

Abstract
N-acetylglutamate (NAG) is a unique enzyme cofactor, essential for liver ureagenesis in mammals while it is the first committed substrate for de novo arginine biosynthesis in microorganisms and plants. The enzyme that produces NAG from glutamate and CoA, NAG synthase (NAGS), is allosterically inhibited by arginine in microorganisms and plants and activated in mammals. This transition of the allosteric effect occurred when tetrapods moved from sea to land. The first mammalian NAGS gene (from mouse) was cloned in 2002 and revealed significant differences from the NAGS ortholog in microorganisms. Almost all NAGS genes possess a C-terminus transferase domain in which the catalytic activity resides and an N-terminus kinase domain where arginine binds. The three-dimensional structure of NAGS shows two distinctly folded domains. The kinase domain binds arginine while the acetyltransferase domain contains the catalytic site. NAGS deficiency in humans leads to hyperammonemia and can be primary, due to mutations in the NAGS gene or secondary due to other mitochondrial aberrations that interfere with the normal function of the same enzyme. For either condition, N-carbamylglutamate (NCG), a stable functional analog of NAG, was found to either restore or improve the deficient urea cycle function.
Keywords: urea cycle, ureagenesis, N-carbamylglutamate, arginine, hyperammonemia, regulation
Historical and evolutionary aspects of NAGS
The existence of N-acetylglutamate synthase (NAGS; EC 2.3.1.1), a liver enzyme that catalyzes formation of N-acetylglutamate (NAG) from glutamate and acetyl coenzyme A was inferred after NAG was identified as an essential cofactor necessary for the function of the urea cycle [1,2]. The gene for mammalian NAGS was identified almost 50 years later, probably due to poor evolutionary conservation of the NAGS protein sequence compared to other urea cycle enzymes.
The urea cycle in mammals evolved from the arginine biosynthesis pathway in microbes; the two systems having in common six enzymes and their substrates and products [3,4]. In microbes, NAGS catalyzes the formation of the first committed intermediate of arginine biosynthesis, while in mammals NAGS produces an essential cofactor for CPSI, the first and rate-limiting enzyme of the urea cycle. Although NAGS was purified from human and rat liver almost three decades ago [5–7], not until 2002 where we able to use bioinformatic tools to identify and subsequently clone mammalian NAGS genes based on limited sequence similarities to Neurospora crassa NAGS [8,9].
Alignments of amino acid sequences from six mammalian NAGS genes revealed three regions with different degrees of sequence conservation: the mitochondrial targeting signal (MTS), the variable segment and the conserved segment (Figure 1). When mouse NAGS preprotein was expressed in insect cells, it was processed at two sites. Cleavage of the MTS resulted in mature NAGS (NAGS-M), while removal of both the MTS and variable segment resulted in conserved NAGS (NAGS-C). Recombinant NAGS-M and NAGS-C are both catalytically active, the affinities of NAGS-M and NAGS-C for substrates are similar while the maximal velocity of NAGS-C is about two-fold higher than that of NAGS-M [10].
Figure 1.

Conservation of mammalian NAGS. Logo representation of the conservation of amino acids among NAGS from human, mouse, rat, dog, horse and cow. The letter size is proportional to the degree of conservation and the color indicates the type of amino acid. Post-translational processing sites found upon expression of mouse NAGS in insect cells are indicated by arrows. MTS - mitochondrial targeting sequence.
Sequencing of genomes from organisms spanning a wide variety of phyla has allowed us to identify NAGS in these organisms based on their similarity to either human and mouse or bacterial NAGS. These searches identified several Xanthomonadales and marine α-proteobacteria with highly similar gene sequence to mammalian NAGS [11]. To confirm that these bacterial sequences were indeed more closely related to mammalian NAGS than to other bacterial NAGS encoding genes, we carried out a phylogenetic analysis of NAGS sequences from 31 organisms including bacteria, fungi, amoebae, plants and vertebrates. Four fungal NAGK were also included in the analysis based on the previously noted similarity between N. crassa NAGK and E. coli NAGS [12]. NAGS and NAGK sequences clustered in two groups. One group contained NAGS from vertebrates, fungi, amoebae, and three species of Xanthomonadales and marine α-proteobacteria, indicating that these genes were more closely related to each other than to other bacterial and plant NAGS clustered in the second group [11]. Examination of two NAGS and NAGK-like gene sequences (argA and argB) in the arginine operon of Xanthomonads, followed by enzymatic assays with their respective purified proteins have revealed that the Xanthomonad argB encoded enzyme can catalyze the first two reactions of arginine biosynthesis. We therefore termed this enzyme N-acetylglutamate synthase-kinase (NAGS-K).
Arginine differentially affects the enzymatic activity of NAGS from plants, microbes and vertebrates. Microbial NAGS (including Xanthomonad NAGS-K that are more similar to mammalian NAGS than other bacterial NAGS) and plant NAGS are inhibited by arginine as part of feedback inhibition of arginine biosynthesis [4,11]. Mammalian NAGS on the other hand, are activated by arginine [5,6,7,10]. This indicates that an allosteric inversion of the effect of arginine on the activity of NAGS enzymes occurred during evolution. In spite of this allosteric inversion, sequence alignments of NAGS proteins show that the arginine binding site has been conserved across all phyla, and site-directed mutagenesis revealed that arginine binds at the same site in Xanthomonas campestris NAGS-K, which is inhibited by arginine, as in mouse NAGS, which is activated by the same amino acid [13]. This suggests that binding of arginine induces conformational changes or alters protein dynamics and results in the opposite effects on enzymatic activity of bacterial and mammalian NAGS as will be discussed in the structural studies described below. To determine when this allosteric inversion occurred during evolution, we examined the effect of arginine on the enzymatic activity of fish and amphibian NAGS. The activity of NAGS from zebrafish (Danio rerio) and pufferfish (Fugu rubripes) was partially inhibited by arginine, while the effect of arginine on the respective activity of NAGS from the amphibians, Xenopus laevis and Xenopus tropicalis, ranged from neutral to slight activation [13]. Therefore, inversion of the allosteric effect of arginine on NAGS occurred in amphibians and coincided with the transition from CPSIII to CPSI and conquest of land by tetrapods. Since mammalian NAGS proteins are activated by arginine, this amino acid could regulate ureagenesis by modulating the activity of NAGS, which then produces variable amounts of NAG for activation of variable number of CPSI molecules. The combined activation of NAGS by arginine and activation of CPSI by NAG forms a double positive feedback loop, which allows rapid and robust regulation of metabolic and signaling pathways [14]. This has resulted in a robust ammonia detoxification system, which effectively protects the central nervous system of land tetrapods from neurotoxic effects of hyperammonemia.
NAGS structure and catalytic and regulatory mechanisms
Until recently, no structure of any NAGS protein was available due to the recalcitrant nature of the protein to crystallization and poor X-ray diffraction quality. Last year, Shi and colleagues were able to crystallize and solve the three dimensional NAGS structure from Neissria gonohorreae (ngNAGS), which is closely related to E. coli and other bacterial NAGS proteins (50–60% sequence similarity), but has low sequence similarity (20–30%) to mammalian NAGS. [15,16]. Four crystal structures, at high resolution (better than 2.5 Å), with the substrates (CoA, AcCoA, L-glutamate and NAG) and the bound allosteric effector (L-arginine), provided insights into the catalytic and arginine regulatory mechanisms of the NAGS enzyme. The coordinates and structural factors have been deposited in the Protein Data Bank (accession codes: 2R8V, 3B8G, 2R98, 3D2P and 3D2M) for public access.
Monomer structure
The monomer of ngNAGS consists of two independently folded domains, an N-terminal amino acid kinase (AAK) domain and a C-terminal N-acetyltransferase (NAT) domain, connected by a 3 amino acid (~10 Å) linker (Fig. 2A). The AAK domain contains two lobes and has a typical AAK fold with a central 8-stranded parallel β sheet sandwiched between α helices (Fig. 2B). The N-terminal lobe forms the inter-subunit interfaces while the C-terminal lobe hosts the arginine binding site. The NAT domain has a typical GCN5-related fold with a central anti-parallel β sheet flanked by α helices. The central β sheet is divided into two arms that form a “V” shape, with substrate (CoA, AcCoA, L-Glutamate or NAG) binding between the two arms (Fig. 2C). The allosteric effector, L-arginine, binds to the C-terminal end of AAK domain at a location that is approximately 25 Å away from the catalytic site. Without bound arginine, the AAK and NAT domains do not interact within a single monomer. However, upon binding of arginine, the NAT domain undergoes marked reorientation relative to the AAK domain allowing them to interact (Fig. 3). This structural model clearly indicates that the catalytic site is located within the NAT domain and rules out the earlier speculation that L-glutamate binds to the AAK domain of the enzyme [17].
Figure 2.

Structure of N. gonorrhoeae NAGS. Ribbon diagram of a monomer (A), and its AAK (B) and NAT (C) domains. Green arrows indicate the direction of strands in β-sheets, α-helices are in red and β-sheets are in green. AcCoA is represented as a stick model.
Figure 3.
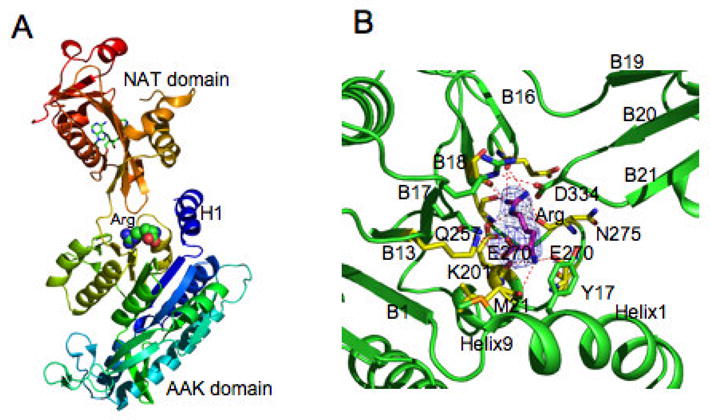
N. gonorrhoeae NAGS L-arginine binding site in the T-state structure. A. Ribbon diagram of T-state monomer. Ribbons are shown in rainbow colors from blue (N-terminal) to red (C-terminal). Bound L-arginine is represented as space-filling models. Bound CoA is shown as green sticks. B. Arginine binding site. Electron density maps (2Fo-Fc) are shown as a blue cage contoured at 1.0σ. Carbon atoms of L-arginine are shown as pink sticks. Carbon atoms of the side chains to interacts with L-arginine directly and indirectly are shown as yellow and green sticks, respectively. Hydrogen bonds between bound ligands and protein are indicated by red dashed lines.
Quaternary structure
The crystal structures indicate that six monomers of ngNAGS assemble together as the minimal functioning unit by forming a hexameric ring via the AAK domains (Fig. 4) with exact 32-point symmetry. The C-terminal NAT domain interacts with the AAK domain from the adjacent subunit as well. Even though the AAK domain is not involved in the catalysis, it is required for maintaining the NAT domain in the correct conformation for optimal catalytic activity.
Figure 4.

Molecular hexamer of N. gonorrhoeae NAGS. Simplified model showing the quaternary interactions of the subunits. Two dimer interfaces across the two-fold axes between AAK domain K1 and K4, and K1 and K5, and one interface across the three-fold axis between the NAT domain S1 and the AAK domain K2 are clearly visible. Different subunits are shown in different colors.
Analytical gel chromatography demonstrated that ngNAGS is a hexamer in solution as in crystal form, regardless of enzyme concentration and binding status of arginine. Thus, there is strong evidence that the basic functional unit for the “classical” version of bacterial NAGS is a hexamer. Our results do not support the earlier studies on NAGS from E. coli, which proposed that a trimer is the smallest active unit and that the enzyme oligomeric states depended on its concentration and the presence of substrates or inhibitor [18]. Since the primary sequences of NAGS from E. coli and N. gonohorreae are highly similar (55% similarity) and all of the key residues for substrate and L-arginine binding are identical, it is likely these homologous proteins function in identical oligomeric states.
The hexameric assembly of ngNAGS creates three different types of interface. Types K1:K5 and K1:K4 (Fig. 4), are created through AAK-AAK domain interactions and are similar to those found in L-arginine sensitive NAGK structures, while type K1:K4 interface is common for both L-arginine sensitive and insensitive NAGK such as E. coli NAGK [19]. The dimer formed through K1:K4 interface interaction is quite rigid and, upon L-arginine binding, it rotates as one unit. The third interface (K2:S1) is formed through AAK-NAT domain interactions and is unique to ngNAGS. Upon L-arginine binding, this interface changes completely, implying its critical role in signal transduction from the L-arginine binding site to the catalytic site.
Substrate binding site
The crystal structures of liganded ngNAGS identified sequence motifs of glutamate and AcCoA binding in bacterial and plant NAGS. Both AcCoA and L-glutamate clearly bind to the NAT domain in the cleft between the two arms (Fig. 2C). Two stretches of amino acid residues (393–398 and 355–369) are involved in binding AcCoA, mainly using main-chain nitrogen or oxygen atoms for hydrogen bonding interactions (Table 1). The sequence Gln364-Glu365-Gly366-Gly367-Tyr368-Gly369, which conforms to the (Arg/Gln)-Xaa-Xaa-Gly-Xaa-(Gly/Ala) motif for AcCoA recognition in the GCN5-related NAT super family [20], is involved in binding the pyrophosphate group of AcCoA. The slightly modified motif of (Arg/Gln)-Xaa-Xaa-(Gly/Ser)-Xaa-Gly can be found in all “classical” bacterial and plant NAGS sequences.
Table 1.
Interactions between AcCoA and protein atoms
| AcCoA atoms | Protein atoms | Distance (Å) |
|---|---|---|
| O | Cys356 N | 3.39 |
| Leu357 N | 3.03 | |
| NBS | Leu357 O | 2.54 |
| OBR | Thr395 OG1 | 2.47 |
| OBM | Val359 N | 3.48 |
| OBK | Gln364 OE1 | 3.03 |
| OBD | Asp365 N | 2.79 |
| Wat58 OH | 2.70 | |
| OBC | Gly369 N | 2.82 |
| War52 OH | 2.72 | |
| OAZ | Glu370 N | 3.24 |
| OAY | Gly367 N | 2.98 |
| Wat71 OH | 2.97 | |
| O5 | Trp398 NE1 | 3.05 |
| O2 | Arg151§ NE | 3.51 |
| O3 | Arg151§ NE | 3.61 |
| OAQ | Lys152§ NZ | 3.62 |
| N1 | Glu397 OE1 | 2.66 |
| N6 | Glu394 O | 3.28 |
residues from the K domain of an adjacent monomer (symmetry operation: −y+1, x−y, z).
A second motif (Trp-Xaa-Xaa-Arg) is also conserved in all “classical” bacterial and plant NAGS sequences. The side chain of Trp398, which is located within the active site near the adenosine and pyrophosphate groups of AcCoA, appears to be important in binding of AcCoA and maintaining it in the correct conformation. Similarly, the side chain of Phe399, which has hydrophobic interaction with the acetyl group of AcCoA, is crucial for “fixing” the acetyl group for the catalytic reaction.
Since the acetyl group of AcCoA is located deep in the protein core, L-glutamate must enter the protein from the opposite side of the protein through a channel that provides the acetyl group of AcCoA with access to the solvent. The channel is formed by H10–H11 and H14–β25 loops, and part of β21 and β22 strands. Within the channel, side chains of residue Arg312, Arg425 and Ser427, and the main chain nitrogen atoms of Cys356 and Leu314 are involved in binding the two carboxyl groups of L-glutamate. The L-glutamate binding residue, Arg312, Arg425 and Ser427, are conserved in all “classical” bacterial and plant NAGS sequences.
L-Arginine binding site
L-Arginine binds to the C-terminal end of AAK domain [16]. The binding site is formed by the central β-sheet (strand B13, B17, and B18), the C-terminal segment of the N-terminal helix, and the loop connecting helix H9 and strand B18 (residues 270–280) (Fig. 3B). The amino acid sequences for the L-arginine binding loop (residue 270–280) is highly conserved across all known NAGS proteins. This universality of the L-arginine binding site is supported by the mutagenesis studies of this region in mouse NAGS and bifunctional X. campestris NAGS/K [13] and Pseudomonas aeruginosa NAGS [21] and NAGK [22].
Catalytic mechanism
The ngNAGS structures clearly rule out a two-step ping-pong mechanism of catalysis. A two-step mechanism uses a cysteine as a bridge to transfer the acetyl group from AcCoA to the amino group of L-glutamate. However, in the crystal structures, the closest cysteine (Cys356) is 8.4 Å away from the sulfur atom of AcCoA, too far for a covalently-linked intermediate. In contrast, the α-amino nitrogen atom of L-glutamate is about 2.5–3.1 Å from the carbon atom of acetyl group, poised to attack the acetyl group directly [15,16]. During the reaction, the carbonyl bond is polarized by two strong hydrogen bonding interactions with the main chain nitrogen atoms of Cys356 and Leu357, and the tetrahedral intermediate is in S-configuration. Upon formation of the reaction products (CoA and NAG), the sulfur atom of CoA moves ~0.9 Å toward the side chain of Ser392 and the side chain of Arg316 moves away from the active site which then allows NAG to dissociate from the protein.
Regulatory mechanism
The allosteric binding site for L-arginine is close to the C-terminal end of the AAK domain and is more than 25 Å away from the active site within the NAT domain. Even though the L-arginine binding site in ngNAGS is similar to the respective site in arginine sensitive NAGK structures [17], the allosteric mechanism in NAGS is likely to be different from that of NAGK where the active site is close to the L-arginine binding site and within the same AAK domain and the inhibition mechanism was proposed to involve the enlargement of the active site upon L-arginine binding [17].
In ngNAGS, arginine binding causes a series of structural rearrangements, including tightening the arginine binding site, reorienting the N-terminal helix, rotating (~4°) and moving the K1:K4 dimer outwards (~5 Å) and ordering the H4–B5 loop to enhance the K1:K5 interface interactions. In addition, the synthase NAT domain is rotated relative to the AAK domain (~109°) to change domain-domain interactions within the subunit (K1:S1) and between subunits (K2:S1), disordering the glutamate binding loops (H10–H11 and H14–B25) and reducing the affinity of the enzyme for glutamate. Arginine binding also induces global conformational changes that increase the diameter of the hexamer by ~10 Å and decreases its height by ~20 Å. Kinetics studies of ngNAGS support the proposed regulatory mechanism of arginine that reduces the affinity of the enzyme for L-glutamate.
Recent kinetic and mutagenesis studies of P. aeruginosa NAGS further support the observations that L-arginine inhibits the NAGS activity by reducing the substrate binding and catalytic efficiency through increasing Km of L-glutamate, decreasing the Vmax, and triggering substrate inhibition by AcCoA [23]. These and mutagenesis studies also demonstrated that the length of inter-domain linker plays an important role in transduction of arginine regulation signals from its binding site in the AAK domain to the active site in the NAT domain.
Vertebrate NAGS structural model
Primary sequence analysis classified all known NAGS into two major groups in a phylogenetic tree [11]. Within the group that contains bacterial and plant NAGS, all active site residues involved in binding substrates and catalysis are conserved. The members of this group are likely to have similar active site structures and quaternary hexamer arrangements. The vertebrate NAGS, along with fungal NAGS and bacterial bifunctional NAGS/K, belong to a different group. The amino acid sequence similarity between these two groups is very low (20–30%), particularly for the NAT domain, implying the NAT domains in two major groups evolved from different ancestor proteins.
NAGS in the vertebrate-containing group, appears to have a similar two-domain structure to ngNAGS, and the CoA binding motif, Gln-Xaa-Xaa-Gly-Xaa-Gly, can be clearly identified in the sequence [24]. However, a sequence alignment between NAGS in the two groups indicates that the key residues in the L-glutamate binding site are not conserved for NAGS in vertebrates. This implies that either NAGS in the vertebrate branch uses different residues for L-glutamate binding or that the L-glutamate binding site is in a different location. The possibility for a shared active site by AAK and NAT domains in the NAGS of vertebrates has been proposed previously [24] and cannot be ruled out until a vertebrate NAGS structure becomes available.
Clinical and therapeutic aspects of NAGS deficiency
Clinical features and diagnosis of NAGS deficiency
NAGS deficiency is the rarest [25] and most-recently identified urea cycle disorder [26]. The age of onset of clinical symptoms is variable and ranges from early neonatal hyperammonemia to presentation in the fifth decade [27–30], with approximately half of all reported cases presenting in the neonatal period [31].
NAGS and CPSI deficiency are indistinguishable by clinical and/or biochemical parameters alone, as both conditions result in decreased flux through the CPSI reaction. Biochemically, plasma ammonia and glutamine are increased, whereas the concentrations of other urea-cycle intermediates are low to normal, and urinary orotic acid is also not elevated [29]. Discrimination between these two proximal urea-cycle disorders can be achieved through hepatic enzymatic studies [32] or molecular diagnostics [33]. The majority of presenting neonates with NAGS deficiency have <5% residual activity [31], and frequently have frameshift or nonsense mutations [31,33]. In contrast, late-onset presentation is associated with greater residual enzyme activity and hypomorphic alleles with single amino acid substitutions [30,31,33]. A minority of patients have been shown to have reduced hepatic NAGS activity, despite failure to detect deleterious mutations by molecular testing [34–36]. Since the enzymatic assay may not be completely reliable, these patients may not have primary NAGS deficiency.
Other conditions with N-acetylglutamate deficiency
A secondary deficiency of NAG may arise from a diminution of available coenzyme A or acetyl-CoA or L-glutamate, or by inhibition of the NAGS reaction [37–40]. A reduction of hepatic N-acetylglutamate has been hypothesized as the mechanism of hyperammonemia in the organic acidemias, such as propionic acidemia, methylmalonic acidemia, and isovaleric acidemia [38–39], as well as in hyperinsulinism-hyperammonemia syndrome [40], and in valproate treatment [37]. Exogenous benzoate may also decrease the intra-mitochondrial NAG concentration [41].
Treatment of NAG deficiency with N-carbamylglutamate
Grisolia et al. [42] determined that derivatives of L-glutamic acid, including N-carbamylglutamate (NCG), could activate the biosynthesis of carbamyl phosphate by CPSI. While the natural CPSI activator, NAG, has the strongest affinity [43], it is a poor pharmacological candidate since it is hydrolyzed in vivo by acyl-amino acid acylase. In contrast, NCG is entirely acylase-resistant [44], and was therefore evaluated as a pharmacological substitute for NAG in patients with NAGS deficiency [28,45–48], where it has been shown to normalize plasma ammonia levels within 8 hours [49]. A diagnostic trial of NCG has been suggested for hyperammonemic newborns with suspected urea cycle disorders, given that a rapid response is not only diagnostic, but presents a life-saving therapeutic option for patients with NAGS deficiency [49]. NCG supplementation has also been used as an adjunct to the treatment of hyperammonemia in propionic acidemia (PA) [50], methylmalonic acidemia (MMA) [51], and hyperinsulinism-hyperammonemia syndrome (HHS) [52].
In patients with NAGS deficiency, we have shown that a 3-day trial of NCG, at a dose of 2.2 g/m2/d (100mg/kg/d if < 25 kg), restores ureagenesis [53, 54]. In order to assess changes in ureagenesis, we used isotope-ratio mass-spectrometry to monitor the in-vivo synthesis of [13C]urea following administration of a single oral dose of [1-13C]acetate. The labeled acetate is rapidly converted to H13CO3, of which a small fraction is consumed by CPSI, and ultimately incorporated into [13C]urea. Following NCG administration, both atom % excess plasma [13C]urea and absolute plasma [13C]urea concentrations (Fig. 5) increase several-fold above the depressed baseline values. Peak plasma [13C]urea concentration reached twice that obtained in normal controls [54]. Mean plasma ammonia decreased to close to normal levels, while plasma urea increased nearly four-fold. Plasma glutamine levels decreased substantially as well.
Figure 5.

Increase over time of isotopic enrichment in plasma [13C] urea (A) and the concentration of plasma [13C] urea (B) in a patient with NAGS deficiency before (○) and after (●) 3-d treatment with N-carbamylglutamate.
We have also performed 3-day trials of NCG in stable patients with severe PA [54] and B12-unresponsive Cbl-B MMA (unpublished data), who all presented with neonatal hyperammonemia. In a cohort of PA patients (unpublished data), we observed statistically significant decreases in ammonia and glutamine following NCG administration, supportive of an increase in nitrogen disposal via urea. Similar results were observed in our single patient with Cbl-B deficiency. NCG also increased ureagenesis in patients with HHS (unpublished) and in healthy adults [55].
In summary, while NAGS deficiency is exceedingly rare, it is the only inherited urea cycle disorder that can be specifically and effectively treated by a drug. Moreover, the same drug (NCG) appears to be beneficial for the treatment of other hyperammonemic conditions and increases the rate of ureagenesis even in healthy individuals, observations that as a whole support the investigation of NCG for the treatment of both inherited and acquired hyperammonemia.
Acknowledgments
This work was supported in part by public health service grants R01DK47870, R01DK064913 and R01DK53761 and K01DK067935 and K01DK076846 from the National Institute of Diabetes and Digestive and Kidney Diseases, grant R01HD058567 from the National Institute of Child Health and Human Development (NICHD), grants U54RR019453 and U54HD061221 from the Rare Disease Center Program of the Office for Rare Disorders, NICHD and the National Center for Research Resources (NCRR), and grants P30HD40677 and P30HD2697 from the Intellectual and Developmental Disabilities Research Centers from the NICHD, and grant M01RR020359 from the General Clinical Research Center program, NCRR National Institutes of Health, Department of Health and Human Services.
Abbreviations
- CPSI
carbamyl phosphate synthetase I
- NAG
N-acetylglutamate
- NAGS
N-acetylglutamate synthase
- NCG
N-carbamylglutamate
Footnotes
Conflict of Interest statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Ljubica Caldovic, Email: lcaldovic@cnmcresearch.org.
Nicholas Ah Mew, Email: nahmew@cnmc.org.
Dashuang Shi, Email: dshi@cnmcresearch.org.
Hiroki Morizono, Email: hmorizono@cnmcresearch.org.
Marc Yudkoff, Email: Yudkoff@email.chop.edu.
Mendel Tuchman, Email: mtuchman@cnmc.org.
References
- 1.Hall LM, Metzenberg RL, Cohen PP. Isolation and characterization of a naturally occurring cofactor of carbamyl phosphate biosynthesis. J Biol Chem. 1958;230:1013–1021. [PubMed] [Google Scholar]
- 2.Shigesada K, Tatibana M. Enzymatic synthesis of acetylglutamate by mammalian liver preparations and its stimulation by arginine. Biochem Biophys Res Commun. 1971;44:1117–1124. doi: 10.1016/s0006-291x(71)80201-2. [DOI] [PubMed] [Google Scholar]
- 3.Caldovic L, Tuchman M. N-acetylglutamate and its role through evolution. Biochem J. 2003;372(Pt 2):279–290. doi: 10.1042/BJ20030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meijer AJ, Lamers WH, Chamuleau RA. Nitrogen metabolism and ornithine cycle function. Physiol Rev. 1990;70:701–748. doi: 10.1152/physrev.1990.70.3.701. [DOI] [PubMed] [Google Scholar]
- 5.Bachmann C, Krahenbuhl S, Colombo JP. Purification and properties of acetyl-CoA:L-glutamate N-acetyltransferase from human liver. Biochem J. 1982;205:123–127. doi: 10.1042/bj2050123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shigesada K, Tatibana M. N-Acetylglutamate synthetase from rat-liver mitochondria. Partial purification and catalytic properties, (1978) Eur J Biochem. 1978;84:285–291. doi: 10.1111/j.1432-1033.1978.tb12167.x. [DOI] [PubMed] [Google Scholar]
- 7.Sonoda T, Tatibana M. Purification of N-acetyl-L-glutamate synthetase from rat liver mitochondria and substrate and activator specificity of the enzyme. J Biol Chem. 1983;258:9839–9844. [PubMed] [Google Scholar]
- 8.Caldovic L, Morizono H, Yu X, Thompson M, Shi D, Gallegos R, Allewell NM, Malamy MH, Tuchman M. Identification, cloning and expression of the mouse N acetylglutamate synthase gene. Biochem J. 2002;364(Pt 3):825–831. doi: 10.1042/BJ20020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caldovic L, Morizono H, Gracia Panglao M, Gallegos R, Yu X, Shi D, Malamy MH, Allewell NM, Tuchman M. Cloning and expression of the human N-acetylglutamate synthase gene. Biochem Biophys Res Commun. 2002;299:581–586. doi: 10.1016/s0006-291x(02)02696-7. [DOI] [PubMed] [Google Scholar]
- 10.Caldovic L, Lopez GY, Haskins N, Panglao M, Shi D, Morizono H, Tuchman M. Biochemical properties of recombinant human and mouse N-acetylglutamate synthase. Mol Genet Metab. 2006;87:226–232. doi: 10.1016/j.ymgme.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Qu Q, Morizono H, Shi D, Tuchman M, Caldovic L. (2007) A novel bifunctional N-acetylglutamate synthase-kinase from Xanthomonas campestris that is closely related to mammalian N-acetylglutamate synthase, BMC. Biochem. 2007;8:4. doi: 10.1186/1471-2091-8-4. ePub Apr 12, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gessert SF, Kim JH, Nargang FE, Weiss RL. A polyprotein precursor of two mitochondrial enzymes in Neurospora crassa. Gene structure and precursor processing. J Biol Chem. 1994;269:8189–8203. [PubMed] [Google Scholar]
- 13.Haskins N, Panglao M, Qu Q, Majumdar H, Cabrera-Luque J, Morizono H, Tuchman M, Caldovic L. Inversion of allosteric effect of arginine on N-acetylglutamate synthase, a molecular marker for evolution of tetrapods. BMC Biochem. 2008;9:24. doi: 10.1186/1471-2091-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandman O, Ferrell JE, Jr, Li R, Meyer T. Interlinked fast and slow positive feedback loops drive reliable cell decisions. Science. 2005;310:496–498. doi: 10.1126/science.1113834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi D, Sagar V, Jin Z, Yu X, Caldovic L, Morizono H, Allewell NM, Tuchman M. The crystal structure of N-acetyl-L-glutamate synthase from Neisseria gonorrhoeae provides insights into mechanisms of catalysis and regulation. J Biol Chem. 2008;283:7176–7184. doi: 10.1074/jbc.M707678200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Min L, Jin Z, Caldovic L, Morizono H, Allewell NM, Tuchman M, Shi D. Mechanism of allosteric inhibition of N-acetyl-L-glutamate synthase by L-arginine. J Biol Chem. 2009;284:4873–4880. doi: 10.1074/jbc.M805348200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramón-Maiques S, Fernández-Murga ML, Gil-Ortiz F, Vagin A, Fita I, Rubio V. Structural bases of feed-back control of arginine biosynthesis, revealed by the structures of two hexameric N-acetylglutamate kinases, from Thermotoga maritima and Pseudomonas aeruginosa. J Mol Biol. 2006;356:695–713. doi: 10.1016/j.jmb.2005.11.079. [DOI] [PubMed] [Google Scholar]
- 18.Marvil DK, Leisinger T. N-Acetylglutamate synthase of Escherichia coli: purification, characterization, and molecular properties. J Biol Chem. 1977;252:3295–3303. [PubMed] [Google Scholar]
- 19.Ramón-Maiques S, Marina A, Gil-Ortiz F, Fita I, Rubio V. Structure of acetylglutamate kinase, a key enzyme for arginine biosynthesis and a prototype for the amino acid kinase enzyme family, during catalysis. Structure. 2002;10:329–342. doi: 10.1016/s0969-2126(02)00721-9. [DOI] [PubMed] [Google Scholar]
- 20.Neuwald AF, Landsman D. (1997) GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem Sci. 1997;22:154–155. doi: 10.1016/s0968-0004(97)01034-7. [DOI] [PubMed] [Google Scholar]
- 21.Sancho-Vaello E, Fernández-Murga ML, Rubio V. Site-directed mutagenesis studies of acetylglutamate synthase delineate the site for the arginine inhibitor. FEBS Lett. 2008;582:1081–1086. doi: 10.1016/j.febslet.2008.02.060. [DOI] [PubMed] [Google Scholar]
- 22.Fernández-Murga ML, Rubio V. Basis of arginine sensitivity of microbial N-acetyl-L-glutamate kinases: mutagenesis and protein engineering study with the Pseudomonas aeruginosa and Escherichia coli enzymes. J Bacteriol. 2008;190:3018–3025. doi: 10.1128/JB.01831-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sancho-Vaello E, Fernández-Murga ML, Rubio V. Mechanism of arginine regulation of acetylglutamate synthase, the first enzyme of arginine synthesis. FEBS Lett. 2009;583:202–206. doi: 10.1016/j.febslet.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Morizono H, Caldovic L, Shi D, Tuchman M. Mammalian N-acetylglutamate synthase. Mol Genet Metab. 2004;81(Suppl 1):S4–S11. doi: 10.1016/j.ymgme.2003.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tuchman M, Lee B, Lichter-Konecki U, Summar ML, Yudkoff M, Cederbaum SD, Kerr DS, Diaz GA, Seashore MR, Lee HS, McCarter RJ, Krischer JP, Batshaw ML. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94:397–402. doi: 10.1016/j.ymgme.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bachmann C, Krahenbuhl S, Colombo JP, Schubiger G, Jaggi KH, Tonz O. N-acetylglutamate synthetase deficiency: a disorder of ammonia detoxication. N Engl J Med. 1981;304:543. doi: 10.1056/NEJM198102263040918. [DOI] [PubMed] [Google Scholar]
- 27.Bachmann C, Brandis M, Weissenbarth-Riedel E, Burghard R, Colombo JP. N-acetylglutamate synthetase deficiency, a second patient. J Inherit Metab Dis. 1988;11:191–193. doi: 10.1007/BF01799871. [DOI] [PubMed] [Google Scholar]
- 28.Guffon N, Vianey-Saban C, Bourgeois J, Rabier D, Colombo JP, Guibaud P. A new neonatal case of N-acetylglutamate synthase deficiency treated by carbamylglutamate. J Inherit Metab Dis. 1995;18:61–65. doi: 10.1007/BF00711374. [DOI] [PubMed] [Google Scholar]
- 29.Caldovic L, Morizono H, Panglao M, Cheng SF, Packman S, Tuchman M. Null mutations in the N-acetylglutamate synthase gene associated with acute neonatal disease and hyperammonemia. Hum Genet. 2003;112:364–368. doi: 10.1007/s00439-003-0909-5. [DOI] [PubMed] [Google Scholar]
- 30.Caldovic L, Morizono H, Panglao MG, Lopez G, Shi D, Summar ML, Tuchman M. Late onset N-acetylglutamate synthase deficiency caused by hypomorphic alleles. Hum Mutat. 2005;25:293–298. doi: 10.1002/humu.20146. [DOI] [PubMed] [Google Scholar]
- 31.Caldovic L, Morizono H, Tuchman M. Mutations and polymorphisms in the human N-acetylglutamate synthase (NAGS) gene. Hum Mutat. 2007;28:754–759. doi: 10.1002/humu.20518. [DOI] [PubMed] [Google Scholar]
- 32.Bachmann C, Colombo JP, Jaggi K. N-acetylglutamate synthetase (NAGS) deficiency: diagnosis, clinical observations and treatment. Adv Exp Med Biol. 1982;153:39–45. doi: 10.1007/978-1-4757-6903-6_6. [DOI] [PubMed] [Google Scholar]
- 33.Haberle J, Schmidt E, Pauli S, Kreuder JG, Plecko B, Galler A, Wermuth B, Harms E, Koch HG. Mutation analysis in patients with N-acetylglutamate synthase deficiency. Hum Mutat. 2003;21:593–597. doi: 10.1002/humu.10216. [DOI] [PubMed] [Google Scholar]
- 34.Vockley J, Walsh-Vockley CM, Lin SP, Tuchman M, Wu TC, Lin CY, Seashore MR. Normal N-acetylglutamate concentration measured in liver from a new patient with N-acetylglutamate synthetase deficiency: Physiologic and biochemical implications. Biochem Med Metab Biol. 1992;47:38–46. doi: 10.1016/0885-4505(92)90006-k. [DOI] [PubMed] [Google Scholar]
- 35.Heckmann M, Wermuth B, Haberle J, Koch HG, Gortner L, Kreuder JG. Misleading diagnosis of partial N-acetylglutamate synthase deficiency based on enzyme measurement corrected by mutation analysis. Acta Paediatr. 2005;94:121–124. doi: 10.1111/j.1651-2227.2005.tb01799.x. [DOI] [PubMed] [Google Scholar]
- 36.Hwu WL, Chien YH, Tang NL, Law LK, Lin CY, Lee NC. Deficiency of the carnitine transporter (OCTN2) with partial N-acetylglutamate synthase (NAGS) deficiency. J Inherit Metab Dis 2007. 2007;30:816. doi: 10.1007/s10545-007-0594-y. Epub Aug 20, 2007. [DOI] [PubMed] [Google Scholar]
- 37.Williams CA, Tiefenbach S, McReynolds JW. Valproic acid-induced hyperammonemia in mentally retarded adults. Neurology. 1984;34:550–553. doi: 10.1212/wnl.34.4.550. [DOI] [PubMed] [Google Scholar]
- 38.Coude FX, Grimber G, Parvy P, Rabier D. Role of N-acetylglutamate and acetyl-CoA in the inhibition of ureagenesis by isovaleric acid in isolated rat hepatocytes. Biochim Biophys Acta. 1983;761:13–16. doi: 10.1016/0304-4165(83)90356-2. [DOI] [PubMed] [Google Scholar]
- 39.Coude FX, Sweetman L, Nyhan WL. Inhibition by propionyl-coenzyme A of N-acetylglutamate synthetase in rat liver mitochondria. A possible explanation for hyperammonemia in propionic and methylmalonic academia. J Clin Invest. 1979;64:1544–1551. doi: 10.1172/JCI109614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stanley CA. Hyperinsulinism/hyperammonemia syndrome: insights into the regulatory role of glutamate dehydrogenase in ammonia metabolism. Mol Genet Metab. 2004;81(Suppl 1):S45–S51. doi: 10.1016/j.ymgme.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 41.O’Connor JE, Costell M, Grisolia S. Carbamyl glutamate prevents the potentiation of ammonia toxicity by sodium benzoate. Eur J Pediatr. 1989;148:540–542. doi: 10.1007/BF00441553. [DOI] [PubMed] [Google Scholar]
- 42.Grisolia S, Cohen PP. The catalytic role of carbamyl glutamate in citrulline biosynthesis. J Biol Chem. 1952;198:561–571. [PubMed] [Google Scholar]
- 43.Fahien LA, Schooler JM, Gehred GA, Cohen PP. Studies on the mechanism of action of acetylglutamate as an activator of carbamyl phosphate synthetase. J Biol Chem. 1964;239:1935–1941. [PubMed] [Google Scholar]
- 44.Kim S, Paik WK, Cohen PP. Ammonia intoxication in rats: protection by N-carbamoyl-L-glutamate plus L-arginine. Proc Natl Acad Sci USA. 1972;69:3530–3533. doi: 10.1073/pnas.69.12.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schubiger G, Bachmann C, Barben P, Colombo JP, Tonz O, Schupbach D. N-acetylglutamate synthetase deficiency: diagnosis, management and follow-up of a rare disorder of ammonia detoxication. Eur J Pediatr. 1991;150:353–356. doi: 10.1007/BF01955939. [DOI] [PubMed] [Google Scholar]
- 46.Hinnie J, Colombo JP, Wermuth B, Dryburgh FJ. N-Acetylglutamate synthetase deficiency responding to carbamylglutamate. J Inherit Metab Dis. 1997;20:839–840. doi: 10.1023/a:1005344507536. [DOI] [PubMed] [Google Scholar]
- 47.Morris AA, Richmond SW, Oddie SJ, Pourfarzam M, Worthington V, Leonard JV. N-acetylglutamate synthetase deficiency: favourable experience with carbamylglutamate. J Inherit Metab Dis. 1998;21:867–868. doi: 10.1023/a:1005478904186. [DOI] [PubMed] [Google Scholar]
- 48.Kuchler G, Rabier D, Poggi-Travert F, Meyer-Gast D, Bardet J, Drouin V, Cadoudal M, Saudubray JM. Therapeutic use of carbamylglutamate in the case of carbamoyl-phosphate synthetase deficiency. J Inherit Metab Dis. 1996;19:220–222. doi: 10.1007/BF01799434. [DOI] [PubMed] [Google Scholar]
- 49.Guffon N, Schiff M, Cheillan D, Wermuth B, Haberle J, Vianey-Saban C. Neonatal hyperammonemia: the N-carbamoyl-L-glutamic acid test. J Pediatr. 2005;147:260–262. doi: 10.1016/j.jpeds.2005.04.059. [DOI] [PubMed] [Google Scholar]
- 50.Gebhardt B, Dittrich S, Parbel S, Vlaho S, Matsika O, Bohles H. N-carbamylglutamate protects patients with decompensated propionic aciduria from hyperammonaemia. J Inherit Metab Dis. 2005;28:241–244. doi: 10.1007/s10545-005-5260-7. [DOI] [PubMed] [Google Scholar]
- 51.Gebhardt B, Vlaho S, Fischer D, Sewell A, Bohles H. N-carbamylglutamate enhances ammonia detoxification in a patient with decompensated methylmalonic aciduria. Mol Genet Metab. 2003;79:303–304. doi: 10.1016/s1096-7192(03)00095-7. [DOI] [PubMed] [Google Scholar]
- 52.De Lonlay P, Benelli C, Fouque F, Ganguly A, Aral B, Dionisi-Vici C, Touati G, Heinrichs C, Rabier D, Kamoun P, Robert JJ, Stanley C, Saudubray JM. Hyperinsulinism and hyperammonemia syndrome: report of twelve unrelated patients. Pediatr Res. 2001;50:353–357. doi: 10.1203/00006450-200109000-00010. [DOI] [PubMed] [Google Scholar]
- 53.Caldovic L, Morizono H, Daikhin Y, Nissim I, McCarter RJ, Yudkoff M, Tuchman M. Restoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamylglutamate. J Pediatr. 2004;145:552–554. doi: 10.1016/j.jpeds.2004.06.047. [DOI] [PubMed] [Google Scholar]
- 54.Tuchman M, Caldovic L, Daikhin Y, Horyn O, Nissim I, Korson M, Burton B, Yudkoff M. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatr Res. 2008;64:213–217. doi: 10.1203/PDR.0b013e318179454b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mew NA, Payan I, Daikhin Y, Nissim I, Nissim I, Tuchman M, Yudkoff M. Effects of a single dose of N-carbamylglutamate on the rate of ureagenesis. Mol Genet Metab. 2009 Jul 14; doi: 10.1016/j.ymgme.2009.07.010. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]