

Abstract
We describe a severe congenital myasthenic syndrome (CMS) caused by two missense mutations in the gene encoding the muscle specific receptor tyrosine kinase (MUSK). The identified MUSK mutations M605I and A727V are both located in the kinase domain of MuSK. Intracellular microelectrode recordings and microscopy studies of the neuromuscular junction conducted in an anconeus muscle biopsy revealed decreased miniature endplate potential amplitudes, reduced endplate size and simplification of secondary synaptic folds, which were consistent with postsynaptic deficit. The study also showed a striking reduction of the endplate potential quantal content, consistent with additional presynaptic failure. Expression studies in MuSK deficient myotubes revealed that A727V, which is located within the catalytic loop of the enzyme, caused severe impairment of agrin-dependent MuSK phosphorylation, aggregation of acetylcholine receptors (AChRs) and interaction of MuSK with Dok-7, an essential intracellular binding protein of MuSK. In contrast, M605I, resulted in only moderate impairment of agrin-dependent MuSK phosphorylation, aggregation of AChRs and interaction of MuSK with Dok-7. There was no impairment of interaction of mutants with either the low-density lipoprotein receptor-related protein, Lrp4 (a co-receptor of agrin) or with the mammalian homolog of the Drosophila tumorous imaginal discs (Tid1). Our findings demonstrate that missense mutations in MUSK can result in a severe form of CMS and indicate that the inability of MuSK mutants to interact with Dok-7, but not with Lrp4 or Tid1, is a major determinant of the pathogenesis of the CMS caused by MUSK mutations.
INTRODUCTION
Several forms of congenital myasthenic syndromes (CMS) have been described to date. These syndromes, which are characterized by varying degrees of muscle weakness due to impaired neuromuscular transmission are classified into presynaptic, synaptic basal lamina-associated and postsynaptic sub groups, depending on which domain of the neuromuscular junction (NMJ) is primarily targeted by the disease (1,2). Representing the most common form of CMS, postsynaptic syndromes typically result from mutations in genes encoding the four adult AChR subunits (CHRNA1, CHRNB1, CHRND and CHRNE) and the intracellular protein rapsyn (RAPSN) (3,4). However, in recent years it has been shown that mutations in genes encoding proteins involved in a critical signal transduction pathway requiring the neurally derived agrin, the muscle specific tyrosine kinase (MuSK) and the intracellular adapter protein Dok-7, can also result in severe forms of CMS (5–7).
The agrin-dependent activation of MuSK through the low-density lipoprotein receptor (LDLR)-related protein (Lrp4) (8,9) is essential for the induction of presynaptic and postsynaptic differentiation, including the clustering of AChRs and rapsyn (10). In addition to agrin, MuSK activation requires phosphorylation of its Tyr553 residue, located within the juxtamembrane consensus recognition site (NPXY), via the phosphotyrosine-binding domain (PTB domain)-containing protein Dok-7 (11). MuSK also binds to the short splice form of the human tumorous imaginal discs (Tid1) protein (12). The dual activation of MuSK extracellularly by agrin and intracellularly by Dok-7 and Tid1 results in the recruitment of several downstream kinases and phosphorylation of the AChR β-subunit, leading to the reorganization of the actin cytoskeleton and AChR clustering (13–15). The fundamental role of the MuSK-signaling pathway is supported by the fact that mice deficient in agrin, MuSK, rapsyn or Dok-7 lack postsynaptic differentiation and die at birth from respiratory failure (11,16–19).
Two heteroallelic MUSK mutations were identified in the first reported case of MUSK-associated CMS (5). One of the mutations, c220insC resulting in frameshift, produced no MuSK expression when assayed in an expression system. The other mutation, a missense (V790M), was associated with diminished expression and stability of MuSK, leading to decreased agrin-dependent AChR clustering. More recently, it was reported that the MuSK V790M mutation also failed to co-immunoprecipitate with Dok-7 in co-transfected 293T cells (11).
Here, we describe a severe form of CMS resulting from two missense mutations (M605I and A727V). These mutations impair the expression and stability of MuSK, diminish agrin-dependent MuSK phosphorylation and AChR clustering, and fail to co-immunoprecipitate with Dok-7 but not with Lrp4 or Tid1. Our findings confirm previous observations and further indicate that disruption of the interaction of MuSK with Dok-7 plays a fundamental role in the pathogenesis of MUSK-associated CMS.
RESULTS
Clinical data
Case report
The patient is a 19-year-old woman who was born full-term to a non-consanguineous couple. At birth she was hypotonic, and her breathing and crying were weak. During her first month of life she experienced respiratory failure and required tracheotomy and mechanical ventilation. Through the course of infancy her weakness continued; however, none of her motor developmental milestones were delayed. She also had intermittent cyanosis resulting from a patent ductus arteriosus and required corrective heart surgery when she was 2-years-old. At the age of four repetitive stimulation of the left axillary nerve at 2 Hz resulted in a 36% decrement of the compound muscle action potential (CMAP) area. No repetitive CMAPs were observed after single nerve stimulation. Serum antibodies against AChR and MuSK were negative. Throughout her life she has been hospitalized several times due to recurrent respiratory infections and, at the age of 12, underwent corrective surgery for severe scoliosis. Her muscle weakness improved after puberty, but worsens significantly before each menstrual period. A recent neurologic examination revealed normal cognition, bilateral ptosis and intact external ocular movements, except for mild reduction of upward gaze. She had facial, bulbar, neck and proximal limb weakness with intact deep tendon reflexes. Finally, she currently requires continuous respiratory support with bi-level positive airway pressure during night sleep, and she has shown moderate improvement with albuterol sulfate, but no response to pyridostigmine bromide, ephedrine or 3,4-diaminopyridine.
Muscle biopsy
To elucidate the nature of the CMS, we performed a biopsy of the right anconeus muscle at the age of five, which included in vitro microelectrode recordings and electron microscopy of the NMJ.
Intracellular microelectrode studies
The most significant finding of the microelectrode recordings was the marked reduction of the amplitudes of miniature endplate potentials (MEPPs) and currents (MEPCs) relative to the controls (Table 1), with normal time constants of the MEPC decay. The quantal content of the nerve-evoked endplate potentials (EPPs) at 1 Hz was also diminished; however, the ratio of EPP quantal content using 20 to 1 Hz stimulation was not different from the controls (Table 1).
Table 1.
Physiological data
| Patient | Controls | |
|---|---|---|
| MEPP amplitude (mV) | 0.43 ± 0.05* (n = 14) | 1.24 ± 0.15 (n = 9) |
| MEPC amplitude (nA) | 1.29 ± 0.05* (n = 9) | 4.55 ± 0.28 (n = 11) |
| MEPC time constant (ms) | 3.14 ± 0.16 (n = 9) | 3.58 ± 0.16 (n = 11) |
| EPP quantal content (1 Hz) | 6.79 ± 1.48† (n = 7) | 12.71 ± 1.60 (n = 18) |
| EPP quantal content (20 Hz/1 Hz) | 1.14 ± 0.11 (n = 11) | 1.00 ± 0.05 (n = 23) |
Values reported as mean ± SEM.
*P < 0.001, Student t-test.
†P< 0.05, Student t-test.
Light microscopy
There was mild random variation in the fiber diameter and type I fiber predominance; however, except for rare necrotic fibers, there were no other histological abnormalities. The acetylcholinesterase (AChE) performed in whole-mount unstained slides revealed that the mean endplate area of 75.6 ± 9.44 µm2 (n = 67) in the patient was markedly reduced compared with the mean endplate area in three age-matched controls of 120.67 ± 6.27 µm2 (n = 235), P < 0.001 (Student's t-test).
Electron microscopy
The study showed that the postsynaptic folds and secondary synaptic clefts were underdeveloped; however, the nerve terminals were normal in size. In addition, at several axon terminals there was a striking decrease in the number of synaptic vesicles. However, this was not a general feature since the number of synaptic vesicles at many nerve terminals was normal or even increased (Fig. 1).
Figure 1.

Ultrastructural findings at the NMJ. (A) An example of a NMJ from the patient demonstrating marked simplification of postsynaptic folds and underdeveloped secondary synaptic clefts (black arrows). In contrast the size of the nerve terminal (asterisk) and the width of the primary synaptic cleft (white arrow) are normal. (B) An example of a NMJ from a control showing normal secondary synaptic clefts (black arrows), nerve terminal (asterisk) and width of the primary synaptic cleft (white arrow). Calibration marks represent 1 µm.
Morphometric analysis
Relative to the controls, the patient showed a marked reduction in the endplate index (EI, length of the postsynaptic membrane/length of the presynaptic membrane) and a trend toward a reduction in the mean number of secondary clefts per micron of primary cleft, although the latter was not statistically significant relative to the controls (Table 2). Similarly, the average axon terminal area and number of synaptic vesicles per area of nerve terminal were not different from controls.
Table 2.
Morphometric data
| Patient | Controls | |
|---|---|---|
| EI ratio | 4.99 ± 0.34a,* (n = 18) | 11.71 ± 2.36 (n = 12) |
| Secondary Clefts Per Primary Cleft Length | 1.45 ± 0.1 (n = 20) | 1.79 ± 0.14 (n = 12) |
| Nerve Terminal Area (um2) | 7.28 ± 1.06 (n = 24) | 7.34 ± 0.93 (n = 12) |
| # of Synaptic Vesicles/um2 | 13.48 ± 4.62 (n = 17) | 16.77 ± 2.77 (n = 12) |
EI, length of the presynaptic membrane/length of the postsynaptic membrane.
aValues reported as mean ± SEM.
*P < 0.05, Student t-test.
Immunohistochemical analysis
Endplates were labeled with rhodamine-tagged alpha-bungarotoxin (α-BGT) and the co-localization of the MuSK protein was visualized using a primary antibody directed against the N-terminal of human MuSK. In comparison with two age-matched controls, the patient showed a marked reduction in the mean surface extension of α-BGT per endplate (2.47 ± 0.36 µm2, n = 21 versus 20.79 ± 2.73 µm2, n = 45; P < 0.001, Student's t-test). The mean surface extension of MuSK per endplate in the patient was also greatly reduced (3.17 ± 0.73 µm2, n = 21 versus 19.29 ± 2.68 µm2, n = 45; P < 0.001, Student's t-test), however, the intensity of MuSK fluorescence in the patient was not different from controls (18.90 ± 1.44, n = 21 versus 17.71 ± 0.74, n = 45; Supplementary Material, Fig. S1). The smaller size of the patient's endplates calculated by immunohistochemistry in comparison with the size of the patient's endplates calculated by the AChE stain may be in part due to the angle of sectioning, which resulted in the visualization of very small fractions of the patient's endplates with α-BGT and the anti-MuSK antibody.
Mutational analysis
DNA sequencing
We first amplified and sequenced all the coding regions and splice junctions of the genes encoding the subunits of the adult AChR and rapsyn. Since we found no mutations in these genes, and the patient had a characteristic ‘limb girdle myasthenia’ phenotype, we continued with the amplification and sequencing of DOK7 and MUSK and found two novel heteroallelic mutations in MUSK. Both mutations are missense in nature and conserved; one occurs in exon 14, M605I, and the other occurs in exon 15, A727V—as referenced from isoform 1 of human MUSK encoded by transcript variant 1 (20). As shown in Figure 2, M605I is located in the N-terminal lobe of the highly conserved tyrosine kinase domain (TKD) of the protein, and A727V is located in the C-terminal lobe of the TKD, within the catalytic loop of the enzyme (21). In addition, DNA from 100 healthy controls was amplified and sequenced to examine the possibility that these mutations may represent rare single nucleotide polymorphisms, but neither mutation was detected in any of the controls.
Figure 2.

Structure of MuSK showing the position of the identified mutations. (A) Schematic diagram displaying the domain organization of rat MuSK and the location of M604I (human M605I) and A726V (human A727V). The catalytic loop (amino acids 722–729), in which mutation A726V resides, is depicted in yellow. The activation loop (residues 742–763) is highlighted in purple. The positions of mutations M604I and A726V in the TKD are highlighted in red. Ig1-3, immunoglobulin-like domains; CRD, cysteine-rich domain; TM, transmembrane helix. (B) Crystal structure of the MuSK cytoplasmic domain (PDB code 1LUF, 21). M604I, replacement of methionine by isoleucine, is located in the third beta-strand of the N-terminal lobe of the TKD. The isoleucine is colored red (M604I). A726V replacement of alanine by valine is located in the catalytic loop in the C-terminal lobe of the TKD. The valine is colored red. Asp-724 and Arg-728, which are essential catalytic residues (catalytic loop), are colored yellow. Tyr-750, Tyr-754 and Tyr-755 (colored magenta) are autophosphorylation sites in the activation loop.
Restriction digest
This assay was based on the fact that M605I causes a gain of a restriction site for SfcI. Digestion of the exon 14 amplicon resulted in an additional 181 DNA base pair (bp) band in the patient and in her unaffected brother and mother, but not in her father, demonstrating that the patient received M605I from her mother and that she shares the mutation with her unaffected younger brother (Fig. 3). The exon 15 mutation A727V was investigated using allele-specific PCR with a set of primers that selectively amplified the mutant allele. The study revealed that the patient received the A727V from her unaffected father (Fig. 3). Overall, the study demonstrated that an individual must carry both mutations to be affected, thus indicating that these mutations are recessive.
Figure 3.
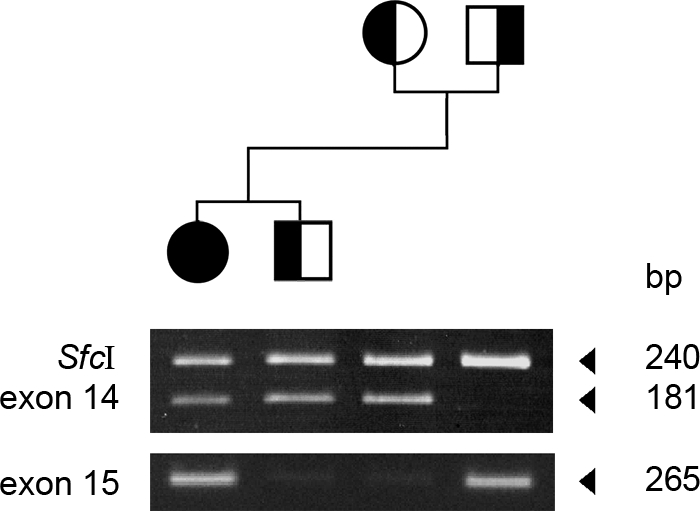
Pedigree of the family and the results of the restriction digest and allele-specific PCR. Because M605I results in a gain of a restriction site, digestion of exon 14 with SfcI results in an additional 181 DNA band in the patient and her unaffected brother and mother, but not in her father. In contrast, allele-specific PCR with a primer designed to amplify selectively the A727V mutant amplifies only the DNA from the patient and her father.
Expression studies
To investigate the effect of the identified MUSK mutations on expression and function of MuSK we engineered the identified MUSK mutants using site-directed mutagenesis of human and mouse full-length MuSK cDNA constructs and expressed them in human heterologous cells and mouse C2C12 and MuSK−/− myotubes.
M605I and A727V result in mild decreased expression without significant change of stability of MuSK
To assess the expression of MUSK mutants in heterologous cells we co-transfected either the human wild-type (WT) or one of the MUSK mutants along with green fluorescent protein (GFP) into HEK 293 cells and evaluated MuSK expression by incubating the whole cell lysate (WCL) with an anti-MuSK antibody. Figure 4A shows a representative example of a total of 12 transfections and demonstrates that the expression of MuSK in cells transfected with M605I- and A727V-constructs is reduced compared with the expression of MuSK in cells transfected with the WT construct. In addition, cells that were exposed to the protein biosynthesis-inhibitor cycloheximide showed that, overall, there were no significant differences in the rate of degradation of MuSK in cells transfected with the WT or MUSK mutant constructs. An example of a total of eight transfection experiments showing MuSK expression in cells exposed to cycloheximide is shown in Figure 4B.
Figure 4.
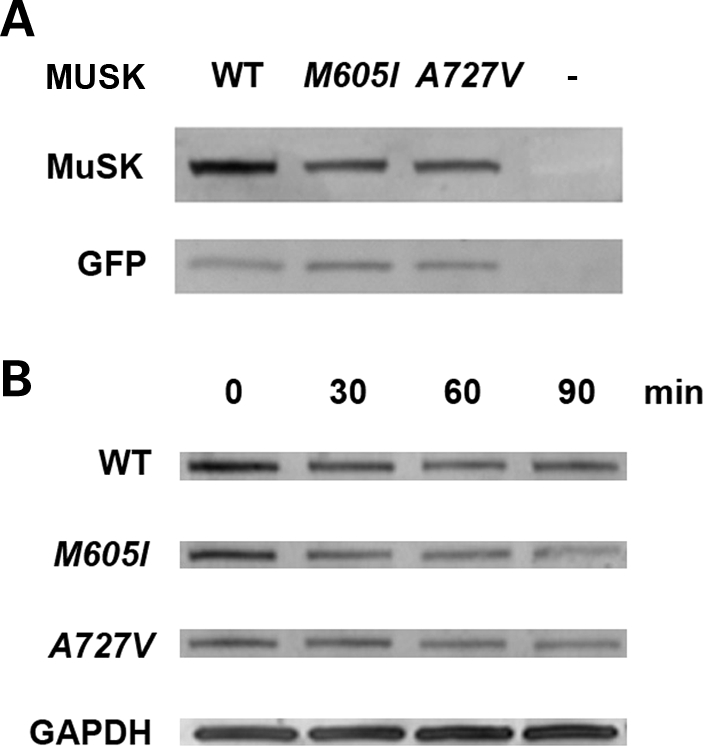
Expression and stability of MuSK mutants in HEK 293 cells. (A) A western blot analysis in cells co-transfected with WT or MUSK mutants and GFP using a polyclonal anti-MuSK goat antibody demonstrates reduced expression of MuSK in cells transfected with M605I and A727V in comparison with cells transfected with the WT construct (82% of WT for M605I and 87% of WT for A727V). The expression of GFP, tested with an anti-GFP polyclonal rabbit antibody, was similar in cells transfected with WT and MUSK mutants. (B) Analysis conducted in cells exposed to cycloheximide at set times intervals: 0, 30, 60 and 90 min, 36 h after transfection with WT or MUSK mutants revealed decreased expression of MuSK in cells transfected with the mutants compared with cells transfected with the WT construct. At time 0, expression of M605I was 87%, whereas A727V was 63% compared with WT. However, there were no major differences in the rate of degradation of MuSK in cells transfected with WT or MUSK mutant constructs. Expression of GAPDH detected with an anti-GAPDH monoclonal antibody showed no reduction of GAPDH expression in all the samples.
M605I and A727V interaction with Dok-7 and MuSK tyrosine phosphorylation
The binding of Dok-7 at the PTB motif of MuSK and MuSK phosphorylation are fundamental steps required for postsynaptic differentiation; hence, we next examined the interaction of mouse MuSK with Dok-7 and the agrin-induced MuSK phosphorylation in MuSK−/− myotubes by co-transfecting plasmids expressing the mouse WT-His-MuSK or either one of the mouse homologous His-MuSK mutant constructs. Cells transfected with the WT construct and exposed to agrin showed strong MuSK–Dok-7 co-immunoprecipitation and positive phosphorylation of MuSK. In contrast, MuSK-M605I inhibited Dok-7 binding by 34% (P < 0.05, Student t-test) and MuSK phosphorylation by 37% (P < 0.05, Student t-test), while MUSK-A727V inhibited Dok-7 binding by 87% (P < 0.05, Student t-test) and MuSK phosphorylation by 73% (P < 0.05, Student t-test). An example of several analyzed gels is shown in Figure 5. Thus, these findings demonstrate that the identified MUSK mutations affect the normal interaction between MuSK and Dok-7 and impair MuSK phosphorylation. However, MUSK-A727V induces much higher inhibition of Dok-7 binding and MuSK phosphorylation than MUSK-M605I likely because MUSK-A727V is located in the catalytic loop of the TKD, whereas MUSK-M605I is located distal to the kinase active site (catalytic loop region) (21) as shown in Figure 2.
Figure 5.
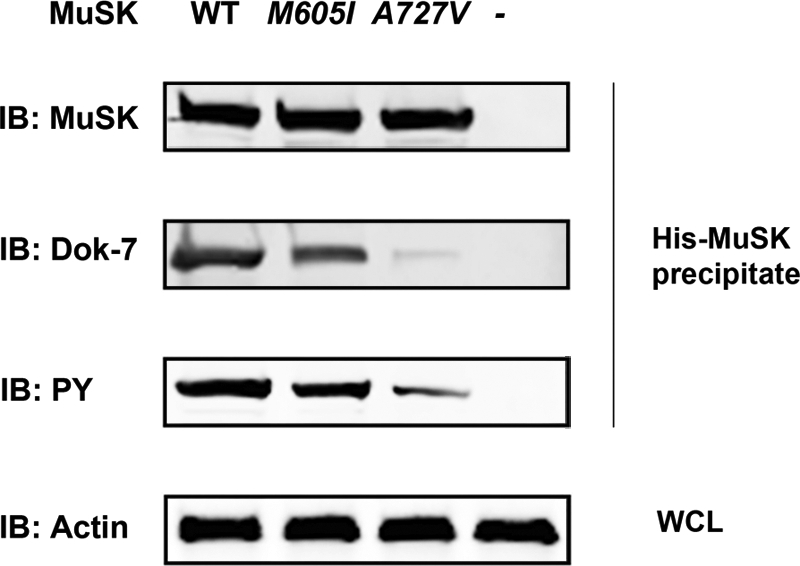
Interaction of MuSK with Dok-7 and MuSK phosphorylation in MuSK−/− myotubes exposed to agrin. The MuSK protein complex was eluted from lysates of MuSK−/− myotubes co-transfected with mouse plasmids expressing either His-WT-MuSK or MuSK mutants using a His SpinTrap affinity column and was subjected to immunoblot (IB) analysis using antibodies against MuSK, Dok-7 and phosphotyrosine (PY). The cells transfected with WT-MuSK showed strong Dok-7 and PY bands indicating that Dok-7 directly interacts with MuSK and induced MuSK tyrosine phosphorylation. In contrast, cells transfected with MuSK-A727V showed very weak Dok-7 and PY bands indicating failure of Dok-7 to interact with the MuSK mutant and to induce phosphorylation of the MuSK protein. The mutant MuSK-M605I showed a moderate interaction with Dok-7 and modest MuSK phosphorylation. The WCL was incubated with a monoclonal antibody directed against β-actin as a positive protein expression control.
M605I and A727V induce deficient clustering of AChRs in MuSK−/− myotubes
Because agrin fails to induce clustering of AChRs in MuSK−/− myotubes (11), we next examined if transfection of mouse WT or MuSK mutants could restore clustering of AChRs in MuSK−/− myotubes exposed to agrin. Figure 6 illustrates a representative example, totaling four transfection experiments, and demonstrates that whereas cells transfected with WT-MuSK show strong clustering of AChRs, cells transfected with MuSK-M605I display reduced clustering of AChRs and cells transfected with MuSK-A727V showed almost no clustering of AChRs.
Figure 6.

Agrin-induced clustering of AChRs in MuSK−/− myotubes transfected with mouse WT- or mutant-MuSK. (A) MuSK−/− myotubes transfected with WT-MuSK showed robust clustering of AChRs. (B) MuSK−/− myotubes transfected with MuSK-M605I showed reduced clustering of AChRs. (C) MuSK−/− myotubes transfected with MuSK-A727V showed almost no clustering of AChRs. (D) Bar-graph showing the number of clusters per field in MuSK−/− myotubes transfected with mouse WT- and mutant-MuSK in four experiments involving four wells per experimental condition and the analysis of 12 fields per group. Similar protein expression for all experimental conditions was verified by Western blot analysis using an antibody against MuSK (Supplementary Material, Fig. S2). Although the difference in clustering between MuSK−/− myotubes transfected with MuSK-M605I and those transfected with WT-MuSK was significant at the P < 0.05 level (Student t-test), the difference in clustering between MuSK−/− myotubes transfected with MuSK-A727V and those transfected with WT-MuSK was significant at the P < 0.001 level (Student t-test). Calibration mark represents 100 µm
M605I and A727V do not prevent interaction between MuSK with Lrp4 or Tid1
Given that agrin does not directly interact with MuSK, but requires the intermediate co-receptor Lrp4 (8,9), and given that Tid1 has been postulated to aggregate receptors independently from agrin (12), we finally examined the interaction between MuSK with Lrp4 and Tid1 in HEK cells, in C2C12 cells and in MuSK−/− myotubes transfected with either WT-MuSK or one of the MuSK mutants. Figure 7 illustrates that the MuSK protein complex eluted from MuSK−/− cells transfected with either mouse His-tagged WT-MuSK or one of the MuSK mutants show equal immunoreactivity to Lrp4 and Tid1. Thus, neither the M605I nor the A727V mutation inhibits the interaction between MuSK and Lrp4 or MuSK and Tid1.
Figure 7.
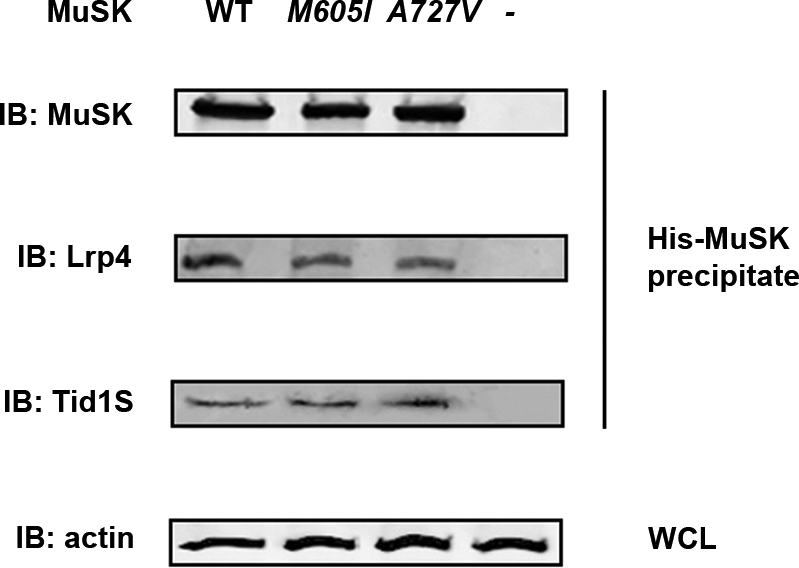
Interaction of mouse MuSK with Lrp4 and Tid1. The His-tagged MuSK protein complex eluted from MuSK−/− cells transfected with His-WT-MUSK or His-MuSK mutants (M605I or A727V) were subject to IB analysis using antibodies against MuSK, Lrp4 and Tid1. The WCL was used to detect β-actin as a loading control. The immunoreactivity to Lrp4 or Tid1 was not different in cells transfected with WT-MuSK or either one of the MuSK mutants. The WCL incubated with a monoclonal antibody against β-actin was consistent with equal amounts of loaded protein.
DISCUSSION
We provide here a complete report of a CMS caused by two missense mutations in MUSK, including a description of the findings from in vitro microelectrode recordings, electron microscopy of the NMJ and the key molecular interactions responsible for this syndrome.
The clinical features of our patient not only resemble those depicted in previously reported cases of CMS associated with MUSK mutations (4,22), but also those described in patients with CMS resulting from mutations in DOK7 (23–27). These clinical features include: weakness and respiratory difficulties with onset at birth or during infancy; no delay of initial motor milestones; and, a pattern of proximal ‘limb girdle’ weakness, which does not associate with significant impairment of external ocular movements. Furthermore, as in many patients with DOK7 mutations, our patient did not improve with either anti-cholinesterase drugs or 3,4-diaminopyridine (23,28), but responded favorably to the sympathomimetic drug albuterol sulfate.
The light-microscope studies which focused on the cholinesterase reactivity demonstrated that our patient had a reduction of the endplate size, a finding that has also been reported in patients with limb girdle myasthenia, and suggestive of incomplete development of endplates (29). Indeed, the electron microscopy studies revealed prominent simplification of the postsynaptic membranes, and the intracellular microelectrode studies showed reduction of amplitudes of miniature synaptic potentials and currents consistent with postsynaptic failure (30). In our patient there was also evidence of presynaptic deficit, considering the decreased EPP quantal content and marked reduction of the number of synaptic vesicles at some of the NMJs, which are suggestive of an increased proportion of immature nerve terminals (31). Thus, our findings are consistent with the interpretation that the patient's MuSK mutants fail to activate a signaling cascade responsible for multiple aspects of synapse formation, including receptor clustering, postsynaptic membrane development and presynaptic differentiation.
As with the previously described human V790M mutation, both M605I and A727V showed reduced expression in HEK cells, however, the ability of each of the mutants to induce AChR aggregation in response to agrin in MuSK−/− cells was quite different. Although A727V almost completely failed to aggregate receptors, the number of clustered receptors induced by M605I was reduced to only about half, when compared with the WT construct. This striking difference between the capability of A727V and M605I to cluster receptors can be readily explained by the fact that A727V, which is located within the catalytic loop of the enzyme (21), results in severe reduction of the agrin-induced autophosphorylation and minimal interaction of MuSK with Dok-7, whereas M605I, which is located in the N-terminal lobe of the TKD of the enzyme, results in only modest reduction of the agrin-induced autophosphorylation and moderate impairment of the MuSK–Dok-7 interaction. Furthermore, the difference can not be explained on the basis of unequal expression or stability of the mutants in HEK cells, since overall we found no major differences between the level of expression and stability of the mutants.
Interestingly, our patient, as with the previously described patient with CMS due to MUSK mutations, is compound heterozygous for two mutations; one that results in moderate reduction of receptor clustering and one that results in severe impairment of clustering. A possible explanation for the relative rarity of this CMS is that carriers of two MUSK mutations that result in only mild defect of MuSK protein function may not express clinical symptoms, whereas the presence of a mutation that results in severe deficiency of MuSK function in each of the alleles is probably not compatible with life. Indeed, this interpretation is supported by a recent study demonstrating that a mouse line carrying a homozygous mouse mutation homologous of V790M (muskv789M/V789M mice) presents no abnormal phenotype, whereas a hemizygous mouse line carrying the V790M allele paired with a null allele (muskv789M/– mice) develops severe weakness (32).
Our expression studies have identified impairment of MuSK phosphorylation and MuSK–Dok-7 interaction as central to the pathogenesis of the CMS described here. MuSK phorphorylation and MuSK–Dok-7 interaction are fundamental steps of synaptic development since tyrosine autophosphorylation radically increases the catalytic activity of MuSK (21) and the recruitment of proteins containing the PTB binding domain, such as Dok-7, is essential for the downstream propagation of signaling events (33).
It appears that spontaneously occurring human mutations in MUSK can impair signal transduction and synaptic differentiation by more than one mechanism. In the case of the two previously described MUSK mutations, 220insC results in complete lack of expression of the protein and V790M impairs binding of MuSK to Dok-7 (11), but does not appear to alter MuSK autophosphorylation (5). In contrast, M605I results in moderate impairment of both phosphorylation and binding to Dok-7. Finally, A727V, by virtue of being located in the catalytic loop of the enzyme, results in drastic impairment of phosphorylation and MuSK–Dok-7 interaction.
Together, these findings suggest that impaired MuSK phosphorylation, which is a feature common to CMS due to MUSK and DOK7 mutations, is a key factor responsible for the abnormal endplate formation and presynaptic differentiation seen in CMS resulting from abnormalities of the agrin-MuSK signal transduction pathway.
We found that the interaction of the patient's MuSK mutants with the co-receptor of agrin Lrp4 was not affected. This was not entirely unexpected since Lrp4 interacts with the extracellular portion of MuSK and the mutations are situated in the TKD of the enzyme located intracellularly. Thus, the abnormal function of the MuSK mutants can not be explained on the basis of an impaired interaction between agrin and MuSK through the co-receptor Lrp4.
Finally, despite the fact that Tid1 interacts with the intracellular portion of MuSK, the patient's mutations do not affect the relationship between MuSK and Tid1. This, and other molecular interactions described in this study may be useful to design a potential therapeutic strategy for some forms of CMS resulting from MuSK mutations.
MATERIALS AND METHODS
Muscle biopsy
Muscle biopsy
A biopsy of the right anconeus muscle was performed under general anesthesia as previously described (34). The specimen was micro-dissected into full-length muscle bundles. Several muscle bundles were frozen by rapid immersion into isopentane cooled with liquid nitrogen. Transversely oriented sections of the frozen material were processed for routine histochemical analysis. A few muscle bundles were fixed in glutaraldehyde and subsequently used for electron microscopy. The rest of the material was further dissected into thin muscle bundles and utilized for microelectrode recordings.
Intracellular microelectrode studies
Recording and analysis of MEPPs, MEPCs and EPPs were performed as previously described (35).
Electron microscopy studies
The ultrastructure of the NMJ was analyzed as previously described (35).
Morphometric analysis of the NMJ
All the morphometric studies were conducted from electron microscopy printouts and were analyzed as previously described (25).
Immunohistochemical analysis
Frozen cryostat tissue sections of 8 µm thickness were fixed on ice with cold acetone for 10 min. The sections were permeabilized with 0.5% Triton X-100 for 30 min at room temperature (RT) and then blocked with 10% goat serum for 1 h at RT. The tissue was incubated overnight at 4°C with a rabbit polyclonal antibody directed against MuSK (1:50; Santa Cruz Biotechnology, Santa Cruz, CA, USA). The following morning, the tissue was labeled for 1.5 h at RT with a goat anti-rabbit IgG FITC secondary antibody (1:200; Santa Cruz Biotechnology) and a rhodamine-conjugated α-BGT counterstain (125 nm; Sigma, St. Louis, MO, USA). The slides were washed and mounted with ProLong Gold Anti-Fade reagent (Invitrogen). The tissue was then visualized using a Nikon E-600 fluorescent microscope (Nikon Instruments Inc., Melville, NY, USA). Quantitative analysis of surface extension and intensity of fluorescence was performed using the imageJ software. Fluorescence intensity was corrected for background intensity and reported in arbitrary units.
Mutational analysis
DNA amplification and sequencing
We amplified and sequenced genomic DNA covering all 15 exons of the human MUSK gene. DNA was extracted from patient blood using the QIAmp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA). Standard amplification techniques were employed and a list of primers is provided in Supplementary Material, Table S1. PCR products were sequenced on an ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA). We assembled and aligned sequences against the reference sequence (downloaded from Genbank; NC_000009, NM_005592) using BioEdit software. All sites showing differences from the reference, whether pathogenic or not, were documented for later analysis. The patient's unaffected parents and brother were genotyped as well. This study was approved by the institutional review board of the University of California, Davis. The patient and her family were informed of their rights and the details of the research, and all signed an informed consent form.
Restriction digest analysis
Digestion of the MuSK M605I amplicon was performed with SfcI restriction enzyme (New England BioLabs, Ipswich, MA, USA) according to the manufacturer's protocol. The cleaved product was run on a 15% TBE acrylamide gel and stained with EtBr for 45 min. The gel was analyzed using an ImageMaster VDS Imager (Molecular Dynamics).
Allele specific PCR
Forward primers were designed for the WT (5′-GTTTGTTCACCGAGATTTAGC-3′) and A727V mutant (5′-GTTTGTTCACCGAGATTTAGT-3′) differing only at the 3′ terminal nucleotide. The same WT reverse primer (5′-CATGGGCCATCCCATAGTAG-3′) was paired with both WT and A727V mutant forward primers with an annealing temperature of 66°C.
Expression studies
Mammalian expression vectors
We purchased a full-length human MUSK cDNA construct (Open Biosystems, Huntsville, AL, USA). The MUSK cDNA clone was PCR amplified and subcloned into a mammalian expression vector (pcDNA4/HisMax TOPO TA, Invitrogen), and this WT MUSK cDNA was then subjected to site-directed mutagenesis in order to create the MUSK M605I and M727V mutations (Quick-Change II, Stratagene, La Jolla, CA, USA). A full-length mouse Musk cDNA clone within a mammalian expression vector (pcDNA3.1 myc-His, Invitrogen) was obtained from Dr Yamanashi (Tokyo Medical and Dental University, Tokyo, Japan). We performed site-directed mutagenesis on this mouse WT construct to generate both Musk mutations detected in our patient. A full-length human, DOK7 cDNA was purchased through Open Biosystems, PCR amplified and subcloned into a mammalian expression vector (pcDNA3.1/Flag TOPO TA, Invitrogen). A Flag-LRP4-mCherry construct cloned into a pMX retroviral vector was kindly provided by Dr Steve Burden (NYU Medical School). A Tid1-pCS2 + MT construct was a gift from Jenny Linnoila (University of Pittsburgh, School of Medicine, Pittsburgh). A pFLAG-CMV-1 vector containing a rat agrin fragment was obtained from Dr Michael Ferns (University of California, Davis).
Cell culture
Human embryonic kidney 293 (HEK293) cells were kindly provided by Dr Tsung-Yu Chen (University of California, Davis). Cultures were grown in Dulbecco's modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), l-glutamine and Penicillin/Streptomycin mix and incubated at 37°C/5% CO2. MuSK−/− cells, generously provided by Dr Lin Mei (Medical College of Georgia, Augusta), were maintained in DMEM supplemented with 15% FBS, 1% pyruvate, 2% chick embryo extract (CEE), gentamycin sulfate and interferon-γ and, incubated at 33°C/10% CO2. Differentiation of myotubes was induced with DMEM containing 10% FBS, 10% horse serum, 1% pyruvate, 0.5% CEE and gentamycin sulfate and incubation at 39°C/10%CO2.
Expression and stability of MUSK mutants in HEK293 cells
Transfection
Each of the human pcDNA4/HisMax MUSK constructs (WT, mutant M605I and mutant A727V) were transfected into HEK293 cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. After transfection, the protein was extracted from the cells using RIPA buffer (Sigma) and Protease Inhibitor Cocktail (Sigma) was added to the WCL to minimize protein degradation.
Expression studies
Cells were co-transfected with GFP to estimate transfection efficiency and proteins were harvested 24 h post-transfection.
Stability studies
The protein biosynthesis-inhibitor cycloheximide (15 µg/ml; Sigma) was added to cells 36 h post-transfection. The cells were harvested at the following exposure times to cycloheximide: 0, 30, 60, 90 min, and the protein was isolated in the same manner as for the expression studies.
SDS–PAGE and Western blot analysis
Protein samples were quantified using a detergent-compatible protein assay kit (Bio-Rad, Hercules, CA, USA). SDS–PAGE and western blot analyses were performed using standard techniques. MuSK C-19 goat polyclonal primary antibody (1:200; Santa Cruz Biotechnology) was used in combination with a secondary donkey anti-goat IgG-HRP antibody (1:5000; Santa Cruz Biotechnology). For the expression studies, the blot was also probed with a primary anti-GFP rabbit polyclonal serum (1:1000; Invitrogen) followed by a polyclonal goat anti-rabbit IgG-HRP secondary antibody (1:2000; Dako North America, Inc., Carpinteria, CA, USA) as a loading control. A mouse anti-GAPDH primary antibody (Zymed, San Francisco, CA, USA) was used in conjunction with a goat anti-mouse IgG-HRP (1:2000; Zymed) as a loading control for the stability studies. The membranes were developed using the ECL + Plus chemiluminescence detection system (Amersham Pharmacia Biotech) and a STORM 860 imager (Molecular Dynamics).
AChR clustering studies in MUSK deficient cells
Transfection
MuSK−/− cells were plated onto 24-well plates containing 12 mm round glass cover slips coated with Matrigel (BD Biosciences, Franklin Lakes, NJ, USA). The mouse WT-MuSK, M605I- and A727V-MuSK constructs were transfected using Lipofectamine reagent with Plus Reagent according to the manufacturer's protocol. At 1 day post-transfection the cells were induced to fusion and fresh differentiation medium was applied daily.
Agrin stimulation of myotubes
Neural agrin was prepared as described elsewhere (36). After 4 days in fusion medium, cells were exposed to ∼0.1 nM agrin conditioned medium or control medium for 24 h prior to cell staining and fixation.
Immunocytochemistry
The cells were exposed to 125 nm rhodamine-conjugated α-BGT for 2 h at 39°C/10% C02, rinsed with PBS and fixed with 4% paraformaldehyde for 10 min. The cover slips were mounted onto microscope slides using ProLong Gold Anti-Fade reagent with DAPI (Invitrogen) and visualized using a Nikon E-600 fluorescent microscope (Nikon Instruments Inc.).
Quantitation of AChR clusters
Myotubes were analyzed using fluorescence microscopy with a 20× lens. Pictures from 12 random fields were taken through a Texas Red filter, and clusters above 5 µm were counted.
Pull-down assay
Transfection
Each of the human WT, M605I and A727V mutant constructs were transfected into HEK293 cells using DharmaFECT1 transfection reagent as described previously (37). The cells were co-transfected with a human DOK7 construct and GFP to verify transfection efficiency. After 48 h of transfection, the transfected and control cells were lysed with RIPA buffer (Sigma) and treated with Protease Cocktail Inhibitor (Sigma).
Histidine affinity column purification analysis
This analysis was based on immobilized metal affinity chromatography as previously described (38,39). Briefly, His SpinTrap columns were used to purify histidine-tagged MuSK proteins from WT-MuSK, A727V and M605I MuSK mutants. The cell pellets were diluted with 1 ml of native conditions binding buffer (20 mm Sodium phosphate, 500 mm NaCl, 20 mm imidazole, pH 7.4), and the cells were enzymatically lysed in 0.2 mg/ml, 20 µg/ml DNAse, 1 mm MgCl2 and 1 mm PMSF.
SDS–PAGE and Western blot analysis
Protein samples were quantified using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). The protein samples were diluted with a sample application buffer [1.0 ml of 0.5 m Tris–HCl (pH 6.8), 1.9 g ultra pure and 10% SDS], separated on a 4–20% SDS–PAGE gradient gel (Bio-Rad) and electroblotted as detailed previously by (40). The polyclonal primary antibodies, goat anti-MuSK and rabbit anti-Dok-7 (Santa Cruz Biotechnology), were diluted 1:1000 in LI-COR Odyssey Blocking Buffer and incubated overnight at 4°C. The secondary antibodies, donkey anti-goat IRDye800cw and goat anti-rabbit IRDye800cw (LI-COR Biosciences, Lincoln, NE, USA), were diluted 1:5000. To normalize for protein content, the housekeeping protein β-actin was visualized in each sample with anti-β-actin monoclonal antibody (1:1000; Santa Cruz Biotechnology) and with a secondary antibody goat anti-mouse rabbit IRDye600cw (1:5000; LI-COR Biosciences). The protein expression was then quantitated using the Odyssey Imaging System (LI-COR).
MuSK structure analysis
The Swiss-Pdb Viewer graphics was used to provide an interface allowing to analyze the position of the identified mutations in the unphosphorylated crystal structure of the cytoplasmic domain of rat MuSK (accession PDB 1LUF).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Muscular Dystrophy Association of America; the National Institutes of Health (Grant R01NS049117-01) the Myasthenia Gravis Foundation of America and the Myasthenia Gravis Foundation of California. Funding to pay the Open Access publication charges for this article was provided by The Myasthenia Gravis Foundation of California.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr Stevan R. Hubbard for his technical assistance and for providing us a diagram with the crystal structure of MuSK. Mary Edwards provided editorial help.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Hantaï D., Richard P., Koenig J., Eymard B. Congenital myasthenic syndromes. Curr. Opin. Neurol. 2004;17:539–551. doi: 10.1097/00019052-200410000-00004. doi:10.1097/00019052-200410000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Engel A.G., Shen X.M., Selcen D., Sine S.M. What have we learned from the congenital myasthenic syndromes. J. Mol. Neurosci. 2010;40:143–153. doi: 10.1007/s12031-009-9229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beeson D., Webster R., Cossins J., Lashley D., Spearman H., Maxwell S., Slater C.R., Newsom-Davis J., Palace J., Vincent A. Congenital myasthenic syndromes and the formation of the neuromuscular junction. Ann. N. Y. Acad. Sci. 2008;1132:99–103. doi: 10.1196/annals.1405.049. doi:10.1196/annals.1405.049. [DOI] [PubMed] [Google Scholar]
- 4.Engel A.G., Shen X.M., Selcen D., Sine S.M. Further observations in congenital myasthenic syndromes. Ann. N. Y. Acad. Sci. 2008;1132:104–113. doi: 10.1196/annals.1405.039. doi:10.1196/annals.1405.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chevessier F., Faraut B., Ravel-Chapuis A., Richard P., Gaudon K., Bauché S., Prioleau C., Herbst R., Goillot E., Ioos C., et al. MUSK, a new target for mutations causing congenital myasthenic syndrome. Hum. Mol. Genet. 2004;13:3229–3240. doi: 10.1093/hmg/ddh333. doi:10.1093/hmg/ddh333. [DOI] [PubMed] [Google Scholar]
- 6.Beeson D., Higuchi O., Palace J., Cossins J., Spearman H., Maxwell S., Newsom-Davis J., Burke G., Fawcett P., Motomura M., et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–1978. doi: 10.1126/science.1130837. doi:10.1126/science.1130837. [DOI] [PubMed] [Google Scholar]
- 7.Huzé C., Bauché S., Richard P., Chevessier F., Goillot E., Gaudon K., Ben Ammar A., Chaboud A., Grosjean I., Lecuyer H.A., et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am. J. Hum. Genet. 2009;85:155–167. doi: 10.1016/j.ajhg.2009.06.015. doi:10.1016/j.ajhg.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim N., Stiegler A.L., Cameron T.O., Hallock P.T., Gomez A.M., Huang J.H., Hubbard S.R., Dustin M.L., Burden S.J. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. doi:10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang B., Luo S., Wang Q., Suzuki T., Xiong W.C., Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–297. doi: 10.1016/j.neuron.2008.10.006. doi:10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim N., Burden S.J. MuSK controls where motor axons grow and form synapses. Nat. Neurosci. 2008;11:19–27. doi: 10.1038/nn2026. doi:10.1038/nn2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okada K., Inoue A., Okada M., Murata Y., Kakuta S., Jigami T., Kubo S., Shiraishi H., Eguchi K., Motomura M., et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. 2006;312:1802–1805. doi: 10.1126/science.1127142. doi:10.1126/science.1127142. [DOI] [PubMed] [Google Scholar]
- 12.Linnoila J., Wang Y., Yao Y., Wang Z.Z. A mammalian homolog of Drosophila tumorous imaginal discs, Tid1, mediates agrin signaling at the neuromuscular junction. Neuron. 2008;60:625–641. doi: 10.1016/j.neuron.2008.09.025. doi:10.1016/j.neuron.2008.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finn A.J., Feng G., Pendergast A.M. Postsynaptic requirement for Abl kinases in assembly of the neuromuscular junction. Nat. Neurosci. 2003;6:717–723. doi: 10.1038/nn1071. doi:10.1038/nn1071. [DOI] [PubMed] [Google Scholar]
- 14.Strochlic L., Cartaud A., Cartaud J. The synaptic muscle-specific kinase (MuSK) complex: new partners, new functions. J. Bioessays. 2005;27:1129–1135. doi: 10.1002/bies.20305. doi:10.1002/bies.20305. [DOI] [PubMed] [Google Scholar]
- 15.Friese M.B., Blagden C.S., Burden S.J. Synaptic differentiation is defective in mice lacking acetylcholine receptor beta-subunit tyrosine phosphorylation. Development. 2007;134:4167–4176. doi: 10.1242/dev.010702. doi:10.1242/dev.010702. [DOI] [PubMed] [Google Scholar]
- 16.Gautam M., Noakes P.G., Mudd J., Nichol M., Chu G.C., Sanes J.R., Merlie J.P. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 1995;377:232–236. doi: 10.1038/377232a0. doi:10.1038/377232a0. [DOI] [PubMed] [Google Scholar]
- 17.DeChiara T.M., Bowen D.C., Valenzuela D.M., Simmons M.V., Poueymirou W.T., Thomas S., Kinetz E., Compton D.L., Rojas E., Park J.S., et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. doi:10.1016/S0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- 18.Gautam M., DeChiara T.M., Glass D.J., Yancopoulos G.D., Sanes J.R. Distinct phenotypes of mutant mice lacking agrin, MuSK, or rapsyn. Brain Res. Dev. Brain Res. 1999;114:171–178. doi: 10.1016/s0165-3806(99)00013-9. doi:10.1016/S0165-3806(99)00013-9. [DOI] [PubMed] [Google Scholar]
- 19.Misgeld T., Kummer T.T., Lichtman J.W., Sanes J.R. Agrin promotes synaptic differentiation by counteracting an inhibitory effect of neurotransmitter. Proc. Natl. Acad. Sci. USA. 2005;102:11088–11093. doi: 10.1073/pnas.0504806102. doi:10.1073/pnas.0504806102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valenzuela D.M., Stitt T.N., DiStefano P.S., Rojas E., Mattsson K., Compton D.L., Nuñez L., Park J.S., Stark J.L., Gies D.R., et al. Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron. 1995;15:573–584. doi: 10.1016/0896-6273(95)90146-9. doi:10.1016/0896-6273(95)90146-9. [DOI] [PubMed] [Google Scholar]
- 21.Till J.H., Becerra M., Watty A., Lu Y., Ma Y., Neubert T.A., Burden S.J., Hubbard S.R. Crystal structure of the MuSK tyrosine kinase: insights into receptor autoregulation. Structure. 2002;10:1187–1196. doi: 10.1016/s0969-2126(02)00814-6. doi:10.1016/S0969-2126(02)00814-6. [DOI] [PubMed] [Google Scholar]
- 22.Mihaylova V., Salih M.A., Mukhtar M.M., Abuzeid H.A., El-Sadig S.M., von der Hagen M., Huebner A., Nürnberg G., Abicht A., Müller J.S., et al. Refinement of the clinical phenotype in musk-related congenital myasthenic syndromes. Neurology. 2009;73:1926–1928. doi: 10.1212/WNL.0b013e3181c3fce9. doi:10.1212/WNL.0b013e3181c3fce9. [DOI] [PubMed] [Google Scholar]
- 23.Müller J.S., Herczegfalvi A., Vilchez J.J., Colomer J., Bachinski L.L., Mihaylova V., Santos M., Schara U., Deschauer M., Shevell M., et al. Phenotypical spectrum of DOK7 mutations in congenital myasthenic syndromes. Brain. 2007;130:1497–1506. doi: 10.1093/brain/awm068. doi:10.1093/brain/awm068. [DOI] [PubMed] [Google Scholar]
- 24.Palace J., Lashley D., Newsom-Davis J., Cossins J., Maxwell S., Kennett R., Jayawant S., Yamanashi Y., Beeson D. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain. 2007;130:1507–1515. doi: 10.1093/brain/awm072. doi:10.1093/brain/awm072. [DOI] [PubMed] [Google Scholar]
- 25.Anderson J.A., Ng J.J., Bowe C., McDonald C., Richman D.P., Wollmann R.L., Maselli R.A. Variable phenotypes associated with mutations in DOK7. Muscle Nerve. 2008;37:448–456. doi: 10.1002/mus.20944. doi:10.1002/mus.20944. [DOI] [PubMed] [Google Scholar]
- 26.Selcen D., Milone M., Shen X.M., Harper C.M., Stans A.A., Wieben E.D., Engel A.G. Dok-7 myasthenia: phenotypic and molecular genetic studies in 16 patients. Ann. Neurol. 2008;64:71–87. doi: 10.1002/ana.21408. doi:10.1002/ana.21408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ben Ammar A., Petit F., Alexandri N., Gaudon K., Bauché S., Rouche A., Gras D., Fournier E., Koenig J., Stojkovic T., et al. Phenotype genotype analysis in 15 patients presenting a congenital myasthenic syndrome due to mutations in DOK7. J. Neurol. 2009 doi: 10.1007/s00415-009-5405-y. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 28.Burke G., Hammans S., Allen D., Arunachalam R., Beeson D. A treatable muscle disease. Pract. Neurol. 2009;9:233–236. doi: 10.1136/jnnp.2009.181966. doi:10.1136/jnnp.2009.181966. [DOI] [PubMed] [Google Scholar]
- 29.Slater C.R., Fawcett P.R., Walls T.J., Lyons P.R., Bailey S.J., Beeson D., Young C., Gardner-Medwin D. Pre- and post-synaptic abnormalities associated with impaired neuromuscular transmission in a group of patients with ‘limb-girdle myasthenia. Brain. 2006;129:2061–2076. doi: 10.1093/brain/awl200. doi:10.1093/brain/awl200. [DOI] [PubMed] [Google Scholar]
- 30.Maselli R.A., Dunne V., Pascual-Pascual S.I., Bowe C., Agius M., Frank R., Wollmann R.L. Rapsyn mutations in myasthenic syndrome due to impaired receptor clustering. Muscle Nerve. 2003;28:293–301. doi: 10.1002/mus.10433. doi:10.1002/mus.10433. [DOI] [PubMed] [Google Scholar]
- 31.Kong L., Wang X., Choe D.W., Polley M., Burnett B.G., Bosch-Marcé M., Griffin J.W., Rich M.M., Sumner C.J. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. doi:10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chevessier F., Girard E., Molgó J., Bartling S., Koenig J., Hantaï D., Witzemann V. A mouse model for congenital myasthenic syndrome due to MuSK mutations reveals defects in structure and function of neuromuscular junctions. Hum. Mol. Genet. 2008;22:3577–3595. doi: 10.1093/hmg/ddn251. doi:10.1093/hmg/ddn251. [DOI] [PubMed] [Google Scholar]
- 33.Pawson T., Nash P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000;14:1027–1047. [PubMed] [Google Scholar]
- 34.Maselli R.A., Mass D.P., Distad B.J., Richman D.P. Anconeus muscle: a human muscle preparation suitable for in-vitro microelectrode studies. Muscle Nerve. 1991;14:1189–1192. doi: 10.1002/mus.880141208. doi:10.1002/mus.880141208. [DOI] [PubMed] [Google Scholar]
- 35.Maselli R.A., Wollman R.L., Leung C., Distad B., Palombi S., Richman D.P., Salazar-Grueso E.F., Roos R.P. Neuromuscular transmission in amyotrophic lateral sclerosis. Muscle Nerve. 1993;16:1193–1203. doi: 10.1002/mus.880161109. doi:10.1002/mus.880161109. [DOI] [PubMed] [Google Scholar]
- 36.Ferns M.J., Campanelli J.T., Hoch W., Scheller R.H., Hall Z. The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron. 1993;11:491–502. doi: 10.1016/0896-6273(93)90153-i. doi:10.1016/0896-6273(93)90153-I. [DOI] [PubMed] [Google Scholar]
- 37.Arredondo J., Chernyavsky A.I., Karaouni A., Jolkosky D.L., Pinkerton K.E., Grando S.A. Receptor-mediated tobacco toxicity: cooperation of the Ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways downstream of alpha7 nicotinic receptor in oral keratinocytes. FASEB J. 2006;20:2093–2101. doi: 10.1096/fj.06-6191com. doi:10.1096/fj.06-6191com. [DOI] [PubMed] [Google Scholar]
- 38.Charlton A., Zachariou M. Immobilized metal ion affinity chromatography of histidine-tagged fusion proteins. Methods Mol. Biol. 2008;421:137–149. doi: 10.1007/978-1-59745-582-4_10. doi:10.1007/978-1-59745-582-4_10. [DOI] [PubMed] [Google Scholar]
- 39.Arredondo J., Chernyavsky A.I., Marubio L.M., Beaudet A.L., Jolkovsky D.L., Pinkerton K.E., Grando S.A. Receptor-mediated tobacco toxicity: regulation of gene expression through alpha3beta2 nicotinic receptor in oral epithelial cells. Am. J. Pathol. 2005;166:597–613. doi: 10.1016/s0002-9440(10)62281-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arredondo J., Chernyavsky A.I., Webber R.J., Grando S.A. Biological effects of SLURP-1 on human keratinocytes. J. Invest. Dermatol. 2005;125:1236–1241. doi: 10.1111/j.0022-202X.2005.23973.x. doi:10.1111/j.0022-202X.2005.23973.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.