

Abstract
The rat parvovirus H-1 (H-1PV) attracts high attention as an anticancer agent, because it is not pathogenic for humans and has oncotropic and oncosuppressive properties. The viral nonstructural NS1 protein is thought to mediate H-1PV cytotoxicity, but its exact contribution to this process remains undefined. In this study, we analyzed the effects of the H-1PV NS1 protein on human cell proliferation and cell viability. We show that NS1 expression is sufficient to induce the accumulation of cells in G2 phase, apoptosis via caspase 9 and 3 activation, and cell lysis. Similarly, cells infected with wild-type H-1PV arrest in G2 phase and undergo apoptosis. Furthermore, we also show that both expression of NS1 and H-1PV infection lead to higher levels of intracellular reactive oxygen species (ROS), associated with DNA double-strand breaks. Antioxidant treatment reduces ROS levels and strongly decreases NS1- and virus-induced DNA damage, cell cycle arrest, and apoptosis, indicating that NS1-induced ROS are important mediators of H-1PV cytotoxicity.
Members of the genus Parvovirus are small, icosahedral, nonenveloped viruses which infect vertebrates. Their single-stranded DNA genome is approximately 5.1 kb and contains two promoters, P4 and P38, which regulate the expression of nonstructural (NS1 and NS2) and capsid (VP1 and VP2) protein-encoding genes, respectively. Several species within the Parvovirus genus, in particular the rat parvovirus H-1 (H-1PV) and its mouse relative, the minute virus of mice (MVM), have attracted high interest for their potential as anticancer agents (7). Indeed, these viruses are not pathogenic for humans and possess intrinsic oncolytic and oncosuppressive properties demonstrated by their ability to infect and kill various human tumor cell cultures of different origins and to inhibit tumorigenesis in various animal models (7).
Rodent parvoviruses replicate preferentially in malignantly transformed cells. One of the reasons accounting for their oncotropism lies with their dependence on host cell proliferation due to their inability to induce quiescent cells to proliferate. Parvovirus replication is strictly dependent on cellular factors which are expressed during the S phase (E2F and cyclin A) and which are highly abundant in fast-dividing cancer cells (3, 9, 14).
It is important to point out that while most normal cells are quite resistant to parvovirus cytotoxicity, they become sensitive as a result of their transformation with various oncogenes (7).
Rodent parvoviruses have been shown to activate several death pathways. In particular, depending on cell type and growth conditions, H-1PV is able to induce apoptosis (35, 42, 46), necrosis (41), or cathepsin B-dependent cell death (16). It is presently unclear why some cells differ in the way they are killed by parvovirus and whether a common trigger initiates these various death processes.
NS1 is thought to be the major effector of parvovirus cytotoxicity. The viral product plays multiple roles in the viral life cycle. Besides driving viral DNA replication, NS1 regulates viral gene expression by modulating its own P4 promoter and activating the P38 promoter that controls the transcription unit encoding capsid proteins (9). Ectopic expression of NS1 from various parvoviruses has been shown to exert cytostatic and cytotoxic effects. For instance, ectopic expression of NS1 from parvovirus B19, a human pathogen, induces cell cycle arrest in G1 and apoptosis in erythroid lineage cells and hepatocytes (27, 28). Similarly, the adeno-associated virus type 2 (AAV-2) Rep 78 protein was shown to induce cell cycle arrest in S phase (43) and apoptosis in a p53-independent manner (45). Rat fibroblast lines stably expressing MVM NS1, under the control of a glucocorticoid-inducible promoter, are arrested in G1, S, and G2 phases of the cell cycle in the absence of typical signs of apoptosis (37, 38) and eventually die within 3 to 5 days from induction (6). It has also been reported that the expression of MVM NS1 correlates with the induction of single-strand DNA breaks, block of cell DNA replication (37), and cytoskeleton structural alterations (33). Whether these changes are direct or indirect effects of NS1 and how they impact on cell cycle or viability are still matters of speculation. In A9 mouse fibroblasts, MVM NS1 was found to bind to the catalytic subunit of the cell protein kinase CKII and alter its substrate specificity, leading in particular to changes in the phosphorylation pattern of the cytoskeleton component tropomyosin (34). While the CKII/NS1 interaction appears to mediate, at least in part, the toxicity of MVM for A9 cells, the role of this complex remains to be confirmed in other parvovirus/cell systems.
It is clear that additional studies are required to elucidate the interrelation between parvoviral NS1 proteins and cell death. Indirect evidence of H-1PV NS1 cytotoxicity was gathered by Rayet et al., who found that both wild-type H-1PV and an H-1PV-derived recombinant vector that does not express viral capsid proteins were able to induce apoptosis in the human promonocytic U937 cell line (42). As the only viral gene products shared by the wild-type and recombinant parvoviruses consist of the NS1 and NS2 proteins, viral cytotoxicity was attributed to either or both of these two polypeptides.
In this study, we asked whether expression of H-1PV NS1 alone is sufficient to exert cytostatic and cytotoxic activities and whether these effects could be traced back to distinct early cell disturbances caused by the viral protein. To address this issue, we established HEK-293 and HeLa stable cell lines that express H-1PV NS1 under a tetracycline-inducible promoter. For the sake of ascertaining their physiological relevance, the activities of isolated NS1 were compared with the ones exerted by the entire wild-type virus. The results show that NS1 expression recapitulates the sequence of events by which full H-1PV induces the death of permissive cells, demonstrating that the protein is indeed the mediator of H-1PV cytotoxicity.
MATERIALS AND METHODS
Cell culture.
HEK-293 T-REx Flp-In cells (Invitrogen), the HEK-293-NS1 stable cell line, HeLa cells, HeLa T-Rex cells (Invitrogen), and HeLa-NS1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% tetracycline-free fetal bovine serum (PAA), 2 mM l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. HEK-293 T-REx Flp-In cells were grown in the presence of 15 μg/ml Blasticidin and 100 μg/ml Zeocin, and HeLa T-Rex cells in the presence of 15 μg/ml Blasticidin, according to the Invitrogen instruction manual. The HEK-293-NS1 stable cell line was maintained in medium containing 15 μg/ml Blasticidin (Invitrogen) and 50 μg/ml hygromycin B (Invitrogen). The HeLa-NS1 stable cell line was maintained in DMEM supplemented with 6 μg/ml Blasticidin and 200 μg/ml Zeocin (Invitrogen).
Plasmid generation.
pcDNA5-NS1-H1 was constructed by subcloning the NS1 gene from pBlueNS1 (pBluescript SK1-NS1) (10) into the pcDNA5/FRT/TO and pcDNA4/TO vectors (Invitrogen) using the BamHI and XhoI restriction sites. pcDNA4/TO-Flag-HA-NS1-H1 was generated by subcloning the Flag- hemagglutinin (HA)-NS1-H1 fragment from pCDNA5/TO-Flag-HA-NS1-H1 into the pcDNA4/TO plasmid (Invitrogen) using the BamHI and XhoI restriction sites. pcDNA5/TO-Flag-HA-NS1-H1 was generated by PCR amplifying the Flag and HA tags from Flag-HA-Ago (26) using the TAG-FOR (5′-GGGGTACCCATCGCCGCCG-3′) and TAG-REV (5′-GGGTACCCCAGCGTAATCGG-3′) primers and subsequently subcloning the fragment into the KpnI cloning site of the pcDNA5-NS1-H1 vector. All the constructed plasmids were confirmed by DNA sequencing.
Establishment of NS1-inducible stable cell lines.
HEK-293 T-REx Flp-In cells were plated in 10-cm culture dishes and then transfected with the inducible expression vector pcDNA5-NS1-H1 and the pOG44 vector containing the Flp recombinase (ratio of 1:9) using Lipofectamine 2000 reagent (Invitrogen), according to the manufacturer's instructions. After 48 h, transfected cells were selected in medium containing 15 μg/ml Blasticidin and 50 μg/ml hygromycin B, changing the medium every 2 to 3 days for 3 weeks. The generated NS1-expressing cell line, representing a pool of different clones, was named 293-NS1. Genomic DNA was extracted from these cells, and the PCR-amplified NS1 gene was completely sequenced to ensure that the integrated gene maintained the wild-type NS1 sequence. NS1 gene expression was induced by adding to the culture medium 1 μg/ml of the tetracycline analogue doxycycline (DOX). Similarly, HeLa-NS1 stable cell lines were generated by transfecting HeLa T-REx cells (Invitrogen) with the pcDNA4/TO-Flag-HA-NS1-H1 construct and selecting transfected cells in medium containing 6 μg/ml Blasticidin and 200 μg/ml Zeocin, according to the manufacturer's handbook. A pool of selected clones was used in this study. Also in this case, induction of NS1 was obtained by adding 1 μg/ml of doxycycline.
Viruses and infections.
Wild-type H-1PV was produced in NB324K cells and purified in iodixanol gradients as previously described (49). Virus titers were determined by plaque assays (12). H-1PV was UV-treated as described by Morita et al. (28) with a total dose of 50 mJ/cm2. Parental 293 T-REx Flp-In and HeLa cells were infected with H-1PV at various multiplicities of infection (MOI; expressed as the number of PFU/cell). In brief, 1 × 106 cells were plated in 10-cm culture dishes and grown overnight. After removal of the media, cells were incubated with 1 ml of a suspension containing the virus in medium without serum or antibiotics for 1 h at 37°C, with gentle shaking of the culture dishes every 10 min. After this infection period, 9 ml of complete medium (DMEM) was added to the dishes.
Protein extractions and Western blot analysis.
Cells grown in the presence or absence of doxycycline or infected with H-1PV were trypsinized and washed with phosphate-buffered saline (PBS). After centrifugation, cell pellets were lysed (20 min on ice) in lysis buffer (1:5 pellet/volume ratio) consisting of 50 mM Tris-HCl at pH 8, 200 mM NaCl, 0.5% NP-40, 1 mM dithiothreitol (DTT), 10% glycerol, and a mix of protease inhibitors (Roche Diagnostics). After centrifugation (16,000 × g for 10 min at 4°C), supernatants were collected, and protein concentrations were measured by bicinchoninic acid (BCA) assay (Thermo Fisher Scientific), according to the manufacturer's instructions. Total cell extracts (20 to 30 μg) were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto a Hybond-P membrane (GE Healthcare). After being blocked for 1 h at room temperature (RT) with a PBS-milk solution (PBS, 0.05% Tween 20, 5% nonfat dry milk) or Tris-buffered saline (TBS)-casein solution (TBS, 0.1% Tween 20, 2% casein), in the case of phospho-Ser139 H2AX, membranes were incubated overnight at 4°C with primary antibodies specific for the following: NS1 (rabbit antiserum anti-SP8, dilution of 1:5,000) (5), N-terminal NS1 and NS2 (1:3,000) (8), NS2 (rabbit antiserum anti-SP6, 1:1,000) (49), β-tubulin (1:5,000; Sigma), E1A (1:1,000; BD Pharmingen), E1B-55K (1:10; a generous gift from Thomas Dobner, Heinrich-Pette-Institute, Hamburg, Germany), E1B-19K (1:1,000; Calbiochem), α-actin (1:10,000; MP Biomedicals), poly(ADP-ribose) polymerase (PARP) and pRB (1:1,000; BD Pharmingen), caspase 9 (1:1,000; Cell Signaling), caspase 3 (1:1,000; Stressgen), cyclin A, cyclin B1, p107, p130, p21, p27, CDC II (1:1,000; Santa Cruz Biotechnology), p53 (1:1,000; Oncogene), and phospho-Ser139 histone H2AX (γ-H2AX) (1:3,000; Lake Placid Biologicals). After the membranes were washed, peroxidase-conjugated goat anti-rabbit or goat anti-mouse antibodies (1:5,000; Santa Cruz Biotechnology) were added for 1 h at RT. The membranes were then washed and visualized with the ECL detection kit (PerkinElmer).
Immunofluorescence.
Cells grown on glass coverslips were fixed with 4% paraformaldehyde in PBS for 10 min and then permeabilized for another 10 min with PBS containing 0.2% Triton X-100. After being washed with PBS, cells were incubated for 20 min with normal goat serum (Santa Cruz Biotechnology) diluted 1:200 in PBS to reduce unspecific binding. After being washed with PBS, cells were incubated with anti-NS1 (monoclonal 3D9 [11]), anti-FLAG (Sigma), or anti-phospho-Ser139 histone H2AX (γ-H2AX; Lake Placid Biologicals) antibodies diluted 1:50, 1:400, and 1:500 in PBS, respectively, for 1 h in a dark humidified chamber. Cells were then washed three times with PBS and incubated for 1 h with Alexa Fluor 594-conjugated goat anti-mouse or Oregon Green 488-conjugated goat anti-rabbit secondary antibodies (Invitrogen) diluted 1:200 in PBS. After being washed five times in PBS, cell nuclei were stained with 10 μg/ml Hoechst 33342 (Sigma) for 2 min at RT. Coverslips were mounted onto glass slides with Elvanol (ICN) and analyzed by fluorescence microscopy (Leica).
Growth curves.
Stably transfected 293-NS1 and parental HEK-293 T-REx Flp-In cells were seeded at the same density onto two different sets of 10-cm dishes (1 × 105 cells/plate). After 24 h, one set of dishes was induced with doxycycline. Every 24 h for a total of 6 to 7 days, cells were trypsinized and counted using a cell counter and analyzer system (Schärfe System). Three independent experiments each in triplicate were performed.
For the doxycycline dose-response experiments in 293-NS1 cells and the cell proliferation analysis of HeLa cells, the xCELLigence system (Roche) was used, according to the manufacturer's instructions. Briefly, 5,000 (293-NS1) or 8,000 (HeLa T-Rex and HeLa-NS1) cells per well were plated in a 96-well E plate. After 24 h, cells were induced with different concentrations of doxycycline (0, 1, 5, 100, and 1,000 ng/ml). Cell growth was monitored in real time for up to 100 h by measuring the cell index impedance every 30 min.
Flow cytometric analyses. (i) Determination of cell cycle distribution and sub-G1 apoptotic cell population.
Cells were trypsinized, spun down for 5 min at 200 × g, washed twice with PBS, and then fixed with 1 ml ice-cold ethanol (70%) added dropwise. Fixed cells were stored for a maximum of 1 week at 4°C before fluorescence-activated cell sorting (FACS) analysis. Cells were centrifuged for 5 min at 200 × g and washed twice with PBS. Pellets were resuspended in PBS containing 100 μg/ml RNase and 25 μg/ml propidium iodide (Sigma). The stained cells were placed on ice for 1 h in the dark, then filtered through a 41-μm nylon mesh, and analyzed for DNA content on a FACSort flow cytometer (Becton Dickinson). A minimum of 20,000 events were acquired and analyzed by using the CellQuest software (Becton Dickinson). Cell cycle distribution analysis was performed using the ModFit cell cycle analysis software package (Verity Software House, Inc.) and the FlowJo flow cytometry analysis software (Tree Star, Inc.).
(ii) Annexin V/To-Pro-3 apoptosis assays.
293-NS1 and HEK-293 parental cells (50,000 cells/ml) were plated onto 6-cm dishes and induced with doxycycline (1 μg/ml) or infected with H-1PV (MOI, 1 PFU/cell). Apoptotic cells were identified by resuspending 1 × 105 cells in 100 μl of annexin V binding buffer containing 5 μl of 10 μg/ml annexin V-fluorescein isothiocyanate (Roche) for 15 min at room temperature. The vital dye To-Pro-3 (Molecular Probes, Inc.) was used to distinguish living cells from dead cells. A minimum of 10,000 events were acquired and analyzed by using the CellQuest software (Becton Dickinson).
(iii) Detection of ROS.
The level of intracellular reactive oxygen species (ROS) was measured using dichlorofluorescein diacetate (DCFH-DA) (25), which is oxidized in the presence of ROS and converted into the highly fluorescent DCF compound (1). Cells were seeded onto 6- or 10-cm dishes and were grown in the presence or absence of doxycycline (293-NS1 and HeLa-NS1) or infected with H-1PV at a MOI of 1 PFU/cell (HeLa) or 10 PFU/cell (parental HEK-293). DCFH-DA (10 μM) was added to the culture medium for 1 h before harvesting the cells. After trypsinization, cells were washed twice with PBS, resuspended in 500 μl of PBS, and directly analyzed by FACS. Rescue experiments were performed by using the antioxidant N-acetyl-l-cysteine (NAC; 5 mM) or glutathione (GSH; 5 and 10 mM), which was added to the culture medium at the time of the NS1 induction or H-1PV infection.
Lactate dehydrogenase (LDH) assay.
Cell lysis was evaluated by measuring the release of lactate dehydrogenase in the medium using the CytoTox 96 nonradioactive cytotoxicity assay (Promega), according to the manufacturer's instructions and the protocol described by Daeffler et al. (12). Briefly, 2,500 cells per well were seeded in 96-well plates in 50 μl of culture medium. After 24 h, cells were infected by adding to the wells 50 μl of medium containing either H-1PV (MOI, 20 to 200 PFU/cell) or treated with 1 μg/ml of doxycycline. LDH activity was measured by using an enzyme-linked immunosorbent assay (ELISA) reader at 492 nm.
MTT activity assay.
For the determination of cell viability, the metabolic activity of mitochondrial dehydrogenases was measured through the ability of these enzymes to convert 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) into a formazan product. MTT assays were performed as described by Daeffler et al. (12), plating 2,500 cells per well.
DNA extraction and Southern blot analysis.
Viral DNA extraction from infected cells and Southern blotting were carried out as described by Daeffler et al. (12), using a 32P-labeled DNA probe corresponding to a EcoRV-EcoRI fragment of H-1PV NS1 gene unit.
Electron microscopy.
293-NS1 cells were grown in the presence or absence of doxycycline in 10-cm dishes. After 72 h, the cells were fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2) at 4°C. After being washed in 50 mM sodium cacodylate buffer, cells were successively postfixed in 2% osmium tetroxide and 0.5% aqueous uranylacetate, dehydrated through ascending alcohol concentrations and propylene oxide, and embedded in Epon. Ultrathin sections of the fixed cells were cut with a Reichert-Jung microtome and examined with a Zeiss EM 10A transmission electron microscope. The magnification indicator was routinely controlled by using a grating replica.
RESULTS
H-1PV NS1 expression induces cell cycle alterations.
In order to study the cellular functions of H-1PV NS1 protein, we have generated a system based on HEK-293 cells, in which NS1 expression is under the control of a tetracycline-inducible cytomegalovirus (CMV) promoter. As shown in Fig. 1A, incubation with the tetracycline analogue doxycycline (DOX; 1 μg/ml) resulted in a striking induction of NS1 expression, as detected by Western blot analysis of cell lysates. On the contrary, NS2 was not detected using NS2-specific antibodies (5) or antibodies recognizing the N terminus region of NS1, which is also contained in the NS2 coding sequence (8) (see Fig. S1 in the supplemental material). This system allows NS1 expression to be experimentally tuned, as apparent from the dependence of the levels of NS1 expression on the concentrations of DOX in the culture medium (Fig. 1B). NS1 was mostly detected in the cell nucleus, showing a punctate staining (Fig. 1C), similar to the one observed upon H-1PV viral infection (11). It should also be stated that NS1-expressing cells were enlarged, especially at the level of their nuclei, compared with non-induced cells or DOX-treated HEK-293 parental cells (Fig. 1C; see also Fig. S2 in the supplemental material). While addition of DOX did not affect the growth of parental cells (Fig. 1D), it arrested the 293-NS1 cells in a dose-dependent manner (Fig. 1D; see also Fig. S3 in the supplemental material). After prolonged expression of NS1 (from 72 h postinduction on), we also observed the appearance of rounded cells easily detaching from the dish surface and of a small number of cells (1 to 3%) containing two or more nuclei (data not shown).
FIG. 1.

The 293-NS1 system allows the tightly controlled induction of H-1PV NS1 expression. The human T-REx Flp-In HEK-293 embryonic kidney cell line expressing the full-length H-1PV NS1 gene under the control of a doxycycline-inducible promoter (293-NS1) was established as described in Materials and Methods. (A) Total protein extracts were prepared from cells treated (induced [ind.]) or not treated (noninduced [non ind.]) with doxycycline (1,000 ng/ml) for 24 h or from cells infected for 24 h with H-1PV (MOI, 20 PFU/cell). Equal amounts of proteins were separated by SDS-PAGE and analyzed for the presence of NS1 protein by Western blot analysis. Actin was used as a loading control. (B) Total protein extracts were prepared from cells either grown in the absence (far right lane) or presence of various doxycycline concentrations, indicated on top of the lanes, and tested for their NS1 content, as described in the legend to panel A. β-tubulin was used as a loading control. (C) Induced and noninduced 293-NS1 cells were grown on coverslips, fixed, and processed for immunofluorescence using anti-NS1 and Alexa Fluor 594 anti-mouse secondary antibodies (red) and Hoechst nuclear staining (blue). The merge picture shows the nuclear localization of NS1. (D) Parental 293 and stably transfected 293-NS1 cell lines were seeded at a low density and cultured in the presence (ind.) or absence (non ind.) of doxycycline (1,000 ng/ml). Cell numbers were measured every 24 h. Mean values resulting from three independent experiments, each performed in triplicate, are given, with standard deviation bars shown.
These results prompted us to investigate whether H-1PV NS1 expression, like that of its MVM and B19 counterparts, would lead to perturbation in the cell cycle. Time course experiments using asynchronous cell populations were performed by collecting samples at different time points after NS1 induction for a total period of 24 h. Flow cytometric analyses revealed cell cycle alteration starting from the time NS1 became detectable by Western blot analysis (after 4 h) (Fig. 2A). We observed an initial and transient increase of the fraction of cells in S phase (reaching a maximum of 60% after 11 h) which was accompanied by a decrease of the cell subpopulation in G1 phase and was followed by a significant accumulation of cells in G2/M phase of the cell cycle (Fig. 2B and C). As we did not observe an increase in mitotic figures by microscopy analysis of NS1-induced cells (data not shown), we assumed that NS1-expressing cells were likely to accumulate in G2 rather than in M phase. Further accumulation of cells in G2 phase was not observed at later time points (48 and 72 h postinduction), although FACS analysis shows that a small fraction of cells contains more than 4N DNA content (see Fig. S4 in the supplemental material). These cell cycle perturbations were not observed in DOX-treated parental 293 cultures, clearly indicating that these events are dependent on the expression of NS1. Similar experiments performed with stable cell lines expressing the H-1PV transcription unit coding for VP1 and VP2 capsid proteins, under the control of the same inducible promoter used for NS1, failed to reveal any significant impact of VPs on the cell cycle (data not shown).
FIG. 2.

NS1 arrests the cell cycle at the G2/M stage. Parental and stably transfected 293-NS1 cells were cultured in medium supplemented (ind.) or not supplemented (non ind.) with doxycycline for 12 or 24 h and processed for Western blot analysis and FACS determination of cell cycle distribution. (A) Western blot analysis for the steady-state levels of NS1 protein. n.i., noninduced. (B) FACS analysis. At least 20,000 events were acquired for each flow cytometric analysis. The percentage of cells in the G1, S, and G2/M phases of the cell cycle was calculated by using the ModFit software. (C) NS1-induced cells were collected at the times indicated and analyzed by flow cytometry, as indicated in the legend to panel A.
We next investigated whether the levels of pivotal cell cycle regulators were modified after NS1 induction. It is known that S phase is controlled by a complex of cyclin-dependent kinase 2 (CDK2) with cyclin A, while G2 phase and G2/M transition are regulated by complexes of cyclin-dependent kinase 1 (cdc2) with cyclin A and cyclin B1. As illustrated in Fig. 3, the above-mentioned sequential accumulation of NS1-expressing cells first in S phase and subsequently in G2 phase indeed correlated with the time-dependent increase of cyclin A, cdc2, and cyclin B1 in these cultures. The activity of the cyclin/cyclin-dependent kinase complexes is regulated by their interaction with cyclin-dependent kinase inhibitors (CKIs). Protein levels of the CKIs p21 and p27 were strongly enhanced upon NS1 induction, suggesting that these two CKIs play a role in the NS1-mediated cell cycle arrest. Interestingly, NS1 had no detectable impact on the protein levels of p53, a key regulator of p21 expression. The retinoblastoma gene family, which includes pRb, p107, and p130, plays a key role in controlling cell cycle progression and in maintaining genome integrity (18, 39). We found that pRB protein levels were not affected upon NS1 induction. On the other hand, the steady-state levels of p107 and p130 were slightly or significantly increased, respectively. Altogether, these results show that the H-1PV NS1 protein has a strong impact on the cell cycle.
FIG. 3.

NS1 expression is associated with differential expression of cell cycle regulatory proteins. Doxycycline was added to the medium (1,000 ng/ml) of 293-NS1 cultures. Whole cellular lysates were prepared every 4 h and subjected to SDS-PAGE fractionation and immunoblotting using the antibodies indicated. Actin was used as a loading control. p.i., postinfection.
NS1 expression leads to cell lysis.
Parvoviruses typically induce the lysis of productively infected cells, favoring the release and spread of progeny viruses. We next investigated whether the viral product was sufficient to eventually induce cell lysis. Parental and 293-NS1 cell lines grown in the presence or absence of DOX were subjected to LDH and MTT assays to determine cell lysis and cell viability, respectively. As shown in Fig. 4, NS1 induction correlated with a marked enhancement of cell lysis (LDH assay), concurrently with a decrease of cell viability (MTT assay). In conclusion, these results provide the first evidence that NS1 expression is sufficient to induce cell lysis.
FIG. 4.

NS1 expression is sufficient to induce cell lysis. Cell lysis and viability were measured by LDH and MTT assays, respectively, in parental 293 and 293-NS1 cultures grown in the absence (−) or presence (+) of doxycycline (1,000 ng/ml) for 48 and 72 h. Average values with standard deviation bars are shown from three independent experiments performed in triplicate.
NS1-expressing 293 cells die through apoptosis.
NS1-expressing cells were then analyzed by flow cytometry for the presence of a sub-G1 DNA content cell population (less than 2N) that is indicative of DNA fragmentation and apoptosis. As shown in Fig. 5A, NS1 induction indeed correlated with the appearance of a sub-G1 population, which dramatically increased over time, reaching more than 50% of the total cell population after 144 h. In contrast, no signs of apoptosis were observed in DOX-treated parental HEK-293 cells. Annexin V staining (Fig. 5B), use of a terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay (data not shown), and electron microscopy analysis (Fig. 5D) confirmed the presence of an apoptotic cell population in NS1-expressing cells. Furthermore, NS1 induction was associated with activation of caspases 9 and 3 and cleavage of PARP (Fig. 5C). In agreement with these observations, addition of the pan-caspase inhibitor zVAD-FMK (10 μM) to the culture medium protected NS1-expressing cells from the induction of apoptosis (Fig. 6A) and cell lysis (Fig. 6B). NS1 expression did not affect the protein levels of E1A and E1B adenovirus proteins that are normally expressed in HEK-293 and are known to play a role in controlling cell cycle and apoptosis (see Fig. S5 in the supplemental material). These results indicate that NS1 alone is sufficient to induce apoptosis in HEK-293 cells through caspase 9 and 3 activation, which eventually results in cell lysis.
FIG. 5.

NS1 induces apoptosis in HEK-293 cells. (A) Cells grown in the presence (induced) or absence (noninduced) of doxycycline were harvested at the indicated times, fixed, labeled with propidium iodide, and analyzed by FACS. At least 15,000 events were analyzed for the presence of DNA fragmentation (sub-G1 DNA content) and subsequently evaluated using the CellQuest software. Values represent the percentages of cells with less than 2N DNA content (sub-G1 cell population). (B) Parental and 293-NS1 cells were grown in medium supplemented or not supplemented with doxycycline. After 72 h, cells were stained with annexin V Fluos/To-Pro-3 and subjected to flow cytometric analysis. Results from a representative experiment are shown. (C) Total protein extracts were prepared from 293-NS1 cells induced (+) and not induced (−) with doxycycline for 72 h and then subjected to SDS-PAGE and analyzed by immunoblotting using antibodies specific for caspase 9, caspase 3, PARP, NS1, and β-tubulin (used as a loading control). Bars indicate active caspases and PARP cleavage products. (D) Electron microscopy picture of an NS1-expressing cell (293-NS1 induced), showing typical features of apoptosis (chromatin condensation and nuclear fragmentation), compared with a control cell (293-NS1 noninduced).
FIG. 6.

The pan-caspase inhibitor zVAD-FMK protects NS1-expressing and H-1PV-infected cells from undergoing apoptosis. 293-NS1 cells grown in medium supplemented (induced) or not supplemented (noninduced) with doxycycline, and HEK-293 cells either mock treated or infected with H-1PV (MOI, 10 PFU/cell), were treated with the pan-caspase inhibitor zVAD-FMK (10 μM) for 72 h. (A, C) Cultures were analyzed by flow cytometry, as described in the legend to Fig. 5, for the determination of the sub-G1 cell population. Results are presented as average values with standard deviation bars from three independent experiments, each performed in triplicate. (B, D) Cell lysis of NS1-expressing and H-1PV-infected cells treated with zVAD-FMK was determined by LDH assay.
H-1PV infection induces cell cycle arrest and apoptosis in HEK-293 cells.
The results shown above prompted us to investigate whether H-1PV infection, similar to NS1 expression, also induces cell cycle arrest and apoptosis in HEK-293 cells. Cells infected with H-1PV (MOI, 10 PFU/cell) were analyzed by flow cytometry for cell cycle distribution and the presence of a sub-G1 apoptotic cell population. Time course experiments revealed that, similar to NS1-expressing cells, virus-infected 293 cells accumulated in G2/M (Table 1; see also Fig. S7 in the supplemental material). In support of the essential role of NS1 in H-1PV-induced cell cycle arrest, UV-irradiated virions that lost the ability to express this protein failed to alter cell cycle distribution (Table 1). Control experiments revealed that H-1PV infection was not associated with a strong reduction of E1A and E1B protein levels, although in this case, a slight decrease of E1B was observed (see Fig. S5 in the supplemental material).
TABLE 1.
FACS analysis of the cell cycle distribution of H-1PV-infected HEK-293 cellsa
| Type of infected cell | Subpopulation size (%) at indicated phase and time postinfection (h) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| G1 |
S |
G2 |
|||||||
| 24 | 48 | 72 | 24 | 48 | 72 | 24 | 48 | 72 | |
| Mock | 46.04 ± 0.33 | 46.52 ± 0.64 | 48.23 ± 1.74 | 33.05 ± 3.48 | 32.6 ± 2.46 | 33.58 ± 2.53 | 20.92 ± 3.46 | 20.87 ± 1.83 | 17.18 ± 0.52 |
| H-1PV | |||||||||
| ΜΟI of 1 PFU/cell | NA | NA | 34.96 ± 0.96 | NA | NA | 21.49 ± 4.46 | NA | NA | 43.54 ± 3.5 |
| ΜΟI of 10 PFU/cell | 19.5 ± 2.28 | 22.75 ± 2.04 | 19.81 ± 0.92 | 23.86 ± 5.91 | 17.59 ± 3.38 | 14.24 ± 3.94 | 56.64 ± 3.67 | 59.66 ± 2.04 | 65.73 ± 5.14 |
| UV-treated H-1PV | |||||||||
| ΜΟI of 10 PFU/cell | 49.14 ± 0.43 | 47.65 ± 1.05 | 45.94 ± 1.33 | 29.41 ± 2.15 | 31.75 ± 2.28 | 35.01 ± 0.4 | 21.45 ± 1.74 | 20.6 ± 1.54 | 19.05 ± 1.32 |
| ΜΟI of 30 PFU/cell | NA | NA | 43.1 ± 2.17 | NA | NA | 36.54 ± 2.43 | NA | NA | 20.36 ± 0.31 |
| ΜΟI of 100 PFU/cell | NA | NA | 44.6 ± 2.24 | NA | NA | 36.31 ± 0.91 | NA | NA | 19.1 ± 1.33 |
Cultures were infected at different MOI and analyzed at the indicated times postinfection. NA = data not available.
From 72 h postinfection onward, a sub-G1 cell population was detected and increased in H-1PV-infected cells in comparison with mock-treated cells, providing evidence that H-1PV infection of HEK-293 cells, as previously shown for NS1 expression, leads to apoptosis (Fig. 6C). Apoptotic death of infected cells was confirmed by annexin V staining and the detection of PARP cleavage (data not shown). Treatment with zVAD-FMK did not affect viral DNA replication or NS1 protein levels (see Fig. S6 in the supplemental material) but significantly reduced the ability of H-1PV to trigger apoptosis (Fig. 6C). As shown before for NS1, addition of zVAD-FMK into the culture medium also prevented cell lysis, indicating that also in this case, cell lysis is a secondary event which follows apoptosis (Fig. 6D).
Reactive oxygen species mediate NS1- as well as H1PV-induced apoptosis.
Reactive oxygen species (ROS) play an important role in the induction of cell death pathways and are known to be involved in the initiation of apoptosis, autophagy, and necrosis (44). This prompted us to investigate whether NS1 expression was associated with intracellular production of ROS to which NS1-induced apoptosis could be traced back. 293-NS1 cells were grown for 24 h in the presence or absence of DOX and further incubated for an additional 30 min with the ROS-sensitive dye DCFH-DA. A significant increase in intracellular levels of ROS was observed in NS1-expressing cells at 24 h (Fig. 7A). Treatment of cells with the antioxidative reagent N-acetyl-l-cysteine (NAC) (2), a precursor of the potent ROS scavenger, reduced glutathione (GSH), significantly reduced ROS levels (Fig. 7A), while it did not effect NS1 protein levels (see Fig. S8A in the supplemental material). NS1-induced apoptosis was also significantly diminished by NAC at 72 h, providing evidence that ROS play an important role in this event (Fig. 7B and C). To assess the physiological relevance of these observations, we next determined whether ROS were also mediators of H-1PV cytotoxicity. Cells were infected with H-1PV, and at 24 h postinfection, DCFH-DA was added to the culture medium. Like NS1-expressing cells, virus-infected cells showed increased levels of intracellular ROS that were counteracted by the addition of NAC (Fig. 8A). Importantly, the treatment of cells with NAC, while not displaying any effect on NS1 protein levels and viral DNA replication (see Fig. S8B in the supplemental material), significantly diminished both H-1PV-induced cell cycle arrest and apoptosis (Fig. 8B and C), highlighting the role of ROS in these events also during normal infection.
FIG. 7.

Involvement of ROS production in NS1-induced apoptosis. (A) The levels of ROS in control 293-NS1 cells (non ind., purple) or cells expressing NS1 in the presence (ind.+NAC, red) or absence (ind., green) of the antioxidant NAC (5 mM) were determined by loading the cells with DCFH-DA, followed by flow cytometric analysis at 24 h postinduction. (B) Representative FACS histograms showing the analysis of apoptotic cells by measuring the sub-G1 population after 72 h of growth under the conditions indicated. (C) FACS data obtained from three independent experiments performed in triplicate. Values shown are the means ± standard deviations.
FIG. 8.

ROS play a key role in the apoptotic pathway induced by H-1PV. Mock-treated parental 293 cells (MOCK) were compared with cells infected with H-1PV (MOI, 10 PFU/cell) in the presence (H-1PV+NAC) or absence (H-1PV) of NAC. Intracellular ROS levels (A), cell cycle distribution, and sub-G1 apoptotic cell population (B and C) were determined at 24 h and 72 h, respectively, as described in the legend to Fig. 7. Data shown in panel C are average values with standard deviation bars resulting from three independent experiments performed in triplicate.
NS1 expression and H-1PV infection are associated with induction of double-strand DNA breaks.
ROS are one of the main endogenous causes of DNA damage (47), particularly double-strand breaks (DSBs). These lesions are known to activate the DNA damage checkpoint, which blocks cells in G2 phase, preventing them from entering mitosis with a damaged genome. If the damage is too severe to be repaired, cell cycle arrest is irreversible, and cells are eliminated through apoptosis. We asked whether accumulation of ROS could induce DNA damages that would eventually lead to the observed G2 block and subsequent apoptosis in H-1PV-infected or NS1-expressing cells. This possibility was tested by measuring the appearance of γ-H2AX, which results from the phosphorylation of histone H2AX at Ser 139 and represents a well-established indicator of the formation of DSBs (22). As shown in Fig. 9A and B, a dramatic increase of γ-H2AX was detected 24 h after H-1PV inoculation, indicating the occurrence of DSBs in infected cells. NS1-expressing cells also showed a similar accumulation of γ-H2AX (Fig. 9A and B), arguing against viral DNA species as responsible for H2AX activation in H-1PV-infected cells and strongly suggesting that the occurrence of DSBs in H-1PV-infected cells is due to the expression of the nonstructural protein NS1. Importantly, NAC was efficient at preventing the induction of DSBs in both H-1PV-infected and NS1-expressing cells, providing a direct link between ROS accumulation, DNA damage, cell cycle arrest, and apoptosis induction (Fig. 9B). Altogether, these results give credit to the present NS1-inducible system by showing that NS1 alone is able to recapitulate all known steps leading to the death of H-1PV-infected cells.
FIG. 9.

H-1PV NS1 induces DNA damages via ROS production. (A) Cells were analyzed for the presence of DNA damages by immunofluorescence using an anti-γ-H2AX antibody at 24 h. NS1-expressing and H-1PV-infected cells display a specific nuclear staining pattern of γ-H2AX foci (see 100× magnification images at the top right corners). Untreated or mock-treated cells were used as a control. (B) Western blot analysis of phosphorylated H2AX levels in NS1-expressing (left) and H-1PV-infected (right) cells. NAC treatment effectively decreased the cellular levels of γ-H2AX. Actin was used as a loading control.
H-1PV cytotoxicity in HeLa cells is also mediated by ROS.
If parvoviruses are to be used as cancer therapeutic agents, it is important to understand their mechanisms of cytotoxicity in cancer cells. To address this point and validate our results in another cell line, we asked whether ROS could also be involved in the cytotoxicity of the virus against HeLa cells, a cell line derived from cervical carcinoma that was shown to be highly sensitive to H-1PV cytotoxicity both in vitro and in vivo (17, 41).
HeLa cells were infected with H-1PV (MOI, 1 PFU/cell), loaded with DCFH-DA (10 μM), and then analyzed by FACS analysis for the presence of ROS. A clear accumulation of ROS was observed in infected cells in comparison with mock-treated cells (Fig. 10A). ROS induction was reduced by treating the infected cells with the ROS scavengers GSH and NAC (Fig. 10A and data not shown). Western blot analysis of cellular extracts from these cells also clearly showed an increase in the γ-H2AX protein levels in infected cells that were counteracted by GSH (Fig. 10B).
FIG. 10.

ROS are important mediators of H-1PV cytotoxicity in HeLa cells. (A) Twenty-four hours after infection with H-1PV (MOI, 1 PFU/cell), HeLa cells were loaded with DCFH-DA (10 μM, 1 h) and analyzed by FACS for ROS production. ROS accumulation was neutralized by the addition of the ROS scavenger GSH (5 mM). (B) Twenty-four and 48 h after infection, cell lysates were prepared and analyzed by Western blotting with the indicated antibodies. (C) Cell cycle distribution (top) and percentage of sub-G1 apoptotic cell population (bottom) of mock-treated versus H-1PV-infected cells grown in the presence or absence of GSH (5 or 10 mM). A minimum of 20,000 cells were acquired and analyzed by FACS as described in Materials and Methods. Bars represent the means with relative standard deviations from three independent experiments, each performed in triplicate.
As in HEK-293, infection of HeLa cells with H1-PV leads to G2 cell cycle arrest and an increase in the sub-G1 cell population (Fig. 10C and D). Both cell cycle arrest and the percentage of the sub-G1 population were significantly reduced by the addition of GSH (Fig. 10C and D). Altogether, these results confirm that ROS are important mediators of H-1PV cytotoxicity.
Then, we investigated whether the sole expression of NS1 would be sufficient for the accumulation of ROS. To this end, we have established a stable HeLa cell line expressing NS1 in a DOX-inducible manner (see Fig. S9A and B in the supplemental material). As reported above for HEK-293, also in HeLa cells, expression of NS1 was associated with a decrease in the growth rate (see Fig. S9C), higher intracellular ROS levels (see Fig S9D), G2 accumulation, and increase in the sub-G1 population (see Fig. S9E and F). Antioxidant treatment was protecting the cells from NS1-mediated apoptosis (see Fig S9G). These results further strengthen the concept that NS1 via ROS accumulation is the major effector of H-1PV cytotoxicity.
DISCUSSION
Manipulation of the cell cycle and induction of apoptosis are common strategies adopted by many viruses in order to take advantage of the host cell and favor their multiplication and spreading. This is exemplified by the present study, which shows that the rat parvovirus H-1PV, similar to other members of the parvovirus genus, can interfere with the cell cycle machinery and induce apoptosis. Given the potential of H-1PV as an oncolytic agent for cancer therapy applications in humans (7), we decided to devise inducible cellular systems, allowing individual viral proteins to be conditionally expressed, and used these systems to dissect the sequence of events which eventually leads to cell death. Our results demonstrate that in HEK-293 and HeLa cells, H-1PV infection triggers a pathway which starts with the intracellular production of ROS and successively leads to double-strand DNA damage, arrest in G2, apoptosis, and lysis and that this whole series of changes is recapitulated upon expression of the nonstructural protein 1 (NS1) of H-1PV, in the absence of any other viral products.
In HEK-293 cells, the first consequence of NS1 expression is a temporary increase in the fraction of cells in S phase, followed by their accumulation in G2 phase. It should, however, be stated that a minor fraction of cells appears to escape this G2 block, as NS1-expressing cultures continue to grow for a few days, although at a significantly reduced rate in comparison with that of noninduced cells, before becoming fully arrested and starting to degenerate (Fig. 1D). A possible explanation for this transient escape is that a threshold level of NS1 is required to induce cell cycle arrest, and cells expressing low levels of the protein may still be able to proliferate until they accumulate critical amounts of NS1. In keeping with this possibility, our immunofluorescence analysis shows variability in the NS1 levels within the induced cell population. In addition, growth inhibition is directly proportional to the amount of doxycycline used for the induction (see Fig. S3 in the supplemental material). Given the correlation between the concentration of doxycycline and the protein levels of NS1 (Fig. 1B), these results further support the hypothesis that growth inhibition is related to the amount of NS1 expressed in the cell.
NS1-induced cell cycle disturbances are associated with alterations in the expression of several cell cycle regulators (Fig. 3), including upregulation of the cyclin kinase inhibitors (CKIs) p21 and p27. Upregulation of p21 by other parvoviruses, namely, MVM (38), B19 (32), and AAV-2 (40, 43), has been reported previously. In B19 infections, NS1, in cooperation with the cellular transcription factor SP1, is able to bind and activate the p21 promoter (32). Whether p21 is also a direct transcriptional target of the H-1PV NS1 protein remains to be determined. We also found a time-dependent accumulation of cyclin A, cdc2, and cyclin B1, which are known to play a major role in S and G2 phases. These data are consistent with the G2 cell cycle arrest observed in NS1-induced cells. Interestingly, we also found that the protein levels of p130 were significantly enhanced together with a milder increase in the protein levels of the other member of the pocket protein family, p107. It is known that these two proteins are activated following double-strand DNA damages and play a key role in maintaining G2 arrest by binding to members of the E2F family of transcription factors and thereby repressing the transcription of genes needed for mitosis (39). Their upregulation, therefore, provides the first hints of the activation of the DNA damage checkpoint pathway in NS1-expressing cells. The cytostatic and cytotoxic activities of the NS1 protein were also confirmed in HeLa cells.
Like NS1-expressing cultures, H-1PV-infected HEK-293 cells are arrested in G2 phase (Table 1; see also Fig. S7 in the supplemental material). Given that parvoviruses require proliferating cells to replicate and express their genome, the fact that H-1PV induces cell cycle arrest (mostly a G2 block) may come as a surprise. While the onset of parvovirus DNA replication and expression may be S dependent, the replication phase of the viral life cycle may keep going on at later stages. In particular, G2 cells may retain some of the features of the S phase (pseudo-S phase) (13), which may perpetuate intracellular conditions favorable to parvovirus replication. In support of this hypothesis, we found that, in HEK-293 cells, the protein levels of cyclin A, an important factor required for parvovirus replication (3, 4), are higher in both H-1PV-infected and NS1-induced cells arrested in G2 than those in control cells (Fig. 3 and data not shown). It is worth noting that other Parvovirus members, such as B19, MVM, and AAV-2, also arrest the cell cycle in G2 (30, 38, 48). The mechanisms involved, however, may be different since the NS1 proteins encoded by these viruses do not show the H-1PV NS1 capacity for specifically blocking cells in G2. Indeed, NS1 of B19 induces a G1 block (28), Rep 78 of AAV-2 arrests the cell cycle at both G1 (45) and S (43) phases, and MVM NS1 appears to affect G1, S, and G2 phases (36-38). Therefore, the contribution of these nonstructural proteins to the G2 cell cycle arrest induced by the full virus remains to be clarified. Other parvoviral components have also been reported to contribute to cell cycle arrest. The AAV-2-induced G2 cell cycle arrest is not mediated by any viral proteins but depends instead on the conformation of the AAV single-strand genome, which contains hairpin structures at both ends. These structures elicit a DNA damage response which activates the G2 checkpoint pathway and triggers cell cycle arrest (40). Similarly, in B19, NS1 is unlikely to be involved in the G2 arrest, since UV-irradiated viruses, which are unable to express NS proteins, retain the capacity to induce this block (29). Furthermore, the inhibition of ATM and ATR kinases (two key regulators of the G2/M DNA damage checkpoint) with caffeine prevents B19 from causing the G2 arrest, suggesting that, like in the case of AAV, the DNA damage response is activated by the B19 genome and is instrumental in blocking cells in G2 (29). Caffeine also inhibits B19 DNA replication, highlighting the importance of the G2 cell cycle arrest for the life cycle of this virus (29). At present, we cannot fully rule out that, besides NS1, other H-1PV components may contribute to the antiproliferative activity of the full virus. However, the primary role of NS1 in the cytostatic activity of H-1PV is supported by our observations, as follows: (i) UV-irradiated H-1PV virions unable to produce NS1 fail to induce cell cycle arrest (Table 1); (ii) ectopic expression of the VP capsid proteins does not alter cell proliferation (data not shown); and (iii) NS1 expression induces a G2 cell cycle arrest similar to the one induced by the virus (Fig. 2 and Table 1; see also Fig. S7 in the supplemental material).
Our results show that after H-1PV infection, HEK-293 and HeLa cells die through apoptosis. Similarly, apoptosis is induced after prolonged expression of NS1 (from 48 h on) in these cells, further substantiating the role of NS1 as the major cytotoxic effector of H-1PV. It appears from LDH assays that NS1-expressing cells eventually become lysed, which may favor virus release and spreading. Cell lysis is an outcome of apoptosis under the in vitro conditions tested, which is apparent from the fact that cell lysis was effectively suppressed by inhibitors of apoptosis.
The G2 cell cycle arrest is often associated with DNA lesions which activate the G2 DNA damage checkpoint. If the damage is too severe to be repaired, cells undergo apoptosis, thereby preventing cells with altered genomes from entering mitosis (23). Both virus-infected and NS1-expressing cells show a dramatic increase in histone H2AX phosphorylation on serine 139 (γ-H2AX), an event that specifically occurs in response to DNA double-strand breaks (22). This leads us to hypothesize that NS1-induced DNA damage may play a key role in triggering the G2 arrest and activating apoptotic pathway. In agreement with this idea, MVM NS1 expression was reported to be associated with the formation of nicks in the cell chromatin (37). The present study clarifies the molecular mechanisms that underlie NS1-dependent DNA damage. Op De Beeck and Caillet-Fauquet proposed that NS1 could directly nick cellular DNA through its endonuclease activity, although direct experimental evidence of this hypothesis has not been provided (37). Our results indicate that NS1 may instead act indirectly to damage cellular DNA. Both H-1PV-infected and NS1-expressing cells accumulate high intracellular levels of ROS, suggesting that these cells are subjected to oxidative stress. ROS are well-established DNA-damaging agents and activators of cell death pathways (44, 47). In support of the role of ROS in NS1-induced DNA damage and cytotoxicity, the antioxidant treatment of NAC or GSH proved able to protect cells from H-1PV- and NS1-induced phosphorylation of histone H2AX and to decrease the fraction of cells undergoing G2/M cell cycle arrest and apoptosis. It is also important to point out that, according to our data, ROS production is an early event in the cytotoxic pathways activated by the virus, which occurs before any evident features of apoptosis are detectable. Other ROS could be also generated as a result of activation of the apoptotic pathway, e.g., as a consequence of mitochondrial dysfunction, reinforcing the cytotoxic activities of the virus. However, as the use of antioxidants at the concentrations used in our experiments did not completely protect the cells from apoptosis, it appears as though other still uncharacterized mechanisms, acting in parallel with ROS, could also account for the NS1- and virus-associated cell death processes.
Altogether, these results lead us to propose the model depicted in Fig. 11, placing H-1PV-induced cell lysis at the end of a pathway triggered by NS1-dependent ROS induction and comprising a G2 block and caspase-dependent apoptosis.
FIG. 11.
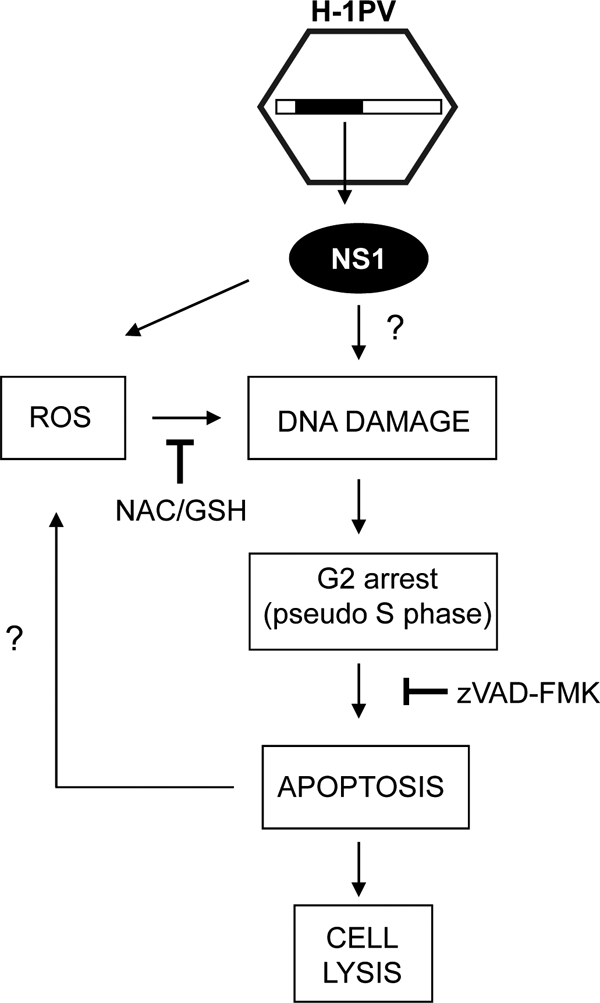
Tentative model of H-1PV cytotoxicity. After parvovirus infection, expression of NS1 triggers an oxidative stress with accumulation of intracellular ROS associated with DNA damage. As a consequence of DNA damage, cells accumulate in G2 phase and then undergo apoptosis and ultimately lysis. NS1 may also induce DNA damage by directly nicking DNA, thereby contributing to the cell cycle arrest. Furthermore, ROS may also be produced during the execution of the apoptotic pathway, reinforcing the cytotoxic activity of the virus.
Other viruses have also been reported to induce oxidative stress in infected cells, including HIV-1 (31), hepatitis C virus (24), Epstein-Barr virus (19), and herpes simplex virus type 1 (21). Viral factors play an active role in the induction of ROS production. While the Vpr protein of HIV induces ROS by increasing H2O2 production (15), the hepatitis C virus core protein and the Epstein-Barr virus nuclear antigen 1 (EBNA-1) stimulate ROS formation in infected cells through the functional inhibition of Hsp60 (20) and the transcriptional activation of NOX2, an enzyme involved in ROS metabolism (19), respectively. We provide evidence that H-1PV induces ROS accumulation as a result of NS1 expression, as the sole expression of this protein is sufficient to trigger oxidative stress. It will be interesting to investigate whether NS1, by means of its transcriptional activity, is able to stimulate the expression of genes involved in ROS production or in the regulation of ROS homeostasis. It could also be possible that the viral protein through physical interaction might deregulate enzymes involved in ROS metabolism. How NS1 induces ROS accumulation and affects in this way the efficiency of productive infection of parvoviruses, and which domains of NS1 are required for this function, are open questions that will be addressed in future studies. The answers to these will help to optimize oncolytic parvovirus vectors for clinical applications for the treatment of various malignancies.
Supplementary Material
Acknowledgments
We thank Barbara Leuchs for H-1PV production, Tiina Marttila for technical assistance, Nathalie Salomé and Michele Vogel for the 3D9 anti-NS1 and SP6 anti-NS2 antibodies, Thomas Dobner for the E1B antibody, and Sandra Caldeira for critical reading of the manuscript.
N.E.-A., J.L., and R.M. were supported by EMBO, INSERM, and Deutscher Akademischer Austausch Dienst (DAAD) fellowships, respectively. This work was partly supported by a grant from the Helmholtz-Gemeinschaft in the frame of the Deutsches Krebsforschungszentrum/Cancéropôle du Grand-Est joint program in Applied Tumor Virology and by an INCa (Institut Nationale du Cancer, France) grant.
Footnotes
Published ahead of print on 7 April 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Armstrong, J. S., and M. Whiteman. 2007. Measurement of reactive oxygen species in cells and mitochondria. Methods Cell Biol. 80:355-377. [DOI] [PubMed] [Google Scholar]
- 2.Atkuri, K. R., J. J. Mantovani, and L. A. Herzenberg. 2007. N-acetylcysteine—a safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharmacol. 7:355-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bashir, T., R. Horlein, J. Rommelaere, and K. Willwand. 2000. Cyclin A activates the DNA polymerase delta-dependent elongation machinery in vitro: a parvovirus DNA replication model. Proc. Natl. Acad. Sci. U. S. A. 97:5522-5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bashir, T., J. Rommelaere, and C. Cziepluch. 2001. In vivo accumulation of cyclin A and cellular replication factors in autonomous parvovirus minute virus of mice-associated replication bodies. J. Virol. 75:4394-4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodendorf, U., C. Cziepluch, J. C. Jauniaux, J. Rommelaere, and N. Salome. 1999. Nuclear export factor CRM1 interacts with nonstructural proteins NS2 from parvovirus minute virus of mice. J. Virol. 73:7769-7779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caillet-Fauquet, P., M. Perros, A. Brandenburger, P. Spegelaere, and J. Rommelaere. 1990. Programmed killing of human cells by means of an inducible clone of parvoviral genes encoding non-structural proteins. EMBO J. 9:2989-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cornelis, J. J., L. Deleu, U. Kock, and J. Rommelaere. 2006. Parvovirus oncosuppression, p. 365-378. In J. R. Kerr, M. E. Bloom, R. M. Linden, and C. R. Parrish (ed.), Parvoviruses. Hodder Arnold E. Ltd., London, United Kingdom.
- 8.Cotmore, S. F., and P. Tattersall. 1986. Organization of nonstructural genes of the autonomous parvovirus minute virus of mice. J. Virol. 58:724-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cotmore, S. F., and P. Tattersall. 2007. Parvoviral host range and cell entry mechanisms. Adv. Virus Res. 70:183-232. [DOI] [PubMed] [Google Scholar]
- 10.Cziepluch, C., E. Kordes, R. Poirey, A. Grewenig, J. Rommelaere, and J. C. Jauniaux. 1998. Identification of a novel cellular TPR-containing protein, SGT, that interacts with the nonstructural protein NS1 of parvovirus H-1. J. Virol. 72:4149-4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cziepluch, C., S. Lampel, A. Grewenig, C. Grund, P. Lichter, and J. Rommelaere. 2000. H-1 parvovirus-associated replication bodies: a distinct virus-induced nuclear structure. J. Virol. 74:4807-4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daeffler, L., R. Horlein, J. Rommelaere, and J. P. Nuesch. 2003. Modulation of minute virus of mice cytotoxic activities through site-directed mutagenesis within the NS coding region. J. Virol. 77:12466-12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davy, C., and J. Doorbar. 2007. G2/M cell cycle arrest in the life cycle of viruses. Virology 368:219-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deleu, L., F. Fuks, D. Spitkovsky, R. Horlein, S. Faisst, and J. Rommelaere. 1998. Opposite transcriptional effects of cyclic AMP-responsive elements in confluent or p27KIP-overexpressing cells versus serum-starved or growing cells. Mol. Cell. Biol. 18:409-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deshmane, S. L., R. Mukerjee, S. Fan, L. Del Valle, C. Michiels, T. Sweet, I. Rom, K. Khalili, J. Rappaport, S. Amini, and B. E. Sawaya. 2009. Activation of the oxidative stress pathway by HIV-1 Vpr leads to induction of hypoxia-inducible factor 1alpha expression. J. Biol. Chem. 284:11364-11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Piazza, M., C. Mader, K. Geletneky, Y. C. M. Herrero, E. Weber, J. Schlehofer, L. Deleu, and J. Rommelaere. 2007. Cytosolic activation of cathepsins mediates parvovirus H-1-induced killing of cisplatin and TRAIL-resistant glioma cells. J. Virol. 81:4186-4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faisst, S., D. Guittard, A. Benner, J. Y. Cesbron, J. R. Schlehofer, J. Rommelaere, and T. Dupressoir. 1998. Dose-dependent regression of HeLa cell-derived tumours in SCID mice after parvovirus H-1 infection. Int. J. Cancer 75:584-589. [DOI] [PubMed] [Google Scholar]
- 18.Genovese, C., D. Trani, M. Caputi, and P. P. Claudio. 2006. Cell cycle control and beyond: emerging roles for the retinoblastoma gene family. Oncogene 25:5201-5209. [DOI] [PubMed] [Google Scholar]
- 19.Gruhne, B., R. Sompallae, D. Marescotti, S. A. Kamranvar, S. Gastaldello, and M. G. Masucci. 2009. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. U. S. A. 106:2313-2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang, S. M., S. J. Kim, J. H. Kim, W. Lee, G. W. Kim, K. H. Lee, K. Y. Choi, and J. W. Oh. 2009. Interaction of hepatitis C virus core protein with Hsp60 triggers the production of reactive oxygen species and enhances TNF-alpha-mediated apoptosis. Cancer Lett. 279:230-237. [DOI] [PubMed] [Google Scholar]
- 21.Kavouras, J. H., E. Prandovszky, K. Valyi-Nagy, S. K. Kovacs, V. Tiwari, M. Kovacs, D. Shukla, and T. Valyi-Nagy. 2007. Herpes simplex virus type 1 infection induces oxidative stress and the release of bioactive lipid peroxidation by-products in mouse P19N neural cell cultures. J. Neurovirol. 13:416-425. [DOI] [PubMed] [Google Scholar]
- 22.Kinner, A., W. Wu, C. Staudt, and G. Iliakis. 2008. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 36:5678-5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lobrich, M., and P. A. Jeggo. 2007. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat. Rev. Cancer 7:861-869. [DOI] [PubMed] [Google Scholar]
- 24.Machida, K., K. T. Cheng, C. K. Lai, K. S. Jeng, V. M. Sung, and M. M. Lai. 2006. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J. Virol. 80:7199-7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathew, R., C. M. Karp, B. Beaudoin, N. Vuong, G. Chen, H. Y. Chen, K. Bray, A. Reddy, G. Bhanot, C. Gelinas, R. S. Dipaola, V. Karantza-Wadsworth, and E. White. 2009. Autophagy suppresses tumorigenesis through elimination of p62. Cell 137:1062-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meister, G., M. Landthaler, L. Peters, P. Y. Chen, H. Urlaub, R. Luhrmann, and T. Tuschl. 2005. Identification of novel argonaute-associated proteins. Curr. Biol. 15:2149-2155. [DOI] [PubMed] [Google Scholar]
- 27.Moffatt, S., N. Yaegashi, K. Tada, N. Tanaka, and K. Sugamura. 1998. Human parvovirus B19 nonstructural (NS1) protein induces apoptosis in erythroid lineage cells. J. Virol. 72:3018-3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morita, E., A. Nakashima, H. Asao, H. Sato, and K. Sugamura. 2003. Human parvovirus B19 nonstructural protein (NS1) induces cell cycle arrest at G1 phase. J. Virol. 77:2915-2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morita, E., and K. Sugamura. 2002. Human parvovirus B19-induced cell cycle arrest and apoptosis. Springer Semin. Immunopathol. 24:187-199. [DOI] [PubMed] [Google Scholar]
- 30.Morita, E., K. Tada, H. Chisaka, H. Asao, H. Sato, N. Yaegashi, and K. Sugamura. 2001. Human parvovirus B19 induces cell cycle arrest at G(2) phase with accumulation of mitotic cyclins. J. Virol. 75:7555-7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura, H., H. Masutani, and J. Yodoi. 2002. Redox imbalance and its control in HIV infection. Antioxid. Redox Signal. 4:455-464. [DOI] [PubMed] [Google Scholar]
- 32.Nakashima, A., E. Morita, S. Saito, and K. Sugamura. 2004. Human Parvovirus B19 nonstructural protein transactivates the p21/WAF1 through Sp1. Virology 329:493-504. [DOI] [PubMed] [Google Scholar]
- 33.Nuesch, J. P., S. Lachmann, and J. Rommelaere. 2005. Selective alterations of the host cell architecture upon infection with parvovirus minute virus of mice. Virology 331:159-174. [DOI] [PubMed] [Google Scholar]
- 34.Nuesch, J. P., and J. Rommelaere. 2007. A viral adaptor protein modulating casein kinase II activity induces cytopathic effects in permissive cells. Proc. Natl. Acad. Sci. U. S. A. 104:12482-12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohshima, T., M. Iwama, Y. Ueno, F. Sugiyama, T. Nakajima, A. Fukamizu, and K. Yagami. 1998. Induction of apoptosis in vitro and in vivo by H-1 parvovirus infection. J. Gen. Virol. 79(Pt. 12):3067-3071. [DOI] [PubMed] [Google Scholar]
- 36.Op De Beeck, A., F. Anouja, S. Mousset, J. Rommelaere, and P. Caillet-Fauquet. 1995. The nonstructural proteins of the autonomous parvovirus minute virus of mice interfere with the cell cycle, inducing accumulation in G2. Cell Growth Differ. 6:781-787. [PubMed] [Google Scholar]
- 37.Op De Beeck, A., and P. Caillet-Fauquet. 1997. The NS1 protein of the autonomous parvovirus minute virus of mice blocks cellular DNA replication: a consequence of lesions to the chromatin? J. Virol. 71:5323-5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Op De Beeck, A., J. Sobczak-Thepot, H. Sirma, F. Bourgain, C. Brechot, and P. Caillet-Fauquet. 2001. NS1- and minute virus of mice-induced cell cycle arrest: involvement of p53 and p21(cip1). J. Virol. 75:11071-11078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plesca, D., M. E. Crosby, D. Gupta, and A. Almasan. 2007. E2F4 function in G2: maintaining G2-arrest to prevent mitotic entry with damaged DNA. Cell Cycle 6:1147-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raj, K., P. Ogston, and P. Beard. 2001. Virus-mediated killing of cells that lack p53 activity. Nature 412:914-917. [DOI] [PubMed] [Google Scholar]
- 41.Ran, Z., B. Rayet, J. Rommelaere, and S. Faisst. 1999. Parvovirus H-1-induced cell death: influence of intracellular NAD consumption on the regulation of necrosis and apoptosis. Virus Res. 65:161-174. [DOI] [PubMed] [Google Scholar]
- 42.Rayet, B., J. A. Lopez-Guerrero, J. Rommelaere, and C. Dinsart. 1998. Induction of programmed cell death by parvovirus H-1 in U937 cells: connection with the tumor necrosis factor alpha signaling pathway. J. Virol. 72:8893-8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saudan, P., J. Vlach, and P. Beard. 2000. Inhibition of S-phase progression by adeno-associated virus Rep78 protein is mediated by hypophosphorylated pRb. EMBO J. 19:4351-4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scherz-Shouval, R., and Z. Elazar. 2007. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 17:422-427. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt, M., S. Afione, and R. M. Kotin. 2000. Adeno-associated virus type 2 Rep78 induces apoptosis through caspase activation independently of p53. J. Virol. 74:9441-9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ueno, Y., T. Harada, H. Iseki, T. Ohshima, F. Sugiyama, and K. Yagami. 2001. Propagation of rat parvovirus in thymic lymphoma cell line C58(NT)d and subsequent appearance of a resistant cell clone after lytic infection. J. Virol. 75:3965-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang, Y. 2008. Bulky DNA lesions induced by reactive oxygen species. Chem. Res. Toxicol. 21:276-281. [DOI] [PubMed] [Google Scholar]
- 48.Winocour, E., M. F. Callaham, and E. Huberman. 1988. Perturbation of the cell cycle by adeno-associated virus. Virology 167:393-399. [PubMed] [Google Scholar]
- 49.Wrzesinski, C., L. Tesfay, N. Salome, J. C. Jauniaux, J. Rommelaere, J. Cornelis, and C. Dinsart. 2003. Chimeric and pseudotyped parvoviruses minimize the contamination of recombinant stocks with replication-competent viruses and identify a DNA sequence that restricts parvovirus H-1 in mouse cells. J. Virol. 77:3851-3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.