

Abstract
There is evidence that reactive oxygen species (ROS) signalling is required for normal increases in glucose uptake during contraction of isolated mouse skeletal muscle, and that AMP-activated protein kinase (AMPK) is involved. The aim of this study was to determine whether ROS signalling is involved in the regulation of glucose disposal and AMPK activation during moderate-intensity exercise in humans. Nine healthy males completed 80 min of cycle ergometry at 62 ± 1% of peak oxygen consumption (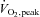 . A 6,6-2H-glucose tracer was infused at rest and during exercise, and in a double-blind randomised cross-over design, N-acetylcysteine (NAC) or saline (CON) was co-infused. NAC was infused at 125 mg kg−1 h−1 for 15 min and then at 25 mg kg−1 h−1 for 20 min before and throughout exercise. NAC infusion elevated plasma NAC and cysteine, and muscle NAC and cysteine concentrations during exercise. Although neither NAC infusion nor exercise significantly affected muscle reduced or oxidised glutathione (GSH or GSSG) concentration (P > 0.05), S-glutathionylation (an indicator of oxidative stress) of a protein band of ∼270 kDa was increased ∼3-fold with contraction and this increase was prevented by NAC infusion. Despite this, exercised-induced increases in tracer determined glucose disposal, plasma lactate, plasma non-esterified fatty acids (NEFAs), and decreases in plasma insulin were not affected by NAC infusion. In addition, skeletal muscle AMPKα and acetyl-CoA carboxylase-β (ACCβ) phosphorylation increased during exercise by ∼3- and ∼6-fold (P < 0.05), respectively, and this was not affected by NAC infusion. Unlike findings in mouse muscle ex vivo, NAC does not attenuate skeletal muscle glucose disposal or AMPK activation during moderate-intensity exercise in humans.
. A 6,6-2H-glucose tracer was infused at rest and during exercise, and in a double-blind randomised cross-over design, N-acetylcysteine (NAC) or saline (CON) was co-infused. NAC was infused at 125 mg kg−1 h−1 for 15 min and then at 25 mg kg−1 h−1 for 20 min before and throughout exercise. NAC infusion elevated plasma NAC and cysteine, and muscle NAC and cysteine concentrations during exercise. Although neither NAC infusion nor exercise significantly affected muscle reduced or oxidised glutathione (GSH or GSSG) concentration (P > 0.05), S-glutathionylation (an indicator of oxidative stress) of a protein band of ∼270 kDa was increased ∼3-fold with contraction and this increase was prevented by NAC infusion. Despite this, exercised-induced increases in tracer determined glucose disposal, plasma lactate, plasma non-esterified fatty acids (NEFAs), and decreases in plasma insulin were not affected by NAC infusion. In addition, skeletal muscle AMPKα and acetyl-CoA carboxylase-β (ACCβ) phosphorylation increased during exercise by ∼3- and ∼6-fold (P < 0.05), respectively, and this was not affected by NAC infusion. Unlike findings in mouse muscle ex vivo, NAC does not attenuate skeletal muscle glucose disposal or AMPK activation during moderate-intensity exercise in humans.
Introduction
Exercise stimulates skeletal muscle glucose uptake by increasing GLUT-4 translocation from intracellular vesicles to the cell membrane (Kennedy et al. 1999) through a mechanism(s) that differs from insulin-stimulated GLUT-4 translocation and glucose uptake (Zisman et al. 2000). However, the pathway(s) through which contraction stimulates muscle glucose uptake is unclear (Rose & Richter, 2005; Merry & McConell, 2009) with evidence for separate and collective contribution of several signalling intermediates including AMP-activated protein kinase (AMPK) (Hayashi et al. 1998), nitric oxide (NO) (Bradley et al. 1999; Ross et al. 2007), calcium–calmodulin-dependent kinase (CaMK) (Witczak et al. 2007), and Akt substrate of 160 kDa and 150 kDa (AS160 and TBC1D1) (Funai & Cartee, 2008, 2009). Interestingly, recent ex vivo evidence suggests that reactive oxygen species (ROS) may also play a role in signalling skeletal muscle contraction-mediated glucose uptake (Sandstrom et al. 2006).
Exposure of isolated skeletal muscle to exogenous ROS increases glucose uptake (Toyoda et al. 2004; Higaki et al. 2008; Jensen et al. 2008). Similarly, the rate of ROS production in skeletal muscle increases with intense contraction ex vivo (Reid et al. 1992a,b;) and in vivo (Sen et al. 1994; Medved et al. 2004b), and causes an acute oxidative shift in cell redox status. By attenuating the increase in ROS production and oxidative stress during contraction of isolated extensor digitorum longus (EDL) muscle with the antioxidant N-acetylcysteine (NAC) Sandstrom et al. (2006) provided the first evidence that ROS signalling during contraction may, in part, mediate glucose uptake via the activation of AMPK.
The AMPK activator 5-amino-imidazole 4-carboxamide ribonucleoside (AICAR) increases skeletal muscle glucose uptake (Merrill et al. 1997), and during contraction the increase in AMPK activity correlates with glucose uptake (Musi et al. 2001; Chen et al. 2003), thus implicating AMPK in the regulation of glucose uptake during contraction. Interestingly, high levels of exogenous ROS increase skeletal muscle AMPK activity (Toyoda et al. 2004; Jensen et al. 2008) and the attenuation of contraction-mediated increases in ROS levels attenuate increases in contraction-mediated AMPK activity (Sandstrom et al. 2006). This suggests that an increase in ROS levels during skeletal muscle contraction may be partially responsible for activating AMPK and thus regulating glucose uptake (Sandstrom et al. 2006). However, dissociations between skeletal muscle AMPK activity and contraction-stimulated glucose uptake have been frequently reported (Derave et al. 2000; Jorgensen et al. 2004; Wadley et al. 2006; McConell et al. 2008). Indeed, AMPK activation does not appear to be required for normal increases in skeletal muscle glucose uptake following short-term exercise training (McConell et al. 2005) in humans and during in vivo exercise in mice (Lee-Young et al. 2009; Maarbjerg et al. 2009). Importantly, at present the role of ROS in the regulation of contraction-stimulated muscle glucose uptake has only been examined in isolated muscle models. In the absence of blood flow, such models depend on diffusion gradients for substrate delivery and clearance, and result in non-uniform delivery of oxygen to all muscle fibres (Allen et al. 2008). Furthermore, ex vivo muscle preparations generally involve supra-maximal highly fatiguing stimulation protocols. These factors may alter contraction-induced ROS production (Allen et al. 2008; Reid, 2008) and signalling of glucose uptake. Therefore, it is important to investigate the role of ROS signalling in the regulation of skeletal muscle glucose uptake during exercise in vivo.
In a series of studies, Medved et al. (2003, 2004a,b) demonstrated that the non-specific anti-oxidant NAC can be safely infused intravenously into humans during prolonged exercise. Moreover, the cysteine-donating properties of NAC promoted the resynthesis of muscle reduced glutathione (GSH) during exercise indicating an attenuation of exercise-induced increases in skeletal muscle oxidative stress (Medved et al. 2004b). Therefore, in the current study, we infused NAC intravenously in humans during prolonged cycle ergometry to determine the role of ROS in the regulation of glucose disposal and AMPK signalling during exercise. We hypothesised that NAC infusion would attenuate the increases in glucose disposal and skeletal muscle AMPK signalling during exercise in humans.
Methods
Participants
Nine healthy recreationally active (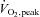 : 51.7 ± 2.3 ml kg−1 min−1) adult males volunteered. The participants’ age, weight and height (mean ±s.e.m.) were 23 ± 2 years, 79.7 ± 3.4 kg and 179 ± 3 cm, respectively. This study was approved by the University of Melbourne Human Ethics Committee, and was conducted in accordance with the Declaration of Helsinki and The Journal of Physiology standards (Drummond, 2009). Participants were informed of the experimental procedures and provided written consent. All participants were non-smokers, were not taking any medication and had no history of cardiovascular, cerebrovascular or respiratory disease.
: 51.7 ± 2.3 ml kg−1 min−1) adult males volunteered. The participants’ age, weight and height (mean ±s.e.m.) were 23 ± 2 years, 79.7 ± 3.4 kg and 179 ± 3 cm, respectively. This study was approved by the University of Melbourne Human Ethics Committee, and was conducted in accordance with the Declaration of Helsinki and The Journal of Physiology standards (Drummond, 2009). Participants were informed of the experimental procedures and provided written consent. All participants were non-smokers, were not taking any medication and had no history of cardiovascular, cerebrovascular or respiratory disease.
Preliminary procedures
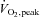 was measured in normal ambient laboratory conditions (∼20°C) using a graded cycle ergometer (electronically braked ergometer; Lode, Groningen, the Netherlands) protocol to voluntary exhaustion with respiratory gas analysis for volume expired (Air flow meter; Vacuumed, Ventura, CA, USA), O2 and CO2 content (S-3A O2 and AMETEK CO2 analyser; Applied Electrochemistry, Sunnyvale, CA, USA). On a separate day, 1–2 weeks before the first experimental trial, participants completed a familiarisation session where they cycled for 20 min at ∼60% of
was measured in normal ambient laboratory conditions (∼20°C) using a graded cycle ergometer (electronically braked ergometer; Lode, Groningen, the Netherlands) protocol to voluntary exhaustion with respiratory gas analysis for volume expired (Air flow meter; Vacuumed, Ventura, CA, USA), O2 and CO2 content (S-3A O2 and AMETEK CO2 analyser; Applied Electrochemistry, Sunnyvale, CA, USA). On a separate day, 1–2 weeks before the first experimental trial, participants completed a familiarisation session where they cycled for 20 min at ∼60% of 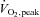 .
.
Experimental trials
The study involved a double-blind randomised cross-over design, with counterbalanced testing order. Experimental trials were conducted at the same time of day and separated by at least 2 weeks. A 24 h food diary was completed prior to the first trial, and this was photocopied and returned to the participants who were asked to follow the same diet prior to the second trial.
Participants fasted overnight and reported to the laboratory at 6.30 am on trial days, having abstained from exercise, caffeine and alcohol for the preceding 24 h. A 22-gauge cannula was inserted into an antecubital forearm vein for the infusion of stable isotope glucose tracer (6,6-2H-glucose; Cambridge Isotope Labratories, MA, USA) and NAC, and another cannula was inserted into the contralateral forearm for blood sampling. An initial blood sample was obtained and then a bolus of 40.2 μmol kg−1 of tracer was administered followed by a 2 h pre-exercise continuous constant infusion (0.39 μmol kg−1 min−1), which was continued through to the end of exercise (McConell et al. 2006). As described previously by Medved et al. (2003), an initial loading dose of either NAC (Parvolex, Faulding Pharmaceuticals; 125 mg kg−1 h−1 in 0.9% saline) or 0.9% saline alone (control, CON) was co-infused intravenously 35 min prior to exercise for 15 min, followed by a constant infusion of saline or NAC (25 mg kg−1 h−1) that continued until the end of exercise. As outlined in Fig. 1, following 35 min of NAC/CON infusion at rest, participants cycled for 80 min at 62 ± 1%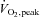 in standard laboratory conditions (∼20°C) with a fan on high setting positioned ∼1 m directly in front of the handlebars. Water was consumed ad libitum.
in standard laboratory conditions (∼20°C) with a fan on high setting positioned ∼1 m directly in front of the handlebars. Water was consumed ad libitum.
Figure 1. Experimental protocol.

See Methods for details. RPE, rating of perceived exertion; HR, heart rate; NAC/Saline 1, infusion of N-acetylcysteine (NAC; 125 mg kg−1 h−1) or saline; NAC/Saline 2, infusion of N-acetylcysteine at lower rate (25 mg kg−1 h−1) or saline.
Experimental trial sample collection and measurements
Heart rate was measured using a heart rate monitor (Polar Favor, Oulu, Finland) and recorded at 10 min intervals during exercise. Expired respiratory gases were sampled for 3 min at t= 10, 30 and 70 min, and volume expired, O2 and CO2 content were measured. Rating of perceived exertion (Borg, 1974) was obtained at 10 min intervals during exercise, and any adverse reactions to the infusions were recorded during the trial as described by Medved et al. (2003, 2004b).
Venous blood samples were obtained at t=−120, −65, −45, −35 and −20 min, and then every 10 min until the end of exercise (Fig. 1). Blood for glucose, per cent enrichment of [6,6-2H]glucose, lactate, insulin and thiol analysis was transferred immediately to tubes containing lithium–heparin. Blood for non-esterified fatty acid (NEFA) analysis was transferred immediately to tubes containing EDTA. All blood tubes were placed on ice until the end of the trial then spun at 3000 g for 20 min and plasma stored at −80°C for later analysis.
For the sampling of muscle, during the pre-exercise infusion period three separate ∼1 cm incisions were made under local anaesthesia above the vastus lateralis of one leg. Muscle samples were then obtained at t= 0, 40 and 80 min (Fig. 1) using the percutaneous needle biopsy technique (distal-to-promimal order, at least 1 cm apart). Muscle samples were frozen in liquid nitrogen while still in the biopsy needle within 6–12 s following the cessation of exercise. Muscle samples were stored in liquid nitrogen for later analysis and were obtained from the contralateral leg during the second trial.
Blood analysis
Plasma lactate was measured using an automated l-lactate oxidase method (YSI 2300 Stat, Yellow Springs, OH, USA) and plasma glucose was determined using an enzymatic fluorometric assay involving NADPH production (Lowry & Passonneau, 1972). Plasma NEFA was measured by an enzymatic colourimetric procedure (NEFA-C test; Wako, Osaka, Japan) and plasma insulin using a human radioimmunoassay kit (Linco Research, St Charles, MO, USA). As described previously (McConell et al. 1994), glucose kinetics were measured using a modified one-pool, non-steady-state model (Steele et al. 1956; Radziuk et al. 1978). We assumed 0.65 as the rapidly mixing portion of the glucose pool and estimated the apparent glucose space as 25% of body weight. Plasma glucose appearance (Ra) and disappearance (Rd) rates were determined from changes in the per cent enrichment of 6,6-2H-glucose and the plasma glucose concentration. Over 95% of tracer-determined Rd is oxidised at power outputs requiring ∼60%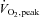 (Jeukendrup et al. 1999).
(Jeukendrup et al. 1999).
For the analysis of plasma total thiols and NAC, 10 μl of 1:10 tributylphosphine (Sigma-Aldrich Chemicals, St Louis, MO, USA) was added to 50 μl of plasma, and following a 30 min incubation on ice, 25 μl of 4-fluoro-7-sulfamoylbenzofurazan (Sigma-Aldrich Chemicals; ADB-F; 5 mg ml−1 in borate buffer: 0.2 m boric acid, 2 mm sodium EDTA, pH 8.0) was added. Samples were then incubated at 50°C for 10 min, 10 μl of 2 m perchloric acid (PCA) was added and they were spun at 13,000 g for 5 min. The supernatant was recovered and 40 μl was injected into a reverse-phase HPLC Gemini column (5 μm C18 110Å, phenomenex) with 0.1 m sodium acetate buffer (pH 4.0) in 10% methanol as a mobile phase, at a flow rate of 1.5 ml min−1 and detection wavelength of 386 nm excitation and 516 nm emission. For the determination of plasma reduced thiols and NAC, tributylphosphine was replaced with H2O and the protocol was repeated. Oxidised thiols were calculated from the difference between total and reduced thiols.
Muscle analysis
Approximately 25 mg of muscle were freeze-dried and ground to a powder. For the determination of muscle glycogen, ∼1 mg of freeze-dried muscle was incubated at 95°C for 2 h in 250 μl of 2 m HCl and then neutralised with 750 μl of 0.67 m NaOH. The extracts were then analysed for glucosyl units using an enzymatic fluorometric assay (Passonneau & Lauderdale, 1974). Muscle metabolites (ATP, creatine, creatine phosphate (PCr) and lactate) were determined by extracting ∼2 mg of freeze-dried muscle in 250 μl of 0.5 m PCA and 1 mm EDTA, and the supernatant analysed using enzymatic fluorometric assays (Lowry & Passonneau, 1972). To account for any non-muscle contamination, muscle metabolites were corrected to the highest muscle total creatine content for each participant and free AMP and ADP were calculated as described previously (Chen et al. 2000).
Extraction for NAC and thiols analysis involved the homogenization of 30 mg of frozen muscle in 300 μl of ice-cold 0.42 m PCA; 40 μl of 2.5 m K2CO3 was then added to neutralize samples before spinning at 13,000 g for 5 min at 4°C and recovering supernatant. The protocol described above for the determination of plasma thiols was then repeated with the muscle supernatant.
For Western blot analysis, 50 mg of frozen muscle were homogenized in ice-cold lysis buffer (10 μl mg−1 tissue; 50 mm Tris-HCl at pH 7.5 containing 1 mm EDTA, 10% v/v glycerol, 1% v/v Triton X-100, 50 mm NaF, 5 mm Na4P2O7, 1 mm DTT, 1 mm phenylmethylsulphonyl floride (PMSF) and 5 μl ml−1 Protease Inhibitor Cocktail (Sigma-Aldrich Chemicals, St Louis, MO, USA), incubated for 20 min on ice and spun at 16,000 g for 20 min at 4°C. The supernatant was recovered and protein concentration was determined using a bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL, USA) with BSA as the standard. The remaining supernatant was solubilised in Laemmli sample buffer (1.5 m Tris-HCl, pH 6.8, 30% glycerole, 10% SDS, 0.6 m DTT, 0.0012% bromophenol blue), heated for 10 min at 100°C and stored at −20°C. For analysis of protein S-glutathionylation, muscle was extracted under non-reducing conditions with lysis and Laemmli sample buffer containing 5 mm and 10 mm of N-ethylmaleimide, respectively, in the absence of DTT. SDS-PAGE was used to separate 80 μg of total protein before transferring to PVDF membrane and blocking in 5% non-fat milk for 1 h at room temperature. Membranes were incubated overnight at 4°C with primary antibodies for 3-nitrotyrosine (Chemicon; Temecula, CA, USA), glutathione (Abcam, Cambridge, UK) and phosphorylation-specific antibodies for ACCβ Ser221 and AMPK Thr172 (Upstate Biotechnology, NY, USA) before binding was detected with rabbit IgG and mouse IgG secondary fluorescent antibodies (Rockland, Gilbertsville, PA, USA). Direct fluorescence was detected and quantified using the Odyssey infrared imaging system (LICOR Biosciences, Lincoln, NB, USA). Membranes were then stripped (2% SDS (w/v) in 25 mm glycine, pH 2.0) and re-probed with primary antibodies for ACCβ (streptavidin) (Rockland), AMPKα and α-tubulin (Cell Signaling Technology, Hertfordshire, UK) to determine total protein levels. Protein phosphorylation was expressed relative to the total protein abundance of the protein of interest.
Statistical analysis
All data are expressed as means ±s.e.m. Results were analysed by SPSS statistical package using two-factor repeated measures ANOVA. To assess the resting effects and because NAC infusion started pre-exercise, the ANOVA was partitioned to assess the effect of NAC during rest (−30 to −10 min) and during exercise (0 to 80 min). If the ANOVA revealed a significant treatment by time interaction, specific differences between mean values were located using the Fisher's least significance difference test. The level of significance was set at P < 0.05.
Results
Respiratory measures, heart rate and rating of perceived exertion
Participants exercised at 62 ± 1%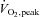 during both trials. Oxygen consumption (2.6 ± 0.1 vs. 2.5 ± 0.2 l min−1), respiratory exchange ratio (RER; 0.90 ± 0.02 vs. 0.91 ± 0.02), heart rate (HR; 154 ± 2 vs. 154 ± 4 beats min−1) and rating of perceived exertion (RPE; 13 ± 1 vs. 14 ± 2) were similar (P > 0.05) in saline (control) vs. NAC trials, respectively.
during both trials. Oxygen consumption (2.6 ± 0.1 vs. 2.5 ± 0.2 l min−1), respiratory exchange ratio (RER; 0.90 ± 0.02 vs. 0.91 ± 0.02), heart rate (HR; 154 ± 2 vs. 154 ± 4 beats min−1) and rating of perceived exertion (RPE; 13 ± 1 vs. 14 ± 2) were similar (P > 0.05) in saline (control) vs. NAC trials, respectively.
Muscle and plasma NAC and adverse reactions
No adverse reactions to either NAC or control infusions were observed. No NAC was detectable in muscle or plasma of control infusion. By the onset of exercise, NAC infusion increased (P < 0.05) plasma NAC and reduced NAC content to 148.2 ± 16.1 μmol and 79.8 ± 18.4 μmol, respectively, and this content was maintained throughout exercise (Fig. 2A). NAC infusion increased the levels of resting muscle NAC and resting muscle reduced NAC to 74.2 ± 22.5 and 46.2 ± 16.4 pmol (mg wet wt)−1, respectively, and these levels remained essentially unchanged during exercise (Fig. 2B).
Figure 2. Plasma and muscle NAC levels.

Plasma (A) and muscle (B) N-acetylcysteine (NAC) during 80 min of steady-state exercise at 62 ± 1%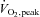 while receiving N-acetylcysteine infusion. N= 8.
while receiving N-acetylcysteine infusion. N= 8.
Plasma cysteine
NAC infusion elevated plasma cysteine (P < 0.01), and tended to increase plasma cystine (P= 0.07; Fig. 3). Plasma cysteine concentration was increased by exercise during NAC (P < 0.05), but not saline infusion (Fig. 3A). Exercise did not affect plasma cystine (Fig. 3B).
Figure 3. Plasma cysteine and cystine levels.

Plasma cysteine (A) and cystine (B) concentration at rest and during 80 min of steady-state exercise at 62 ± 1%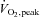 while receiving either saline (CON) or saline +N-acetylcysteine (NAC) infusion. N= 8, #P < 0.05 vs. CON, *P < 0.05 vs. t= 0 of same treatment.
while receiving either saline (CON) or saline +N-acetylcysteine (NAC) infusion. N= 8, #P < 0.05 vs. CON, *P < 0.05 vs. t= 0 of same treatment.
Plasma lactate, NEFA and insulin
During exercise plasma insulin concentration decreased (P < 0.01) and plasma NEFA and lactate concentration increased (P < 0.05) to a similar extent in the two trials (Fig. 4).
Figure 4. Plasma lactate, non-esterified free fatty acids and insulin concentration.

Plasma lactate (A), non-esterified free fatty acids (NEFA; B) and insulin (C) concentration at rest and during 80 min of steady-state exercise at 62 ± 1%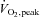 while receiving either saline or saline +N-acetylcysteine (NAC) infusion. N= 9.
while receiving either saline or saline +N-acetylcysteine (NAC) infusion. N= 9.
Glucose kinetics
Plasma glucose concentration was not affected by NAC infusion or exercise (Fig. 5A; P < 0.05). The increase in glucose appearance (Ra) and glucose disappearance (Rd) were not affected by NAC infusion (Fig. 5). The pattern of the glucose clearance rate (CR) was very similar to the glucose Rd results and therefore has not been presented. This is not surprising given the similarity of the plasma glucose concentrations in the two trials. Glucose Ra, and Rd increased with exercise (P < 0.05) and this increase was not influenced by NAC infusion (Fig. 5).
Figure 5. Plasma glucose, rate of glucose appearance and rate of glucose disappearance.

Plasma glucose (A), rate of glucose appearance (Glucose Ra; B) and rate of glucose disappearance (Glucose Rd; C) at rest and during 80 min of steady-state exercise at 62 ± 1%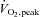 while receiving either saline or saline +N-acetylcysteine (NAC) infusion. N= 9.
while receiving either saline or saline +N-acetylcysteine (NAC) infusion. N= 9.
Muscle thiols
NAC infusion increased muscle cysteine at rest and during exercise (P < 0.05), but cystine was only increased during exercise (P < 0.05; Fig. 6B and D). Muscle cysteine was increased with exercise at 40 min and cystine at 40 and 80 min but only during NAC infusion (P < 0.05; Fig. 6B and D). Muscle GSH, GSSG or GSSG/GSH ratio were not affected by exercise or NAC infusion (Fig. 6A, C and E).
Figure 6. Muscle reduced glutathione, cysteine, oxidised glutathione, cystine and GSSG/GSH ratio.

Muscle reduced glutathione (GSH; A), cysteine (B), oxidised glutathione (GSSG; C), cystine (D) and GSSG/GSH ratio (E) at rest and during 80 min of steady-state exercise at 62 ± 1%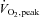 while receiving either saline (CON) or saline +N-acetylcysteine (NAC) infusion. N= 9, #P < 0.05 vs. CON, *P < 0.05 vs. t= 0 min of same treatment, †P < 0.05 vs. t= 40 min of same treatment.
while receiving either saline (CON) or saline +N-acetylcysteine (NAC) infusion. N= 9, #P < 0.05 vs. CON, *P < 0.05 vs. t= 0 min of same treatment, †P < 0.05 vs. t= 40 min of same treatment.
S-Glutathionylation and tyrosine nitration
Exercise increased muscle S-glutathionylation of a protein band of approximately 270 kDa (Fig. 7A) by ∼3-fold (P < 0.05). NAC infusion prevented the exercise-induced increase in S-glutathionylation of this protein band. The nature of this protein band is currently being investigated. Muscle tyrosine nitration was not significantly affected by exercise or NAC infusion (Fig. 7B).
Figure 7. Muscle protein S-glutathionylation and tyrosine nitration.

Muscle protein S-glutathionylation (A) and tyrosine nitration (B) at rest and during 80 min of steady-state exercise at 62 ± 1%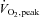 saline (CON) or saline +N-acetylcysteine (NAC) infusion. Western blots are representative for one participant from each trial at each timepoint. N= 9, #P < 0.05 vs. CON, *P < 0.05 vs. t= 0 min of same treatment.
saline (CON) or saline +N-acetylcysteine (NAC) infusion. Western blots are representative for one participant from each trial at each timepoint. N= 9, #P < 0.05 vs. CON, *P < 0.05 vs. t= 0 min of same treatment.
Muscle metabolites
NAC infusion had no affect on resting muscle metabolite concentrations (Table 1). Exercise did not affect muscle ATP concentration (P= 0.20), but resulted in an increase in muscle lactate, creatine content and calculated free ADP, free AMP and free AMP/ATP ratio (P < 0.05), and a reduction in muscle PCr and glycogen content with no significant differences between treatments (Table 1).
Table 1.
Muscle metabolites at rest and during exercise at 62 ± 1%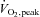 while receiving saline (CON) or saline +N-acetylcysteine (NAC) infusion
while receiving saline (CON) or saline +N-acetylcysteine (NAC) infusion
| Metabolite | Trial | 0 min | 40 min | 80 min |
|---|---|---|---|---|
| Muscle lactate (mmol (kg dry wt)−1) | CON § | 3.4 ± 0.7 | 21.7 ± 4.5 | 19.0 ± 6.1 |
| NAC § | 4.2 ± 0.5 | 12.8 ± 2.9 | 18.2 ± 3.8 | |
| PCr (mmol (kg dry wt)−1) | CON § | 94.8 ± 3.8 | 63.4 ± 5.5 | 67.2 ± 4.8 |
| NAC § | 95.6 ± 4.2 | 72.2 ± 4.5 | 67.4 ± 7.6 | |
| Cr (mmol (kg dry wt)−1) | CON § | 46.6 ± 1.7 | 78.0 ± 3.6 | 74.3 ± 4.2 |
| NAC § | 45.8 ± 2.6 | 69.2 ± 7.1 | 74.1 ± 6.4 | |
| ATP (mmol (kg dry wt)−1) | CON | 25.9 ± 0.3 | 25.2 ± 1.0 | 25.2 ± 0.8 |
| NAC | 26.2 ± 0.7 | 25.6 ± 1.1 | 23.7 ± 1.4 | |
| Free AMP (mmol (kg dry wt)−1) | CON § | 0.6 ± 0.1 | 3.2 ± 0.6 | 2.5 ± 0.4 |
| NAC § | 0.6 ± 0.1 | 2.5 ± 0.9 | 2.5 ± 0.9 | |
| Free ADP (μmol (kg dry wt)−1) | CON § | 120.8 ± 5.2 | 266.6 ± 25.1 | 239.9 ± 21.6 |
| NAC § | 115.0 ± 9.2 | 219.5 ± 34.2 | 218.1 ± 42.0 | |
| Free AMP:ATP | CON § | 0.02 ± 0.00 | 0.13 ± 0.03 | 0.09 ± 0.01 |
| NAC § | 0.02 ± 0.00 | 0.11 ± 0.05 | 0.11 ± 0.03 | |
| Muscle glycogen (mmol (kg dry wt)−1) | CON § | 373.6 ± 18.7 | 216.9 ± 16.9 | 177.8 ± 26.5 |
| NAC § | 348.5 ± 24.1 | 266.1 ± 23.6 | 206.8 ± 33.7 |
P < 0.05 for time effect, N= 9, PCr, creatine phosphate; Cr, creatine.
AMPKα and ACCβ phosphorylation
Exercise increased skeletal muscle AMPKα Thr172 and ACCβ Ser221 phosphorylation by ∼3- and ∼6-fold, respectively, and these increases were not affected by NAC infusion (Fig. 8A and B).
Figure 8. Muscle AMPK Thr172 and ACCβ Ser221 phosphorylation.

Muscle AMPK Thr172 phosphorylation (A) and ACCβ Ser221 phosphorylation (B) at rest and during 80 min of steady-state exercise at 62 ± 1%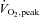 while receiving either saline or saline +N-acetylcysteine (NAC) infusion. Western blots are representative for one participant from each trial at each timepoint. N= 9, §P < 0.05 for time effect.
while receiving either saline or saline +N-acetylcysteine (NAC) infusion. Western blots are representative for one participant from each trial at each timepoint. N= 9, §P < 0.05 for time effect.
Discussion
The major finding of this study was that the systemic infusion of the antioxidant N-acetylcysteine (NAC) in humans did not affect glucose disposal during prolonged moderate-intensity exercise. In addition we show that skeletal muscle AMPK signalling during exercise is also unaffected by NAC infusion. Interestingly, we provide evidence in humans that skeletal muscle S-glutathionylation is increased during moderate-intensity exercise, despite no significant changes in muscle GSH and GSSG, and that NAC infusion prevented the increase in S-glutathionylation of a protein band at ∼270 kDa during exercise.
The antioxidant NAC has been shown previously to attenuate glucose uptake in isolated skeletal muscles contracted ex vivo (Sandstrom et al. 2006; Merry et al. 2009). In contrast, we show in this study that NAC infusion does not affect glucose disposal during exercise in humans (Fig. 5), despite preventing exercise-induced increases in muscle S-glutathionylation (the post-translational addition of glutathione to a specific cysteine residual of a protein which is promoted by oxidative stress (Dalle-Donne et al. 2009)) (Fig. 7). Furthermore, other measures of substrate utilisation during exercise such as respiratory exchange ratio (RER), muscle glycogen use, muscle metabolites, plasma lactate and plasma NEFA concentration were also not affected by NAC infusion (Table 1 and Fig. 4). This suggests that ROS signalling may not be essential for the regulation of skeletal muscle glucose uptake during moderate-intensity exercise in humans. However, although NAC infusion prevented S-glutathionylation, it does not exclude the possibility that the concentration of NAC in the muscle was insufficient to prevent all ROS signalling events. Therefore, it may have been the case that muscle NAC-enhanced cysteine levels were too low to prevent ROS effects on glucose uptake. Indeed, very high (20 mm) concentrations of NAC are used to attenuate skeletal muscle contraction-stimulated glucose uptake ex vivo (Sandstrom et al. 2006).
Our finding that NAC treatment also did not affect AMPK signalling during exercise in humans is in contrast to the previously reported finding that NAC similarly attenuates contraction-stimulated glucose uptake and AMPK activity during ex vivo skeletal muscle contraction (Sandstrom et al. 2006). High concentrations of exogenous ROS lower cell energy levels (AMP/ATP and creatine/PCr ratio) (Leon et al. 2004), activate AMPK and increase skeletal muscle glucose uptake (Toyoda et al. 2004). However, NAC infusion did not affect moderate-intensity exercise-induced lowering of cell energy in the present study, suggesting that the increase in ROS levels during moderate-intensity exercise was not sufficient to alter cell energy status (Table 1). It is possible that ex vivo muscle incubation conditions of non-uniform oxygen delivery, and highly fatiguing contraction protocols, promote higher than physiological ROS levels in skeletal muscle (Reid, 2001) which may alter cell energy balance and therefore activate AMPK. Therefore, it may be that during high-intensity exhaustive exercise/contractions when ROS production is greatly elevated and substantially increases skeletal muscle oxidative stress (Reid, 2001), ROS signalling may be involved in regulating skeletal muscle glucose uptake via AMPK.
A novel finding of this study was that muscle S-glutathionylation of a protein band at ∼270 kDa (Fig. 7A) was increased with moderate intensity exercise, and this increase was prevented by NAC infusion. Protein S-glutathionylation has been shown to be involved in redox-related regulation of cellular processes from protein folding (Demasi et al. 2008) to energy metabolism (Cotgreave et al. 2002); however, targets of S-glutathionylation during skeletal muscle contraction are yet to be investigated. The nature of the protein band at ∼270 kDa has not yet been conclusively identified. Regardless, these data provide some evidence that moderate-intensity exercise caused an increase in muscle oxidative stress and this increase was prevented by the infusion of the antioxidant NAC. Since oxidative stress causes the rapid oxidation of GSH to GSSG, the GSSG/GSH ratio is commonly used as a marker of oxidative stress (Powers & Jackson, 2008). However, it is only during moderately high (Medved et al. 2004b) (>70%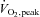 ) and strenuous (Svensson et al. 2002; Medved et al. 2004b; Zhang et al. 2007) (>80%
) and strenuous (Svensson et al. 2002; Medved et al. 2004b; Zhang et al. 2007) (>80%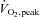 ) exercise in humans, and exercise to exhaustion in rats (Sen et al. 1994; Gomez-Cabrera et al. 2005) that ROS levels increase enough to cause detectable depletion of muscle GSH and/or increases in GSSG. Therefore, not surprisingly, and as reported previously (Ji et al. 1992; Sahlin et al. 1992), we found that muscle GSH and GSSG levels were not affected by moderate-intensity exercise. In support, skeletal muscle nitric oxide production is increased during contraction (Balon & Nadler, 1994) and NO can interact with superoxide to form peroxynitrite (ONOO−; Halliwell, 1989), which at high concentrations promotes tyrosine nitration (Halliwell, 1989). Here we report that tyrosine nitration was not increased during exercise or influenced by NAC (Fig. 7). This supports our finding of only a modest oxidative shift in muscle redox during moderate-intensity exercise.
) exercise in humans, and exercise to exhaustion in rats (Sen et al. 1994; Gomez-Cabrera et al. 2005) that ROS levels increase enough to cause detectable depletion of muscle GSH and/or increases in GSSG. Therefore, not surprisingly, and as reported previously (Ji et al. 1992; Sahlin et al. 1992), we found that muscle GSH and GSSG levels were not affected by moderate-intensity exercise. In support, skeletal muscle nitric oxide production is increased during contraction (Balon & Nadler, 1994) and NO can interact with superoxide to form peroxynitrite (ONOO−; Halliwell, 1989), which at high concentrations promotes tyrosine nitration (Halliwell, 1989). Here we report that tyrosine nitration was not increased during exercise or influenced by NAC (Fig. 7). This supports our finding of only a modest oxidative shift in muscle redox during moderate-intensity exercise.
Although NAC can directly scavenge ROS (Aruoma et al. 1989), the primary antioxidant properties of NAC are derived from its rapid deacetylation to cysteine (Deneke, 2000), a precursor to GSH synthesis (Sen et al. 1992). The increased bioavailability of cysteine enhances antioxidant defences by promoting the regeneration of GSH (Sen et al. 1992) and reduces ROS by direct scavenging (Cotgreave, 1997). As reported previously (Medved et al. 2004b), systemic infusion of NAC into humans during exercise elevates skeletal muscle NAC content and increases plasma and muscle cysteine concentration (Fig. 2). However, NAC infusion did not affect skeletal muscle GSH or GSSG/GSH ratio. In agreement, NAC generally only increases muscle GSH availability under conditions where muscle GSH oxidation is elevated (Sandstrom et al. 2006), such as during strenuous exercise/muscle contraction (Medved et al. 2004b; Sandstrom et al. 2006). Therefore, it is likely that NAC did not affect muscle GSH or GSSG concentration in the current study because GSH was not depleted by moderate-intensity exercise.
In conclusion, although skeletal muscle glutathione balance was not affected by moderate-intensity exercise, S-glutathionylation of a protein band of ∼270 kDa was increased and this increase was prevented by NAC infusion. Since glucose disposal during exercise was not attenuated by NAC, this study provides evidence to suggest that small to moderate increases in ROS levels during moderate-intensity exercise in humans may not be involved in the regulation of skeletal muscle glucose disposal or AMPK signalling. This provides evidence to suggest previous results obtained using intense ex vivo contractions may not always be relevant to normal prolonged exercise.
Acknowledgments
The authors would like to thank Professor Michael McKenna for his expert advice, Miss Eloise Bradley and Miss Sophie Yeo for their excellent technical assistance and the participants for their commitment.
Glossary
Abbreviations
- ACCβ
acetyl-CoA carboxylase-β
- AICAR
5-amino-imidazole 4-carboxamide ribonucleoside
- CaMK
calcium/calmodulin-dependent kinase
- GSH
reduced glutathione
- GSSG
oxidised glutathione
- NAC
N-acetylcysteine
- NEFA
non-esterified fatty acids
- PVDF
polyvinylidene difluoride membrane
- Ra
appearance rate
- Rd
disappearance rate
- ROS
reactive oxygen species
Author contributions
T.L.M., M.H. and G.K.M. contributed to the conception and design of experiments. T.L.M., G.D.W., C.G.S., A.P.G., S.R., M.H. and G.K.M. contributed to the execution, analysis and interpretation of experiments. T.L.M. wrote the initial draft of the manuscript and G.D.W., M.H. and G.M. contributed to writing and revising the manuscript. All authors approved the final version of the manuscript.
Author's present address
G.K. McConell: Institute of Sport, Exercise and Active Living (ISEAL) and Biomedical and Health Sciences, Victoria University, Footscray, Victoria, Australia.
References
- Allen DG, Lamb GD, Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol Rev. 2008;88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med. 1989;6:593–597. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- Balon TW, Nadler JL. Nitric oxide release is present from incubated skeletal muscle preparations. J Appl Physiol. 1994;77:2519–2521. doi: 10.1152/jappl.1994.77.6.2519. [DOI] [PubMed] [Google Scholar]
- Borg GA. Perceived exertion. Exerc Sport Sci Rev. 1974;2:131–153. [PubMed] [Google Scholar]
- Bradley SJ, Kingwell BA, McConell GK. Nitric oxide synthase inhibition reduces leg glucose uptake but not blood flow during dynamic exercise in humans. Diabetes. 1999;48:1815–1821. doi: 10.2337/diabetes.48.9.1815. [DOI] [PubMed] [Google Scholar]
- Chen Z-P, McConell GK, Michell BJ, Snow RJ, Canny BJ, Kemp BE. AMPK signalling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. Am J Physiol Endocrinol Metab. 2000;279:E1202–E1206. doi: 10.1152/ajpendo.2000.279.5.E1202. [DOI] [PubMed] [Google Scholar]
- Chen Z-P, Stephens TJ, Murthy S, Canny BJ, Hargreaves M, Witters LA, Kemp BE, McConell GK. Effect of exercise intensity on skeletal muscle AMPK signalling in humans. Diabetes. 2003;52:2205–2212. doi: 10.2337/diabetes.52.9.2205. [DOI] [PubMed] [Google Scholar]
- Cotgreave IA. N-acetylcysteine: pharmacological considerations and experimental and clinical applications. Adv Pharmacol. 1997;38:205–227. [PubMed] [Google Scholar]
- Cotgreave IA, Gerdes R, Schuppe-Koistinen I, Lind C. S-glutathionylation of glyceraldehyde-3-phosphate dehydrogenase: role of thiol oxidation and catalysis by glutaredoxin. Methods Enzymol. 2002;348:175–182. doi: 10.1016/s0076-6879(02)48636-3. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Demasi M, Piassa Filho GM, Castro LM, Ferreira JC, Rioli V, Ferro ES. Oligomerization of the cysteinyl-rich oligopeptidase EP24.15 is triggered by S-glutathionylation. Free Radic Biol Med. 2008;44:1180–1190. doi: 10.1016/j.freeradbiomed.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Deneke SM. Thiol-based antioxidants. Curr Top Cell Regul. 2000;36:151–180. doi: 10.1016/s0070-2137(01)80007-8. [DOI] [PubMed] [Google Scholar]
- Derave W, Ai H, Ihlemann J, Witters LA, Kristiansen S, Richter EA, Ploug T. Dissociation of AMP-activated protein kinase activation and glucose transport in contracting slow-twitch muscle. Diabetes. 2000;49:1281–1287. doi: 10.2337/diabetes.49.8.1281. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funai K, Cartee GD. Contraction-stimulated glucose transport in rat skeletal muscle is sustained despite reversal of increased PAS-phosphorylation of AS160 and TBC1D1. J Appl Physiol. 2008;105:1788–1795. doi: 10.1152/japplphysiol.90838.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funai K, Cartee GD. Inhibition of contraction-stimulated AMPK inhibits contraction-stimulated increases in PAS-TBC1D1 and glucose transport without altering PAS-AS160 in rat skeletal muscle. Diabetes. 2009;58:1096–1104. doi: 10.2337/db08-1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Cabrera MC, Borras C, Pallardo FV, Sastre J, Ji LL, Vina J. Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J Physiol. 2005;567:113–120. doi: 10.1113/jphysiol.2004.080564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B. Free radicals, reactive oxygen species and human disease: a critical evaluation with special reference to atherosclerosis. Br J Exp Pathol. 1989;70:737–757. [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ. Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes. 1998;47:1369–1373. doi: 10.2337/diab.47.8.1369. [DOI] [PubMed] [Google Scholar]
- Higaki Y, Mikami T, Fujii N, Hirshman MF, Koyama K, Seino T, Tanaka K, Goodyear LJ. Oxidative stress stimulates skeletal muscle glucose uptake through a phosphatidylinositol 3-kinase-dependent pathway. Am J Physiol Endocrinol Metab. 2008;294:E889–E897. doi: 10.1152/ajpendo.00150.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TE, Schjerling P, Viollet B, Wojtaszewski JF, Richter EA. AMPKα1 activation is required for stimulation of glucose uptake by twitch contraction, but not by H2O2, in mouse skeletal muscle. PLoS ONE. 2008;3:e2102. doi: 10.1371/journal.pone.0002102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeukendrup AE, Raben A, Gijsen A, Stegen JH, Brouns F, Saris WH, Wagenmakers AJ. Glucose kinetics during prolonged exercise in highly trained human subjects: effect of glucose ingestion. J Physiol. 1999;515:579–589. doi: 10.1111/j.1469-7793.1999.579ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji LL, Fu R, Mitchell EW. Glutathione and antioxidant enzymes in skeletal muscle: effects of fibre type and exercise intensity. J Appl Physiol. 1992;73:1854–1859. doi: 10.1152/jappl.1992.73.5.1854. [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JFP. Knockout of the α2 but not α1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- Kennedy JW, Hirshman MF, Gervino EV, Ocel JV, Forse RA, Hoenig SJ, Aronson D, Goodyear LJ, Horton ES. Acute exercise induces GLUT4 translocation in skeletal muscle of normal human subjects and subjects with type 2 diabetes. Diabetes. 1999;48:1192–1197. doi: 10.2337/diabetes.48.5.1192. [DOI] [PubMed] [Google Scholar]
- Lee-Young RS, Griffee SR, Lynes SE, Bracy DP, Ayala JE, McGuinness OP, Wasserman DH. Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. J Biol Chem. 2009;284:23925–23934. doi: 10.1074/jbc.M109.021048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon H, Atkinson LL, Sawicka J, Strynadka K, Lopaschuk GD, Schulz R. Pyruvate prevents cardiac dysfunction and AMP-activated protein kinase activation by hydrogen peroxide in isolated rat hearts. Can J Physiol Pharmacol. 2004;82:409–416. doi: 10.1139/y04-050. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Passonneau JV. A Flexible System of Enzymatic Analysis. New York: Academic; 1972. [Google Scholar]
- McConell G, Fabris S, Proietto J, Hargreaves M. Effect of carbohydrate ingestion on glucose kinetics during exercise. J Appl Physiol. 1994;77:1537–1541. doi: 10.1152/jappl.1994.77.3.1537. [DOI] [PubMed] [Google Scholar]
- McConell GK, Huynh NN, Lee-Young RS, Canny BJ, Wadley GD. L-Arginine infusion increases glucose clearance during prolonged exercise in humans. Am J Physiol Endocrinol Metab. 2006;290:E60–E66. doi: 10.1152/ajpendo.00263.2005. [DOI] [PubMed] [Google Scholar]
- McConell GK, Lee-Young RS, Chen Z-P, Stepto NK, Huynh NN, Stephens TJ, Canny BJ, Kemp BE. Short-term exercise training in humans reduces AMPK signalling during prolonged exercise independent of muscle glycogen. J Physiol. 2005;568:665–676. doi: 10.1113/jphysiol.2005.089839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConell GK, Manimmanakorn A, Lee-Young RS, Kemp BE, Linden KC, Wadley GD. Differential attenuation of AMPK activation during acute exercise following exercise training or AICAR treatment. J Appl Physiol. 2008;105:1422–1427. doi: 10.1152/japplphysiol.01371.2007. [DOI] [PubMed] [Google Scholar]
- Maarbjerg SJ, Jorgensen SB, Rose AJ, Jeppesen J, Jensen TE, Treebak JT, Birk JB, Schjerling P, Wojtaszewski JF, Richter EA. Genetic impairment of α2-AMPK signaling does not reduce muscle glucose uptake during treadmill exercise in mice. Am J Physiol Endocrinol Metab. 2009;297:E924–E934. doi: 10.1152/ajpendo.90653.2008. [DOI] [PubMed] [Google Scholar]
- Medved I, Brown MJ, Bjorksten AR, Leppik JA, Sostaric S, McKenna MJ. N-acetylcysteine infusion alters blood redox status but not time to fatigue during intense exercise in humans. J Appl Physiol. 2003;94:1572–1582. doi: 10.1152/japplphysiol.00884.2002. [DOI] [PubMed] [Google Scholar]
- Medved I, Brown MJ, Bjorksten AR, McKenna MJ. Effects of intravenous N-acetylcysteine infusion on time to fatigue and potassium regulation during prolonged cycling exercise. J Appl Physiol. 2004a;96:211–217. doi: 10.1152/japplphysiol.00458.2003. [DOI] [PubMed] [Google Scholar]
- Medved I, Brown MJ, Bjorksten AR, Murphy KT, Petersen AC, Sostaric S, Gong X, McKenna MJ. N-acetylcysteine enhances muscle cysteine and glutathione availability and attenuates fatigue during prolonged exercise in endurance-trained individuals. J Appl Physiol. 2004b;97:1477–1485. doi: 10.1152/japplphysiol.00371.2004. [DOI] [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol Endocrinol Metab. 1997;273:E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- Merry TL, McConell GK. Skeletal muscle glucose uptake during exercise: a focus on reactive oxygen species and nitric oxide signaling. IUBMB Life. 2009;61:479–484. doi: 10.1002/iub.179. [DOI] [PubMed] [Google Scholar]
- Merry TL, Steinberg GR, Lynch GS, McConell GK. Skeletal muscle glucose uptake during contraction is regulated by nitric oxide and ROS independently of AMPK. Am J Physiol Endocrinol Metab. 2009;298:E577–E585. doi: 10.1152/ajpendo.00239.2009. [DOI] [PubMed] [Google Scholar]
- Musi N, Hayashi T, Fujii N, Hirshman MF, Witters LA, Goodyear LJ. AMP-activated protein kinase activity and glucose uptake in rat skeletal muscle. Am J Physiol Endocrinol Metab. 2001;280:E677–E684. doi: 10.1152/ajpendo.2001.280.5.E677. [DOI] [PubMed] [Google Scholar]
- Passonneau JV, Lauderdale VR. A comparison of three methods of glycogen measurement in tissues. Anal Biochem. 1974;60:405–412. doi: 10.1016/0003-2697(74)90248-6. [DOI] [PubMed] [Google Scholar]
- Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radziuk J, Norwich KH, Vranic M. Experimental validation of measurements of glucose turnover in nonsteady state. Am J Physiol Endocrinol Metab. 1978;234:E84–E93. doi: 10.1152/ajpendo.1978.234.1.E84. [DOI] [PubMed] [Google Scholar]
- Reid MB. Invited review: Redox modulation of skeletal muscle contraction: what we know and what we don’t. J Appl Physiol. 2001;90:724–731. doi: 10.1152/jappl.2001.90.2.724. [DOI] [PubMed] [Google Scholar]
- Reid MB. Free radicals and muscle fatigue: of ROS, canaries, and the IOC. Free Radic Biol Med. 2008;44:169–179. doi: 10.1016/j.freeradbiomed.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Reid MB, Haack KE, Franchek KM, Valberg PA, Kobzik L, West MS. Reactive oxygen in skeletal muscle. I. Intracellular oxidant kinetics and fatigue in vitro. J Appl Physiol. 1992a;73:1797–1804. doi: 10.1152/jappl.1992.73.5.1797. [DOI] [PubMed] [Google Scholar]
- Reid MB, Shoji T, Moody MR, Entman ML. Reactive oxygen in skeletal muscle. II. Extracellular release of free radicals. J Appl Physiol. 1992b;73:1805–1809. doi: 10.1152/jappl.1992.73.5.1805. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Richter EA. Skeletal muscle glucose uptake during exercise: how is it regulated? Physiology (Bethesda) 2005;20:260–270. doi: 10.1152/physiol.00012.2005. [DOI] [PubMed] [Google Scholar]
- Ross RM, Wadley GD, Clark MG, Rattigan S, McConell GK. Local nitric oxide synthase inhibition reduces skeletal muscle glucose uptake but not capillary blood flow during in situ muscle contraction in rats. Diabetes. 2007;56:2885–2892. doi: 10.2337/db07-0745. [DOI] [PubMed] [Google Scholar]
- Sahlin K, Cizinsky S, Warholm M, Hoberg J. Repetitive static muscle contractions in humans – a trigger of metabolic and oxidative stress? Eur J Appl Physiol Occup Physiol. 1992;64:228–236. doi: 10.1007/BF00626285. [DOI] [PubMed] [Google Scholar]
- Sandstrom ME, Zhang SJ, Bruton J, Silva JP, Reid MB, Westerblad H, Katz A. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J Physiol. 2006;575:251–262. doi: 10.1113/jphysiol.2006.110601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen CK, Atalay M, Hanninen O. Exercise-induced oxidative stress: glutathione supplementation and deficiency. J Appl Physiol. 1994;77:2177–2187. doi: 10.1152/jappl.1994.77.5.2177. [DOI] [PubMed] [Google Scholar]
- Sen CK, Marin E, Kretzschmar M, Hanninen O. Skeletal muscle and liver glutathione homeostasis in response to training, exercise, and immobilization. J Appl Physiol. 1992;73:1265–1272. doi: 10.1152/jappl.1992.73.4.1265. [DOI] [PubMed] [Google Scholar]
- Steele R, Wall JS, De Bodo RC, Altszuler N. Measurement of size and turnover rate of body glucose pool by the isotope dilution method. Am J Physiol. 1956;187:15–24. doi: 10.1152/ajplegacy.1956.187.1.15. [DOI] [PubMed] [Google Scholar]
- Svensson MB, Ekblom B, Cotgreave IA, Norman B, Sjoberg B, Ekblom O, Sjodin B, Sjodin A. Adaptive stress response of glutathione and uric acid metabolism in man following controlled exercise and diet. Acta Physiol Scand. 2002;176:43–56. doi: 10.1046/j.1365-201X.2002.01008.x. [DOI] [PubMed] [Google Scholar]
- Toyoda T, Hayashi T, Miyamoto L, Yonemitsu S, Nakano M, Tanaka S, Ebihara K, Masuzaki H, Hosoda K, Inoue G, Otaka A, Sato K, Fushiki T, Nakao K. Possible involvement of the α1 isoform of 5′AMP-activated protein kinase in oxidative stress-stimulated glucose transport in skeletal muscle. Am J Physiol Endocrinol Metab. 2004;287:E166–E173. doi: 10.1152/ajpendo.00487.2003. [DOI] [PubMed] [Google Scholar]
- Wadley GD, Lee-Young RS, Canny BJ, Wasuntarawat C, Chen ZP, Hargreaves M, Kemp BE, McConell GK. Effect of exercise intensity and hypoxia on skeletal muscle AMPK signalling and substrate metabolism in humans. Am J Physiol Endocrinol Metab. 2006;290:E694–E702. doi: 10.1152/ajpendo.00464.2005. [DOI] [PubMed] [Google Scholar]
- Witczak CA, Fujii N, Hirshman MF, Goodyear LJ. Ca2+/calmodulin-dependent protein kinase kinase-α regulates skeletal muscle glucose uptake independent of AMP-activated protein kinase and Akt activation. Diabetes. 2007;56:1403–1409. doi: 10.2337/db06-1230. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Sandstrom ME, Lanner JT, Thorell A, Westerblad H, Katz A. Activation of aconitase in mouse fast-twitch skeletal muscle during contraction-mediated oxidative stress. Am J Physiol Cell Physiol. 2007;293:C1154–C1159. doi: 10.1152/ajpcell.00110.2007. [DOI] [PubMed] [Google Scholar]
- Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF, Virkamaki A, Goodyear LJ, Kahn CR, Kahn BB. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med. 2000;6:924–928. doi: 10.1038/78693. [DOI] [PubMed] [Google Scholar]