

Abstract
Coxsackievirus B2 (CVB2), one of six human pathogens of the group B coxsackieviruses within the enterovirus genus of Picornaviridae, causes a wide spectrum of human diseases ranging from mild upper respiratory illnesses to myocarditis and meningitis. The CVB2 prototype strain Ohio-1 (CVB2O) was originally isolated from a patient with summer grippe in the 1950s. Later on, CVB2O was adapted to cytolytic replication in rhabdomyosarcoma (RD) cells. Here, we present analyses of the correlation between the adaptive mutations of this RD variant and the cytolytic infection in RD cells. Using reverse genetics, we identified a single amino acid change within the exposed region of the VP1 protein (glutamine to lysine at position 164) as the determinant for the acquired cytolytic trait. Moreover, this cytolytic virus induced apoptosis, including caspase activation and DNA degradation, in RD cells. These findings contribute to our understanding of the host cell adaptation process of CVB2O and provide a valuable tool for further studies of virus-host interactions.
Virus infections depend on complex interactions between viral and cellular proteins. Consequently, the nature of these interactions has important implications for viral cell type specificity, tissue tropism, and pathogenesis. Group B coxsackieviruses (CVB1 to CVB6), members of the genus Enterovirus within the family of Picornaviridae, are human pathogens that cause a broad spectrum of diseases, ranging from mild upper respiratory illnesses to more severe infections of the central nervous system, heart, and pancreas (61). These viruses have also been associated with certain chronic muscle diseases and myocardial infarction (2, 3, 12, 13, 22).
The positive single-stranded RNA genome (approximately 7,500 nucleotides in length) of CVBs is encapsidated within a small T=1, icosahedral shell (30 nm in diameter) comprised of repeating identical subunits made up of four structural proteins (VP1 to VP4). Parts of VP1, VP2, and VP3 are exposed on the outer surface of the capsid, whereas VP4 is positioned on the interior. The virion morphology is characterized by a star-shaped mesa at each 5-fold icosahedral symmetry axis, surrounded by a narrow depression referred to as the “canyon” (69). All six serotypes of CVB can use the coxsackie and adenovirus receptor (CAR) for cell attachment and entry (9, 55, 82). Some strains of CVB1, -3, and -5 also use decay accelerating factor ([DAF] CD55) for initial attachment to the host cell; however, binding to DAF alone is insufficient to permit entry into the cell (10, 54, 76).
Picornaviruses are generally characterized by their cytolytic nature in cell culture. However, several in vivo and in vitro studies have shown that some picornaviruses, e.g., poliovirus, Theiler's murine encephalomyelitis virus, foot-and-mouth disease virus, CVB3, CVB4, and CVB5, may also establish persistent, noncytolytic infections (4, 29, 35, 39, 62, 74). Recently, it has been shown that the diverse outcomes of picornaviral infections may depend on interactions between the virus and the apoptotic machinery of the infected cell (14, 30, 71). Several picornaviral proteins have been identified as inducers of an apoptotic response, including viral capsid proteins VP1, VP2, and VP3, as well as nonstructural proteins 2A and 3C (7, 20, 32, 33, 42, 50, 63). In addition, antiapoptotic activity has been assigned to the nonstructural proteins 2B and 3A (16, 59).
Picornaviruses have the potential to adapt rapidly to new host environments. Virus features affecting adaptability include high mutation rates, short replication times, large populations, and frequent incidences of recombination (25-27, 53). Consequently, picornaviruses exist as genetically heterogenous populations, referred to as viral quasispecies (25, 26).
Previously, the CVB2 prototype strain Ohio-1 (CVB2O) was adapted to cytolytic replication in rhabdomyosarcoma (RD) cells (66). Two amino acid changes were identified in the capsid-coding region, and one was identified in the 2C-coding region of the adapted virus. Further characterization of the virus-host interaction showed that the infection was not affected by anti-DAF antibodies, indicating the use of an alternative receptor.
In this study, the amino acid substitutions associated with the adaptation of CVB2O to cytolytic infection of RD cells were evaluated. Site-directed mutagenesis studies showed that a single amino acid change in the VP1 capsid protein was responsible for the cytolytic RD phenotype. In addition, as indicated by caspase activation and DNA degradation, the apoptotic pathway was activated in RD cells infected by the cytolytic virus.
MATERIALS AND METHODS
Cells and viruses.
Cultures of a local variant of green monkey kidney (GMK) cells, human epithelial (HEp-2) cells, and human RD cells, obtained from the American Tissue Culture Collection (ATCC), were maintained in Dulbecco's modified Eagles medium (DMEM) supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 10% newborn calf serum (NCS) at 37°C in 7.5% CO2. The prototype strain CVB2 Ohio-1 (VR-29; ATCC) (56, 67) and the CVB2ORD variant (66) were propagated in GMK and RD cells, respectively, as previously described (66).
Flow cytometry.
The flow cytometry procedure has been described previously (66). RD cells were stained with an anti-CAR (RmcB) antibody (37) (hybridoma kindly provided by L. Philipson and R. Pettersson, Karolinska Institute, Sweden; also available as ATCC CRL-2379) or an anti-DAF (BRIC110) antibody (Cymbus Biotechnologies). A monoclonal mouse IgG1 antibody (X0931; Dako) was used as a negative control. After 1 h of incubation at 4°C, the cells were stained with a secondary R-phycoerythrin-conjugated polyclonal rabbit anti-mouse antibody (R0439; Dako). Data were acquired using a FACSCalibur (Becton Dickinson) and analyzed with CellQuest, version 3.3, software (Becton Dickinson).
Infectious viral cDNA clones.
The complete CVB2O genome was amplified and cloned into the pCR-Script Direct SK(+) vector (Stratagene) by using the AscI and NotI restriction enzyme cleavage sites, as previously described (Fig. 1A) (49, 67). In this pCVB2Owt (where wt is wild type) construct, the individual CVB2ORD substitutions in the protein VP1 (I to F and Q to K) and 2C (K to R) were introduced by site-specific mutagenesis (Fig. 1). The PflFI and SexAI sites were used to generate the single-amino acid mutant pCVB2OVP1I118F clone (vVP1I118F) encoding the VP1 I118F substitution (names of viruses derived from the infectious cDNA clones are given in parenthesis). The SexAI and EcoRI restriction sites were used to construct the pCVB2OVP1Q164K clone (vVP1Q164K), while EcoRI and BamHI sites were used to produce the pCVB2O2CK185R clone (v2CK185R). The CVB2O constructs were propagated in Escherichia coli, isolated, and verified by sequencing.
FIG. 1.

Schematic overview of the CVB2O genome organization and the construction of CVB2O variants containing adaptive mutations. (A) A representation of the CVB2O genome is depicted at the top, with boxes delineating viral genes as well as approximate positions of the relevant restriction enzyme sites used to construct infectious viral cDNA clones. Below, the location of amino acid substitutions in the CVB2ORD genome are indicated next to the VP1- and 2C-encoding genes; the first letter refers to the amino acid residue of the CVB2O protein and the number indicates the amino acid position in the individual protein. Infectious CVB2O cDNA clone variants, as inserted in the pCR-Script Direct SK+ vector, are shown in the lower part of the figure. The designation of constructed cDNA clones and simplified names of viruses generated from these clones (given within parentheses) are used throughout the article. (B) A surface-rendered view of a pentameric subunit of the viral capsid showing the location of the exposed CVB2O substitution that evolved during propagation in RD cells. The entire capsid of the closely related CVB3 (PDB 1COV) (58) is shown at the top with five protomers related by an icosahedral 5-fold symmetry axis colored to denote proteins VP1 (blue), VP2 (green), and VP3 (red), whereas the remaining virus surface is shown in shades of gray. Below is an enlarged view of the pentamer showing the location of the surface-exposed amino acid change of CVB2O VP1 (Q164K; cyan and encircled) given as the equivalent CVB3 VP1 residue 160, based on multiple sequence alignment (ClustalW) (79). The icosahedral asymmetric unit of the virus is indicated by the triangular boundary.
Generation of viruses from infectious viral cDNA clones.
Viruses were generated by transfection (Lipofectamine 2000; Invitrogen) of 2.5 μg of the prototype (pCVB2Owt) or mutant (pCVB2OVP1I118F, pCVB2OVP1Q164K, and pCVB2O2CK185R) DNA into RD cells according to manufacturer's protocol. At 5 days posttransfection, the virus from transfected cells was released by three freeze-thaw cycles and further propagated by one subsequent passage in RD cells as described previously (57). Viral RNA was extracted from infected cell cultures (QIAamp viral RNA mini kit; Qiagen), reverse transcribed (Superscript III; Invitrogen), and PCR amplified (PicoMaxx high fidelity PCR system; Stratagene) using virus-specific primers. PCR amplicons were visualized on an agarose gel, purified (QIAquick gel extraction kit; Qiagen), and sequenced. Virus titers were determined by 50% tissue culture infectious dose (TCID50) assays in GMK cells, according to standard procedures (34).
Virus infection.
Cells were infected with the different CVB2O variants according to standard procedures (57). Briefly, subconfluent monolayers of RD cells grown in 25-cm2 flasks were inoculated with clone-derived viruses at a multiplicity of infection (MOI) ranging from 1 to 100 TCID50s/cell. After a 1-h adsorption at room temperature, the inoculum was removed, and cells were washed three times to remove unbound virus before addition of DMEM supplemented with 2 mM l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. Incubation was continued for 7 days or until cytopathic effect (CPE) was observed.
In order to determine the number of mutations that were introduced in the vVP1Q164K genome during serial passages in RD cells, infected cells were incubated until complete CPE was observed. These samples were subjected to three freeze-thaw cycles and further passaged five times in RD cells at an apparently high MOI (in two parallel experiments). After the fifth passage, the viral progeny was sequenced.
For studies of virus production during repeated noncytolytic infections, RD cells infected with CVB2Owt were detached by EDTA treatment and passaged every fourth day at a 1/3 dilution. The titer of virus released into the medium of each passage was measured by the TCID50 method in GMK cells. After the 10th passage, the viral progeny was sequenced.
Release of CVB2Owt from RD cells.
Confluent RD cells cultured in 24-well plates were infected with CVB2Owt at an MOI of 1 TCID50/cell. In order to assess both the intra- and extracellular virus production, samples (only medium or medium together with cells) were frozen at different time points postinfection (p.i.), and virus titers were determined after three cycles of freezing and thawing (to release intracellular viruses) by a TCID50 assay in GMK cells.
Quantitation of viral infection.
The replication of viral plus-strand RNA in RD cells infected with the CVB2O variants was determined by real-time PCR, according to established procedures (43). Confluent RD cells in 24-well plates were infected with the different CVB2O variants at an MOI of 1 TCID50/cell as described above. Infected cell samples were harvested at 0 h and 96 h p.i. and subjected to three freeze-thaw cycles. Viral RNA was extracted as described above, followed by reverse transcription of extracted RNA using an Applied Biosystems TaqMan reverse transcriptase kit and random hexamers. Reverse transcribed viral cDNA was quantified using real-time PCR and a Power SYBR green Master Mix in accordance with the manufacturer's instructions (Applied Biosystems). The CVB2-specific primers were designed to amplify a 147-bp product from nucleotide positions 454 to 601 in the genomic 5′ untranslated region (UTR) region. The final PCR mixture contained 100 nM (each) forward (5′-GCCCCTGAATGCGGCTAAT-3′) and reverse (5′-ATTGTCACCATAAGCAGCCA-3′) primers, 10 μl of Power SYBR green PCR Master Mix (Applied Biosystems), and 1 μl of cDNA. Samples were analyzed in triplicates in a final volume of 20 μl. Thermal cycling was performed with an ABI Prism 7500 SDS machine (Applied Biosystems) and universal cycling conditions (2 min at 50°C, 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C). Nuclease-free water was used as a negative control. Cycle threshold (CT) values were determined by automated threshold analysis included in the ABI sequence detection software, version 1.3.1. Dissociation curves were recorded after each run. The difference between the copy numbers of cDNA (i.e., viral RNA) detected at 0 h and 96 h p.i. was presented as the fold change (2CT,0h−CT,96h) under the assumption of 100% amplification efficiency according to the 2−ΔΔCT method (52). Furthermore, virus production in these samples (at 0 h and 96 h p.i.) was measured by the TCID50 method in GMK cells.
Plaque formation assay.
The plaque phenotype of the CVB2O variants was determined by utilizing a gum tragacanth overlay as previously described (24). Briefly, the CVB2O variants were titrated by 10-fold dilutions onto confluent monolayers of RD cells in six-well plates. Following adsorption for 1 h at 37°C, the inoculum was aspirated and replaced with an overlay medium, which consisted of 0.8% (wt/vol) gum tragacanth (Sigma), DMEM, 1% NCS, 2 mM l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. RD cells were stained at 96 h p.i. with a crystal violet-ethanol solution for visualization of plaques.
Single-step growth kinetics.
Confluent monolayers of RD cells cultured in 24-well plates were infected with virus at an MOI of 1 TCID50/cell as described above. For assessing virus growth, samples were frozen at different time points p.i., and virus titers were determined by a TCID50 assay in GMK cells.
Immunofluorescence staining of virus-infected cells.
For immunofluorescence assays, monolayers of RD cells cultured on Lab-TEK II chamber glass slides (Nalge Nunc International) were infected with the CVB2O variants at an MOI of 1 TCID50/cell. Alternatively, to analyze expression of CVB2 antigens during repeated noncytolytic infection, a fraction of CVB2Owt-infected RD cells from passages 1, 5, and 10 were transferred and cultured on glass slides. At indicated time points (see Fig. 3C and 4B), the cells were fixed with 4% formaldehyde for 30 min at 4°C, washed twice, and stained with a monoclonal mouse anti-CVB2 antibody (MAB946; Chemicon International, Inc.) for 1 h at room temperature. For analyses of apoptosis, the fixed cells were incubated with a polyclonal antibody against cleaved/activated caspase-3 ([CC-3] 9661; Cell Signaling Technology). After the slides were washed, a secondary goat anti-mouse or goat anti-rabbit antibody labeled with Alexa Fluor 488 (A11001 or A11034; Molecular probes Inc.) was added for 1 h at room temperature. Finally, the slides were mounted with Vectashield (Immunkemi, Sweden) containing 4′,6-diamidino-2-phenylindole (DAPI). Images were captured with an epifluorescence microscope.
FIG. 3.

Characterization of CVB2O infection in RD cells. (A) Uninfected RD cells (control) and RD cells infected with CVB2Owt, vVP1I118F, vVP1Q164K, and v2CK185R at an MOI of 1 TCID50/cell. Cells were visualized by light microscopy (96 h p.i.). Bar, 100 μm. (B) Plaque morphology of the CVB2O variants (96 h p.i.). Plaques were visualized by crystal violet staining of RD cells. (C) Immunostaining of RD cells infected with the different CVB2O viruses (MOI of 1). Viruses were detected (24 h p.i.) with an anti-CVB2 monoclonal antibody and a secondary antibody labeled with Alexa Fluor 488 (green). The cellular nuclei were visualized with DAPI (blue). Bar, 50 μm. (D) Quantitation of viral genome replication. RD cells were infected with CVB2O virus variants (MOI of 1) and the amount of positive-sense RNA was quantified by real-time PCR (0 h and 96 h p.i.). The amount of viral RNA was determined by the cycle threshold (CT) value and is presented as the fold change (mean ± SEM) of viral RNA for triplicate samples (from 0 h to 96 h p.i.). (E) CVB2O progeny production in RD cells measured by titration. RD cells were infected (MOI of 1), and the number of infectious viral particles was determined by the TCID50 method for bound viruses (0 h p.i.) and virus progeny (96 h p.i.). Results are presented as the mean ± SEM (n = 3).
DNA fragmentation assay.
DNA fragmentation in CVB2O-infected and uninfected RD cells was analyzed as described previously (80). Briefly, EDTA-treated RD cells were lysed with 0.2% Triton X-100 and 1 mM EDTA in 10 mM Tris-HCl, pH 7.4. The nucleic acid components were separated by centrifugation (20,000 × g for 10 min at 4°C) and collected from the supernatant fraction by precipitation. The purified nucleic acids were dissolved in 1 mM EDTA-10 mM Tris-HCl (pH 7.4) and treated with RNase A (10 μg/ml) for 30 min at 37°C. DNA fragmentation was analyzed by electrophoresis in 1% agarose gels. RD cells treated with 0.5 μM staurosporine ([STS] S4400; Sigma-Aldrich), a broad-spectrum protein kinase inhibitor, were used as a positive control.
Western blot analysis.
Protein extraction was performed as described previously (8). Virus-infected, uninfected, and STS-treated RD cells were detached by EDTA treatment and collected together with the floating cell material by centrifugation. The pellet was washed with phosphate-buffered saline (PBS) and resuspended in lysis buffer (2% SDS, 35 mM β-mercaptoethanol, 50 mM Tris-HCl, pH 6.8) supplemented, immediately before use, with 1 mM phenylmethylsulfonyl fluoride and a Complete Mini protease inhibitor cocktail (Roche). The obtained lysate was incubated in boiling water for 10 min and sonicated. The total protein concentration was determined with a spectrophotometer (NanoDrop ND-1000; Saveen Werner). The protein samples (5 to 40 μg) were fractionated by SDS-PAGE (12 or 14% polyacrylamide) and electroblotted onto a 0.45-μm-pore-size polyvinylidene difluoride membrane (Amersham). Nonspecific sites were blocked with 5% nonfat dry milk and 0.1% Tween-20 in Tris-buffered saline (TBS) for 1 h at room temperature and incubated overnight with primary antibodies diluted in TBS with 5% bovine serum albumin (BSA) and 0.1% Tween-20. The blocking solution was also used for the dilution of secondary antibodies. For Western blot analyses, the following primary antibodies were used: anti-caspase-3 (9665; Cell Signaling Technology), anti-Bid (2002; Cell Signaling Technology), anti-caspase-8 (551242; BD Bioscience), anti-caspase-9 (ab28131; Abcam), and anti-actin (ab8227; Abcam). Immunoreactive proteins were visualized by using secondary antibodies conjugated with horseradish peroxidase (P0448 and P0260; Dako) and a chemiluminescence detection system (ECL; Amersham).
In vivo infection assay.
Male A/JOlaHsd mice with the H-2a locus (Harlan, United Kingdom) and weighing 15 to 17 g were housed three per cage in sterile pathogen-free conditions and supplied with sterile water and commercial food pellets (Topdovo, Trnava, Slovak Republic). Groups of mice (4 weeks of age) were injected intraperitoneally with CVB2O (2 × 106 TCID50s/mouse), a corresponding amount of CVB2ORD, or PBS (control) as described previously (11). Mice were sacrificed at day 5 postinoculation, a time point selected based on previous in vivo studies of coxsackieviruses (87, 88). Pancreas and heart tissue specimens were fixed in 4% formalin, embedded in paraffin, and sectioned (5- to 7-μm thickness) for subsequent staining with hematoxylin and eosin. Pancreatic tissues were subjected to three freeze-thaw cycles and homogenized in PBS to an ∼10% organ suspension. After removal of cell debris by centrifugation and addition of antibiotics (200 U/ml penicillin and 0.2 mg/ml streptomycin), the virus titer was determined by the TCID50 assay in HEp-2 cells. The animal study was conducted according to directives of the European Commission and approved by the State Veterinary and Food Administration of the Slovak Republic.
Prediction of amino acid changes in the VP1 protein.
The locations of CVB2ORD-specific amino acid substitutions were mapped based on multiple sequence alignment (ClustalW) (79) with the closely related CVB3 (Protein Data Bank [PDB] accession number 1COV) (58) that identified the equivalent CVB3 residues. The X-ray crystal structure of the CVB3 capsid was then modeled and visualized using Chimera (64, 73).
Statistical analysis.
Data are presented as means ± standard errors of the mean (SEM) of triplicate observations. One-way analysis of variance (ANOVA) followed by Bonferroni's test was used for multiple comparisons, and a P value of <0.05 was considered statistically significant.
RESULTS
A surface-exposed VP1 substitution in CVB2O controls cytolysis.
CVB2 is an enterovirus that causes CPE in susceptible CAR-expressing cells such as GMK and HeLa (55). A previous study has shown that by serial passages in RD cells, CVB2O can be transformed from a noncytolytic virus to a cytolytic variant, CVB2ORD (66). Sequence analysis revealed that the genome of the RD-adapted virus contained three nonsynonymous mutations, one in the 2C-encoding gene (K to R at amino acid position 185) and two in the gene that codes for the capsid protein VP1 (I to F at position 118 and Q to K at position 164) (Fig. 1A). The structural proteins of CVB2 and CVB3 share a high amino acid sequence identity (77%). Therefore, the published tertiary structure of CVB3 (58) was used to model the capsid locations of amino acid substitutions unique to the CVB2ORD variant. The highly conserved VP1 residue (Q) (67) was mapped to be exposed (αB helix) on the surface of the capsid, whereas the amino acid at position 118 (I) was positioned in proximity to the hydrophobic pocket below the canyon floor surrounding the 5-fold axis of the virion (βD strand) (Fig. 1B) (66).
To characterize how individual mutations were associated with the cytolytic RD phenotype, three different CVB2O variants were constructed by employing a reverse genetics approach (Fig. 1A). The substitutions in the VP1 protein (I118F and Q164K) and 2C protein (K185R) where introduced individually into a full-length CVB2O cDNA clone. Viruses were derived from cDNA clones in RD cells that expressed DAF but without evident expression of CAR (Fig. 2A). Sequencing of progeny virus populations showed that the mutations were individually tolerated and also that no other substitutions or silent mutations were introduced during viral replication. In order to determine the yield of infectious viral particles produced in RD cells after transfection, the different CVB2O variants were titrated on GMK cells (Fig. 2B), a cell line where both the parental and the RD variant of CVB2O cause cytolysis. Similar virus titers (105.0 to 105.9 TCID50s/ml) were obtained for CVB2Owt and the three CVB2O variants (96 h p.i.). Hence, both the wild-type CVB2O virus and the CVB2O single mutants were able to replicate with similar efficiency in RD cells.
FIG. 2.

Generation of virus from cDNA clones in RD cells. (A) Flow cytometric analysis of CAR and DAF expression on RD cells. For detection of CAR and DAF, anti-CAR (RmcB) and anti-DAF (BRIC110) monoclonal antibodies were used (black histograms). A mouse IgG1 antibody was used as a negative control (gray histograms). (B) Total virus yield (extracellular and intracellular) determined for indicated viral variants by a TCID50 assay in GMK cells. Viruses were generated from infectious cDNA clones by transfection and one passage in RD cells (96 h p.i.). Error bars represent mean ± SEM of triplicate samples.
To investigate the cellular effects caused by viruses derived from viral cDNA clones, RD cells were infected at an MOI of 1 TCID50/cell. The mutant virus expressing the surface-exposed amino acid substitution (vVP1Q164K) induced partial cell lysis at 72 h p.i. and complete CPE at 96 h p.i. (Fig. 3A). In addition, CVB2ORD encoding a combination of all three substitutions induced cytolysis by 48 h p.i. (data not shown). In contrast, CVB2Owt, vVP1I118F, or v2CK185R virus did not induce CPE in RD cells (Fig. 3A). No differences in CPE were observed when cells were infected with vVP1Q164K or CVB2Owt at a higher MOI (100 TCID50s/cell) (data not shown). The association between the VP1 Q164K substitution and cytolytic infection was verified by a plaque assay, where only the Q164K-expressing variant formed visible plaques in the RD cell monolayers at 96 h p.i. (Fig. 3B). In order to analyze the accumulation of mutations in the vVP1Q164K genome during replication in RD cells, the virus was passaged five times in these cells and then sequenced. Sequence analyses of two independent passages revealed that new mutations had been introduced in the 5′ UTR (C to A at nucleotide position 655) or in the VP3 protein (Y to F at amino acid position 107). However, it is worth noting that the Q164K substitution in the VP1 protein was retained, indicating that this mutation is conserved during replication in RD cells.
An immunofluorescence assay was employed to further characterize the CVB2O infection. Uninfected and CVB2O-infected RD cells were stained with a CVB2-specific antibody (24 h p.i.). Surprisingly, the production of viral antigens was equally efficient in cells infected with each one of the CVB2O variants, regardless of their respective propensities to cause cytolysis (Fig. 3C). At a later stage of the infection (72 h p.i.), a majority of RD cells subjected to virus infection showed various degrees of staining (data not shown).
The viral positive-sense RNAs of viruses adsorbed to RD cells (0 h) as well as those of synthesized progeny viruses (96 h p.i.) were quantified by real-time PCR (Fig. 3D). This quantification showed that the amount of viral RNA increased approximately 800-fold for the cytolytic vVP1Q164K virus. A significant increase of viral plus-strand RNA (100- to 400-fold) was also detected at 96 h p.i. for the noncytolytic viruses (CVB2Owt, vVP1I118F, and v2CK185R). Further, the production of infectious particles was determined by titration in GMK cells. All CVB2O variants showed similar binding characteristics to RD cells (approximately 104 TCID50s/ml) and produced similar amounts of viral progeny (approximately 106 to 107 TCID50s/ml) during the following 4 days (Fig. 3E).
Previously, it has been shown that several types of CVB can establish a persistent infection in RD cells (4, 29). To further characterize the noncytolytic CVB2O infection, RD cells infected with CVB2Owt were repeatedly passaged. After 10 passages, no signs of CPE could be observed (Fig. 4A) although infection was verified by detection of CVB2 antigens and by a continuous production of infectious progeny (105 to 107 TCID50s/ml at 96 h p.i. of each passage) (Fig. 4B and C). Sequence analysis of this progeny virus showed that none of the RD-adaptive mutations were introduced during RD cell passages. However, due to the canonical propensity of the viral RNA-dependent RNA polymerase, other mutations were observed in the 5′ UTR (C to T at nucleotide position 391), the VP2 protein (R to C at amino acid position 40), the VP1 protein (T to M at amino acid position 93), and the 3D protein (a silent mutation from a T to a C at amino acid position 199). In addition, detailed analysis of the CVB2Owt infection in RD cells showed that the virus progeny was first released at 12 to 16 h p.i. (Fig. 4D). These analyses demonstrated that the CVB2 infection was maintained during serial passages of RD cells and that the virus was released from the RD cells without signs of CPE.
FIG. 4.

RD cells persistently infected with CVB2Owt. (A) RD cells persistently infected with CVB2Owt at passage 1 (P1), P5, and P10. Successive passages were performed every fourth day. Bar, 100 μm. (B) Immunofluorescence micrographs of persistently infected RD cells. Infected RD cells from passages 1, 5, and 10 were cultured in chamber slides and fixed after 24 h. Viral protein expression was analyzed with a mouse anti-CVB2 monoclonal antibody, followed by a secondary antibody labeled with Alexa Fluor 488 (green). Nuclei were visualized with DAPI (blue). Bar, 50 μm. (C) RD cells infected with CVB2Owt (MOI of 1) were cultured, and successive 1/3 passages were performed every fourth day. The release of progeny virus during noncytolytic infection was quantified at passages 1 to 10 by endpoint titration in GMK cells (mean ± SEM; n = 3). (D) Release of virus progeny from RD cells infected with CVB2Owt (MOI of 1). Extracellular (viruses present in the medium) and total (extra- and intracellular) virus yields were determined at different time points after infection by endpoint titration in GMK cells. Error bars represent the SEM (n = 3).
Cytolytic and noncytolytic CVB2O viruses replicate with equal efficiencies.
In order to compare the replication kinetics of CVB2Owt and vVP1Q164K in RD cells, one-step growth curves were established. In this analysis, the wild-type virus and the vVP1Q164K variant showed comparable replication kinetics (Fig. 5). Only minor differences at the initial phase of the infection (2 h p.i.) and at the plateau of the growth curve (96 h p.i.) were detected. Conclusively, the growth kinetics analysis did not reveal any major differences between the cytolytic and noncytolytic virus.
FIG. 5.
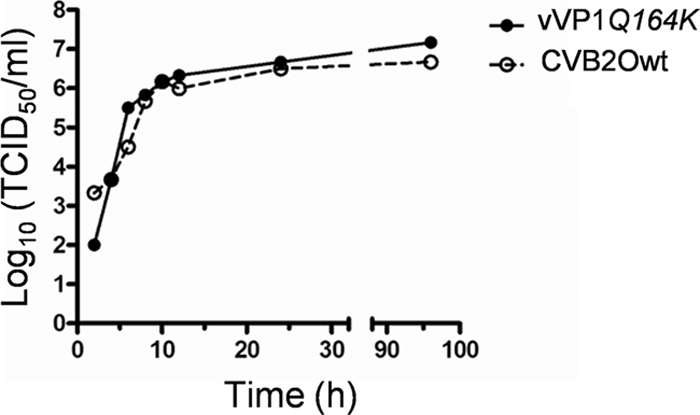
Growth kinetics of CVB2Owt and vVP1Q164K in RD cells. RD cells were infected at an MOI of 1 TCID50/cell. Total virus yields were determined at different time points after infection by endpoint titration in GMK cells. The results shown are representative of three independent experiments.
The VP1 Q164K substitution induces an apoptotic response.
Picornaviruses have been shown to manipulate the apoptotic response of their host cells, a characteristic that is most likely associated with the pathology of these viruses (14, 30, 71). To investigate whether apoptosis plays a role during cytolytic and noncytolytic CVB2O infection in RD cells, a number of apoptotic hallmarks were analyzed. As a first step, we evaluated the level of DNA fragmentation in cells infected with virus. Nuclear DNA extracted from virus-infected cells was compared with DNA isolated from uninfected cells and cells treated with a chemical apoptosis inducer, STS (18). At 72 h p.i., degradation of DNA was observed in RD cells infected with the cytolytic vVP1Q164K, a result comparable to that of STS-treated RD cells (Fig. 6A). In contrast, no evident fragmentation was detected in cells infected with the noncytolytic parental CVB2O virus.
FIG. 6.

Apoptotic status of RD cells infected with CVB2Owt and vVP1Q164K. RD cells were infected with the CVB2O viruses at an MOI of 1 and incubated at 37°C. (A) At the indicated times postinfection, cells were harvested, and DNA was extracted and resolved by electrophoresis in a 1% agarose gel as described in Materials and Methods. Uninfected RD cells (−) and cells treated with STS for 12 h (+) were used as negative and positive controls, respectively. M, molecular weight marker. (B) At the indicated times postinfection, RD cell lysates were prepared, and equal amounts of proteins (5 to 40 μg) were assayed for caspase-3, caspase-9, caspase-8, and Bid by Western blotting. Uninfected RD cells and cells treated with STS for 8 h were used as negative and positive controls, respectively, while expression of actin was used as a control of equal protein loading. Activated forms of Bid and caspase-3 are shown after longer durations of exposure than the unprocessed fragment. The molecular masses of the active fragments are indicated in the right margin. The results shown are representative of three independent experiments. (C) RD cells infected with CVB2Owt and vVP1Q164K were fixed at 24 h postinfection and analyzed with a polyclonal antibody against cleaved/activated caspase-3 (CC-3). CC-3 was visualized with a secondary antibody conjugated with Alexa Fluor 488 (green), and nuclei of RD cells were visualized with DAPI (blue). (D) Induction of apoptosis determined by caspase-3 activation. The percentage of CC-3-labeled RD cells from each sample was estimated from >300 randomly counted nuclei. Error bars represent the SEM (n = 3). A P value of <0.001 (***) was statistically significant for vVP1Q164K versus CVB2Owt or control cells. NS, not significant (P > 0.05).
Cellular caspases, a family of cysteine proteases, are key regulators of apoptosis. They are expressed as inactive proenzymes that become activated by specific cleavage in response to apoptotic stimuli (21, 68). To further examine the CVB2O-induced apoptotic response in more detail, cell lysates from RD cells infected with noncytolytic CVB2Owt and cytolytic vVP1Q164K virus for 12, 24, and 48 h were assayed for caspase activation by immunoblotting. The Western blotting results were validated with both positive and negative controls, consisting of STS-treated and uninfected RD cells, respectively. In the analysis of caspase-3, one of the key executors in the apoptotic pathway, only the 35-kDa proform was detected in RD cells infected with CVB2Owt (Fig. 6B). In contrast, the cytolytic CVB2O virus induced processing of procaspase-3, as shown by protein bands corresponding to the active enzymes (17 and 19 kDa). To further characterize the apoptotic status, two upstream caspases (caspase-8 and caspase-9) were analyzed. The active forms of caspase-8 (36 and 40 kDa) and caspase-9 (35 and 37 kDa) were detected in RD cells infected with vVP1Q164K. However, only the precursors of caspase-8 and caspase-9 were detected in cells infected with the noncytolytic CVB2O virus. Surprisingly, at 48 h p.i., a downregulation of procaspase-8 was observed in these cells.
The Bid protein, a proapoptotic protein of the Bcl-2 family, is an important regulator of apoptosis. Upon activation by the initiator caspase-8, the active form of Bid translocates to the mitochondria where it triggers Bax activation, which, in turn, is followed by cytochrome c efflux into the cytosol (46). To further assess the CVB2O-triggered apoptosis in RD cells, the processing of Bid was determined. The proform of Bid (22 kDa) was clearly processed into its active form (15 kDa) in RD cells infected with the cytolytic vVP1Q164K while no activation was observed for CVB2Owt-infected cells (Fig. 6B).
The association between apoptosis and the cytolytic infection (vVP1Q164K) in RD cells was also quantified by immunofluorescence based on caspase-3 activation. Only a small fraction of uninfected (∼1%) and CVB2Owt-infected (∼3.6%) RD cells showed staining of active caspase-3 (Fig. 6C and D). However, a significantly higher percentage (∼12%) of cells infected with the vVP1Q164K mutant stained positively for cleaved caspase-3 (24 h p.i.).
Taken together, these results suggested that the VP1 substitution Q164K in CVB2O was associated with apoptosis in RD cells via a number of key players of the apoptotic cascade.
CVB2ORD causes pancreatic inflammation in mice.
CVBs are known to have pancreatic and myocardial tropism in murine models (31, 36, 38, 83, 86). In order to investigate the relevance of the cytolytic RD phenotype (CVB2ORD) in a murine model, A/J mice susceptible to CVB infection (83, 87, 88) were inoculated intraperitoneally with CVB2O or CVB2ORD. At day 5 p.i., CVB2ORD-infected mice showed mild to intense perivascular lymphocyte infiltration with focal vasculopathy (i.e., inflammation observed as bleb formation of endothelial cells in blood vessels) in the exocrine pancreas (Fig. 7A). In contrast, no visible signs of infection were observed in mice infected with CVB2O. Moreover, neither CVB2O nor CVB2ORD induced histopathological changes in the endocrine pancreas or in the heart of infected mice. Titrations of the pancreatic tissue showed that the viral titer was higher in the pancreas from mice subjected to the CVB2ORD virus variant (Fig. 7B).
FIG. 7.

Histological analysis of pancreatic tissue from mice infected with CVB2O or CVB2ORD. Male A/J mice were intraperitoneally injected with CVB2O (dose of 2 × 106 TCID50s), CVB2ORD (dose of 2 × 106 TCID50s), or PBS (control). At day 5 postinoculation, the pancreata were formalin fixed and paraffin embedded, and tissue sections were stained with hematoxylin and eosin. Arrows indicate pancreatic tissue regions with mild to intense exocrine and perivascular lymphocyte infiltration with focal vasculopathy. Representative histopathological sections are shown. Original magnification, ×20. (B) CVB2O and CVB2ORD titers in murine pancreas at 5 days after inoculation. No viral titer was measurable in pancreas of control mice. Titers were determined by endpoint titration in HEp-2 cells. Error bars represent the SEM (n = 3).
DISCUSSION
In the present study, we examined specific amino acid changes associated with adaptation of CVB2O to cytolytic infection in RD cells. Our results showed that a single amino acid change on the capsid surface of CVB2O transforms the virus from a noncytolytic variant to a virus causing cytolysis. The characterization of the viral infection suggested that the CVB2O-induced cytolysis was associated with an apoptotic response (Fig. 8).
FIG. 8.

Schematic illustration of the CVB2O-induced apoptotic response in RD cells. Cytolytic CVB2O infection (vVP1Q164K) of RD cells results in activation of both the extrinsic (procaspase-8) and intrinsic (procaspase-9) pathways. These pathways are interconnected by Bid. When Bid is proteolytically activated by caspase-8, it translocates to the mitochondria and induces cytochrome c release. This, in turn, causes a successive activation of caspase-9 and caspase-3. The activated apoptotic signal eventually results in apoptotic cell death. In contrast, CVB2Owt causes a persistent infection in RD cells without detectable signs of apoptosis.
Previously published results have shown that CVB2O has the capacity to adapt to cytolytic replication in RD cells and that this novel property was associated with three nonsynonymous mutations (66). The results from reverse genetics studies with the cloned single mutants presented here demonstrated that a single surface-exposed amino acid change (Q164K) in the VP1 capsid protein of this virus is sufficient for the transformation to a cytolytic phenotype. This observation supports the view that specific capsid residues influence picornaviral cell type specificity and tissue tropism (1, 6, 15, 39, 44, 45, 62).
In detailed studies of the virus replication in RD cells by real-time PCR and titration on permissive GMK cells, the different cDNA clone-derived CVB2O variants showed similar properties regarding genome replication and virus progeny production. The productive infection of the cytolytic virus and noncytolytic CVB2O variants was further confirmed by immunofluorescence studies. This study also showed, together with the single-step growth curve and the observed onset of CPE, that there is a lag phase between the initial phase of viral replication and the CPE. The same phenomenon has previously been observed for another picornavirus, the Ljungan virus of the Parechovirus genus (28, 80). Possibly, this delay of CPE is a consequence of a virus that is not yet completely adapted to its host cell. However, the mechanism(s) involved in this delay of the CPE in RD cells infected with the cytolytic CVB2 variant remains to be elucidated. Taken together, the CVB2Owt and all CVB2O mutants were able to replicate in RD cells, a property that was not linked to cytolysis.
The nonstructural 2C protein of picornaviruses is a multifunctional protein with reported activities, including guidance of viral replication complexes to cytoplasmic membranes, enzymatic nucleotide triphosphatase activity, and involvement in virion assembly (48, 65, 78, 84). Results from analyses of the CVB2O mutant expressing a single 2C substitution (K185R) presented here suggested that this genetic change was not an essential determinant for the cytolytic phenotype of CVB2ORD. However, infection of RD cells with CVB2ORD, expressing all three adaptive substitutions, resulted in an earlier onset of cytolysis than infection with vVP1Q164K. Thus, the additional substitution in VP1 (I118F) together with the amino acid change of 2C contributed to the cytolytic phenotype in RD cells by a mechanism that remains to be elucidated.
Previously, it has been shown that CVB can establish persistent infections in RD cells (4, 29). Monitoring of repeated passages of the wild-type CVB2O in RD cells revealed a continuous release of infectious progeny although no signs of CPE were observed. The ability of the CVB2O wild-type strain to replicate in RD cells without evident signs of CPE has, to our knowledge, never been characterized before. Studies of poliovirus have shown that persistent infections may be established when HEp-2 cells are subjected to virus at a very low MOI (62). In contrast, the CVB2O infection of RD cells seems to be independent of virus dosage since these cells remained persistently infected even when they were exposed to a very high viral dose (MOI of 100). The release of noncytolytic CVB2Owt is possibly facilitated by viral proteins such as nonstructural protein 2B, which has been shown to modify membrane permeability (23, 60, 85). Conclusively, these results suggest that CVB2O established a persistent infection in cultured RD cells. This may have implications for CVB2 infections in vivo where muscle cells possibly serve as virus reservoirs. Indeed, CVB2 RNA has been detected in muscle tissue of patients with chronic muscle diseases (3, 12, 13, 22).
The virus-host cell system represented by the different CVB2O variants and the RD cells provided a well-defined model system for studies of persistent and cytolytic CVB2O infection. This model system also made it possible to examine whether apoptosis played a role during CVB2O infection in these cells. Theiler's murine encephalomyelitis virus is highly cytolytic in permissive BHK-21 cells, causing rapid cell destruction without signs of apoptosis; however, in less permissive cells, virus growth is markedly reduced, and viral replication is accompanied by induction of apoptosis (40, 41). Other picornaviruses, including CVB3, enterovirus 71, foot-and-mouth disease virus, poliovirus and avian encephalomyelitis virus, have also been shown to interact with the cellular apoptotic pathway (17, 19, 47, 50, 51, 63, 81). Although one-step growth analysis demonstrated that RD cells were equally susceptible to replication of the cytolytic and persistent CVB2O variants, major disparities were revealed when the apoptotic status of infected cells was examined. Prior to cell cytolysis, distinctive apoptotic hallmarks, i.e., extensive DNA degradation and activation of caspase-8, caspase-9, and caspase-3, were observed in RD cells infected with the cytolytic CVB2O (vVP1Q164K) variant. In addition, the infection was accompanied by an activation of Bid, an activation previously described for RD cells infected with enterovirus 71 (19). Conversely, RD cells persistently infected with the parental CVB2O virus showed no signs of DNA degradation and caspase activation. In conclusion, these data add to the increasing knowledge of the interplay between picornaviruses and the cellular apoptotic pathway during infection.
Several reports have suggested that the structural proteins of picornaviruses are involved in the induction of apoptosis (33, 50, 63). For example, the VP1 protein of foot-and-mouth disease virus has been shown to activate a proapototic response by binding to integrins and by deactivation of the Akt signaling pathway (63). In other experiments, the VP3 protein of avian encephalomyelitis virus colocalized with mitochondria (50), whereas the VP2 protein of CVB3 interacted with a proapoptotic factor, called Siva (33). Hence, picornaviral structural proteins are associated with induction of apoptosis by different mechanisms. Whether any of these mechanisms are involved in the apoptotic response associated with the VP1 protein of CVB2O remains to be elucidated.
Although induction of apoptosis in RD cells was linked to caspase activation, the involvement of other apoptotic mechanisms cannot be excluded. For example, the large quantities of viral products that accompany a poliovirus infection result in drastic rearrangement of the endoplasmic reticulum (ER) (72). ER-mediated stress resulting in activation of apoptosis has previously been reported for cells infected with a virus of the Flaviviridae family, the Japanese encephalitis virus (77). In addition, a previous study has shown that caspase inhibition does not prevent cell death of CVB3-infected HeLa cells (17). Thus, apoptotic activity is certainly not the only mechanism that can induce cell death and cytolysis during picornavirus infections. For example, the multifunctional 2A and 3C proteins are involved in the breakdown process of the cytoskeleton at the time of cell lysis (5, 42, 75). Consequently, cell lysis induced by a virus infection is probably a result of several different intracellular processes.
CVB infections cause myocarditis and pancreatitis (31, 38). As suggested by serological studies, these viruses also appear to be associated with insulin-dependent diabetes mellitus (38). Several experimental murine models have been developed to study CVB infections. In this report, a comparative study in a mouse model indicated that the cytolytic CVB2ORD variant induced inflammation in the exocrine pancreas while no signs of inflammation were observed in pancreatic tissue of mice infected with the parental CVB2O strain. This pancreatic virulence is consistent with other studies of CVB infections in mice, which have shown that virus-induced inflammation was located to the exocrine pancreas and not the endocrine tissue (70). However, these initial observations, including the possible association between the cytolytic RD phenotype and the increased propensity to induce pancreatic inflammation in mice, need to be explored further.
Knowledge about virus-host cell interactions is important in order to elucidate mechanisms by which the virus causes damage and to enable development of new antiviral treatments. The present study illustrates the adaptive potential of picornaviruses, where a single capsid substitution in CVB2O played a pivotal role for both cytolytic infection and induction of an apoptotic response in RD cells. In the absence of this amino acid change, CVB2O established a persistent, noncytolytic infection without signs of apoptosis. Thus, this study shows that the fate of the infected cell depends on a complex balance between the host and the virus and that this balance can be disrupted by a single substitution.
Acknowledgments
We thank Marlene Norrby for outstanding technical assistance and Kjell Edman for valuable discussions.
Figure 1B was produced using the UCSF Chimera package from the Computer Graphics Laboratory, University of California, San Francisco (supported by NIH P41 RR-01081). This work was also supported by grants from the Swedish Knowledge Foundation, the University of Kalmar, the Helge Ax:son Johnsons Foundation, the Sparbanken Kronan Foundation, and the Slovak Ministry of Health (MZSR code.2005/23-SZU-01).
Footnotes
Published ahead of print on 7 April 2010.
REFERENCES
- 1.Al-Hello, H., P. Ylipaasto, T. Smura, E. Rieder, T. Hovi, and M. Roivainen. 2009. Amino acids of Coxsackie B5 virus are critical for infection of the murine insulinoma cell line, MIN-6. J. Med. Virol. 81:296-304. [DOI] [PubMed] [Google Scholar]
- 2.Andreoletti, L., L. Venteo, F. Douche-Aourik, F. Canas, G. Lorin de la Grandmaison, J. Jacques, H. Moret, N. Jovenin, J. F. Mosnier, M. Matta, S. Duband, M. Pluot, B. Pozzetto, and T. Bourlet. 2007. Active coxsackieviral B infection is associated with disruption of dystrophin in endomyocardial tissue of patients who died suddenly of acute myocardial infarction. J. Am. Coll. Cardiol. 50:2207-2214. [DOI] [PubMed] [Google Scholar]
- 3.Archard, L. C., N. E. Bowles, P. O. Behan, E. J. Bell, and D. Doyle. 1988. Postviral fatigue syndrome: persistence of enterovirus RNA in muscle and elevated creatine kinase. J. R Soc Med. 81:326-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Argo, E., B. Gimenez, and P. Cash. 1992. Non-cytopathic infection of rhabdomyosarcoma cells by coxsackie B5 virus. Arch. Virol. 126:215-229. [DOI] [PubMed] [Google Scholar]
- 5.Badorff, C., G. H. Lee, B. J. Lamphear, M. E. Martone, K. P. Campbell, R. E. Rhoads, and K. U. Knowlton. 1999. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat. Med. 5:320-326. [DOI] [PubMed] [Google Scholar]
- 6.Bae, Y. S., and J. W. Yoon. 1993. Determination of diabetogenicity attributable to a single amino acid, Ala776, on the polyprotein of encephalomyocarditis virus. Diabetes 42:435-443. [DOI] [PubMed] [Google Scholar]
- 7.Barco, A., E. Feduchi, and L. Carrasco. 2000. Poliovirus protease 3C(pro) kills cells by apoptosis. Virology 266:352-360. [DOI] [PubMed] [Google Scholar]
- 8.Belov, G. A., L. I. Romanova, E. A. Tolskaya, M. S. Kolesnikova, Y. A. Lazebnik, and V. I. Agol. 2003. The major apoptotic pathway activated and suppressed by poliovirus. J. Virol. 77:45-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergelson, J. M., J. A. Cunningham, G. Droguett, E. A. Kurt-Jones, A. Krithivas, J. S. Hong, M. S. Horwitz, R. L. Crowell, and R. W. Finberg. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320-1323. [DOI] [PubMed] [Google Scholar]
- 10.Bergelson, J. M., J. G. Mohanty, R. L. Crowell, N. F. St John, D. M. Lublin, and R. W. Finberg. 1995. Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55). J. Virol. 69:1903-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bopegamage, S., M. Borsanyiova, A. Vargova, A. Petrovicova, M. Benkovicova, and P. Gomolcak. 2003. Coxsackievirus infection of mice. I. Viral kinetics and histopathological changes in mice experimentally infected with coxsackieviruses B3 and B4 by oral route. Acta Virol. 47:245-251. [PubMed] [Google Scholar]
- 12.Bowles, N. E., V. Dubowitz, C. A. Sewry, and L. C. Archard. 1987. Dermatomyositis, polymyositis, and Coxsackie-B-virus infection. Lancet 1:1004-1007. [DOI] [PubMed] [Google Scholar]
- 13.Bowles, N. E., P. J. Richardson, E. G. Olsen, and L. C. Archard. 1986. Detection of Coxsackie-B-virus-specific RNA sequences in myocardial biopsy samples from patients with myocarditis and dilated cardiomyopathy. Lancet 1:1120-1123. [DOI] [PubMed] [Google Scholar]
- 14.Buenz, E. J., and C. L. Howe. 2006. Picornaviruses and cell death. Trends Microbiol. 14:28-36. [DOI] [PubMed] [Google Scholar]
- 15.Caggana, M., P. Chan, and A. Ramsingh. 1993. Identification of a single amino acid residue in the capsid protein VP1 of coxsackievirus B4 that determines the virulent phenotype. J. Virol. 67:4797-4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campanella, M., A. S. de Jong, K. W. Lanke, W. J. Melchers, P. H. Willems, P. Pinton, R. Rizzuto, and F. J. van Kuppeveld. 2004. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J. Biol. Chem. 279:18440-18450. [DOI] [PubMed] [Google Scholar]
- 17.Carthy, C. M., D. J. Granville, K. A. Watson, D. R. Anderson, J. E. Wilson, D. Yang, D. W. Hunt, and B. M. McManus. 1998. Caspase activation and specific cleavage of substrates after coxsackievirus B3-induced cytopathic effect in HeLa cells. J. Virol. 72:7669-7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chae, H. J., J. S. Kang, J. O. Byun, K. S. Han, D. U. Kim, S. M. Oh, H. M. Kim, S. W. Chae, and H. R. Kim. 2000. Molecular mechanism of staurosporine-induced apoptosis in osteoblasts. Pharmacol. Res. 42:373-381. [DOI] [PubMed] [Google Scholar]
- 19.Chang, S. C., J. Y. Lin, L. Y. Lo, M. L. Li, and S. R. Shih. 2004. Diverse apoptotic pathways in enterovirus 71-infected cells. J. Neurovirol. 10:338-349. [DOI] [PubMed] [Google Scholar]
- 20.Chau, D. H., J. Yuan, H. Zhang, P. Cheung, T. Lim, Z. Liu, A. Sall, and D. Yang. 2007. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 12:513-524. [DOI] [PubMed] [Google Scholar]
- 21.Cohen, G. M. 1997. Caspases: the executioners of apoptosis. Biochem. J. 326:1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cunningham, L., N. E. Bowles, R. J. Lane, V. Dubowitz, and L. C. Archard. 1990. Persistence of enteroviral RNA in chronic fatigue syndrome is associated with the abnormal production of equal amounts of positive and negative strands of enteroviral RNA. J. Gen. Virol. 71:1399-1402. [DOI] [PubMed] [Google Scholar]
- 23.de Jong, A. S., W. J. Melchers, D. H. Glaudemans, P. H. Willems, and F. J. van Kuppeveld. 2004. Mutational analysis of different regions in the coxsackievirus 2B protein: requirements for homo-multimerization, membrane permeabilization, subcellular localization, and virus replication. J. Biol. Chem. 279:19924-19935. [DOI] [PubMed] [Google Scholar]
- 24.Dobos, P. 1976. Use of gum tragacanth overlay, applied at room temperature, in the plaque assay of fish and other animal viruses. J. Clin. Microbiol. 3:373-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Domingo, E., C. K. Biebricher, M. Eigen, and J. J. Holland. 2001. Quasispecies and RNA virus evolution: principles and consequences. Landes Bioscience, Austin, TX.
- 26.Domingo, E., E. Martinez-Salas, F. Sobrino, J. C. de la Torre, A. Portela, J. Ortin, C. Lopez-Galindez, P. Perez-Brena, N. Villanueva, R. Najera, et al. 1985. The quasispecies (extremely heterogeneous) nature of viral RNA genome populations: biological relevance—a review. Gene 40:1-8. [DOI] [PubMed] [Google Scholar]
- 27.Drake, J. W., and J. J. Holland. 1999. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. U. S. A. 96:13910-13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ekstrom, J. O., C. Tolf, C. Fahlgren, E. S. Johansson, G. Arbrandt, B. Niklasson, K. A. Edman, and A. M. Lindberg. 2007. Replication of Ljungan virus in cell culture: the genomic 5′-end, infectious cDNA clones and host cell response to viral infections. Virus Res. 130:129-139. [DOI] [PubMed] [Google Scholar]
- 29.Frisk, G., M. A. Lindberg, and H. Diderholm. 1999. Persistence of coxsackievirus B4 infection in rhabdomyosarcoma cells for 30 months. Brief report. Arch. Virol. 144:2239-2245. [DOI] [PubMed] [Google Scholar]
- 30.Galluzzi, L., C. Brenner, E. Morselli, Z. Touat, and G. Kroemer. 2008. Viral control of mitochondrial apoptosis. PLoS Pathog. 4:e1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gauntt, C., and S. Huber. 2003. Coxsackievirus experimental heart diseases. Front Biosci. 8:e23-35. [DOI] [PubMed] [Google Scholar]
- 32.Goldstaub, D., A. Gradi, Z. Bercovitch, Z. Grosmann, Y. Nophar, S. Luria, N. Sonenberg, and C. Kahana. 2000. Poliovirus 2A protease induces apoptotic cell death. Mol. Cell. Biol. 20:1271-1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henke, A., H. Launhardt, K. Klement, A. Stelzner, R. Zell, and T. Munder. 2000. Apoptosis in coxsackievirus B3-caused diseases: interaction between the capsid protein VP2 and the proapoptotic protein Siva. J. Virol. 74:4284-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hierholzer, J. C., and R. A. Killington. 1996. Quantitation of virus, p. 35-37. In B. W. J. Mahy and H. O. Kangro (ed.), Virology methods manual. Academic Press, London, United Kingdom.
- 35.Horsington, J., and Z. Zhang. 2007. Consistent change in the B-C loop of VP2 observed in foot-and-mouth disease virus from persistently infected cattle: implications for association with persistence. Virus Res. 125:114-118. [DOI] [PubMed] [Google Scholar]
- 36.Horwitz, M. S., A. Ilic, C. Fine, E. Rodriguez, and N. Sarvetnick. 2003. Coxsackievirus-mediated hyperglycemia is enhanced by reinfection and this occurs independent of T cells. Virology 314:510-520. [DOI] [PubMed] [Google Scholar]
- 37.Hsu, K. H., K. Lonberg-Holm, B. Alstein, and R. L. Crowell. 1988. A monoclonal antibody specific for the cellular receptor for the group B coxsackieviruses. J. Virol. 62:1647-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huber, S., and A. I. Ramsingh. 2004. Coxsackievirus-induced pancreatitis. Viral Immunol. 17:358-369. [DOI] [PubMed] [Google Scholar]
- 39.Jarousse, N., R. A. Grant, J. M. Hogle, L. Zhang, A. Senkowski, R. P. Roos, T. Michiels, M. Brahic, and A. McAllister. 1994. A single amino acid change determines persistence of a chimeric Theiler's virus. J. Virol. 68:3364-3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jelachich, M. L., and H. L. Lipton. 1999. Restricted Theiler's murine encephalomyelitis virus infection in murine macrophages induces apoptosis. J. Gen. Virol. 80:1701-1705. [DOI] [PubMed] [Google Scholar]
- 41.Jelachich, M. L., and H. L. Lipton. 1996. Theiler's murine encephalomyelitis virus kills restrictive but not permissive cells by apoptosis. J. Virol. 70:6856-6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joachims, M., K. S. Harris, and D. Etchison. 1995. Poliovirus protease 3C mediates cleavage of microtubule-associated protein 4. Virology 211:451-461. [DOI] [PubMed] [Google Scholar]
- 43.Jonsson, N., M. Gullberg, and A. M. Lindberg. 2009. Real-time polymerase chain reaction as a rapid and efficient alternative to estimation of picornavirus titers by tissue culture infectious dose 50% or plaque forming units. Microbiol. Immunol. 53:149-154. [DOI] [PubMed] [Google Scholar]
- 44.Jun, H. S., Y. Kang, A. L. Notkins, and J. W. Yoon. 1997. Gain or loss of diabetogenicity resulting from a single point mutation in recombinant encephalomyocarditis virus. J. Virol. 71:9782-9785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knowlton, K. U., E. S. Jeon, N. Berkley, R. Wessely, and S. Huber. 1996. A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J. Virol. 70:7811-7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kruidering, M., and G. I. Evan. 2000. Caspase-8 in apoptosis: the beginning of “the end”? IUBMB Life 50:85-90. [DOI] [PubMed] [Google Scholar]
- 47.Kuo, R. L., S. H. Kung, Y. Y. Hsu, and W. T. Liu. 2002. Infection with enterovirus 71 or expression of its 2A protease induces apoptotic cell death. J. Gen. Virol. 83:1367-1376. [DOI] [PubMed] [Google Scholar]
- 48.Li, J. P., and D. Baltimore. 1990. An intragenic revertant of a poliovirus 2C mutant has an uncoating defect. J. Virol. 64:1102-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindberg, A. M., C. Polacek, and S. Johansson. 1997. Amplification and cloning of complete enterovirus genomes by long distance PCR. J. Virol. Methods 65:191-199. [DOI] [PubMed] [Google Scholar]
- 50.Liu, J., T. Wei, and J. Kwang. 2002. Avian encephalomyelitis virus induces apoptosis via major structural protein VP3. Virology 300:39-49. [DOI] [PubMed] [Google Scholar]
- 51.Liu, J., T. Wei, and J. Kwang. 2004. Avian encephalomyelitis virus nonstructural protein 2C induces apoptosis by activating cytochrome c/caspase-9 pathway. Virology 318:169-182. [DOI] [PubMed] [Google Scholar]
- 52.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔCT) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 53.Lukashev, A. N. 2005. Role of recombination in evolution of enteroviruses. Rev. Med. Virol. 15:157-167. [DOI] [PubMed] [Google Scholar]
- 54.Martino, T. A., M. Petric, M. Brown, K. Aitken, C. J. Gauntt, C. D. Richardson, L. H. Chow, and P. P. Liu. 1998. Cardiovirulent coxsackieviruses and the decay-accelerating factor (CD55) receptor. Virology 244:302-314. [DOI] [PubMed] [Google Scholar]
- 55.Martino, T. A., M. Petric, H. Weingartl, J. M. Bergelson, M. A. Opavsky, C. D. Richardson, J. F. Modlin, R. W. Finberg, K. C. Kain, N. Willis, C. J. Gauntt, and P. P. Liu. 2000. The coxsackie-adenovirus receptor (CAR) is used by reference strains and clinical isolates representing all six serotypes of coxsackievirus group B and by swine vesicular disease virus. Virology 271:99-108. [DOI] [PubMed] [Google Scholar]
- 56.Melnick, J. L., N. Ledinko, et al. 1950. Ohio strains of a virus pathogenic for infant mice, Coxsackie group; simultaneous occurrence with poliomyelitis virus in patients with summer grippe. J. Exp. Med. 91:185-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Minor, P. 1985. Growth, assay and purification of picornaviruses, p. 25-41. In B. Mahy (ed.), Virology, a practical approach. IRL Press, Oxford, United Kingdom.
- 58.Muckelbauer, J. K., M. Kremer, I. Minor, G. Diana, F. J. Dutko, J. Groarke, D. C. Pevear, and M. G. Rossmann. 1995. The structure of coxsackievirus B3 at 3.5 Å resolution. Structure 3:653-667. [DOI] [PubMed] [Google Scholar]
- 59.Neznanov, N., A. Kondratova, K. M. Chumakov, B. Angres, B. Zhumabayeva, V. I. Agol, and A. V. Gudkov. 2001. Poliovirus protein 3A inhibits tumor necrosis factor (TNF)-induced apoptosis by eliminating the TNF receptor from the cell surface. J. Virol. 75:10409-10420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nieva, J. L., A. Agirre, S. Nir, and L. Carrasco. 2003. Mechanisms of membrane permeabilization by picornavirus 2B viroporin. FEBS Lett. 552:68-73. [DOI] [PubMed] [Google Scholar]
- 61.Pallansch, M. A., and R. P. Roos. 2001. Enteroviruses: polioviruses, coxsackieviruses, echoviruses and newer enteroviruses, p. 723-775. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 62.Pelletier, I., G. Duncan, and F. Colbere-Garapin. 1998. One amino acid change on the capsid surface of poliovirus Sabin 1 allows the establishment of persistent infections in HEp-2c cell cultures. Virology 241:1-13. [DOI] [PubMed] [Google Scholar]
- 63.Peng, J. M., S. M. Liang, and C. M. Liang. 2004. VP1 of foot-and-mouth disease virus induces apoptosis via the Akt signaling pathway. J. Biol. Chem. 279:52168-52174. [DOI] [PubMed] [Google Scholar]
- 64.Pettersen, E. F., T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng, and T. E. Ferrin. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605-1612. [DOI] [PubMed] [Google Scholar]
- 65.Pfister, T., and E. Wimmer. 1999. Characterization of the nucleoside triphosphatase activity of poliovirus protein 2C reveals a mechanism by which guanidine inhibits poliovirus replication. J. Biol. Chem. 274:6992-7001. [DOI] [PubMed] [Google Scholar]
- 66.Polacek, C., J. O. Ekstrom, A. Lundgren, and A. M. Lindberg. 2005. Cytolytic replication of coxsackievirus B2 in CAR-deficient rhabdomyosarcoma cells. Virus Res. 113:107-115. [DOI] [PubMed] [Google Scholar]
- 67.Polacek, C., A. Lundgren, A. Andersson, and A. M. Lindberg. 1999. Genomic and phylogenetic characterization of coxsackievirus B2 prototype strain Ohio-1. Virus Res. 59:229-238. [DOI] [PubMed] [Google Scholar]
- 68.Pop, C., and G. S. Salvesen. 2009. Human caspases: activation, specificity, and regulation. J. Biol. Chem. 284:21777-21781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Racaniello, V. R. 2001. Picornaviridae: the viruses and their replication, p. 685-722. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 70.Ramsingh, A. I. 1997. Coxsackieviruses and pancreatitis. Front Biosci. 2:e53-62. [DOI] [PubMed] [Google Scholar]
- 71.Roulston, A., R. C. Marcellus, and P. E. Branton. 1999. Viruses and apoptosis. Annu. Rev. Microbiol. 53:577-628. [DOI] [PubMed] [Google Scholar]
- 72.Rust, R. C., L. Landmann, R. Gosert, B. L. Tang, W. Hong, H. P. Hauri, D. Egger, and K. Bienz. 2001. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 75:9808-9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sanner, M. F., A. J. Olson, and J. C. Spehner. 1996. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers 38:305-320. [DOI] [PubMed] [Google Scholar]
- 74.Schmidtke, M., H. C. Selinka, A. Heim, B. Jahn, M. Tonew, R. Kandolf, A. Stelzner, and R. Zell. 2000. Attachment of coxsackievirus B3 variants to various cell lines: mapping of phenotypic differences to capsid protein VP1. Virology 275:77-88. [DOI] [PubMed] [Google Scholar]
- 75.Seipelt, J., H. D. Liebig, W. Sommergruber, C. Gerner, and E. Kuechler. 2000. 2A proteinase of human rhinovirus cleaves cytokeratin 8 in infected HeLa cells. J. Biol. Chem. 275:20084-20089. [DOI] [PubMed] [Google Scholar]
- 76.Shafren, D. R., R. C. Bates, M. V. Agrez, R. L. Herd, G. F. Burns, and R. D. Barry. 1995. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J. Virol. 69:3873-3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Su, H. L., C. L. Liao, and Y. L. Lin. 2002. Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J. Virol. 76:4162-4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Teterina, N. L., D. Egger, K. Bienz, D. M. Brown, B. L. Semler, and E. Ehrenfeld. 2001. Requirements for assembly of poliovirus replication complexes and negative-strand RNA synthesis. J. Virol. 75:3841-3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tolf, C., M. Gullberg, J. O. Ekstrom, N. Jonsson, and A. Michael Lindberg. 2009. Identification of amino acid residues of Ljungan virus VP0 and VP1 associated with cytolytic replication in cultured cells. Arch. Virol. 154:1271-1284. [DOI] [PubMed] [Google Scholar]
- 81.Tolskaya, E. A., L. I. Romanova, M. S. Kolesnikova, T. A. Ivannikova, E. A. Smirnova, N. T. Raikhlin, and V. I. Agol. 1995. Apoptosis-inducing and apoptosis-preventing functions of poliovirus. J. Virol. 69:1181-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tomko, R. P., R. Xu, and L. Philipson. 1997. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. U. S. A. 94:3352-3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tracy, S., K. Hofling, S. Pirruccello, P. H. Lane, S. M. Reyna, and C. J. Gauntt. 2000. Group B coxsackievirus myocarditis and pancreatitis: connection between viral virulence phenotypes in mice. J. Med. Virol. 62:70-81. [DOI] [PubMed] [Google Scholar]
- 84.Vance, L. M., N. Moscufo, M. Chow, and B. A. Heinz. 1997. Poliovirus 2C region functions during encapsidation of viral RNA. J. Virol. 71:8759-8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.van Kuppeveld, F. J., J. G. Hoenderop, R. L. Smeets, P. H. Willems, H. B. Dijkman, J. M. Galama, and W. J. Melchers. 1997. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. 16:3519-3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Woodruff, J. F. 1980. Viral myocarditis. A review. Am. J. Pathol. 101:425-484. [PMC free article] [PubMed] [Google Scholar]
- 87.Yang, D., J. Yu, Z. Luo, C. M. Carthy, J. E. Wilson, Z. Liu, and B. M. McManus. 1999. Viral myocarditis: identification of five differentially expressed genes in coxsackievirus B3-infected mouse heart. Circ Res. 84:704-712. [DOI] [PubMed] [Google Scholar]
- 88.Yu, J. Z., J. E. Wilson, S. M. Wood, R. Kandolf, K. Klingel, D. Yang, and B. M. McManus. 1999. Secondary heterotypic versus homotypic infection by Coxsackie B group viruses: impact on early and late histopathological lesions and virus genome prominence. Cardiovasc. Pathol. 8:93-102. [DOI] [PubMed] [Google Scholar]