycin sulfate (Meiji Seika Kaisha, Ltd.) at 37°C under 5% CO2 and used for the preparation of viruses, transfection with infectious cDNA clones, and plaque assays.
The virulent type 1 PV strain Mahoney [PV1(M)OM], derived from an infectious cDNA clone, pOM1 (39), and the type 3 PV strain Leon were employed in this study. The viruses recovered from the cells transfected with the RNAs of pOMΔ0.8, pOMΔ1.8, pOMΔP1, pOMΔP1Δ2A, pOM-EGFPΔP1, pOM-EGFPΔP1Δ2A, pOME, and pOMEΔ2A were designated OMΔ0.8, OMΔ1.8, OMΔP1, OMΔP1Δ2A, OM-EGFPΔP1, OM-EGFPΔP1Δ2A, OME, and OMEΔ2A, respectively. For providing the PV P1 capsid precursor for P1-defective genomes, a recombinant vaccinia virus (VV-P1) was used (5).
Filtrated ascites fluid of an anti-Mahoney mouse monoclonal antibody (7m008) and an anti-Leon mouse monoclonal antibody (Thai p34-120) were used for the neutralizing assay.
Construction of recombinant cDNAs.
pOMΔ0.8 was constructed with a deletion of 816 nucleotides in pOM1 from nucleotide (nt) 1663 to nt 2478 (Fig. 1). Similarly, pOMΔ1.8 had a 1,782-nucleotide deletion in pOM1 from nt 1175 to nt 2956, pOMΔP1 had a 2,628-nucleotide deletion in pOM1 from nt 746 to nt 3373, pOMΔP1Δ2A had a 3,072-nucleotide deletion in pOM1 from nt 746 to nt 3817, and pOMΔP1Δ2AΔ2B had a 3,367-nucleotide deletion in pOM1 from nt 746 to nt 4112. All these plasmids except pOMΔ1.8 were constructed by PCR using KOD Plus DNA polymerase (Toyobo). pOMΔ1.8 was constructed from pOM1 digested by NruI (nt 1172) and SnaBI (nt 2954) and self-ligated.
FIG. 1.

PV genomic constructions. (A) Construction of PV and recombinant PVs. (B) Junctional amino acid sequences of pOM-EGFPΔP1, pOM-EGFPΔP1Δ2A, pOME, and pOMEΔ2A. The nucleic acid sequence (CCCGGG) recognized by SmaI, corresponding to the amino acid sequence Pro Gly, is shown by underlines. The inserted EMCV IRES are shown by the striped horizontal bars. Closed triangles indicate the sites of cleavage by 2Apro, and open triangles indicate the sites of cleavage by 3Cpro or 3CDpro. Nucleotide numbers of the deleted fragments are shown at the deletion positions.
pOM-EGFPΔP1 was constructed by PCR, subcloning into pBluescript II KS(+), and recombination. Briefly, the fragment from nt 1 to nt 735 of pOM1 which had an XhoI restriction site downstream of nt 735 was digested by KpnI and XhoI and inserted into the equivalent sites of pBluescript II KS(+). This plasmid was designated pBS(1). A fragment amplified from pEGFP-N1 (Clontech) by PCR using a sense primer (MunI>EGFP; 5′-CCCAATTGTATCATAATGGTGAGCAAGGCG-3′) and an anti-sense primer (EGFP<SmaI; 5′-TCCCCCGGGCTTGTACAGCTCGT-3′) was digested by MunI and SmaI and inserted into the equivalent sites of pBS(1). This plasmid was designated pBS(2). A fragment from nt 3365 to nt 4252 of pOM1 which had a SmaI restriction site just upstream of nt 3365 was digested by SmaI and SpeI (nt 3982 of pOM1) and inserted into the equivalent sites of pBS(2). This plasmid was designated pBS(3). pBS(3) was digested by KpnI and inserted into the equivalent sites of pOM1 (nt 66 and nt 3660). pOM-EGFPΔP1Δ2A was similarly constructed except for the final recombination sites (KpnI [nt 66 of pOM1] and SpeI [nt 3982 of pOM1]).
pOME was constructed by PCR from pOM1 and a plasmid which contains the IRES of EMCV. A fragment containing the PV IRES and P1 coding region with a termination codon which had an EcoRI restriction site just downstream of the termination codon was digested by Bpu1102I (nt 285 of pOM1) and EcoRI (fragment 1). The other fragment contained an EMCV IRES-related region (nt 214 to nt 852 of EMCV cDNA) which had an EcoRI restriction site just upstream of nt 214 of EMCV cDNA and a SmaI restriction site downstream of nt 852 of EMCV cDNA. The sequence of the junction was 5′-(EMCV IRES)-ATG GCC ACA ACC ATG GAA CCC GGG-(SmaI restriction site)-3′. This fragment was digested by EcoRI and SmaI (fragment 2), and fragment 1 and fragment 2 were inserted between the Bpu1102I and SmaI sites of pBluescript II SK(+). This plasmid was designated pBS(4). pBS(4) was digested by Bpu1102I and SmaI (fragment 3). A fragment containing the PV P2-P3 coding region which had a SmaI restriction site just upstream of the sequence required for PV 2Apro digestion between PV P1 and 2Apro was digested by SmaI and BglII (nt 5601 of pOM1) (fragment 4). The sequence of the junction between the SmaI restriction site and the 2A coding sequence was 5′-(SmaI restriction site) ACC TAC (2Apro coding sequence)-3′. Fragments 3 and 4 were inserted between the Bpu1102I and BglII sites of pOM1, and the final plasmid was designated pOME. pOMEΔ2A was constructed similarly to pOME. The sequence of the junction between the SmaI restriction site and 2B coding sequence was 5′-(SmaI recognition site) GAA GCC ATG GAA CAA (2B coding sequence)-3′.
Nucleotide sequences derived from PCR fragments were analyzed using a BigDye terminator cycle sequencing kit (version 3 or 3.1) (Applied Biosystems) with an ABI Prism 310 genetic analyzer (Applied Biosystems) or ABI Prism 3100-Avant genetic analyzer (Applied Biosystems).
RNA transfection.
RNA transcripts were synthesized from PvuI-linearized cDNAs using an AmpliScribe T7 high-yield transcription kit (Epicentre Biotechnologies) and digested with RNase-free DNase I. AGMK cells on a 6-cm dish (Falcon) were transfected with 1 to 3 μg of RNA by a DEAE-dextran method (16). The cultures were harvested at 42 h [for PV1(M)OM and OME] or 75 h (for OMEΔ2A) after the transfection. OMEΔ2A was serially passaged through AGMK cells two times.
Reverse transcription (RT)-PCR.
RNA was extracted from PV1(M)OM, OME, OMEΔ2A, OM-EGFPΔP1, and OM-EGFPΔP1Δ2A with chloroform containing phenol and isoamyl alcohol and reverse transcribed with Superscript II transcriptase (Invitrogen) using an anti-sense primer (PV5699<5719 or PV4784<4805). PCR was then performed with KOD Plus (Toyobo) using a sense primer [PV(M)2955>2923] and an anti-sense primer [PV(M)4232<4252] for PV1(M)OM, OME, and OMEΔ2A or using a sense primer (PV577>597) and an anti-sense primer (PV4512<4431) for OM-EGFPΔP1 and OM-EGFPΔP1Δ2A. The PCR products were analyzed by agarose gel electrophoresis. For sequencing of the products, DNA was extracted from the gels using GenElute Minus ethidium bromide spin columns (Sigma).
Slot blot analysis.
Monolayers of HeLa cells (4.4 × 105 cells/well) in 6-well plates were transfected with the in vitro-synthesized RNA from each plasmid by the DEAE-dextran method or infected with the viruses. At the time points indicated below, cytoplasmic RNA was extracted from the transfected cells using Isogen (Nippon Gene Co., Ltd.) dissolved in 40 μl of H2O. One-fourth was mixed with 30 μl of denaturation buffer (66% [vol/vol] formamide and 7.8% [vol/vol] formaldehyde) in 3-(N-morpholino)propanesulfonic acid (MOPS) buffer (26 mM MOPS [pH 7.0], 6.5 mM sodium acetate, and 1.3 mM EDTA) and denatured at 65°C for 5 min, followed by chilling on ice. An equal volume of 20× SSC (333 mM NaCl and 333 mM sodium citrate tribasic dehydrate) was added. In each slot of a slot blotting apparatus (Bio-Dot SF; Bio-Rad Laboratories, Inc.), 10 μl of RNA solution was applied, and the RNA was immobilized on a nylon filter (Hybond-N; GE Healthcare UK Ltd.) and cross-linked by a UV cross-linker (UV Stratalinker1800; Stratagene). The filters were hybridized to a labeled cDNA corresponding to nt 1 to nt 742 of the viral RNA (43) or to exon 7 of glycerol-3-phosphate dehydrogenase (GAPDH) mRNA using AlkPhos Direct (GE Healthcare UK Ltd.) according to the manufacturer's instructions. The probes were detected by chemiluminescence using CDP-Star (GE Healthcare UK Ltd.).
Encapsidation of PV replicons, serial passaging, and purification.
The encapsidation and serial passaging of PV replicons using VV-P1 have been described previously (5), and basically the same method was adopted here. Briefly, HeLa S3 cells were infected with 5 PFU of VV-P1, which expresses the PV capsid precursor protein P1, per cell. At 2 h postinfection, the cells were transfected by the DEAE-dextran method with in vitro-transcribed RNA. The cultures were harvested at 24 h posttransfection by three successive freeze-thaws, sonicated, and clarified by low-speed centrifugation at 14,000 × g for 20 min. For serial passage of the encapsidated replicons and generation of virus stocks, HeLa S3 cells were first infected with 10 to 20 PFU of VV-P1 per cell. At 2 h postinfection, the cells were infected with passage 1 encapsidated replicons. The cultures were harvested at 16 h after PV infection by three successive freeze-thaws, sonicated, and clarified by low-speed centrifugation at 14,000 × g for 20 min. The supernatant was then stored at −80°C or used immediately for additional passages by the same procedure.
For purifying the P1-null PV particles, the supernatant was lysed with 1% sodium dodecyl sulfate and centrifuged in a Beckman type 45 rotor at 35,000 rpm for 2.5 h. The supernatant was discarded, and the pellet was washed under the same conditions in 0.1 M phosphate buffer (pH 7.35) for an additional 2.5 h. The pellet was then resuspended in serum-free DMEM, filtrated, and stored at −80°C.
Titration of viruses.
The numbers of PFU in AGMK cells were determined by the plaque assay, and the numbers of infectious units (IU) were determined by counting fluorescence-positive cells. Units of viral RNA (U) were adopted for the infection with P1-null viruses. For the measurement of PFU, AGMK cells on 6-cm dishes were inoculated with the viral suspension and then incubated at 37°C for 2 to 5 days for the observation of plaques. To measure the units of viral RNA of OMΔ0.8, OMΔ1.8, OMΔP1, and OMΔP1Δ2A, viral RNA was extracted from the suspensions with chloroform containing phenol and isoamyl alcohol. The units were measured using a LightCycler system (Roche Diagnostics) according to the manufacturer's instructions. As 1.1 × 108 U/ml was equivalent to 2.1 × 109 PFU/ml for PV1(M)OM, we adopted 5.2 × 101 U/cell, which was supposed to be equivalent to 1,000 PFU/cell (multiplicity of infection [MOI] of 1,000) of PV1(M)OM for the infection with OMΔ0.8, OMΔ1.8, OMΔP1, and OMΔP1Δ2A. For the measurement of IU of OM-EGFPΔP1 and OM-EGFPΔP1Δ2A, fluorescence-positive cells were counted under an inverted fluorescence microscope (DM6000B; Leica Microsystems) at 24 h postinfection in HeLa cells. The amount of virus leading to one fluorescence-positive cell was defined as 1 IU. Comparing the units of RNA with the infectious units of fluorescence-positive cells, 5.3 × 109 U/ml was equivalent to 7.1 × 109 IU/ml for an OM-EGFPΔP1 stock and 4.2 × 109 U/ml was equivalent to 1.1 × 109 IU/ml for an OM-EGFPΔP1Δ2A stock. Based on the data, we considered that the units of RNA were not significantly different from the infectious units for OM-EGFPΔP1 and OM-EGFPΔP1Δ2A.
Observation of CPE.
HeLa S3 cells in 16-well Lab-Tek chamber slides (Nalge Nunc International K.K.) were infected with PVs. Eight, 18, or 24 h after the incubation with PVs at 37°C, the cells were observed under an inverted microscope.
Neutralization assay.
The viral suspension (50 μl) was mixed with filtrated ascites fluid (50 μl) containing the antibody and incubated at 37°C for 1 h. The virus-antibody mixture was overlaid on HeLa S3 cells in 16-well chamber slides and incubated at room temperature for 20 min and then at 37°C for 30 min. The mixture was then replaced with DMEM supplemented with 5% NCS. The cells were observed 18 h after the incubation at 37°C under an inverted microscope.
TUNEL assay.
A direct terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay was performed using an in situ cell death detection kit (Roche Diagnostics), and cells were observed under a confocal laser scanning microscope (LSM510; Carl Zeiss MicroImaging Co. Ltd.). Actinomycin D (10 μg/ml) was added for an apoptosis-positive control.
RESULTS
2Apro is not required for full-length dc PV to form progeny virus.
2Apro is known to be cytotoxic, and a 2Apro-deficient PV vector might be desirable. To examine whether 2Apro is needed to form progeny virus, cDNAs of the full-length dc pOME and pOMEΔ2A with the backbone of the PV type 1 Mahoney strain (pOM1) (Fig. 1A and B) were constructed and the ability to form progeny viruses was examined. HeLa cells were transfected with the RNA of pOM1, pOME, and pOMEΔ2A and examined for a CPE. RNA derived from pOM1, pOME, and pOMEΔ2A was not degraded as assessed by gel electrophoresis (data not shown). All these RNAs induced a CPE that led to cell death, whereas no CPE was observed in the mock-transfected cells (data not shown). The supernatants also induced a CPE in HeLa cells. These results show that pOM1, pOME, and pOMEΔ2A can form progeny viruses, called PV1(M)OM, OME, and OMEΔ2A, respectively. This suggests that 2Apro is not essential to form progeny virus for full-length dc PV.
To confirm that the OME and OMEΔ2A suspensions do not contain IRES deletion revertants, the sizes of the fragments encompassing VP1 and 2B were examined by PCR (data not shown). The PV1(M)OM, OME, and OMEΔ2A suspensions produced appropriate fragments, that is, 1.3-kb, 1.9-kb, and 1.5-kb fragments, respectively. The sequences of the PCR fragments were analyzed, and they were not mutated (data not shown). These results mean that IRES deletion revertants were not detected in the OME and OMEΔ2A suspensions under the experimental conditions and that it is highly probable that these suspensions do not contain IRES deletion revertants. This suggests that OME and OMEΔ2A can proliferate by themselves.
To ascertain that the CPE was actually induced by PV, a neutralization assay was performed using a monoclonal antibody for the virulent type 1 Mahoney strain (7m008) and a monoclonal antibody for the virulent type 3 Leon strain (Thai p34-120). HeLa cells were infected with PV1(M)OM, OME, OMEΔ2A, or Leon at an MOI of 10 in the presence or absence of 7m008 or Thai p34-120 and examined for a CPE (Fig. 2). In the absence of the antibodies, all the strains induced a CPE. In the presence of 7m008, only Leon caused a CPE. In contrast, in the presence of Thai p34-120, all the strains except Leon had a CPE. These results suggest that PV1(M)OM, OME, and OMEΔ2A are type 1 strains, and the CPE was induced by PV.
FIG. 2.

Neutralization assay of PV1(M)OM, OME, OMEΔ2A, and Leon with anti-PV type 1 or type 2 monoclonal antibody. HeLa cells were mock infected (e, j, and o) or infected with PV1(M)OM (a, f, and k), OME (b, g, and l), OMEΔ2A (c, h, and m), or Leon (d, i, and n) in the absence (k to o) or presence of an anti-PV type 1 Mahoney monoclonal antibody (7m008, a to e) or anti-PV type 2 Leon monoclonal antibody (Thai p34-120, f to j). Eighteen hours after the infection, the CPE was observed under a microscope. Bar, 50 μm.
To define the characteristics of the viral strains, a plaque assay was performed using AGMK cells (Fig. 3). PV1(M)OM produced large plaques, OME moderate-sized plaques, and OMEΔ2A the smallest plaques. The extremely small plaques of OMEΔ2A may relate to its slow replication rate.
FIG. 3.

Plaque phenotypes of PV1(M)OM, OME, and OMEΔ2A. AGMK cells were infected with PV1(M)OM (a), OME (b), and OMEΔ2A (c). The cells were fixed 2 days after the infection with PV1(M)OM, and OME and 4 days after the infection with OMEΔ2A.
To compare these strains further, HeLa cells were infected at an MOI of 10 and cell morphology was observed 8 or 24 h after the infection (Fig. 4). All the viruses had a CPE (Fig. 4A). No CPE was observed in the mock-infected cells. To quantify the rate of CPE-expressing cells, the percentages of round-shaped cells, detached cells, and cells with membrane blebbing among all cells were analyzed, and the kinetics are shown in Fig. 4B. OMEΔ2A had a lesser CPE than did OME or PV1(M)OM. OME exhibited a similar level of CPE to PV1(M)OM, although OME had slightly slower kinetics than PV1(M)OM. The results were reproducible. These results suggest that in speed of viral replication and/or the ability to induce CPE, the strains rank as OMEΔ2A < OME ≒ PV1(M)OM. They also suggest that 2Apro contributes to efficient viral replication and/or the induction of a CPE. When the cells were infected at a higher MOI (MOI of 50), the kinetics were essentially unchanged (data not shown). This result suggests that an MOI of 10 was high enough to assess the kinetics of CPE expression. Only OMEΔ2A induced morphological changes typical of apoptosis, namely, cell membrane blebbing, in a significant proportion of cells (Fig. 4Ac, Ae, and 4Cs). This result indicates that OMEΔ2A may induce apoptosis.
FIG. 4.

CPE in cells infected with or without PV1(M)OM, OME, and OMEΔ2A. (A) HeLa cells were mock infected (d and f) or infected with PV1(M)OM (a), OME (b), or OMEΔ2A (c and e) at an MOI of 10. Eight (a to d) or 24 h (e and f) after the infection (h.a.i.), morphological changes were observed under a microscope. Bar, 50 μm. (B) Percentages of CPE expression among all cells. (C) Rates of membrane blebbing among CPE-expressing cells. Average rates in two to three microscopic fields in one experiment were plotted. The rates were examined 0, 2, 4, 6, 8, 10, 12, and 24 h after the infection. Blue lines with squares indicate results for PV1(M)OM, pink lines with triangles indicate results for OME, and orange lines with circles indicate results for OMEΔ2A.
2Apro-deficient full-length dc PV induces apoptosis.
To confirm that OMEΔ2A induces apoptosis, a direct TUNEL assay was performed in cells infected with or without PV1(M)OM or OMEΔ2A or treated with actinomycin D, which induces apoptosis (Fig. 5). Uninfected samples and PV1(M)OM-infected samples contained few fluorescence-positive cells. On the other hand, actinomycin D-treated samples and OMEΔ2A-infected samples included many fluorescence-positive cells. These results suggest that OMEΔ2A induces apoptosis. This raises the possibility that 2Apro is important to prevent typical apoptosis in PV-infected cells.
FIG. 5.

Apoptosis in cells infected with OMEΔ2A, detected by the TUNEL assay. HeLa cells were mock infected (d, h, and l), infected with PV1(M)OM (a, e, and i) or OMEΔ2A (b, f, and j), or treated without (d, h, and l) or with (c, g, and k) actinomycin D (Act D). Seven hours after the infection with PV1(M)OM, 8 h after the treatment with actinomycin D, and 21 h after the infection with OMEΔ2A, the cells were TUNEL stained. TUNEL-positive cells were fluorescent. Fluorescence (FL) images are shown at the top (a to d), bright-field (BF) images are shown in the middle (e to h), and merged (FL+BF) images are shown at the bottom (i to l). Bar, 50 μm.
P1-null PV RNA deficient in the 2Apro coding region can replicate in cells.
It has been reported that the PV P1 coding region is not required for RNA replication and the formation of progeny virus in P1-expressing cells (4, 37). To further define which parts of the PV genome are necessary for RNA replication, deletion mutants were prepared (Fig. 1A) and RNA replication activity was examined by slot blotting (Fig. 6A). RNAs derived from pOM1, pOMΔ0.8, pOMΔ1.8, pOMΔP1, pOMΔP1Δ2A, and pOMΔP1Δ2AΔ2B were not degraded as assessed by gel electrophoresis (data not shown). The RNAs were introduced into HeLa cells, and the cells were collected 2 and 8 h after the transfection. Cell lysates were used for slot blotting. When a probe for PV IRES RNA was used, all the RNAs except for pOMΔP1Δ2AΔ2B RNA were increased at 8 h compared to the levels at 2 h after the transfection. The amounts of GAPDH RNA hardly changed up to 8 h after the transfection. The results were reproducible. These results suggest that the 2Apro coding region is not needed for the RNA replication of P1-null PV and that 2B is necessary for the PV replicon activity.
FIG. 6.
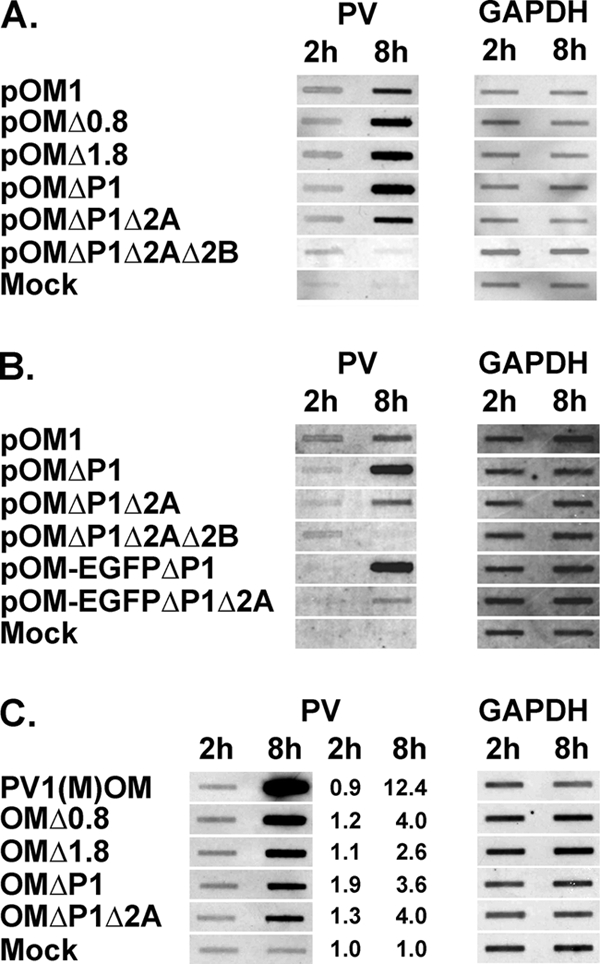
Slot blot analysis of cells after transfection with or without synthesized viral RNA or after infection with or without viruses. (A) HeLa cells were transfected with or without (Mock) RNAs of pOM1, pOMΔ0.8, pOMΔ1.8, pOMΔP1, pOMΔP1Δ2A, or pOMΔP1Δ2AΔ2B, and cell lysates were collected 2 and 8 h later. The RNAs were detected with probes for the PV IRES sequence or GAPDH sequence. (B) A similar experiment was performed using the RNAs of pOM1, pOMΔP1, pOMΔP1Δ2A, pOMΔP1Δ2AΔ2B, pOM-EGFPΔP1, and pOM-EGFPΔP1Δ2A. (C) HeLa cells were infected with or without (Mock) PV1(M)OM, OMΔ0.8, OMΔ1.8, OMΔP1, or OMΔP1Δ2A, and cell lysates were collected 2 and 8 h later. The RNAs were detected with probes for the PV IRES sequence or GAPDH sequence. Relative amounts of PV RNA corrected to the amount of GAPDH RNA are shown.
The 2Apro coding region is not required for P1-null PV to form progeny virus in P1-expressing cells.
Next, the ability to form progeny virus was examined. The RNAs of pOM1, pOMΔ0.8, pOMΔ1.8, pOMΔP1, pOMΔP1Δ2A, and pOMΔP1Δ2AΔ2B were introduced into P1-expressing cells, and the supernatant was recovered after freezing and thawing. HeLa cells were covered with medium containing the supernatants and examined for a CPE 24 h later (data not shown). All the supernatants except those of pOMΔP1Δ2AΔ2B RNA and mock transfectants had a CPE. These results suggest that the RNAs of pOMΔ0.8, pOMΔ1.8, pOMΔP1, and pOMΔP1Δ2A can form progeny viruses, OMΔ0.8, OMΔ1.8, OMΔP1, and OMΔP1Δ2A, respectively, whereas the pOMΔP1Δ2AΔ2B RNA cannot. The quality and sizes of the viral RNA genomes were confirmed by agarose gel electrophoresis (data not shown). To adjust the titers among these viruses, the copy numbers of the PV RNA strands in the virus-containing supernatants were examined by quantitative real-time PCR and the units of viral RNA were determined. To examine the CPE expression, HeLa cells were infected at 5.2 × 101 U/cell and examined for a CPE up to 24 h after the infection (Fig. 7), and the rates of round cells, detached cells, and cells with membrane blebbing among all cells were analyzed (Fig. 7B). Similar to the previous results, all the viruses had a CPE. P1-null PV had slower kinetics than PV1(M)OM. OMΔ0.8, OMΔ1.8, and OMΔP1 showed similar numbers of CPE-expressing cells, although the kinetics of OMΔP1 until 10 h after the infection was faster than that of other P1-null PVs. OMΔP1Δ2A reproducibly had a lower rate of CPE than other P1-null PVs until 24 h after the infection. This result suggests that OMΔP1Δ2A is less able to induce a CPE or else takes longer and that 2Apro plays an important role in inducing a CPE. OMΔP1, significantly, started to cause membrane blebbing typical of apoptosis from 2 h after the infection, and the proportion of CPE-expressing cells was relatively high at early time points (Fig. 7C). This result indicates the possibility that OMΔP1 induces apoptosis. Compared with other P1-null PVs, OMΔP1Δ2A showed at least about a two-times-higher rate of membrane blebbing among CPE-expressing cells at 24 h after infection (OMΔ0.8, 12%; OMΔ1.8, 4.7%; OMΔP1, 7.9%; and OMΔP1Δ2A, 20%) (Fig. 7C). This indicates that OMΔP1Δ2A may induce apoptosis at a relatively low efficiency.
FIG. 7.

CPE in the cells infected with PV1(M)OM, OMΔ0.8, OMΔ1.8, OMΔP1, and OMΔP1Δ2A. (A) HeLa cells were mock infected (f) or infected with PV1(M)OM (a), OMΔ0.8 (b), OMΔ1.8 (c), OMΔP1 (d), or OMΔP1Δ2A (e) at an MOI of 1,000, and cell morphology was observed 24 h later under a microscope. Bar, 50 μm. (B) Percentages of CPE expression among all cells. (C) Rates of membrane blebbing among CPE-expressing cells. Average rates in two to four microscopic fields in one experiment were plotted. The rates were examined at 2, 6, 10, and 24 h after the infection. Blue lines with filled squares indicate results for PV1(M)OM, green lines with triangles indicate results for OMΔ0.8, violet lines with inverted triangles indicate results for OMΔ1.8, orange lines with rhombuses indicate results for OMΔP1, pink lines with circles indicate results for OMΔP1Δ2A, and moss-green lines with open squares indicate results for mock-infected cells.
2Apro coding region-deficient P1-null PV vector expresses foreign genes.
Because OMΔP1Δ2A can form progeny virus, 2Apro may not be essential for P1-null PV to express foreign genes. To examine whether OMΔP1 and OMΔP1Δ2A can express foreign genes, an EGFP coding sequence was inserted into the region of pOMΔP1 and pOMΔP1Δ2A with P1 deleted (Fig. 1A and B). The new constructs were designated pOM-EGFPΔP1 and pOM-EGFPΔP1Δ2A, respectively. RNAs derived from pOM1, pOMΔP1, pOMΔP1Δ2A, pOMΔP1Δ2AΔ2B, pOM-EGFPΔP1, and pOM-EGFPΔP1Δ2A were not degraded as assessed by gel electrophoresis (data not shown). The RNAs were introduced into HeLa cells, and the cells collected 2 and 8 h after the transfection. The cell lysate was used for slot blotting (Fig. 6B). When a probe for PV IRES RNA was used, all the RNAs except for pOMΔP1Δ2AΔ2B RNA increased at 8 h after the transfection compared to the levels at 2 h. pOM-EGFPΔP1 RNA increased more than pOM-EGFPΔP1Δ2A RNA. The amounts of GAPDH RNA hardly changed up to 8 h after the transfection. The results were reproducible. These results suggest that the 2Apro coding region is not required for the RNA replication of P1-null PV with EGFP inserted, although deletion of the 2Apro coding region results in a lower rate of RNA replication. When a probe for PV IRES RNA was used, the RNAs of pOMΔP1 and pOMΔP1Δ2A reproducibly increased to levels similar to the RNAs of pOM-EGFPΔP1 and pOM-EGFPΔP1Δ2A, respectively. These results suggest that the insertion of EGFP does not significantly affect PV RNA replication. Next, the RNAs of pOM-EGFPΔP1 and pOM-EGFPΔP1Δ2A were introduced into P1-expressing cells and the supernatants were recovered after freezing and thawing. The viral particles, OM-EGFPΔP1 and OM-EGFPΔP1Δ2A, respectively, proliferated in P1-expressing cells and were then purified. HeLa cells were covered with the purified virus at an MOI of 100, and the fluorescence of EGFP was observed under the fluorescence microscope 24 h after the infection (Fig. 8). Fluorescence-positive cells were observed in the OM-EGFPΔP1- and OM-EGFPΔP1Δ2A-infected samples, whereas no positive cells were observed in the mock-infected sample. These results suggest that OMΔP1 and OMΔP1Δ2A can express EGFP and that 2Apro is not essential for P1-null PV to express EGFP.
FIG. 8.

P1-null PV vector with or without the 2Apro coding region expresses EGFP. HeLa cells were mock infected (c, f, and i) or infected with OM-EGFPΔP1 (a, d, and g) or OM-EGFPΔP1Δ2A (b, e, and h) and observed 24 h later under a fluorescence microscope. Upper panels show fluorescence (FL) images (a to c), panels in the middle show bright-field (BF) images (d to f), and lower panels show merged (FL+BF) images (g to i). Bar, 100 μm.
In their ability to express EGFP 24 h after the infection, the strains ranked reproducibly as OM-EGFPΔP1Δ2A < OM-EGFPΔP1 (the average rates of fluorescence positivity among all cells in three microscopic fields in one experiment were 23% for OM-EGFPΔP1 and 9.7% for OM-EGFPΔP1Δ2A). This suggests that the defect in 2Apro decreases the expression of EGFP and/or the speed of viral replication. OM-EGFPΔP1Δ2A had a CPE, as did OM-EGFPΔP1, but only OM-EGFPΔP1Δ2A reproducibly induced morphological changes typical of apoptosis in a significant proportion of the CPE-expressing cells (the average rates of membrane blebbing among CPE-expressing cells were 10% for OM-EGFPΔP1 and 37% for OM-EGFPΔP1Δ2A). The result implies that 2Apro masks apoptosis in a significant proportion of cells. In the speed with which they induced a CPE, the strains reproducibly ranked as OM-EGFPΔP1 ≒ OM-EGFPΔP1Δ2A (the average rates of CPE expression among all cells were 27% for OM-EGFPΔP1 and 26% for OM-EGFPΔP1Δ2A). This suggests that 2Apro has little or no effect on inducing a CPE. The quality and sizes of the RNA genomes of OM-EGFPΔP1 and OM-EGFPΔP1Δ2A were confirmed by Northern blotting (data not shown). The genomic stability of OM-EGFPΔP1 and OM-EGFPΔP1Δ2A was examined by general RT-PCR, but no deletion was detected even after 20 passages (data not shown). These results suggest that both OM-EGFPΔP1 and OM-EGFPΔP1Δ2A are genetically stable for at least 20 passages.
DISCUSSION
2Apro has been thought important for PV replication. Here, we reveal that 2Apro is not required for PV replication and 2Apro-deficient PV with or without the capsid coding region can produce progeny viruses. This shows the possibility of realizing a vector that expresses a longer foreign gene and is less toxic.
Molla et al. reported that dc PV cDNA without the 2Apro coding region (pT7PVE2B) does not produce a viable virus (30). In contrast, RNA of pOMEΔ2A, which has a structure similar to that of pT7PVE2B, resulted in productive although inefficient replication. This may be due to a difference in the junctional sequences between P1 and 2B. In the case of pT7PVE2B, 2B is translated directly from the second EMCV IRES and methionine is added to the N terminus of 2B. On the other hand, in the case of pOMEΔ2A, the additional N-terminal sequences of 2B can be processed by 3Cpro or 3CDpro. Moreover, pT7PVE2B contains EMCV IRES up to AUG834, whereas pOMEΔ2A contains it up to +18 nt downstream of AUG834. Ribosomal initiation complexes attach directly to AUG834, and initiation does not involve scanning (22, 35). The interaction positions for ribosomal initiation complexes are up to +17 nt downstream of AUG834 (23). It is possible that the difference leads to modification of the 2B activity and/or relatively low efficiency of the initiation of the second cistron translation.
OMEΔ2A caused apoptosis, whereas neither PV1(M)OM nor OME did. Our results show the possibility that OMΔP1, OMΔP1Δ2A, and OM-EGFPΔP1Δ2A induce apoptosis in a significant proportion of cells. Calandria et al. reported that independently expressed 2Apro and 3Cpro induced apoptosis by mechanisms involving caspase activation (10). On the other hand, Burgon et al. reported that a 2A N32D mutation independently caused cells to die by apoptosis much earlier than wild-type-infected cells (9). This is consistent with the hypothesis that a wild-type function of the 2Apro protein is to inhibit apoptosis and cause a canonical necrotic CPE late in infection, perhaps directly or indirectly leading to the aberrant cleavage of procaspase-9, and that this activity is abrogated by the 2A N32D mutation. Together with our results, it seems likely that the apoptotic cell death induced in the cells infected with 2A-deficient or P1-deficient PV occurs via caspase-dependent apoptosis and that the expression of apoptosis depends on a subtle balance of relating proteins, such as 2Apro, 3Cpro, and procaspase-9, at each time point after the infection. It is highly possible that the balance of viral proteins differs depending on whether the virus is monocistronic or dicistronic. 2Apro activity may differ fundamentally depending on whether this protein is expressed individually or in the context of the viral infection.
It has been reported that PV was replication competent upon the replacement of its P1 region with a foreign gene in P1-expressing cells (4, 37). In these cases, an insert of about 2.9 kilobases (carcinoembryonic antigen [CEA]) was the longest. DIs with a 1,212-base deletion at the longest in the P1 region have been detected (25). We revealed that PV can produce progeny viruses with the entire P1 and 2Apro coding region deleted in P1-expressing cells. This construct lacks the longest region. We confirmed that PV can express EGFP without the P1 and 2Apro coding regions and that the insertion of EGFP does not significantly affect viral RNA replication. Moreover, these viruses are genetically stable. These results raise the possibility that a longer foreign gene than CEA can be expressed using the PV vector.
OMΔP1Δ2A had a lesser CPE than OMΔ0.8, OMΔ1.8, OMΔP1, and PV1(M)OM at 5.2 × 101 U/cell, which was supposed to be equivalent to an MOI of 1,000 of PV1(M)OM. To correct the units of viral RNA genomes as infectious units, HeLa cells were infected with OMΔ0.8, OMΔ1.8, OMΔP1, and OMΔP1Δ2A at 5.2 × 101 U/cell or with PV1(M)OM at an MOI of 10, the cells were collected 2 and 8 h later, and cell lysates were used for slot blotting (Fig. 6C). When a probe for PV IRES RNA was used, all the P1-null viruses had increased at almost the same rates at 8 h compared to the levels at 2 h after the infection. PV1(M)OM increased much more than P1-null viruses. These results suggest that 5.2 × 101 U/cell for P1-null viruses results in similar RNA replication activities under these conditions. The viral titers/cell might not be high enough to infect all the cells because the cells infected at 5.2 × 101 U/cell contained more RNA than those infected at 5.2 U/cell (data not shown); it is likely that the units of the RNA genomes of OMΔ0.8, OMΔ1.8, OMΔP1, and OMΔP1Δ2A were almost proportional to these RNA replication activities. Consequently, it was confirmed that OMΔP1Δ2A is less toxic or takes a longer time to have a CPE even though its RNA replication activity is similar to those of OMΔ0.8, OMΔ1.8, and OMΔP1.
Regarding the slot blot analysis of the cells transfected with synthesized viral RNA, the RNAs of pOMΔP1Δ2A and pOM-EGFPΔP1Δ2A showed less RNA replication activity than those of pOMΔP1 and pOM-EGFPΔP1, respectively (Fig. 6B). It seems likely that the defect in 2Apro suppresses the RNA replication activity. In terms of the relevant effect of 2Apro, viral replication speed, and/or the ability to induce a CPE, the ranking was OMEΔ2A < OME and OMΔP1Δ2A < OMΔP1, whereas OM-EGFPΔP1 ≒ OM-EGFPΔP1Δ2A. In the ability to express EGFP, the ranking was OM-EGFPΔP1Δ2A < OM-EGFPΔP1. These results may be also because the defect in 2Apro suppresses the viral replication activity and/or the expression of foreign mRNA.
Acknowledgments
We are grateful to T. Matano, N. Kamoshita, A. Yanagiya, and N. Matsuda for suggestions and discussions. We also thank A. Ohmura for technical support and E. Suzuki for help in preparing the manuscript. We are grateful to C. D. Morrow for generously providing VV-P1.
This work was supported in part by Grants-in-Aid for Advanced Medical Science Research by Ministry of Education, Culture, Sports, Science and Technology (MEXT), a Grant-in-Aid for Scientific Research on Priority Areas, a Grant-in-Aid for Scientific Research (S), a Grant-in-Aid for Scientific Research on Priority Areas, a Grant-in-Aid for Young Scientists (B), a Health Labor Sciences Research Grant, special coordination funds for promoting Science and Technology, contracted research allowance “Research and Development in a New Converting Field Based on Nanotechnology and Materials Science” by MEXT, and The Naito Foundation.
Footnotes
Published ahead of print on 14 April 2010.
The authors have paid a fee to allow immediate free access to this article.
REFERENCES
- 1.Agol, V. I., G. A. Belov, K. Bienz, D. Egger, M. S. Kolesnikova, N. T. Raikhlin, L. I. Romanova, E. A. Smirnova, and E. A. Tolskaya. 1998. Two types of death of poliovirus-infected cells: caspase involvement in the apoptosis but not cytopathic effect. Virology 252:343-353. [DOI] [PubMed] [Google Scholar]
- 2.Agol, V. I., G. A. Belov, K. Bienz, D. Egger, M. S. Kolesnikova, L. I. Romanova, L. V. Sladkova, and E. A. Tolskaya. 2000. Competing death programs in poliovirus-infected cells: commitment switch in the middle of the infectious cycle. J. Virol. 74:5534-5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Almstead, L. L., and P. Sarnow. 2007. Inhibition of U snRNP assembly by a virus-encoded proteinase. Genes Dev. 21:1086-1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ansardi, D. C., Z. Moldoveanu, D. C. Porter, D. E. Walker, R. M. Conry, A. F. LoBuglio, S. McPherson, and C. D. Morrow. 1994. Characterization of poliovirus replicons encoding carcinoembryonic antigen. Cancer Res. 54:6359-6364. [PubMed] [Google Scholar]
- 5.Ansardi, D. C., D. C. Porter, and C. D. Morrow. 1993. Complementation of a poliovirus defective genome by a recombinant vaccinia virus which provides poliovirus P1 capsid precursor in trans. J. Virol. 67:3684-3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belov, G. A., L. I. Romanova, E. A. Tolskaya, M. S. Kolesnikova, Y. A. Lazebnik, and V. I. Agol. 2003. The major apoptotic pathway activated and suppressed by poliovirus. J. Virol. 77:45-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bodian, D. 1955. Emerging concept of poliomyelitis infection. Science 122:105-108. [DOI] [PubMed] [Google Scholar]
- 8.Borman, A. M., R. Kirchweger, E. Ziegler, R. E. Rhoads, T. Skern, and K. M. Kean. 1997. elF4G and its proteolytic cleavage products: effect on initiation of protein synthesis from capped, uncapped, and IRES-containing mRNAs. RNA 3:186-196. [PMC free article] [PubMed] [Google Scholar]
- 9.Burgon, T. B., J. A. Jenkins, S. B. Deitz, J. F. Spagnolo, and K. Kirkegaard. 2009. Bypass suppression of small-plaque phenotypes by a mutation in poliovirus 2A that enhances apoptosis. J. Virol. 83:10129-10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calandria, C., A. Irurzun, A. Barco, and L. Carrasco. 2004. Individual expression of poliovirus 2Apro and 3Cpro induces activation of caspase-3 and PARP cleavage in HeLa cells. Virus Res. 104:39-49. [DOI] [PubMed] [Google Scholar]
- 11.Castello, A., J. M. Izquierdo, E. Welnowska, and L. Carrasco. 2009. RNA nuclear export is blocked by poliovirus 2A protease and is concomitant with nucleoporin cleavage. J. Cell Sci. 122:3799-3809. [DOI] [PubMed] [Google Scholar]
- 12.Cole, C. N. 1975. Defective interfering (di) particles of poliovirus. Prog. Med. Virol. 20:180-207. [PubMed] [Google Scholar]
- 13.Cole, C. N., D. Smoler, E. Wimmer, and D. Baltimore. 1971. Defective interfering particles of poliovirus. I. Isolation and physical properties. J. Virol. 7:478-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collis, P. S., B. J. O'Donnell, D. J. Barton, J. A. Rogers, and J. B. Flanegan. 1992. Replication of poliovirus RNA and subgenomic RNA transcripts in transfected cells. J. Virol. 66:6480-6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Etchison, D., S. C. Milburn, I. Edery, N. Sonenberg, and J. W. Hershey. 1982. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J. Biol. Chem. 257:14806-14810. [PubMed] [Google Scholar]
- 16.Hagino-Yamagishi, K., and A. Nomoto. 1989. In vitro construction of poliovirus defective interfering particles. J. Virol. 63:5386-5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hambidge, S. J., and P. Sarnow. 1992. Translational enhancement of the poliovirus 5′ noncoding region mediated by virus-encoded polypeptide 2A. Proc. Natl. Acad. Sci. U. S. A. 89:10272-10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunt, S. L., T. Skern, H. D. Liebig, E. Kuechler, and R. J. Jackson. 1999. Rhinovirus 2A proteinase mediated stimulation of rhinovirus RNA translation is additive to the stimulation effected by cellular RNA binding proteins. Virus Res. 62:119-128. [DOI] [PubMed] [Google Scholar]
- 19.Jang, S. K., M. V. Davies, R. J. Kaufman, and E. Wimmer. 1989. Initiation of protein synthesis by internal entry of ribosomes into the 5′ nontranslated region of encephalomyocarditis virus RNA in vivo. J. Virol. 63:1651-1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jang, S. K., H. G. Krausslich, M. J. Nicklin, G. M. Duke, A. C. Palmenberg, and E. Wimmer. 1988. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 62:2636-2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kajigaya, S., H. Arakawa, S. Kuge, T. Koi, N. Imura, and A. Nomoto. 1985. Isolation and characterization of defective-interfering particles of poliovirus Sabin 1 strain. Virology 142:307-316. [DOI] [PubMed] [Google Scholar]
- 22.Kaminski, A., M. T. Howell, and R. J. Jackson. 1990. Initiation of encephalomyocarditis virus RNA translation: the authentic initiation site is not selected by a scanning mechanism. EMBO J. 9:3753-3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolupaeva, V. G., I. B. Lomakin, T. V. Pestova, and C. U. Hellen. 2003. Eukaryotic initiation factors 4G and 4A mediate conformational changes downstream of the initiation codon of the encephalomyocarditis virus internal ribosomal entry site. Mol. Cell. Biol. 23:687-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuechler, E., J. Seipelt, H. D. Liebig, and W. Sommergruber. 2002. Picornavirus proteinase-mediated shutoff of host cell translation: direct cleavage of a cellular initiation factor, p. 301-311. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 25.Kuge, S., I. Saito, and A. Nomoto. 1986. Primary structure of poliovirus defective-interfering particle genomes and possible generation mechanisms of the particles. J. Mol. Biol. 192:473-487. [DOI] [PubMed] [Google Scholar]
- 26.Lawson, M. A., and B. L. Semler. 1990. Picornavirus protein processing—enzymes, substrates, and genetic regulation. Curr. Top. Microbiol. Immunol. 161:49-87. [PubMed] [Google Scholar]
- 27.Li, X., H. H. Lu, S. Mueller, and E. Wimmer. 2001. The C-terminal residues of poliovirus proteinase 2A(pro) are critical for viral RNA replication but not for cis- or trans-proteolytic cleavage. J. Gen. Virol. 82:397-408. [DOI] [PubMed] [Google Scholar]
- 28.Lundquist, R. E., M. Sullivan, and J. V. Maizel, Jr. 1979. Characterization of a new isolate of poliovirus defective interfering particles. Cell 18:759-769. [DOI] [PubMed] [Google Scholar]
- 29.Molla, A., S. K. Jang, A. V. Paul, Q. Reuer, and E. Wimmer. 1992. Cardioviral internal ribosomal entry site is functional in a genetically engineered dicistronic poliovirus. Nature 356:255-257. [DOI] [PubMed] [Google Scholar]
- 30.Molla, A., A. V. Paul, M. Schmid, S. K. Jang, and E. Wimmer. 1993. Studies on dicistronic polioviruses implicate viral proteinase 2Apro in RNA replication. Virology 196:739-747. [DOI] [PubMed] [Google Scholar]
- 31.Nomoto, A., B. Detjen, R. Pozzatti, and E. Wimmer. 1977. The location of the polio genome protein in viral RNAs and its implication for RNA synthesis. Nature 268:208-213. [DOI] [PubMed] [Google Scholar]
- 32.Omata, T., H. Horie, S. Kuge, N. Imura, and A. Nomoto. 1986. Mapping and sequencing of RNAs without recourse to molecular cloning: application to RNAs of the Sabin 1 strain of poliovirus and its defective interfering particles. J. Biochem. 99:207-217. [DOI] [PubMed] [Google Scholar]
- 33.Pelletier, J., and N. Sonenberg. 1989. Internal binding of eucaryotic ribosomes on poliovirus RNA: translation in HeLa cell extracts. J. Virol. 63:441-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pelletier, J., and N. Sonenberg. 1988. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334:320-325. [DOI] [PubMed] [Google Scholar]
- 35.Pestova, T. V., C. U. Hellen, and I. N. Shatsky. 1996. Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol. Cell. Biol. 16:6859-6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porter, D. C., D. C. Ansardi, W. S. Choi, and C. D. Morrow. 1993. Encapsidation of genetically engineered poliovirus minireplicons which express human immunodeficiency virus type 1 Gag and Pol proteins upon infection. J. Virol. 67:3712-3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porter, D. C., L. R. Melsen, R. W. Compans, and C. D. Morrow. 1996. Release of virus-like particles from cells infected with poliovirus replicons which express human immunodeficiency virus type 1 Gag. J. Virol. 70:2643-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sabin, A. B. 1956. Pathogenesis of poliomyelitis: reappraisal in the light of new data. Science 123:1151-1157. [DOI] [PubMed] [Google Scholar]
- 39.Shiroki, K., T. Ishii, T. Aoki, M. Kobashi, S. Ohka, and A. Nomoto. 1995. A new cis-acting element for RNA replication within the 5′ noncoding region of poliovirus type 1 RNA. J. Virol. 69:6825-6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tobin, G. J., D. C. Young, and J. B. Flanegan. 1989. Self-catalyzed linkage of poliovirus terminal protein VPg to poliovirus RNA. Cell 59:511-519. [DOI] [PubMed] [Google Scholar]
- 41.Tolskaya, E. A., L. I. Romanova, M. S. Kolesnikova, T. A. Ivannikova, E. A. Smirnova, N. T. Raikhlin, and V. I. Agol. 1995. Apoptosis-inducing and apoptosis-preventing functions of poliovirus. J. Virol. 69:1181-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weidman, M. K., R. Sharma, S. Raychaudhuri, P. Kundu, W. Tsai, and A. Dasgupta. 2003. The interaction of cytoplasmic RNA viruses with the nucleus. Virus Res. 95:75-85. [DOI] [PubMed] [Google Scholar]
- 43.Yanagiya, A., Q. Jia, S. Ohka, H. Horie, and A. Nomoto. 2005. Blockade of the poliovirus-induced cytopathic effect in neural cells by monoclonal antibody against poliovirus or the human poliovirus receptor. J. Virol. 79:1523-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yogo, Y., and E. Wimmer. 1972. Polyadenylic acid at the 3′-terminus of poliovirus RNA. Proc. Natl. Acad. Sci. U. S. A. 69:1877-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ziegler, E., A. M. Borman, F. G. Deliat, H. D. Liebig, D. Jugovic, K. M. Kean, T. Skern, and E. Kuechler. 1995. Picornavirus 2A proteinase-mediated stimulation of internal initiation of translation is dependent on enzymatic activity and the cleavage products of cellular proteins. Virology 213:549-557. [DOI] [PubMed] [Google Scholar]