

Abstract
Receptor protein tyrosine phosphatase α (RPTPα) is the mitotic activator of the protein tyrosine kinase Src. RPTPα serine hyperphosphorylation was proposed to mediate mitotic activation of Src. We raised phosphospecific antibodies to the two main serine phosphorylation sites, and we discovered that RPTPα Ser204 was almost completely dephosphorylated in mitotic NIH 3T3 and HeLa cells, whereas Ser180 and Tyr789 phosphorylation were only marginally reduced in mitosis. Concomitantly, Src pTyr527 and pTyr416 were dephosphorylated, resulting in 2.3-fold activation of Src in mitosis. Using inhibitors and knockdown experiments, we demonstrated that dephosphorylation of RPTPα pSer204 in mitosis was mediated by PP2A. Mutation of Ser204 to Ala did not activate RPTPα, and intrinsic catalytic activity of RPTPα was not affected in mitosis. Interestingly, binding of endogenous Src to RPTPα was induced in mitosis. GRB2 binding to RPTPα, which was proposed to compete with Src binding to RPTPα, was only modestly reduced in mitosis, which could not account for enhanced Src binding. Moreover, we demonstrate that Src bound to mutant RPTPα-Y789F, lacking the GRB2 binding site, and mutant Src with an impaired Src homology 2 (SH2) domain bound to RPTPα, illustrating that Src binding to RPTPα is not mediated by a pTyr-SH2 interaction. Mutation of RPTPα Ser204 to Asp, mimicking phosphorylation, reduced coimmunoprecipitation with Src, suggesting that phosphorylation of Ser204 prohibits binding to Src. Based on our results, we propose a new model for mitotic activation of Src in which PP2A-mediated dephosphorylation of RPTPα pSer204 facilitates Src binding, leading to RPTPα-mediated dephosphorylation of Src pTyr527 and pTyr416 and hence modest activation of Src.
Protein tyrosine phosphatases (PTPs) are responsible for dephosphorylation of the phosphotyrosyl residues. The human genome contains approximately 100 genes that encode members of the four PTP families, and most of them have mouse orthologues (2, 48). According to their subcellular localization, the classical PTPs, encoded by less than half of the total PTP genes, are divided into two subfamilies: cytoplasmic and receptor protein tyrosine phosphatases (RPTPs). The majority of the RPTPs contain, besides a variable extracellular domain and a transmembrane domain, two highly homologous phosphatase domains (27), with the membrane-proximal domain comprising most of the catalytic activity (33).
RPTPα is a typical RPTP with a small, highly glycosylated extracellular domain (13). RPTPα function is regulated by many mechanisms, including proteolysis (18), oxidation (55), dimerization (7, 23, 24, 47, 52), and phosphorylation of serine and tyrosine residues (16, 17, 49). RPTPα is broadly expressed in many cell types, and over the years, RPTPα has been shown to be involved in a number of signaling mechanisms, including neuronal (15) and skeletal muscle (34) cell differentiation, neurite elongation (8, 9, 56), insulin receptor signaling downregulation (3, 28, 30, 31, 35), insulin secretion (25), activation of voltage-gated potassium channel Kv1.2 (51), long-term potentiation in hippocampal neurons (32, 38), matrix-dependent force transduction (53), and cell spreading and migration (21, 45, 57).
The majority of the roles played in these cellular processes involve RPTPα's ability to activate the proto-oncogenes Src and Fyn by dephosphorylating their C-terminal inhibitory phosphotyrosine (5, 15, 39, 45, 61). Normally, this phosphotyrosine (pTyr527 in chicken Src) binds to the Src homology 2 (SH2) domain, keeping the protein in an inactive closed conformation. A displacement mechanism was proposed for RPTPα-mediated Src activation in which pTyr789 of RPTPα is required to bind the SH2 domain of Src before RPTPα dephosphorylates Tyr527 (58). This model is the subject of debate since other studies show that RPTPα lacking Tyr789 is still able to dephosphorylate and activate Src (12, 26, 29, 56). In normal cells, Src reaches its activation peak during mitosis (4, 11, 40, 42), and with the help of overexpressing cells, it was shown that this activation is triggered mainly by RPTPα. The model that emerged is that RPTPα is activated in mitosis due to serine hyperphosphorylation and detaches from the GRB2 scaffolding protein (59, 60) that normally binds most of the pTyr789 of RPTPα via its SH2 domain (14, 17, 46). Two serine phosphorylation sites were mapped in the juxtamembrane domain of RPTPα, Ser180 and Ser204 (49). The kinases that were found responsible for their phosphorylation were protein kinase C delta (PKCdelta) (10) and CaMKIIalpha (9), but there is no clear evidence that these kinases are activated in mitosis. We set out to investigate the role of serine phosphorylation of RPTPα in mitotic activation of Src.
We generated phosphospecific antibodies and show that RPTPα pSer204, but not pSer180, is dephosphorylated in mitotic NIH 3T3 and HeLa cells, concomitantly with activation of Src. Selective inhibitors suggested that PP2A was the phosphatase that dephosphorylated pSer204. RNA interference (RNAi)-mediated knockdown of the catalytic subunit of PP2A demonstrated that indeed PP2A was responsible for mitotic dephosphorylation of RPTPα pSer204. It is noteworthy that PP2A is known to be activated in mitosis. Intrinsic PTP activities of RPTPα were similar in unsynchronized and mitotic cells, and mutation of Ser204 did not activate RPTPα in in vitro PTP assays. Yet, Src binding to RPTPα was induced in mitotic NIH 3T3 cells and RPTPα-S204D with a phosphomimicking mutation at Ser204 coimmunoprecipitated less efficiently with Src. Based on our results, we propose a mechanism for mitotic activation of Src that is triggered by dephosphorylation of RPTPα pSer204, resulting in enhanced affinity for Src and subsequent dephosphorylation and activation of Src.
MATERIALS AND METHODS
Materials and antibodies.
12CA5 antihemagglutinin (anti-HA) tag and 327 anti-Src and 5478AP anti-RPTPα antibodies were prepared as previously described (15, 17). The anti-RPTPα 1951AP antibody was obtained following the same procedure as that for the 5478AP antibody. Briefly, rabbits were immunized with the bacterially expressed cytoplasmic domain of RPTPα fused with glutathione S-transferase (GST). The polyclonal antiserum was first cleared of anti-GST antibodies by using a GST affinity column, and then the anti-RPTPα antibodies were purified using a second affinity column loaded with GST-PTPα protein. Anti-RPTPα-pY789 and anti-Src-npY527 were from Cell Signaling. Anti-Src-pY418 was from Biosource, and anti-rabbit and anti-mouse secondary antibodies were from BD Biosciences. Polyethylenimine (PEI), nocodazole, paclitaxel, and glutathione-Sepharose were from Sigma Life Science. Okadaic acid, calyculin A, and tautomycin were from Calbiochem. Total protein concentration in the lysates was detected using a bicinchoninic acid (BCA) kit (Sigma).
DNA constructs.
The constructs used for the expression of HA-RPTPα wild type (WT) (15) and HA-RPTPα Y789F, Src WT, and Src Y527F were previously described (17). HA-RPTPα S180A, HA-RPTPα S204A, and HA-RPTPα S180A/S204A were generated by PCR-mediated site-directed mutagenesis using HA-RPTPα WT as template and the following oligonucleotides: 5′-AGTCATTCCAACGCTTTCCGCCTGTCA-3′ for S180A and 5′-GCCAGGTCCCCAGCCACCAACAGGAAG-3′ for S204A. Src R175L was generated using the oligonucleotide 5′-GACCTTCCTCGTGCTGGAGAGTGAGACC-3′ and PCR. The constructs were verified by sequencing.
Phospho-specific antibodies.
Anti-phospho-Ser180 and anti-phospho-Ser204 antibodies were raised against two synthetic peptides corresponding to the known sequences containing the serine phosphorylation sites in RPTPα. Extra N-terminal cysteines were added to the peptides to allow coupling of the peptides to the carrier (in this case keyhole limpet hemocyanin). Anti-phospho-Ser180 antibody was raised against CSNpSFRLSNG peptide, and anti-phospho-Ser204 antibody was raised against CSPpSTNRKYP. The production of the peptides, the immunization of the rabbits, and the affinity purification of the antibodies were carried out by Eurogentec.
Cell culture, mitotic arrest, and FACS analysis.
For the experiments described in this study, we used NIH 3T3, HEK293, COS1, and SYF cells. HEK293 and COS1 cells were grown in DF medium supplemented with 7.5% fetal calf serum (FCS). NIH 3T3 cells were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 7.5% newborn calf serum (NCS). SYF cells were grown in DMEM supplemented with 7.5% FCS. The mitotic arrest of NIH 3T3 cells was achieved after nocodazole treatment. NIH 3T3 cells (70 to 80% confluent) were treated with 0.6 μg/ml nocodazole and grown for another 12 to 14 h. Finally the mitotic cells were harvested by rinsing the monolayer repeatedly with a stream of medium or by mitotic shake-off. The collected cells were centrifuged and washed twice with phosphate-buffered saline (PBS) solution. Part of the cells was replated and harvested at different time points. The remaining cells were either used for fluorescence-activated cell sorting (FACS) analysis or lysed. The FACS analysis was performed on a Becton Dickinson FACSCalibur cell sorter. The cell cycle distribution was determined by quantifying the amount of propidium iodide (PI) incorporated in the nuclei. The cells were fixed in 70% ice-cold ethanol and kept at 4°C for at least 12 h. After fixation, PBS solution was added to the tubes and the cells were centrifuged. Afterwards the cells were washed once more with PBS solution and centrifuged. The supernatant was removed, and the cells were resuspended in a small volume of PBS solution containing 250 μg/ml RNase and 1 μg/ml PI. The cells were incubated for 15 min at 37°C and then kept at 4°C until the analysis was performed.
SYF cells were transfected with FuGene6 (Roche) according to the protocol provided by the manufacturer. HEK293 and COS1 cells were transiently transfected with empty vector or HA-RPTPα using polyethylenimine (PEI). After transfection, the cells were grown for 16 h in serum-containing medium and then the medium was exchanged with serum-free medium and the cells were grown for an additional 24 h. The cells were lysed for 20 min on ice in cell lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EGTA, 1.5 mM MgCl2, 1% Triton X-100, 10% glycerol, 5 mM NaF, 5 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, and 1 μg/ml aprotinin). The lysates were collected using a rubber policeman and centrifuged for 10 min at 13,000 rpm. Samples from the lysates were collected and boiled after being mixed with equal volumes of 2× SDS sample buffer (125 mM Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 2% β-mercaptoethanol, and 0.04% bromophenol blue) and resolved on 7.5% SDS-PAGE gels.
NIH 3T3 cells were transfected with small interfering RNAs (siRNAs) against PP2Acα or control siRNAs with Dharmafect according to the manufacturer's protocols (Dharmacon). After 24 h, the medium was replaced with regular growth medium and the cells were grown for an additional 24 h. Subsequently, the cells were treated with nocodazole (600 ng/ml) for 12 h and harvested by mitotic shake-off (mitotic cells) or they were harvested directly (unsynchronized cells).
Immunoprecipitation and immunoblotting.
Nocodazole-treated or untreated NIH 3T3 cells were lysed for 20 min on ice in RIPA buffer containing 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10 mM Na2HPO4, 5 mM EDTA, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 10% glycerol, 5 mM NaF, 5 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, and 1 μg/ml aprotinin. For immunoprecipitation of RPTPα, the lysates were first incubated with the anti-RPTPα antibody (5478AP) for 1 h at 4°C and then with protein A Sepharose for 1 h. For Src immunoprecipitation, the lysates were incubated for 1 h at 4°C with anti-Src monoclonal antibody 327 cross-linked to protein A Sepharose. The immunoprecipitates were extensively washed with RIPA buffer. Each immunoprecipitate was divided into two equal fractions of which one was immunoblotted using anti-Src antibody and the other was subjected to kinase assay. The fraction used for kinase assay was washed once in kinase buffer before the assay.
Kinase assays.
Src kinase assays were performed in 40 μl kinase reaction buffer (50 mM HEPES, pH 7.5, and 10 mM MgCl2), containing 10 μCi [γ-32P]ATP and 3.5 μg acid-denatured enolase. Reaction mixtures were incubated at 30°C for 30 min, and reactions were stopped by the addition of 2× SDS sample buffer and resolved by 7.5% SDS-PAGE. Results were visualized by autoradiography. Similarly Src Y527F was used to phosphorylate myelin basic protein (MBP) for use as substrate in RPTPα phosphatase assays.
Phosphatase assays.
The phosphatase assays were performed following the instructions for the protein tyrosine phosphatase assay system from New England Biolabs. Endogenous RPTPα from NIH 3T3 cells and overexpressed RPTPα from transfected COS1 cells were immunoprecipitated as mentioned above. After washing, the beads were incubated with the lysates of unsynchronized or mitotic NIH 3T3 cells in cell lysis buffer for 1 h at 30°C. The beads were washed four times with HNTG buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 0.1% Triton X-100) and boiled in 2× SDS sample buffer.
The variations in activity of RPTPα and mutants were determined in vitro using phosphorylated MBP or Src as substrate. Overexpressed RPTPα was immunoprecipitated from transfected COS1 cells as previously described. The beads were washed three times with HNTG buffer and one time with phosphatase buffer (20 mM morpholineethanesulfonic acid [MES], pH 6, 150 mM NaCl, 2.5 mM dithiothreitol [DTT], 1 mM EDTA, and 1 mg/ml bovine serum albumin [BSA]). Half of the immunoprecipitated RPTPα was used for the phosphatase assay, and the other half was used for monitoring the amount of RPTPα in the samples. The reactions were performed in a total volume of 40 μl containing immunoprecipitated RPTPα, phosphatase buffer, and 3 μg phosphorylated MBP. The mixtures were incubated for 15 min at 30°C, and the reaction was stopped by adding 200 μl of 20% trichloroacetic acid (TCA) to each tube. Time courses were determined with wild-type RPTPα up to 1 h, and the PTP reaction was linear until 20 min (data not shown). The tubes were kept on ice for 5 min and then centrifuged for 10 min at 12,000 × g and 4°C. Two hundred microliters from each tube was added to vials containing scintillation fluid, and the samples were measured in a scintillation counter. The activity of endogenous RPTPα from unsynchronized and mitotic NIH 3T3 cells was detected similarly. The only difference was that after immunoprecipitation of RPTPα the beads were washed three times with RIPA buffer prior to phosphatase buffer washing.
When Src was used as substrate, RPTPα was immunoprecipitated from transfected COS1 cells as described above and the beads were incubated for 15 min at 30°C with beads containing Src immunoprecipitated from transfected SYF cells. The reactions were conducted in a buffer containing 50 mM HEPES, pH 7.4, 150 mM NaCl, 2 mM DTT, and 1 mM EDTA. The phosphorylation levels of Src Tyr416 and Tyr527 were used as readout for RPTPα activity.
RESULTS
Specificity of anti-pSer180 and anti-pSer204 antibodies.
To study RPTPα phosphorylation in mitosis, we generated antibodies against phosphorylated peptides derived from the mouse RPTPα protein sequence corresponding to Ser180 and Ser204. To test the specificity of these antibodies, HEK293 cells were transfected with vectors coding for HA-tagged wild-type RPTPα and mutants containing Ser-to-Ala mutations. Both antibodies recognized wild-type RPTPα, and mutations of individual phosphorylation sites of RPTPα abolished binding. Further, the antibodies did not bind to RPTPα after blocking with the phosphopeptides that were used for immunization. The nonphosphorylated peptides had no effect (Fig. 1 A and B). The pSer180 and pSer204 antibodies were specific for their respective phosphorylation sites in RPTPα.
FIG. 1.
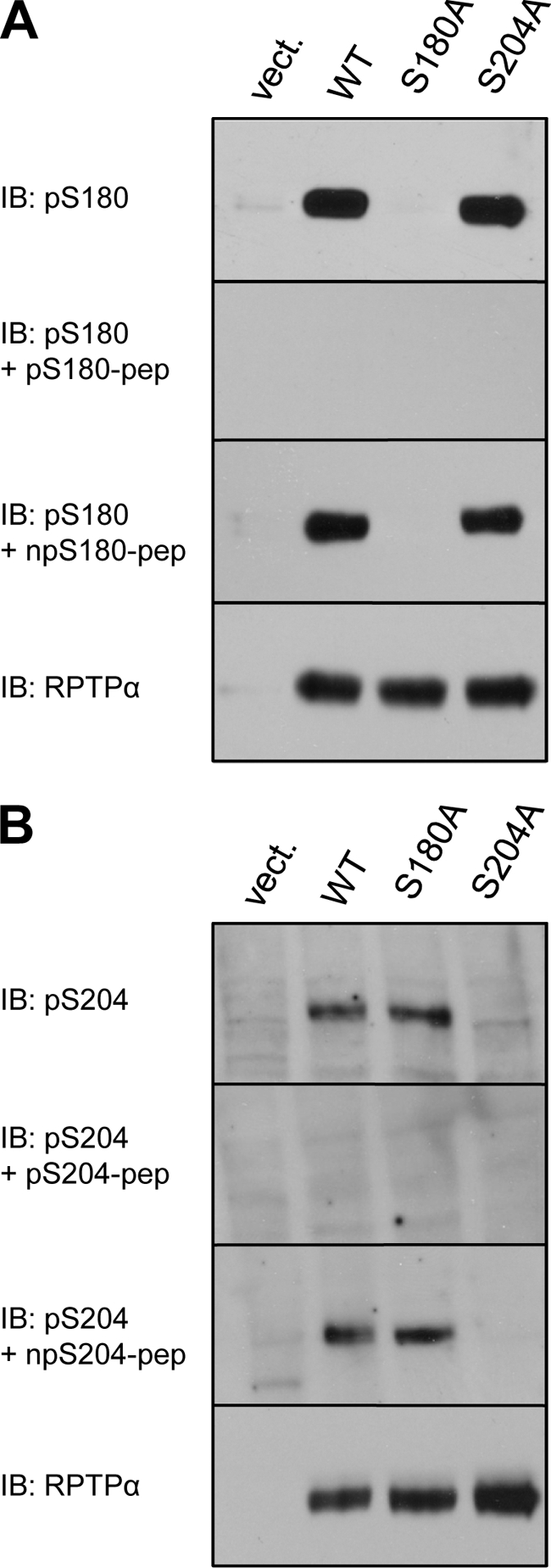
Specificity of anti-pS180 and anti-pS204 antibodies. HEK293 cells were transiently transfected with empty vector, WT HA-RPTPα, HA-RPTPα S180A, or HA-RPTPα S204A. The cells were lysed, and the lysates were fractionated on 7.5% SDS-polyacrylamide gels, transferred to polyvinylidene difluoride (PVDF) membranes, and immunoblotted with anti-pS180 (A) or anti-pS204 (B) antibodies. As indicated, the antibodies were used alone or together with the phospho- or nonphosphopeptides against which the antibodies were raised. The total levels of HA-RPTPα in the lysates were probed with anti-RPTPα antibody and shown in the bottom panels (A and B). IB, immunoblot.
Mitotic dephosphorylation of RPTPα.
Previously it was suggested that RPTPα serine phosphorylation is increased after mitotic arrest (60). Using the pSer180 and pSer204 antibodies and the commercially available pTyr789 antibody, we followed the phosphorylation state of RPTPα in the cell cycle. NIH 3T3 cells were arrested with nocodazole and released from mitotic block. The cells were lysed, and RPTPα was immunoprecipitated with affinity-purified anti-RPTPα serum (5478). In control, unsynchronized cells, all three RPTPα phosphorylation sites were phosphorylated (Fig. 2 A) After mitotic arrest, pSer204 was almost completely dephosphorylated whereas pSer180 and pTyr789 were less affected. RPTPα Ser204 phosphorylation increased rapidly upon release from the mitotic block and reached the levels of unsynchronized cells 3 h after release. Phosphorylation of Ser180 presented a slower recovery after the cells were released, and Tyr789 phosphorylation recovered 1 to 3 h after release (Fig. 2A). FACS analysis was used to monitor cell cycle distribution. The efficiency of the nocodazole-induced mitotic block was high, since more than 90% of the cells were mitotic. The cells were viable, and after removal of nocodazole and washing with PBS, the cells reentered the cell cycle (Fig. 2A).
FIG. 2.
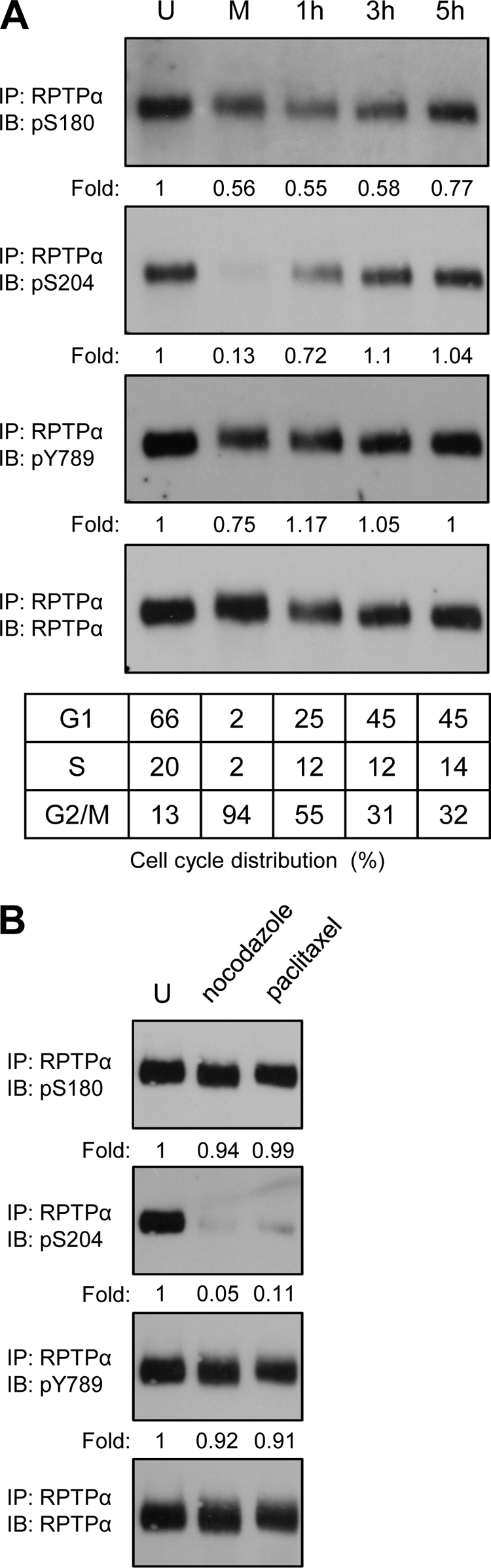
Mitotic dephosphorylation of RPTPα. (A) Endogenous RPTPα was immunoprecipitated from NIH 3T3 lysates (0.5 mg total protein) from unsynchronized (U), mitotic (M), and replated cells after mitotic arrest (1 h, 3 h, and 5 h). The immunoprecipitates were boiled in reducing sample buffer, and the samples were run on a 7.5% SDS-polyacrylamide gel. The proteins were transferred to PVDF membranes, and the membranes were probed with anti-pS204 antibody and subsequently, after stripping, with anti-pS180, anti-pY789, and anti-RPTPα. The cell cycle distribution (percent) of the NIH 3T3 cells used to prepare each sample is shown in the table beneath. (B) Unsynchronized NIH 3T3 cells and cells obtained by mitotic shake-off after nocodazole and paclitaxel treatment were lysed, and endogenous RPTPα was immunoprecipitated from approximately 1 mg total protein/sample. The samples were processed as described for panel A. The blots were quantified, and the phosphorylation levels were normalized for the total amount of RPTPα (bottom panel) and expressed as the ratio to the phosphorylation levels in the unsynchronized cells. The values are presented under each lane. Western blot quantification was performed using Quantity One software (Bio-Rad). All the experiments were repeated at least three times with similar results, and representative experiments are presented in this figure. IP, immunoprecipitation; IB, immunoblot.
Nocodazole interferes with the polymerization of microtubules. To investigate whether dephosphorylation of RPTPα pSer204 is a mitotic effect and not an artifact of nocodazole treatment, we used paclitaxel, which arrests NIH 3T3 cells in mitosis by stabilizing microtubules. After a 14-h paclitaxel treatment, the cells were harvested by mitotic shake-off and lysed and RPTPα was immunoprecipitated. The results were similar for the nocodazole- and paclitaxel-treated cells for all three RPTPα phosphorylation sites. Both types of mitotic arrest induced drastic dephosphorylation of RPTPα pSer204 but not pSer180 or pTyr789 (Fig. 2B).
Mitotic Src activation.
To investigate Src tyrosine phosphorylation in the cell cycle, the same lysates that were used for analysis of the phosphorylation state of RPTPα (Fig. 2A) were analyzed by immunoblotting using antibodies for the autophosphorylation site of Src, pTyr416, and for the dephosphorylated form of the C-terminal phosphorylation site, npTyr527. In mitotic cells, the levels of dephosphorylated Src Tyr527 were highly increased and decreased within 1 h to the levels of the unsynchronized cells after release from mitotic arrest (Fig. 3A). Surprisingly, in the mitotically arrested cells the levels of Src pTyr416 were reduced as well and they increased slowly (1 to 3 h) after release from the mitotic block to the level in unsynchronized cells (Fig. 3A).
FIG. 3.

Mitotic phosphorylation and activation of Src. (A) A fraction of the lysates used for RPTPα immunoprecipitation (Fig. 2A) was boiled in SDS sample buffer, and the samples were run on a 7.5% SDS-polyacrylamide gel. The proteins were transferred to PVDF membranes, and the membranes were probed with anti-pY416 antibody and subsequently, after stripping, with anti-npY527 and anti-Src. (B) Endogenous Src was immunoprecipitated with cross-linked antibodies to protein A beads from unsynchronized and mitotic NIH 3T3 cells. Half of the immunoprecipitate was subjected to an in vitro kinase assay, using enolase as substrate. The other half was used for immunoblotting with anti-Src antibody followed by enhanced chemiluminescence (ECL) (bottom panel). The amount of incorporated phosphate was visualized by autoradiography (top panel). The positions of enolase and Src are indicated by arrows. WCLs, whole-cell lysates; IB, immunoblot; IP, immunoprecipitation.
We observed that in the mitotically arrested cells Src pTyr527 was dephosphorylated, the first step in activation of Src. Phosphorylation of Src Tyr416, which is necessary for full activation, was reduced in mitosis. To understand the cumulative effect of Tyr416 and Tyr527 dephosphorylation, we assayed in vitro kinase activity of Src from unsynchronized and mitotically arrested NIH 3T3 cells by using acid-denatured enolase as a substrate (Fig. 3B). We found that immunoprecipitated Src from mitotically arrested cells was 2.3 times more active than Src from unsynchronized cells, which is consistent with previous results (60). Hence, the net result of pTyr527 and pTyr416 dephosphorylation in mitosis is a modest increase in Src kinase activity.
RPTPα and Src phosphorylation in HeLa cells.
To see if RPTPα pSer204 mitotic dephosphorylation is a general mechanism, we investigated the phosphorylation state of human RPTPα in HeLa cells. Unsynchronized and nocodazole-arrested cells were lysed, and half of the lysates were used for immunoprecipitation of RPTPα, while the other half were used for immunoprecipitation of Src. Immunoblotting with the phosphospecific antibodies indicated that phosphorylation of RPTPα Ser180 and Tyr789 was not significantly affected, whereas Ser204 was completely dephosphorylated (Fig. 4A). In the case of Src, both pTyr416 and pTyr527 levels were decreased in the mitotically arrested HeLa cells (Fig. 4B). These results demonstrate that mitotic dephosphorylation of RPTPα pSer204 and concomitant dephosphorylation of Src pTyr416 and pTyr527 were similar in mouse NIH 3T3 cells and human HeLa cells.
FIG. 4.
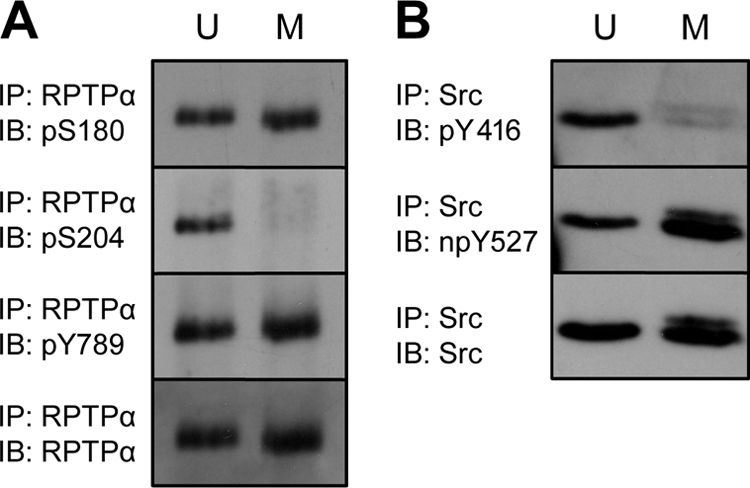
RPTPα and Src phosphorylation in HeLa cells. Unsynchronized (U) and mitotic (M) HeLa cell lysates (1 mg total protein) were split in half. One half was used to immunoprecipitate endogenous RPTPα (A). The samples and the Western blots were prepared as described for Fig. 2A. (B) The second half was used for Src immunoprecipitation. Cross-linked Src antibodies were used for this purpose. The immunoprecipitates were fractionated by 7.5% SDS-PAGE, and after transfer the membranes were probed with anti-pY416 antibody and subsequently, after stripping, with anti-npY527 and anti-Src. The experiment was performed at least three times with similar results, and a representative experiment is shown here. IP, immunoprecipitation; IB, immunoblot.
PP2A dephosphorylates RPTPα in vitro and in vivo.
RPTPα pSer204 was almost completely dephosphorylated in mitotically arrested NIH 3T3 cells (Fig. 2A and B) and HeLa cells (Fig. 4A), whereas pSer180 levels were much less affected under these conditions. We set out to identify the phosphatase(s) responsible for mitotic dephosphorylation of pSer204. The main serine phosphatases that are activated in mitosis are PP1, PP2A, and Cdc25 (50). In vitro phosphatase assays were performed to assess which of these phosphatases dephosphorylated RPTPα pSer204. RPTPα was immunoprecipitated from NIH 3T3 cell lysates, and the immunoprecipitates were incubated with fresh NIH 3T3 lysates, resulting in dephosphorylation of pSer204 compared to control RPTPα that was not incubated with lysate (Fig. 5A). Na3VO4 (V) did not affect RPTPα serine dephosphorylation, whereas NaF blocked pSer204 dephosphorylation almost completely (Fig. 5A). These results indicate that Cdc25, a cysteine-based dual-specificity phosphatase that is sensitive to Na3VO4, was not responsible for RPTPα dephosphorylation. However, the pSer204 phosphatase(s) was NaF sensitive.
FIG. 5.

PP2A dephosphorylates RPTPα in vitro and in vivo. (A to D) Endogenous RPTPα was immunoprecipitated from unsynchronized NIH 3T3 cells (A and B). The immunoprecipitates were incubated with fresh lysates of unsynchronized (U) or mitotic (M) NIH 3T3 cells in the presence of orthovanadate (V), sodium fluoride (NaF), or a cocktail of PP1/PP2A inhibitors containing okadaic acid, calyculin A, and tautomycin (100 nM each). After the reactions were terminated, the proteins were separated by 7.5% SDS-PAGE and transferred to PVDF membranes. The blots were probed with anti-pS204 antibody and subsequently, after stripping, with anti-RPTPα antibody. HA-RPTPα overexpressed in COS1 cells was immunoprecipitated and incubated with unsynchronized (C) or mitotic (D) NIH 3T3 lysates in the presence of increasing amounts of the PP1/PP2A inhibitors as indicated. As a control, immunoprecipitated HA-RPTPα was incubated with cell lysis buffer (CLB). The lysates were removed, and the samples were processed as for panel A. Finally, the blots were probed with anti-pS204 antibody and subsequently, after stripping, with anti-HA tag antibody. (E) NIH 3T3 cells arrested with nocodazole were treated with 100 nM okadaic acid (OA). RPTPα was immunoprecipitated and blotted, and the blots were probed with pS180, pS204, and RPTPα antibodies. These experiments have been done at least three times, and representative blots are shown. (F) siRNA-mediated knockdown of PP2Acα in unsynchronized (U) or mitotic (M) NIH 3T3 cells was done as described in Materials and Methods. Efficiency of knockdown was monitored by blotting using a PP2Acα-specific antibody and equal loading using an actin antibody (bottom two panels). RPTPα was immunoprecipitated, blotted, and probed using pS180, pS204, and RPTPα antibodies, as indicated. IP, immunoprecipitation; IB, immunoblot.
To test whether the pSer204 phosphatase activity was regulated during the cell cycle, RPTPα immunoprecipitates were incubated with lysates of unsynchronized or nocodazole-arrested NIH 3T3 cells. Both lysates readily dephosphorylated RPTPα pSer204 (Fig. 5B). Addition of a cocktail of PP1/PP2A inhibitors (okadaic acid, calyculin A, and tautomycin) inhibited dephosphorylation of pSer204 (Fig. 5B). It appeared that the inhibitor cocktail was less efficient in inhibiting the phosphatases in the mitotic lysates, suggesting that the phosphatase activity was higher in lysates from mitotic cells than in lysates from unsynchronized cells.
To investigate which of the two serine phosphatases, PP1 and PP2A, was responsible for RPTPα dephosphorylation, in vitro phosphatase assays were done in the presence of increasing concentrations of okadaic acid, calyculin A, or tautomycin. Okadaic acid, an inhibitor with higher specificity for PP2A than for PP1 (50% inhibitory concentration [IC50] for PP2A, 0.1 nM; IC50 for PP1, 10 to 15 nM), blocked dephosphorylation of RPTPα already at 10 nM. Tautomycin, an inhibitor with higher efficiency for PP1 (IC50 for PP2A, 10 nM; IC50 for PP1, 1 nM), blocked dephosphorylation only at 1,000 nM. Calyculin A (IC50 for PP2A, 0.5 to 1 nM; IC50 for PP1, 2 nM) had an intermediate effect (Fig. 5C). Based on these results, we conclude that PP2A is likely to be the main RPTPα pSer204 phosphatase in NIH 3T3 cells. Similar results were obtained with lysates from nocodazole-arrested cells (Fig. 5D), in that okadaic acid and calyculin A were the most potent inhibitors of serine phosphatase activity. Higher concentrations of inhibitors were required to fully block phosphatase activity in the lysates of mitotically arrested cells, compared to unsynchronized cells, suggesting that total pSer204 phosphatase activity was elevated in mitotic cells.
To confirm that PP2A is the phosphatase responsible for the mitotic dephosphorylation of pSer204 in vivo, mitotically arrested NIH 3T3 cells were treated with okadaic acid for 14 h. Endogenous RPTPα was immunoprecipitated, and pSer180 and pSer204 levels were probed. RPTPα Ser204 phosphorylation was greatly reduced in mitotic cells compared to that in unsynchronized cells, and okadaic acid completely reversed this effect. pSer204 levels in okadaic acid-treated mitotic cells were as high as those in the unsynchronized cells (Fig. 5E). Interestingly, pSer180 levels were hardly affected following the okadaic acid treatment.
We used siRNAs against the catalytic subunit of PP2A (PP2Acα) to knock down PP2A and assess Ser204 and Ser180 levels in mitotic and unsynchronized cells. The PP2A siRNAs clearly reduced PP2Acα expression, whereas control siRNAs did not affect PP2Acα expression. RPTPα Ser204 phosphorylation was reduced in mitotic cells compared to unsynchronized cells that were treated with the control siRNAs as expected. Interestingly, siRNA-mediated knockdown of PP2A reduced pSer204 dephosphorylation in mitotic cells, and even in unsynchronized cells, Ser204 phosphorylation appeared to be somewhat elevated compared to the control siRNA-treated cells (Fig. 5F). Ser180 phosphorylation was hardly affected by PP2A knockdown (Fig. 5F), suggesting that Ser180 dephosphorylation is mediated by different phosphatases. The pharmacological inhibitors together with the knockdown experiments demonstrate that PP2A is the phosphatase responsible for pSer204 dephosphorylation in mitosis.
RPTPα catalytic activity is not significantly influenced by serine phosphorylation.
We have shown that RPTPα Ser204 is almost completely dephosphorylated in mitotic cells (Fig. 2) concomitantly with Src activation (Fig. 3). Hence, dephosphorylation of RPTPα pSer204 might activate intrinsic RPTPα catalytic activity. To investigate this, we assayed phosphatase activity of RPTPα serine mutants in vitro. HA-tagged wild-type RPTPα, RPTPα serine mutants, and a C433S mutant were expressed in COS1 cells and immunoprecipitated, and PTP activity was determined using phosphorylated MBP as a substrate. Time courses of the PTP assays were performed using wild-type RPTPα, showing that substrate dephosphorylation increased linearly in the first 20 min of the assay (data not shown). To ensure that measurements were done in the linear range, PTP assays were routinely stopped and evaluated after 15 min (Fig. 6). Mutation of Ser204 did not activate PTP activity, nor did mutation of Ser180, whereas mutation of the catalytic Cys433 rendered RPTPα inactive (Fig. 6A). Expression of RPTPα and mutants was monitored by immunoblotting (Fig. 6B). The PTP activity of RPTPα from mitotic cells was not significantly enhanced compared to RPTPα activity from unsynchronized cells in in vitro phosphatase assays (Fig. 6C). We also tested the ability of RPTPα Ser mutants to dephosphorylate Src in vitro. RPTPα and mutants dephosphorylated Src pTyr527 to similar extents and had minimal to no detectable activity toward Src pTyr416 (Fig. 6D). As expected, only the catalytically inactive RPTPα C433S did not dephosphorylate Src pTyr527. These results demonstrate that dephosphorylation of pSer204 in mitosis did not enhance intrinsic catalytic activity of RPTPα.
FIG. 6.

RPTPα catalytic activity is not significantly influenced by serine phosphorylation. (A) WT HA-RPTPα, HA-RPTPα C433S, and serine mutants of HA-RPTPα (S180A, S204A, and S204D) were immunoprecipitated from transfected COS1 cells, and their ability to release [32P]phosphate from phosphorylated MBP was detected. The combined results from three independent experiments are depicted here. (B) For one of the experiments, the serine phosphorylation levels for each overexpressed protein are shown. (C) Endogenous RPTPα was immunoprecipitated from lysates of unsynchronized (U) and mitotic (M) NIH 3T3 cells (1 mg total protein). For the negative control (N), the anti-RPTPα antibody was not added during immunoprecipitation. The ability to dephosphorylate MBP is depicted in the graph. Each bar represents the average of three independent experiments ± standard deviation. The phosphatase activity of WT HA-RPTPα (A) and endogenous RPTPα from unsynchronized cells (C) was set to 100%, and the other data were calculated relative to these values. (D) HA-tagged RPTPα WT and the mutants indicated were immunoprecipitated from transfected COS1 cells and incubated with Src immunoprecipitated from transfected SYF cells. After incubation the samples were boiled in SDS sample buffer and resolved by 7.5% SDS-PAGE. The blots were probed for Src phosphorylation as well as for the amounts of HA-RPTPα present in each reaction. Ser phosphorylation levels of RPTPα are also shown. Src phosphorylation levels were quantified and normalized for the total amount of Src and expressed as the ratio to the phosphorylation levels of Src incubated with immunoprecipitates from empty-vector-transfected cells. The values are presented under each corresponding sample. IP, immunoprecipitation; IB, immunoblot.
Phosphorylation of RPTPα Ser204 prohibits Src binding.
Binding of Src to RPTPα is an important determinant in Src activation. To examine the mechanism underlying Src activation by RPTPα in mitosis, we investigated coimmunoprecipitation of endogenous Src with RPTPα in unsynchronized and mitotic NIH 3T3 cells. Clearly, binding of Src to RPTPα was induced in mitotic cells (Fig. 7A), concomitantly with Ser204 dephosphorylation. pSer180 and pTyr789 levels were determined in the same samples and were hardly affected in mitotic cells compared to unsynchronized cells as observed before (Fig. 2A and 7A).
FIG. 7.

Src binding to RPTPα is induced in mitosis by RPTPα pSer204 dephosphorylation, independently of GRB2. (A) Unsynchronized (U) and mitotic (M) cells were lysed (3 mg total protein), and endogenous RPTPα was immunoprecipitated. The immunoprecipitates were fractionated on a 12.5% SDS-polyacrylamide gel and tested for coimmunoprecipitated Src and GRB2. Phosphorylation of immunoprecipitated RPTPα was also probed. Src and the heavy chain (HC) of the antibody are indicated by arrows. The amount of coimmunoprecipitated GRB2 was quantified and normalized for the total amount of GRB2 in the lysates. The input levels of Src and GRB2 in the lysates (3% of the total lysate run on the same gel) are shown. (B) Src and RPTPα WT, Y789F mutant, or empty vector were cotransfected into SYF cells. The cells were lysed, and Src was immunoprecipitated with cross-linked anti-Src antibodies. The samples were fractionated on a 7.5% SDS-polyacrylamide gel, transferred to PVDF membranes, and immunoblotted with anti-RPTPα serum (top panel) and anti-Src monoclonal antibody (MAb) 327 (middle panel). Whole-cell lysates were monitored for HA-RPTPα expression (bottom panel). (C) As in panel B, except that mutant Src R175L, with a mutation that impairs the ability of the SH2 domain to bind to pTyr residues in target proteins, was cotransfected. (D) Unsynchronized (U) and mitotic (M) NIH 3T3 cells were treated with 100 nM okadaic acid (OA) or left untreated. The samples were processed as for panel A. To reduce the signal from the antibody heavy chain, the blot showing the amount of coimmunoprecipitated Src was probed with horseradish peroxidase (HRP)-coupled protein A, which was less sensitive and hence did not allow detection of the slower-migrating Src band. (E) SYF cells were cotransfected with Src and HA-RPTPα WT, mutants (S204A and S204D), or empty vector (vect.). Coimmunoprecipitation of (mutant) RPTPα with Src was detected as described for panel B. The experiments were repeated at least three times with similar results, and representative experiments are shown here. WCLs, whole-cell lysates; IP, immunoprecipitation; IB, immunoblot.
Previously Zheng et al. (58) suggested that RPTPα-mediated Src activation requires phosphorylation of RPTPα Tyr789 and displacement of the adaptor protein GRB2, which is bound to pTyr789. We found that GRB2 binding to RPTPα was only modestly reduced in mitosis (23% reduction compared to unsynchronized cells), which cannot account for enhanced Src binding and activation (Fig. 7A). Interestingly, mutation of Ser180, Ser204, or both did not affect GRB2 binding in pulldown assays (data not shown), indicating that serine phosphorylation of RPTPα does not affect GRB2 binding. Several reports showed that RPTPα can activate Src in the absence of the Tyr789 phosphorylation site (12, 29, 56). To determine definitively whether Src can bind to RPTPα in the absence of pTyr789, we immunoprecipitated Src from SYF cells cotransfected with Src and RPTPα WT or Y789F mutant. We found that mutant RPTPα-Y798F coimmunoprecipitated with Src, albeit to a lesser extent than did wild-type RPTPα (Fig. 7B). To further address the RPTPα-Src interaction, we introduced a point mutation in the SH2 domain (R175L), which abolishes the ability of the SH2 domain to bind to pTyr. Mutant Src-R175L still bound wild-type RPTPα and RPTPα-Y789F (Fig. 7C). These results clearly demonstrate that the interaction between RPTPα and Src is not mediated by the SH2 domain binding to RPTPα pTyr789.
Next, we investigated whether Ser204 phosphorylation has a decisive role in the Src-RPTPα interaction. Okadaic acid treatment completely restored the phosphorylation of RPTPα Ser204 in the mitotically arrested cells. Concomitantly with Ser204 rephosphorylation, Src binding to mitotic RPTPα was lost completely (Fig. 7D), indicating that Ser204 phosphorylation prohibits Src binding. Okadaic acid treatment did not significantly affect RPTPα Ser204 or Ser180 phosphorylation in unsynchronized NIH 3T3 cells, and Src binding to RPTPα was not detected in unsynchronized cells under these circumstances (Fig. 7D).
We further investigated the effect of Ser204 phosphorylation on RPTPα binding to Src. SYF cells were cotransfected with Src and RPTPα or Ser mutants, the cells were lysed, and Src was immunoprecipitated. Subsequently, the immunoprecipitates were probed for coimmunoprecipitated RPTPα. RPTPα-S204A bound to Src to a similar extent as did WT RPTPα (Fig. 7E). Given our hypothesis that phosphorylation of Ser204 prohibits Src binding, we expected to find enhanced binding of RPTPα-S204A to Src, compared to WT RPTPα. Under these overexpression conditions, RPTPα-S204A binding to Src apparently was maximal. Binding of RPTPα-S204D with a phosphomimicking mutation replacing Ser204 was reduced, varying from experiment to experiment by 30 to 50% (Fig. 7E). Binding of Src to RPTPα-S204D was not completely abolished. However, substitution for Ser by an Asp residue mimics pSer to some extent, although not completely. The reduction in binding of RPTPα-S204D to Src is consistent with our conclusion that phosphorylation of Ser204 inhibits RPTPα binding to Src.
Based on our results, we conclude that RPTPα Ser204 phosphorylation is reduced in mitosis and suggest that this allows Src to bind to mitotic RPTPα, resulting in dephosphorylation of Src pTyr527 and pTyr416.
DISCUSSION
In this study, we explored how endogenous RPTPα in NIH 3T3 and HeLa cells is regulated by serine phosphorylation in mitosis and how these mitotic changes reflect the ability of RPTPα to activate Src. We developed phosphospecific antibodies directed at the known serine phosphorylation sites of RPTPα (Ser180 and Ser204) (49). In contrast to the model of Zheng et al., in which serine hyperphosphorylation drives RPTPα activation (59, 60), we demonstrate that Ser204 was almost completely dephosphorylated in mitosis. Using inhibitors and RNAi-mediated knockdown, we identified PP2A as the pSer204 phosphatase in vitro and in vivo (Fig. 5). It appeared that PP2A was already active in unsynchronized cells, and the moderate increase in PP2A activity in mitosis perhaps cannot account for the complete dephosphorylation of pSer204. We believe that dephosphorylation of pSer204 in mitosis is the net result of the combined effects of activation of PP2A and inactivation of the responsible kinase. Several kinases may phosphorylate Ser204, among which may be protein kinase A (PKA), because (i) the flanking sequences of Ser204 form a consensus PKA phosphorylation site (36), (ii) PKA phosphorylates Ser204 in vitro (data not shown), (iii) pharmacological PKA inhibitors reduced Ser204 kinase activity in NIH 3T3 cell lysates (data not shown), and (iv) cyclic AMP (cAMP) levels and hence PKA kinase activity are decreased in mitosis (1, 19). Additional experiments are required to definitively establish the identity of the Ser204 kinase.
We and others have shown previously that serine phosphorylation may regulate RPTPα activity directly. PKC-mediated enhanced serine phosphorylation of RPTPα leads to activation of RPTPα, and dephosphorylation of RPTPα in vitro reduces its activity to prestimulation levels (16). Bacterially expressed RPTPα fusion protein phosphorylated in vitro using CaMKIIalpha enhanced RPTPα catalytic activity, which appeared to be mediated mostly by Ser180 (9). Here, we demonstrate that mutation of Ser180 and Ser204 did not significantly affect intrinsic catalytic activity of RPTPα when MBP or Src was used as a substrate. Moreover, the activity of endogenous nonphospho-Ser204 RPTPα from mitotic NIH 3T3 cells was not significantly different from the activity of RPTPα from unsynchronized cells (Fig. 6). These results indicate that mitotic activation of Src is unlikely to be caused by changes in the intrinsic catalytic activity of RPTPα.
When looking at endogenous Src phosphorylation in mitotic cells, we noticed that not only Tyr527 but also Tyr416 was dephosphorylated. Previous studies have shown that overexpressed RPTPα dephosphorylates both tyrosine residues (15, 20, 61). RPTPα has similar activity toward phosphorylated peptides coding for these two Src tyrosine phosphorylation sites (37). Dephosphorylation of both pTyr416 and pTyr527 led to a modest net increase in Src activity (∼2.3-fold) in mitosis (Fig. 3B), which is consistent with previously reported mitotic Src activation (60). Moreover, RPTPα overexpression results in modest activation of Src as well (15, 58, 61). Mutation of Tyr527 in Src results in full activation of Src (∼20-fold-higher activity than that of wild-type Src) (22), concomitantly with autophosphorylation of Src on Tyr416. Apparently, dephosphorylation of both pTyr527 and pTyr416 prevents inadvertent overactivation of Src.
Here, we demonstrate for the first time using endogenously expressed proteins that Src binding to RPTPα is induced in mitosis. Previously, detection of Src binding to RPTPα relied on overexpression of Src and RPTPα (60). Binding of Src to RPTPα was suggested to be mediated by binding of the SH2 domain to pTyr789 in RPTPα. The SH2 domain has suboptimal affinity for phosphorylated Tyr789 (the affinity for a peptide encoding the C terminus of RPTPα, AFSDpYANFK, being more than 10 times weaker than the affinity for the optimal peptide sequence pYEEI of SH2) (43, 44). Phosphorylation of RPTPα Tyr789 forms a consensus binding site for GRB2, and it is hard to imagine that Src and GRB2 compete for binding to this site. Nevertheless, the FAK/p130Cas/Src complex dissociates in mitosis following serine phosphorylation of FAK (54), increasing the amount of mitotic Src that can bind to RPTPα. GRB2 binding is only marginally reduced in mitotic NIH 3T3 cells (Fig. 7A). Tyr789 is essential for GRB2 binding but appears to be dispensable for Src binding (Fig. 7B), consistent with several studies showing that RPTPα lacking Tyr789 is still able to dephosphorylate and activate Src (12, 26, 29, 56). Moreover, we demonstrate that a functional SH2 domain is dispensable for binding to RPTPα. Hence, it is unlikely that the underlying mechanism for mitotic activation of Src involves competition for binding of Src and GRB2 to RPTPα pTyr789. Our data suggest that Src binding is mediated by a different region(s) of RPTPα and that phosphorylation of Ser204 prohibits binding. Close to Ser204 in RPTPα is a putative SH3 binding site, RKYPPLP (residues 207 to 213). RPTPα is constitutively phosphorylated on Ser204, and this could impair access of SH3 domains to this region. We observed enhanced binding of RPTPα to Src in mitosis, and mitotic dephosphorylation of Ser204 may open up the SH3 binding site, thus triggering binding to Src. Mutation of Ser204 to Ala, prohibiting phosphorylation of this site, did not affect GRB2 binding (data not shown) or Src binding (Fig. 7E). Src binding to the RPTPα Ser204 phosphomimicking mutant (S204D) was reduced in coimmunoprecipitation assays (Fig. 7E), corroborating the hypothesis that phosphorylation of Ser204 prohibits Src binding.
The RPTPα Ser204 phosphorylation site is positioned in the proximity of the wedge structure, an element shown to play an important role in RPTPα dimerization (6). Phosphorylation or dephosphorylation of Ser204 might influence the architecture of the juxtamembrane domain. Therefore, we also considered that RPTPα dimerization might be affected by pSer204 dephosphorylation, resulting in enhanced RPTPα activity and subsequent Src dephosphorylation. However, we did not observe any differences between RPTPα and serine mutants in coimmunoprecipitation and accessibility assays (52) to assess differences in dimerization or quaternary structure of RPTPα (data not shown). However, Ser204 dephosphorylation might have subtle effects on dimerization (e.g., on stability of the dimers), which might not be detected in the standard dimerization assays.
We cannot exclude the possibility that serine/threonine phosphorylation of Src has a role in mitotic activation of Src as well. Src is phosphorylated on Thr34, Thr46, and Ser72 in mitosis by Cdc2, which leads to an increase in exposure of pTyr527 (reviewed in reference 41). Serine/threonine phosphorylation of mitotic Src causes reduced electrophoretic mobility. There is no obvious difference in coimmunoprecipitation of the slower-migrating forms of Src with RPTPα (Fig. 7A), making it unlikely that serine/threonine phosphorylation of Src has a decisive role in the Src-RPTPα interaction.
Our data led us to propose a model for mitotic activation of Src (Fig. 8). In interphase, Ser180, Ser204, and Tyr789 are phosphorylated. In mitosis, PP2A is activated, resulting in pSer204 dephosphorylation, while phosphorylation of Ser180 and Tyr789 is hardly affected. This results in Src binding to RPTPα without significant effects on GRB2 binding. Subsequently, both pTyr416 and pTyr527 in Src are dephosphorylated, resulting in modest activation of Src. After release from mitosis, Ser204 is phosphorylated again, leading to release of Src and subsequent phosphorylation of Tyr527. This model is different from the model proposed by Zheng and Shalloway (59, 60) at several points. Zheng and Shalloway suggested that serine phosphorylation was enhanced in mitosis. In contrast, we demonstrate that pSer204 is dephosphorylated in mitosis. They proposed as the underlying mechanism an increase in intrinsic RPTPα catalytic activity. We found no evidence for activation of RPTPα catalytic activity in mitosis, and the catalytic activity of mutant RPTPα that cannot be phosphorylated was not significantly different from that of wild-type RPTPα. Finally, we did not find evidence for a displacement mechanism in which GRB2, bound to pTyr789, is displaced by Src. Yet, we demonstrate that binding of Src to RPTPα was induced in mitosis, which we believe mediates the increase in Src activity. The discrepancy between our data and the results of Zheng and collaborators (59, 60) regarding the mitotic RPTPα phosphorylation may be caused by the different setups of the experiments. We followed the phosphorylation of endogenous RPTPα, while Zheng et al. used RPTPα-overexpressing NIH 3T3 cells for most of their experiments. RPTPα overexpression in NIH 3T3 cells reportedly results in metabolic changes (29) which may affect cell behavior. Importantly, the molecular ratios between RPTPα, Src, and the other proteins involved in this mechanism are crucial, and overexpression of one of these factors will severely impact on the balance of these factors.
FIG. 8.

RPTPα-mediated activation of Src in mitosis. RPTPα is phosphorylated constitutively on Ser180, Ser204, and Tyr789. At interphase, phosphorylation of Ser204 prohibits binding of Src, and as a result, Src is not activated. In mitosis, pSer204 is dephosphorylated due to activation of PP2A. Nonphospho-Ser204 binds Src through yet-to-be-identified regions in Src and RPTPα while remaining phosphorylated on Tyr789 and bound to the adaptor protein GRB2. Src binding to RPTPα in mitosis leads to dephosphorylation of pTyr527 and pTyr416 in Src, resulting in modest activation of Src kinase activity. Following release from mitosis, Ser204 is rapidly phosphorylated and Src phosphorylation reverts.
In conclusion, we propose a new model for mitotic activation of Src by RPTPα in mitosis. Dephosphorylation of RPTPα pSer204 leads to Src binding. Whether Src binds directly to RPTPα close to dephosphorylated Ser204 remains to be determined. Concomitant dephosphorylation of pTyr527 and pTyr416 results in modest activation of Src, prohibiting Src from displaying its oncogenic potential.
Acknowledgments
We thank Rob Klompmaker (University Medical Center Utrecht) for technical assistance with FACS analysis and Mathieu Bollen and Nick Tonks for their advice and suggestions.
This study was supported by a Netherlands Proteomics Center grant.
Footnotes
Published ahead of print on 12 April 2010.
REFERENCES
- 1.Abell, C. W., and T. M. Monahan. 1973. The role of adenosine 3′,5′-cyclic monophosphate in the regulation of mammalian cell division. J. Cell Biol. 59:549-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alonso, A., J. Sasin, N. Bottini, I. Friedberg, A. Osterman, A. Godzik, T. Hunter, J. Dixon, and T. Mustelin. 2004. Protein tyrosine phosphatases in the human genome. Cell 117:699-711. [DOI] [PubMed] [Google Scholar]
- 3.Andersen, J. N., A. Elson, R. Lammers, J. Romer, J. T. Clausen, K. B. Moller, and N. P. Moller. 2001. Comparative study of protein tyrosine phosphatase-epsilon isoforms: membrane localization confers specificity in cellular signalling. Biochem. J. 354:581-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bagrodia, S., I. Chackalaparampil, T. E. Kmiecik, and D. Shalloway. 1991. Altered tyrosine 527 phosphorylation and mitotic activation of p60c-src. Nature 349:172-175. [DOI] [PubMed] [Google Scholar]
- 5.Bhandari, V., K. L. Lim, and C. J. Pallen. 1998. Physical and functional interactions between receptor-like protein-tyrosine phosphatase alpha and p59fyn. J. Biol. Chem. 273:8691-8698. [DOI] [PubMed] [Google Scholar]
- 6.Bilwes, A. M., J. den Hertog, T. Hunter, and J. P. Noel. 1996. Structural basis for inhibition of receptor protein-tyrosine phosphatase-alpha by dimerization. Nature 382:555-559. [DOI] [PubMed] [Google Scholar]
- 7.Blanchetot, C., L. G. Tertoolen, and J. den Hertog. 2002. Regulation of receptor protein-tyrosine phosphatase alpha by oxidative stress. EMBO J. 21:493-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bodrikov, V., I. Leshchyns'ka, V. Sytnyk, J. Overvoorde, J. den Hertog, and M. Schachner. 2005. RPTPα is essential for NCAM-mediated p59fyn activation and neurite elongation. J. Cell Biol. 168:127-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bodrikov, V., V. Sytnyk, I. Leshchyns'ka, J. den Hertog, and M. Schachner. 2008. NCAM induces CaMKIIalpha-mediated RPTPα phosphorylation to enhance its catalytic activity and neurite outgrowth. J. Cell Biol. 182:1185-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brandt, D. T., A. Goerke, M. Heuer, M. Gimona, M. Leitges, E. Kremmer, R. Lammers, H. Haller, and H. Mischak. 2003. Protein kinase C delta induces Src kinase activity via activation of the protein tyrosine phosphatase PTP alpha. J. Biol. Chem. 278:34073-34078. [DOI] [PubMed] [Google Scholar]
- 11.Chackalaparampil, I., and D. Shalloway. 1988. Altered phosphorylation and activation of pp60c-src during fibroblast mitosis. Cell 52:801-810. [DOI] [PubMed] [Google Scholar]
- 12.Chen, M., S. C. Chen, and C. J. Pallen. 2006. Integrin-induced tyrosine phosphorylation of protein-tyrosine phosphatase-alpha is required for cytoskeletal reorganization and cell migration. J. Biol. Chem. 281:11972-11980. [DOI] [PubMed] [Google Scholar]
- 13.Daum, G., S. Regenass, J. Sap, J. Schlessinger, and E. H. Fischer. 1994. Multiple forms of the human tyrosine phosphatase RPTP alpha. Isozymes and differences in glycosylation. J. Biol. Chem. 269:10524-10528. [PubMed] [Google Scholar]
- 14.den Hertog, J., and T. Hunter. 1996. Tight association of GRB2 with receptor protein-tyrosine phosphatase alpha is mediated by the SH2 and C-terminal SH3 domains. EMBO J. 15:3016-3027. [PMC free article] [PubMed] [Google Scholar]
- 15.den Hertog, J., C. E. Pals, M. P. Peppelenbosch, L. G. Tertoolen, S. W. de Laat, and W. Kruijer. 1993. Receptor protein tyrosine phosphatase alpha activates pp60c-src and is involved in neuronal differentiation. EMBO J. 12:3789-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.den Hertog, J., J. Sap, C. E. Pals, J. Schlessinger, and W. Kruijer. 1995. Stimulation of receptor protein-tyrosine phosphatase alpha activity and phosphorylation by phorbol ester. Cell Growth Differ. 6:303-307. [PubMed] [Google Scholar]
- 17.den Hertog, J., S. Tracy, and T. Hunter. 1994. Phosphorylation of receptor protein-tyrosine phosphatase alpha on Tyr789, a binding site for the SH3-SH2-SH3 adaptor protein GRB-2 in vivo. EMBO J. 13:3020-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gil-Henn, H., G. Volohonsky, and A. Elson. 2001. Regulation of protein-tyrosine phosphatases alpha and epsilon by calpain-mediated proteolytic cleavage. J. Biol. Chem. 276:31772-31779. [DOI] [PubMed] [Google Scholar]
- 19.Grieco, D., A. Porcellini, E. V. Avvedimento, and M. E. Gottesman. 1996. Requirement for cAMP-PKA pathway activation by M phase-promoting factor in the transition from mitosis to interphase. Science 271:1718-1723. [DOI] [PubMed] [Google Scholar]
- 20.Harder, K. W., N. P. Moller, J. W. Peacock, and F. R. Jirik. 1998. Protein-tyrosine phosphatase alpha regulates Src family kinases and alters cell-substratum adhesion. J. Biol. Chem. 273:31890-31900. [DOI] [PubMed] [Google Scholar]
- 21.Herrera Abreu, M. T., P. C. Penton, V. Kwok, E. Vachon, D. Shalloway, L. Vidali, W. Lee, C. A. McCulloch, and G. P. Downey. 2008. Tyrosine phosphatase PTPalpha regulates focal adhesion remodeling through Rac1 activation. Am. J. Physiol. Cell Physiol. 294:C931-C944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunter, T. 1987. A tail of two src's: mutatis mutandis. Cell 49:1-4. [DOI] [PubMed] [Google Scholar]
- 23.Jiang, G., J. den Hertog, and T. Hunter. 2000. Receptor-like protein tyrosine phosphatase alpha homodimerizes on the cell surface. Mol. Cell. Biol. 20:5917-5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang, G., J. den Hertog, J. Su, J. Noel, J. Sap, and T. Hunter. 1999. Dimerization inhibits the activity of receptor-like protein-tyrosine phosphatase-alpha. Nature 401:606-610. [DOI] [PubMed] [Google Scholar]
- 25.Kapp, K., E. Metzinger, M. Kellerer, H. U. Haring, and R. Lammers. 2003. The protein tyrosine phosphatase alpha modifies insulin secretion in INS-1E cells. Biochem. Biophys. Res. Commun. 311:361-364. [DOI] [PubMed] [Google Scholar]
- 26.Kapp, K., J. Siemens, P. Weyrich, J. B. Schulz, H. U. Haring, and R. Lammers. 2007. Extracellular domain splice variants of a transforming protein tyrosine phosphatase alpha mutant differentially activate Src-kinase dependent focus formation. Genes Cells 12:63-73. [DOI] [PubMed] [Google Scholar]
- 27.Krueger, N. X., M. Streuli, and H. Saito. 1990. Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J. 9:3241-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lacasa, D., N. Boute, and T. Issad. 2005. Interaction of the insulin receptor with the receptor-like protein tyrosine phosphatases PTPalpha and PTPepsilon in living cells. Mol. Pharmacol. 67:1206-1213. [DOI] [PubMed] [Google Scholar]
- 29.Lammers, R., M. M. Lerch, and A. Ullrich. 2000. The carboxyl-terminal tyrosine residue of protein-tyrosine phosphatase alpha mediates association with focal adhesion plaques. J. Biol. Chem. 275:3391-3396. [DOI] [PubMed] [Google Scholar]
- 30.Lammers, R., N. P. Moller, and A. Ullrich. 1997. The transmembrane protein tyrosine phosphatase alpha dephosphorylates the insulin receptor in intact cells. FEBS Lett. 404:37-40. [DOI] [PubMed] [Google Scholar]
- 31.Lammers, R., N. P. Moller, and A. Ullrich. 1998. Mutant forms of the protein tyrosine phosphatase alpha show differential activities towards intracellular substrates. Biochem. Biophys. Res. Commun. 242:32-38. [DOI] [PubMed] [Google Scholar]
- 32.Lei, G., S. Xue, N. Chery, Q. Liu, J. Xu, C. L. Kwan, Y. P. Fu, Y. M. Lu, M. Liu, K. W. Harder, and X. M. Yu. 2002. Gain control of N-methyl-D-aspartate receptor activity by receptor-like protein tyrosine phosphatase alpha. EMBO J. 21:2977-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim, K. L., D. S. Lai, M. B. Kalousek, Y. Wang, and C. J. Pallen. 1997. Kinetic analysis of two closely related receptor-like protein-tyrosine-phosphatases, PTP alpha and PTP epsilon. Eur. J. Biochem. 245:693-700. [DOI] [PubMed] [Google Scholar]
- 34.Lu, H., P. Shah, D. Ennis, G. Shinder, J. Sap, H. Le-Tien, and I. G. Fantus. 2002. The differentiation of skeletal muscle cells involves a protein-tyrosine phosphatase-alpha-mediated C-Src signaling pathway. J. Biol. Chem. 277:46687-46695. [DOI] [PubMed] [Google Scholar]
- 35.Moller, N. P., K. B. Moller, R. Lammers, A. Kharitonenkov, E. Hoppe, F. C. Wiberg, I. Sures, and A. Ullrich. 1995. Selective down-regulation of the insulin receptor signal by protein-tyrosine phosphatases alpha and epsilon. J. Biol. Chem. 270:23126-23131. [DOI] [PubMed] [Google Scholar]
- 36.Neuberger, G., G. Schneider, and F. Eisenhaber. 2007. pkaPS: prediction of protein kinase A phosphorylation sites with the simplified kinase-substrate binding model. Biol. Direct 2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng, D. H., M. D. Jabali, A. Maiti, P. Borodchak, K. W. Harder, T. Brocker, B. Malissen, F. R. Jirik, and P. Johnson. 1997. CD45 and RPTPα display different protein tyrosine phosphatase activities in T lymphocytes. Biochem. J. 327:867-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petrone, A., F. Battaglia, C. Wang, A. Dusa, J. Su, D. Zagzag, R. Bianchi, P. Casaccia-Bonnefil, O. Arancio, and J. Sap. 2003. Receptor protein tyrosine phosphatase alpha is essential for hippocampal neuronal migration and long-term potentiation. EMBO J. 22:4121-4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ponniah, S., D. Z. Wang, K. L. Lim, and C. J. Pallen. 1999. Targeted disruption of the tyrosine phosphatase PTPalpha leads to constitutive downregulation of the kinases Src and Fyn. Curr. Biol. 9:535-538. [DOI] [PubMed] [Google Scholar]
- 40.Roche, S., S. Fumagalli, and S. A. Courtneidge. 1995. Requirement for Src family protein tyrosine kinases in G2 for fibroblast cell division. Science 269:1567-1569. [DOI] [PubMed] [Google Scholar]
- 41.Roskoski, R., Jr. 2005. Src kinase regulation by phosphorylation and dephosphorylation. Biochem. Biophys. Res. Commun. 331:1-14. [DOI] [PubMed] [Google Scholar]
- 42.Shenoy, S., J. K. Choi, S. Bagrodia, T. D. Copeland, J. L. Maller, and D. Shalloway. 1989. Purified maturation promoting factor phosphorylates pp60c-src at the sites phosphorylated during fibroblast mitosis. Cell 57:763-774. [DOI] [PubMed] [Google Scholar]
- 43.Songyang, Z., and L. C. Cantley. 1995. Recognition and specificity in protein tyrosine kinase-mediated signalling. Trends Biochem. Sci. 20:470-475. [DOI] [PubMed] [Google Scholar]
- 44.Sonnenburg, E. D., A. Bilwes, T. Hunter, and J. P. Noel. 2003. The structure of the membrane distal phosphatase domain of RPTPα reveals interdomain flexibility and an SH2 domain interaction region. Biochemistry 42:7904-7914. [DOI] [PubMed] [Google Scholar]
- 45.Su, J., M. Muranjan, and J. Sap. 1999. Receptor protein tyrosine phosphatase alpha activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr. Biol. 9:505-511. [DOI] [PubMed] [Google Scholar]
- 46.Su, J., L. T. Yang, and J. Sap. 1996. Association between receptor protein-tyrosine phosphatase RPTPα and the Grb2 adaptor. Dual Src homology (SH) 2/SH3 domain requirement and functional consequences. J. Biol. Chem. 271:28086-28096. [DOI] [PubMed] [Google Scholar]
- 47.Tertoolen, L. G., C. Blanchetot, G. Jiang, J. Overvoorde, T. W. Gadella, Jr., T. Hunter, and J. den Hertog. 2001. Dimerization of receptor protein-tyrosine phosphatase alpha in living cells. BMC Cell Biol. 2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tonks, N. K. 2006. Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7:833-846. [DOI] [PubMed] [Google Scholar]
- 49.Tracy, S., P. van der Geer, and T. Hunter. 1995. The receptor-like protein-tyrosine phosphatase, RPTP alpha, is phosphorylated by protein kinase C on two serines close to the inner face of the plasma membrane. J. Biol. Chem. 270:10587-10594. [DOI] [PubMed] [Google Scholar]
- 50.Trinkle-Mulcahy, L., and A. I. Lamond. 2006. Mitotic phosphatases: no longer silent partners. Curr. Opin. Cell Biol. 18:623-631. [DOI] [PubMed] [Google Scholar]
- 51.Tsai, W., A. D. Morielli, T. G. Cachero, and E. G. Peralta. 1999. Receptor protein tyrosine phosphatase alpha participates in the m1 muscarinic acetylcholine receptor-dependent regulation of Kv1.2 channel activity. EMBO J. 18:109-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van der Wijk, T., C. Blanchetot, J. Overvoorde, and J. den Hertog. 2003. Redox-regulated rotational coupling of receptor protein-tyrosine phosphatase alpha dimers. J. Biol. Chem. 278:13968-13974. [DOI] [PubMed] [Google Scholar]
- 53.von Wichert, G., G. Jiang, A. Kostic, K. De Vos, J. Sap, and M. P. Sheetz. 2003. RPTP-alpha acts as a transducer of mechanical force on alphav/beta3-integrin-cytoskeleton linkages. J. Cell Biol. 161:143-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamakita, Y., G. Totsukawa, S. Yamashiro, D. Fry, X. Zhang, S. K. Hanks, and F. Matsumura. 1999. Dissociation of FAK/p130(CAS)/c-Src complex during mitosis: role of mitosis-specific serine phosphorylation of FAK. J. Cell Biol. 144:315-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang, J., A. Groen, S. Lemeer, A. Jans, M. Slijper, S. M. Roe, J. den Hertog, and D. Barford. 2007. Reversible oxidation of the membrane distal domain of receptor PTPalpha is mediated by a cyclic sulfenamide. Biochemistry 46:709-719. [DOI] [PubMed] [Google Scholar]
- 56.Yang, L. T., K. Alexandropoulos, and J. Sap. 2002. c-SRC mediates neurite outgrowth through recruitment of Crk to the scaffolding protein Sin/Efs without altering the kinetics of ERK activation. J. Biol. Chem. 277:17406-17414. [DOI] [PubMed] [Google Scholar]
- 57.Zeng, L., X. Si, W. P. Yu, H. T. Le, K. P. Ng, R. M. Teng, K. Ryan, D. Z. Wang, S. Ponniah, and C. J. Pallen. 2003. PTP alpha regulates integrin-stimulated FAK autophosphorylation and cytoskeletal rearrangement in cell spreading and migration. J. Cell Biol. 160:137-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng, X. M., R. J. Resnick, and D. Shalloway. 2000. A phosphotyrosine displacement mechanism for activation of Src by PTPalpha. EMBO J. 19:964-978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng, X. M., R. J. Resnick, and D. Shalloway. 2002. Mitotic activation of protein-tyrosine phosphatase alpha and regulation of its Src-mediated transforming activity by its sites of protein kinase C phosphorylation. J. Biol. Chem. 277:21922-21929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng, X. M., and D. Shalloway. 2001. Two mechanisms activate PTPalpha during mitosis. EMBO J. 20:6037-6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng, X. M., Y. Wang, and C. J. Pallen. 1992. Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature 359:336-339. [DOI] [PubMed] [Google Scholar]