

Abstract
The activation of the protein kinase Raf at the cell membrane is a critical step in cell signaling during development, but the mechanisms that regulate Raf activity remain incompletely defined. We previously demonstrated that the C. elegans cgr-1 gene encodes a CRAL/TRIO domain-containing protein that is a critical modulator of Ras-dependent cell fate specification during C. elegans development. Here we identify the mammalian α-tocopherol associated protein-1 (TAP-1) as a functional orthologue of cgr-1. TAP-1 mRNA was expressed in many tissues, and TAP-1 protein colocalized with Ras and Raf at the cell membrane. Reducing TAP-1 expression by RNA interference increased Ras/ERK signaling in multiple cell types. These functional studies demonstrate that CRAL/TRIO domain proteins play a conserved role in regulating Ras signaling. Biochemical analyses indicated that TAP-1 operates at the level of Raf, since TAP-1 function negatively regulated the amount of Raf-1 recruited to GTP-bound Ras at the cell membrane. TAP-1 plays a significant physiological role in controlling cell division, since reducing TAP-1 expression increased the oncogenic capacity of Ras transformed human cancer cell lines. These studies identify TAP-1 as a critical modulator of Ras-mediated cellular signaling.
Keywords: Ras signaling pathway, Raf, CRAL/TRIO, GOLD, TAP-1, CGR-1, Cancer
Introduction
Cell fates are established during development by spatially controlled activation of signal transduction pathways that regulate transcription factors. The receptor tyrosine kinase (RTK)/Ras/extracellular-regulated kinase (ERK) signaling pathway is used reiteratively during animal development to specify a wide range of cell fates. The activation of Raf is a critical step in this signaling pathway, but the molecular requirements for Raf activation still remain largely unknown. Following ligand binding by Receptor Tyrosine Kinases, Raf binds to GTP-loaded Ras at the cell membrane and initiates a series of biochemical reactions. Activated Raf phosphorylates and activates the mitogen activated ERK kinases, MEK1 and MEK2, which in turn phosphorylate and activate the extracellular signal activated kinases, ERK1 and ERK2. Phosphorylated ERK translocates to the nucleus and affects gene expression through the regulation of transcription factor phosphorylation (Kolch, 2005). Identifying and characterizing the proteins involved in this signaling pathway has important implications for human health, since mutations leading to the constitutive activation of Raf have been widely implicated in cellular oncogenesis (Bos, 1989; Ulku and Der, 2003).
In C. elegans, this signaling pathway has been extensively characterized during development of the hermaphrodite vulva, an epidermal structure for egg laying and sperm entry (Kornfeld, 1997; Greenwald, 1997). In third larval stage hermaphrodites, six ventral epidermal blast cells called P3.p - P8.p (Pn.p cells) lie along the anterior-posterior axis. These Pn.p cells are an equivalence group, since each cell can adopt the 1° vulval cell fate (eight descendents), the 2° vulval cell fate (seven descendents), or the non-vulval 3° cell fate (two descendents). During vulval induction, the anchor cell of the somatic gonad secretes LIN-3 ligand, an epidermal growth factor (EGF) homolog, thereby activating the LET-23/RTK on the adjacent P6.p cell. Activated LET-23 recruits the SEM-5 adaptor (GRB2) and the LET-341 guanine nucleotide exchange factor (SOS), followed by sequential activation of the GTPase LET-60 (RAS) and the protein kinases LIN-45 (RAF), MEK-2 (MEK), and MPK-1 (ERK). Activated MPK-1 phosphorylates substrates including the LIN-1 ETS transcription factor, thereby promoting the 1° fate in P6.p. P6.p signals P5.p and P7.p to adopt 2° fates by activating a lateral signal involving LIN-12/Notch. The 22 descendents of P5.p, P6.p and P7.p invaginate and differentiate to form the vulval structure. P3.p, P4.p and P8.p receive neither signal and therefore adopt 3° fates. Mutations that reduce activation of RTK/Ras/ERK signaling cause a vulvaless (Vul) phenotype, whereas mutations that constitutively activate this pathway cause more than three Pn.p cells to adopt the 1° or 2° vulval cell fate, resulting in a multivulval (Muv) phenotype characterized by ectopic patches of vulval tissue.
Genetic screens for C. elegans mutants have led to the discovery of conserved proteins that play functionally significant roles in Ras-ERK signaling (Kornfeld et al, 1995; Moghal and Sternberg, 2003). We identified the CRAL/TRIO and GOLD domain suppressor of activated ras (cgr-1) as a suppressor of the Muv phenotype caused by a mutation that constitutively actives Ras. Genetic analysis indicated that cgr-1 functions downstream or in parallel to ras, and upstream of the lin-1 ETS domain transcription factor (Goldstein et al, 2006). Precisely where cgr-1 acts within this pathway is unknown. CRAL/TRIO domains form hydrophobic binding pockets that bind small hydrophobic ligands (Bateman et al, 2000; Sha et al, 1998; Stocker et al, 2002; Stocker and Baumann, 2003; Panagabko et al, 2003). GOLD (GOLgi Dynamics) domains were identified by protein alignments and are of unknown function (Stocker et al, 2002; Anantharman and Aravind, 2002).
Utilizing proteomic alignment techniques, we identified three human proteins with an identical domain structure to CGR-1: TAP-1, TAP-2 and TAP-3. The prototypic family member, α-tocopherol associated protein (TAP-1), was identified by its affinity for α-tocopherol (Vitamin E) in vitro. Human TAP-2 and TAP-3 are 77% and 70% identical to TAP-1 at the protein level, and all three mammalian proteins are ∼30% identical to CGR-1 (Goldstein et al, 2006; Kempna et al, 2003). TAP-1 was originally described as Supernatant Protein Factor (SPF) based upon its ability to promote the conversion of squalene to lanosterol by liver microsomes, suggestive of roles in cholesterol metabolism, although the relevance of this interaction in vivo is unknown (Zimmer et al, 2000; Tchen and Bloch, 1957; Shibata et al, 2001). The biological functions of the TAP proteins are not well defined, and the relevance of the identified hydrophobic binding partners remains enigmatic. We hypothesized that the TAP proteins represent functional orthologues of CGR-1 and regulate signaling downstream of Ras activation. Accordingly, we demonstrate that human TAP-1 regulates signaling in vertebrate cells, acting at a previously uncharacterized step of the Ras pathway by inhibiting the ability of Raf to translocate to activated Ras at the cell membrane. These studies also reveal an in vivo function for the TAP family proteins and identify a new regulator of Ras-mediated signaling.
Materials and Methods
Cells, plasmids and antibodies
All cells were purchased from the American Type Culture Collection (Rockville, MD). HeLa, COS-7, and MDA-MB-231 cells were maintained in Dulbecco's Modified Eagles Media supplemented with 10% heat inactivated FCS, 2 mM L-glutamine, 10 mM sodium pyruvate, and 50 mM 2-mercaptoethanol. A549 cells were maintained in Ham's F12K medium with 2 mM-glutamine adjusted to contain 1.5 g/l sodium bicarbonate and 10% heat inactivated FCS. Mouse TAP-1 cDNA was obtained from ATCC. Wild-type full length TAP-1 was expressed in pcDNA3.1Zeo+ (Invitrogen, Carlsbad, CA). Plasmids encoding human wild-type and mutant GFP-tagged H-Ras and K-Ras were a kind gift of John Hancock (Apolloni et al, 2000). For biochemical analyses, rabbit anti-Raf-1 and rabbit anti-transferrin receptor were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), anti-pan-Ras was purchased from ECB Biosciences, and anti-phosphoERK, anti-phosphoMEK, anti-ERK and anti-MEK were purchased from Cell Signaling Technologies (Danvers, MA). Horseradish peroxidase-conjugated secondary antibodies were purchased from Pierce (Rockford, IL).
Expression of TAP mRNA
RNA was extracted from mouse brain, heart, kidney, lung, spleen, thymus and ES cell cultures using an RNA extraction kit (Qiagen, Valencia, CA). cDNA was generated using oligo-dT primed Superscript II Reverse Transcriptase reactions from each tissue. Primers designed to specifically amplify TAP-1, TAP-2 or TAP-3 were then used to PCR amplify the input cDNA. Following ethidium bromide stained gel fractionation, the identity of DNA products from lung tissue was confirmed by sequencing.
Confocal Microscopy
Cells were transfected for 24 hours with Flag-TAP-1 and GFP-tagged Ras or GFP-tagged Raf-1 expression plasmids with lipofectamine 2000 (Invitrogen) and cultured for a further 24 hours in complete DMEM media. Cells were placed in 4% paraformaldehyde, permeablized in 0.15% Triton X-100, and blocked in 1% BSA. Cells were stained with Cy3 conjugated anti-Flag (clone M2, Sigma, St. Louis, MO), washed and mounted for imaging in Slowfade (Molecular Probes, Eugene, OR). Digitized two-color fluorescence images were obtained with an inverted Plan Apochromat ×63/1.4 oil objective on a Zeiss LSM 510 Confocal Microscope.
RNA interference of TAP-1
Human TAP-1 RNAi duplexes were purchased from Dharmacon (Lafayette, CO). Duplexes; A. Sense GAACACAGCGUGCAGAUUUUU, Antisense 5′P-UCUCGCACAUAAUACUUCCUU; B. Sense GGAAGGAGGUUUUACUGAAUU, Antisense 5′P-UUCAGUAAAACCUCCUUCCUU; C. Sense GGAAGUAUUAUGUGCGAGAUU; Antisense 5′P-UCUCGCACAUAAUACUUCCUU; Control sense; UAGCGACUAAACACAUCAA. Cells cultured in OptiMEM (Invitrogen) were transfected with100 nM of RNAi duplex using Oligofectamine Reagent (Invitrogen), incubated overnight, transferred into complete DMEM media for 48 hours, and harvested for analysis or transfected with expression plasmids or analyzed for anchorage independent growth. Alpha-tocopherol (Sigma, St. Louis, MO) was solublized in ethanol, dried under Nitrogen, dissolved in DMSO, incubated with fetal calf serum for 30 minutes at 37°C, and added to cell cultures. Cells were exposed to α-tocopherol or DMSO as a control for the final 24 hours of culture.
Biochemical Analyses
Cells were lysed in Nonidet P-40 lysis buffer (150 mM NaCl, 50 mM Tris-HCl, pH 8.0, 25 mM NaF, 1mM sodium orthovanadate, 0.5% Nonidet P-40) containing protease inhibitors (Roche Complete Mini, Indianapolis, IN) and clarified at 13,000 rpm for 10 minutes at 4°C. Immunoprecipitations were performed for 1h at 4°C, captured with protein-G Sepharose for a further hour at 4°C, and washed extensively in Nonidet P-40 lysis buffer. GST-Ras binding assays were performed using a GST-fusion protein containing the Ras binding domain of Raf that was expressed in E. coli and purified upon Glutathione-Sepharose beads. Purified protein was added to cell lysates, incubated for 1 hour at 4°C, and washed extensively in Nonidet-P40 buffer. Immunoprecipitations and cell lysates were resolved by SDS-PAGE, Western Blotted onto polyvinylidene difluoride membranes (Millipore, Bedford, MA), blocked in Tris Buffered Saline-0.1% Tween (TBS-T) containing 3% non-fat dried milk, and immunoprobed in TBS-T with 1% non-fat dried milk. Blots were stripped for 30min at 55°C (100 mM 2-ME, 2% SDS, 62.5 mM Tris-HCl, pH 6.8) before reprobing. Proteins were detected with horseradish peroxidase-conjugated secondary Abs and enhanced chemiluminescence (Pierce).
Fractionation
Pelleted cells were resuspended in 2 ml ice-cold hypotonic lysis buffer (10 mM Tris-HCl (pH 7.5), 1mM MgCl2, 1 mM sodium orthovanadate) containing protease inhibitors (10 μg/ml aprotinin, 10μg/ml leupeptin, 10 μg/ml pepstatin, 1mM EDTA, 1mM PMSF) added immediately before use. After 30 min incubation on ice, lysis was facilitated by Dounce homogenization for 12 strokes on ice, and lysates were clarified to remove nuclear material and intact cells. Supernatants were centrifuged at 100,000 × g for one hour at 4°C. The pellet (P100) fraction was solublized in Nonidet P-40 lysis buffer containing protease inhibitors.
Cell transfections and reporter assays
Cells were transfected with Lipofectamine 2000 (Invitrogen). For reporter assays cells were transfected so that each well received 100 ng pFR-luciferase reporter plasmid, 200 ng pFA-Elk2 expression plasmid, and 100 ng CMV-β-galactosidase expression plasmid (Lemercier et al, 2000). Cells were harvested after 19 hours, and luciferase activity and β-galactosidase activity were measured on a TR717 microplate luminometer using the Luciferase Assay System (Promega, Madison, WI) and the Galactolight Plus System (Tropix, Bedford, MA) according to the manufacturers' protocols.
Anchorage-Independent Growth Assay
MDA-MB-231 cells treated with RNA duplexes were plated at 2000 cells per 10cm2 tissue culture dish in complete DMEM media containing 0.33%(w/v) agar as described (Valenzuela and Groffen, 1986). Cells were incubated at 37°C in 5% CO2 for twelve days, and colonies containing >20 cells were counted.
Results
TAP-1 was expressed in multiple tissues
Mammalian genomes contain three TAP genes as defined by the presence of sequences encoding a CRAL/TRIO domain amino-terminal to a GOLD domain (Figure 1A). These are located as gene clusters on chromosomes 22 and 11 in humans and mice, respectively. To determine the expression profiles of the TAP genes, we performed a qualitative RT-PCR analysis in a variety of mouse tissues, utilizing primers specific to each isoform (Figure 1B). All three TAP genes were expressed in one or more tissues. TAP-1 was the most widely expressed, being present in four of the seven tissues examined, including brain, kidney, lung, and spleen. TAP-2 was detected in the lung and thymus, and TAP-3 was detected solely in the lung. Due to its widespread expression, we chose to focus on TAP-1 as a potential regulator of Ras-ERK signaling.
Figure 1. Expression profiles of TAP-1, TAP-2, and TAP-3.
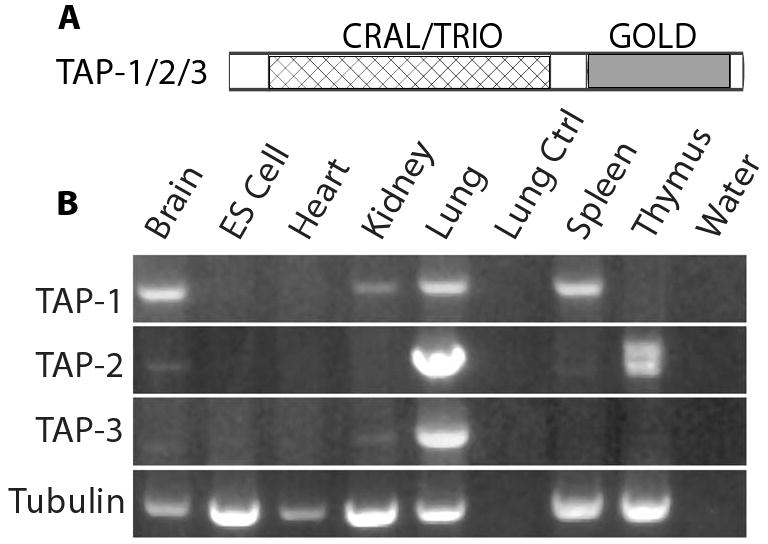
A. The domain structure of TAP proteins. B. RNA purified from mouse tissues (brain, heart, kidney, lung, spleen and thymus) and mouse ES cells was used to generate cDNA. Bands are ethidium bromide stained fragments of DNA that were amplified from the cDNA using primers designed to amplify TAP-1, TAP-2 and TAP-3. Primers that specifically amplified tubulin were used as a control for the input cDNA, and this experiment demonstrated the presence of amplifiable RNA in each tissue sample. To control for non-specific amplification, we analyzed control samples that contained RNA but no cDNA (Lung Ctrl), or contained no RNA (water). TAP-1 was detected in brain, kidney, lung and spleen, TAP-2 was detected in lung and thymus, and TAP-3 was detected only in lung. The TAP-1 and TAP-3 primers generated a single band in all cases, whereas the TAP-2 primers generated two bands from thymus tissue that may result from alternatively spliced transcripts.
TAP-1 colocalized with Ras and Raf at the cell membrane
We analyzed the subcellular localization of TAP-1 in COS-7 cells due to their easily distinguishable cellular morphology. Flag-Tagged TAP-1 was expressed in COS-7 cells and visualized by immunostaining with an anti-Flag monoclonal antibody. The cytosol contained a significant amount of diffusely localized TAP-1 staining, whereas the membrane of the cell contained brightly stained patches interspersed with regions of little TAP-1 staining (Figure 2A, D). To determine the relationship between TAP-1 localization and Ras-ERK signaling proteins, we co-expressed Flag-tagged TAP-1 with GFP tagged K-Ras or H-Ras. Both K-Ras and H-Ras displayed areas of bright staining at the cell membrane (Figure 2B, E) that colocalized with TAP-1 (Figure 2C, F). Because these imaging experiments have limited resolution, these results do not provide evidence that TAP-1 is physically associated with Ras. Furthermore, these results do not directly establish the subcellular localization of endogenous TAP-1 and Ras, since TAP-1 and Ras proteins were over-expressed and epitope tagged to permit visualization.
Figure 2. TAP-1 colocalized with Ras and Raf-1 at the plasma membrane.
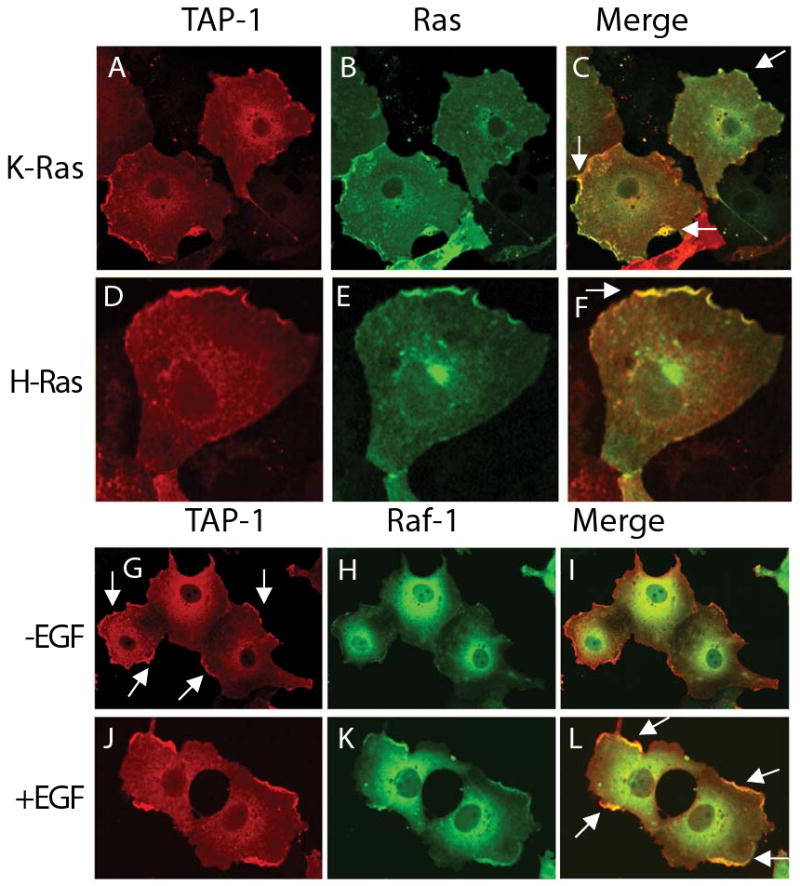
Flag-TAP-1 and GFP-K-Ras (A-C), GFP-H-Ras (D-F) or GFP-Raf-1 (G-L) were expressed in COS-7 cells and visualized by confocal microscopy. Panels illustrate 1 to 3 representative cells. Cells were either untreated (A-I) or treated with 20 ng/ml EGF for 5 minutes (J, K, L). Yellow in the merged images represents patches of colocalization between Flag-TAP-1 (red) and GFP-Ras or GFP-Raf (green). Arrows indicate patches of Flag-TAP-1 at the cell membrane (G), or patches of colocalization of TAP-1 with GFP-Ras (C, F) or Raf (L) at the cell membrane.
To determine if TAP-1 staining is affected by ligand activation of the signaling pathway, we visualized TAP-1 staining in cells treated with EGF. TAP-1 staining was not affected by EGF treatment, indicating that TAP-1 localization at the membrane occurs in a ligand independent manner (Figure 2G, J). In contrast, a GFP-tagged form of Raf-1 was recruited to the cell membrane following treatment with EGF (Figure 2H, K), indicating that the over-expressed and epitope tagged Raf displays normal localization and ligand responsiveness. When Raf is recruited to the membrane, it colocalized with the membrane pool of TAP-1 (Figure 2L). These results support the model that TAP-1 may operate at the cell membrane to affect Ras-ERK signaling.
Endogenous TAP-1 negatively regulated Ras-ERK signaling in cancer cell lines
Having established that TAP-1 colocalizes with Ras and Raf, we wanted to determine if endogenous TAP-1 regulates Ras signaling. We identified that the MDA-MB-231 breast carcinoma and A549 lung carcinoma cancer cell lines express measurable levels of TAP-1 transcript. These cell lines contain mutationally activated oncogenic K-Ras and thus provide a sensitized background to examine the effects of TAP-1 gene silencing (Valenzuela and Groffen, 1986; Gilhooly and Rose, 1999).
We identified three mutually exclusive RNA duplexes that significantly reduced the level of TAP-1 transcripts in MDA-MB-231 cells (Figure 3A). These effects were observed in comparison to control cells treated with no RNA (data not shown) and control cells treated with a dsRNA that has a scrambled sequence (Figure 3A) – this treatment controls for physical effects that might be caused by transfecting small RNA molecules. Treatment with RNA duplexes directed against TAP-1 also reduced the level of transfected TAP-1 protein (Figure 3B). Because this TAP-1 protein was over-expressed and epitope tagged, these results do not directly demonstrate the extent of the effect on endogenous TAP-1 protein. To analyze the function of endogenous TAP-1 in the Ras signaling pathway, we measured the effect of reducing TAP-1 gene expression upon the level of phosphorylation of endogenous ERK. Reducing TAP-1 gene expression increased ERK phosphorylation but did not affect the level of total ERK (Figure 3C). Because this effect was observed with three mutually exclusive RNA duplexes, the effect is likely caused by specifically reducing TAP-1 gene expression and is unlikely to be mediated by off-target effects.
Figure 3. Endogenous TAP-1 affected Ras signaling in MDA-MB-231 breast carcinoma cells.
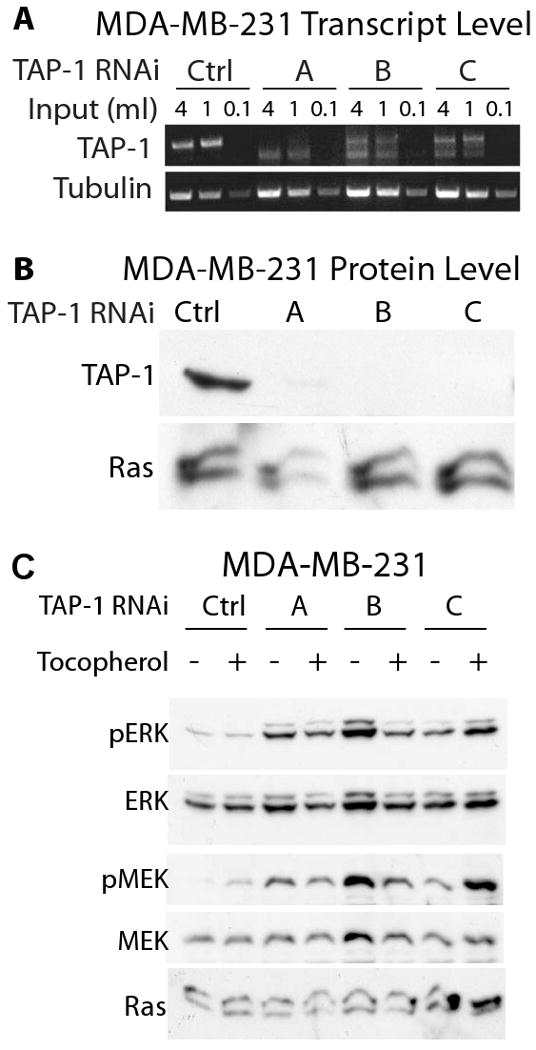
MDA-MB-231 cells were transfected with a non-targeting RNA duplex (Ctrl) or the TAP-1 specific RNA duplexes (A, B, C). A. TAP-1 and tubulin transcript levels were assessed by RT-PCR. Both TAP-1 and tubulin transcript levels were proportional to the cDNA input, demonstrating the semi-quantitative nature of the assay. TAP-1 transcript levels were significantly reduced by the specific targeting duplex, compared to the control non-targeting duplex. B. MDA-MB-231 cells were transfected with a non-targeting RNA duplex or the TAP-1 specific RNA duplexes. After 48 hours cells were transfected with a plasmid expressing Flag-tagged human TAP-1, and exogenous TAP-1 expression was monitored 48 hours later with anti-Flag immunoblotting. Protein loading was confirmed with anti-Ras immunoblotting. All three TAP-1 RNA duplexes caused a striking reduction of TAP-1 protein levels. C. Lysates were immunoprobed for phosphorylated ERK (pERK), total ERK (ERK), phosphorylated MEK (pMEK), total MEK (MEK), and Ras. α-tocopherol-treated cells were exposed to 100 ug/ml of α-tocopherol; this treatment did not affect ERK phosphorylation.
To determine if TAP-1 regulates ERK signaling in additional cell types, we examined the A549 lung carcinoma cell line. Treatment with two mutually exclusive RNA duplexes reduced the levels of TAP-1 transcripts (Figure 4A). Reducing TAP-1 gene expression also increased the level of phosphorylated ERK in A549 carcinoma cells (Figure 4B). HeLa cells transfected with an activated form of K-Ras provide a sensitized system to examine Ras-ERK signaling, and we demonstrated that HeLa cells express endogenous TAP-1 transcripts that can be silenced with RNA duplexes (Figure 5A). Reducing TAP-1 gene expression increased the level of phosphorylated ERK in HeLa cells expressing an activated form of K-Ras, but had minimal effects on the level of phosphorylated ERK in HeLa cells expressing wild-type K-Ras (Figure 5B). In this experimental system, the effects of reducing TAP-1 activity were evident only in cells with an activated mutant form of Ras. In contrast to the MDA-MB-231 and A549 cell lines, we noted that Ras signaling in HeLa cells was sensitive to the α-tocopherol. Notably, α-tocopherol inhibited ERK phosphorylation in HeLa cells expressing oncogenic K-Ras, such that cells treated with 100 ug/ml α-tocopherol displayed lower levels of ERK phosphorylation than untreated cells (Figure 5B). Importantly, reducing TAP-1 gene expression abrogated this effect, making ERK phosphorylation independent of α-tocopherol treatment. These findings indicate that Ras, TAP-1 and α-tocopherol may functionally interact in specific cellular environments.
Figure 4. Endogenous TAP-1 affected phosphorylation of ERK in A549 lung carcinoma cells.
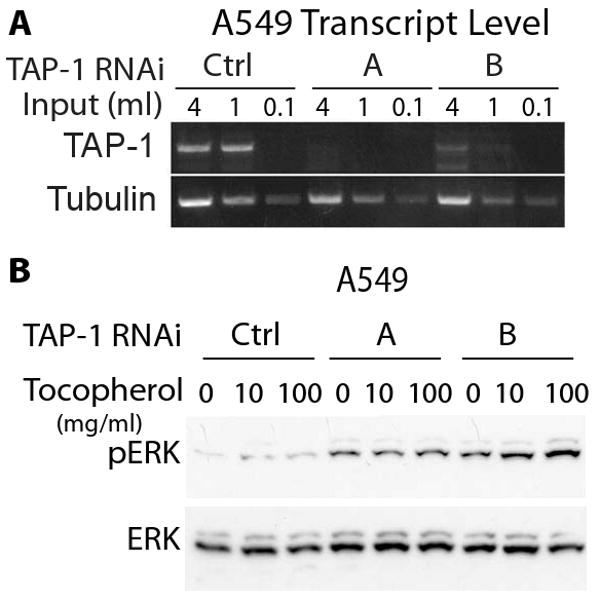
A549 cells were transfected with a non-targeting RNA duplex (Ctrl) or the TAP-1 specific RNA duplexes (A, B). A. TAP-1 and tubulin transcript levels were assessed by RT-PCR. Both TAP-1 and tubulin transcript levels were proportional to the cDNA input, demonstrating the semi-quantitative nature of the assay. TAP-1 transcript levels were significantly reduced by the specific targeting duplexes, compared to the control non-targeting duplex. B. Cell lysates were immunoprobed for endogenous phosphorylated ERK (pERK) and total ERK (ERK). Reducing the level of TAP-1 transcripts caused an increase in the level of phosphorylated ERK and did not affect the level of total ERK. Cells were exposed to 0, 10, or 100 ug/ml of α-tocopherol; this treatment did not affect ERK phosphorylation.
Figure 5. Endogenous TAP-1 affected oncogenic Ras signaling in HeLa cells.
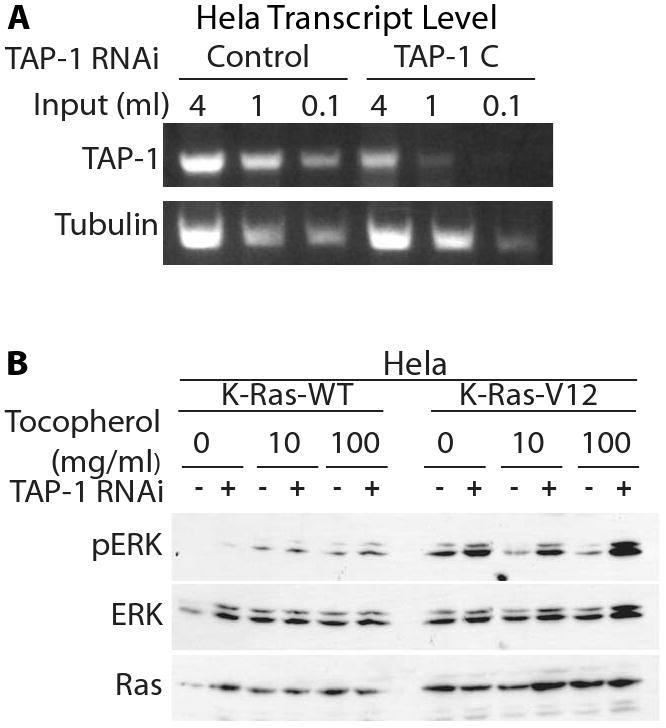
A. A non-targeting RNA duplex (Control) or the TAP-1 specific RNA duplex (TAP-1 [C]) were transfected into HeLa cells. TAP-1 and tubulin transcript levels were assessed by RT-PCR. Both TAP-1 and tubulin transcript levels were proportional to the cDNA input, demonstrating the semi-quantitative nature of the assay. TAP-1 transcript levels were significantly reduced by the specific targeting duplex, but not by the control non-targeting duplex. B. HeLa cells were transfected with a control RNA duplex (-), or with TAP-1 specific RNA duplex C (+). After 48 hours, cells were transfected with plasmids expressing wild-type or oncogenically mutated K-Ras. After 24 hours, cells were exposed to α-tocopherol. Lysates were immunoprobed as indicated.
The following evidence indicates that treatment with dsRNA directed against TAP-1 caused a substantial and specific reduction of TAP-1 gene activity; the treatment reduced the levels of tap-1 mRNA and epitope-tagged TAP-1 protein, and three different, non-overlapping dsRNA molecules directed against TAP-1 caused similar alterations in ERK phosphorylation. Importantly, our studies show that reducing endogenous TAP-1 gene expression increased Ras-ERK signaling in three different types of cells with mutationally activated Ras, demonstrating that these effects are not specific to a single cell type: HeLa cells with exogenously expressed oncogenic K-Ras, breast carcinoma cells and lung carcinoma cells.
TAP-1 regulated the recruitment of Raf to membrane-bound Ras
Having identified an important functional role for endogenous TAP-1, we performed a biochemical analysis to further delineate the position in the signaling pathway affected by TAP-1. To analyze the effect of TAP-1 on the function of Raf, we performed three independent assays. MEK is phosphorylated by Raf following the recruitment of Raf to the plasma membrane by GTP-bound Ras. Therefore, phosphorylation of MEK is a relevant and specific indication of Raf activity. MEK phosphorylation was monitored using an antibody that specifically recognizes phosphorylated MEK. Reducing the level of TAP-1 gene expression increased phosphorylation of MEK, indicating that endogenous TAP-1 negatively regulates the kinase activity of Raf (Figure 3C). To determine if TAP-1 affects Raf-1 recruitment to the plasma membrane, we performed a cellular fractionation using high speed centrifugation and measured the amount of Raf-1 present in the membrane-enriched pellet and in the cytosol-enriched supernatant. An increased amount of Raf-1 was associated with the membrane fraction of cells with reduced levels of TAP-1 gene expression (Figure 6A). Further, an increased amount of MEK was associated with the membrane, suggestive that reducing the level of TAP-1 gene expression increases the levels of functional Ras-ERK signaling complexes at the cell membrane. Conversely the amount of Ras associated with the membrane fraction was unaffected by the reduction of TAP-1 gene expression, indicating that the accumulation of Raf-1 and MEK is not due to an increase in the amount of Ras at the membrane.
Figure 6. Reducing TAP-1 gene expression increased Raf-1 association to Ras.
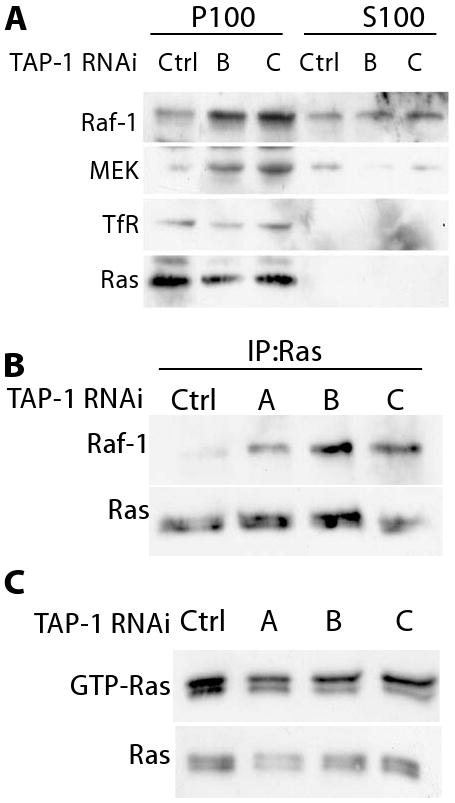
MDA-MB-231 cells were transfected with a control RNA duplex or with one of three different TAP-1 RNA duplexes that reduce endogenous TAP-1 gene expression. A. Cells were fractionated, and aliquots of the membrane (P100) and cytosolic (S100) fractions were immunoprobed as indicated. The fractionation was confirmed by analyzing the transferrin receptor (TfR), which was only detected in the membrane fraction. B. Ras association with Raf-1 was assessed by immunoprecipitating Ras and immunoprobing for Raf-1. C. The fraction of GTP-loaded Ras was determined by affinity chromatography with a GST fusion protein containing the Ras-binding domain of Raf-1 (upper panel), and compared to the total level of Ras in the lysate (lower panel).
To determine if reducing the level of TAP-1 gene expression increased the association of Raf with Ras, we directly examined the amount of Raf-1 associated with Ras by coimmunoprecipitation. The coimmunoprecipitation was performed with a commercially available anti-Ras antibody, and Western blots were used to demonstrate that the antibody recognizes endogenous Ras protein (Figure 6). A comparison of cells treated with control dsRNA and dsRNA directed against TAP-1 showed that reducing the level of TAP-1 gene expression increased the association of Raf-1 with Ras (Figure 6B). The total level of Raf-1 association with Ras might be enhanced by an increase in the fraction of Ras that is GTP-loaded, or by an increased propensity of Raf-1 to associate with GTP-loaded Ras. To distinguish between these possibilities, we assessed the fraction of Ras that is GTP-loaded. Reducing the level of TAP-1 gene expression did not increase the fraction of Ras that is GTP-loaded (Figure 6C). Together these results indicate that endogenous TAP-1 reduces the activity of Raf-1 by reducing the propensity of Raf-1 to bind GTP-loaded Ras at the cell membrane.
TAP-1 regulated cell proliferation
The biochemical analyses of Ras, Raf, MEK and ERK demonstrated that TAP-1 modulates this signaling pathway. To determine the significance of modulation by TAP-1, we analyzed two downstream effects of Ras signaling, transcription and cell proliferation. Transcription controlled by Ras-ERK signaling was monitored using a reporter assay that measures luciferase activity driven by ERK-mediated phosphorylation of the Elk-1 transcription factor. Reducing the level of TAP-1 gene expression in MDA-MB-231 cells enhanced the activation of an Elk-1-luciferase reporter construct 2 to 3 fold (Figure 7A). These results demonstrate that endogenous TAP-1 is necessary for normal levels of Ras-ERK signaling, and the effects on transcriptional control can be readily observed.
Figure 7. Reducing TAP-1 gene expression increased Ras-ERK signaling activity.
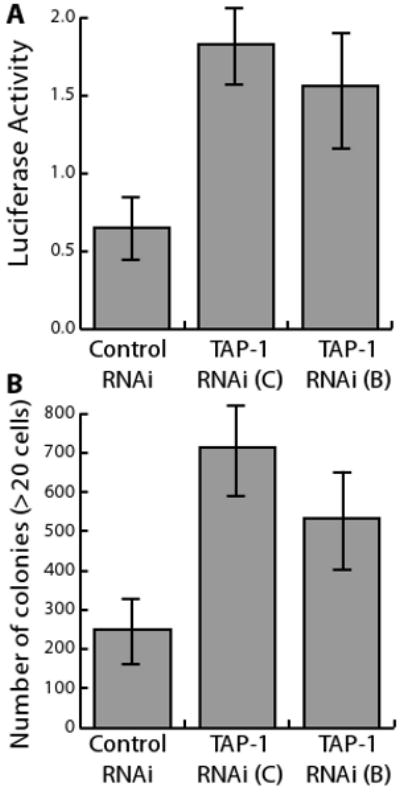
MDA-MB-231 cells were transfected with a control RNA duplex, or with one of two different TAP-1 RNA duplexes that reduce endogenous TAP-1 gene expression. A. After 48 hours, cells were transfected with Elk1-luciferase expression plasmids, and signaling activity was assessed after 48 hours. B. After 48 hours, cells were plated in soft agar, and the number of colonies was determined after 12 days. Error bars are standard deviations of three transfections conducted in parallel.
Oncogenic Ras signaling increases the ability of cells to form colonies in semi-solid media (Clark et al, 1995). We took advantage of this cellular assay to determine if changes in Ras activity arising from decreases in TAP-1 gene expression are functionally significant. Reducing TAP-1 gene expression increased the number of colonies formed in soft agar by approximately 3-fold (Figure 7B). These results suggest that endogenous TAP-1 suppresses the oncogenic capacity of cancer cells in vitro.
Discussion
TAP-1 regulates Raf-1 availability to GTP-loaded Ras at the cell membrane
Despite intense investigation into the Ras/Raf/ERK pathway, the biochemical events required for efficient Raf activation still remain enigmatic. Here we demonstrate that a process that was known to be required for efficient Raf activation, the ability of Raf to translocate from the cellular cytosol to the membrane microenvironment, is regulated by TAP-1. The best-characterized aspect of Raf activation is its physical interaction with Ras. Within the cytoplasm, Raf is maintained in an auto-inhibitory conformation mediated by the interaction of the C1 amino-terminal domain with the carboxy-terminal kinase domain (Cutler et al, 1998). 14-3-3 proteins binding to phosphorylated serine residues of Raf may stabilize this conformation, which can be altered by the activity of the PP2A phosphatase, either before or following recruitment to the cell membrane (Ory et al, 2003). Recently the CNK adaptor protein has been identified as a critical regulator of Raf activity; CNK may work in concert with the modulator KSR to regulate Raf activity through undefined allosteric mechanisms operating at the cell membrane (Douziech et al, 2006). The Ras Binding Domain (RBD) of Raf specifically binds GTP-loaded Ras and is situated in tandem with a cysteine rich domain that provides a second Ras binding site (Wellbrock et al, 2004; Chong et al, 2003; Fabian et al, 1994; Brtva et al, 1995). Once bound to Ras, Raf must adopt a state of increased kinase activity, a process that is poorly understood but which likely involves a number of allosteric changes and post-translational modifications (Wellbrock et al, 2004; Douziech et al, 2006). Because it is clear that the interaction with Ras is essential for Raf activation, understanding the mechanisms that enable Raf to access Ras at the cellular membrane is of critical importance. However, the mechanisms that regulate the equilibrium of Raf distribution between the membrane and cytosolic compartments are not well defined, and this represents a significant gap in our understanding of Raf biology.
Reducing the expression of TAP-1 in human cancer cell lines enhances Ras pathway signaling, demonstrated by the increased activity of a Ras-dependent transcriptional reporter and by the increased phosphorylation of two downstream kinases, MEK and ERK. Importantly, decreasing the expression of TAP-1 increased the recruitment of Raf-1 to the cell membrane and increased the amount of Raf-1 physically associated with Ras, independent of Ras GTP-loading. We conclude that endogenous TAP-1 modulates Ras pathway signaling by regulating the ability of Raf-1 to bind GTP-Ras at the cell membrane. This role of TAP-1 is functionally significant, as reducing TAP-1 expression increased Ras-dependent colony formation of the MDA-MB-231 human cancer cell line in soft agar. Thus, these studies reveal that TAP-1 functionally modulates Ras signaling and identify the regulation of an important step of Ras/Raf signaling.
A physiological function for the TAP family of proteins
CGR-1 was discovered as a regulator of Ras-mediated vulval cell fate specification in C. elegans (Goldstein et al, 2006). We have identified TAP-1 to be a functional orthologue of CGR-1, consistent with the hypothesis that CRAL/TRIO and GOLD domain containing proteins contribute to the regulation of the Ras signaling pathway. While genetic studies of C. elegans identified the function of CGR-1 in Ras signaling, they were unable to define precisely where CGR-1 exerted its effect. Here we advance this understanding by identifying the precise point at which this family of CRAL/TRIO and GOLD domain containing proteins affects Ras signaling, by demonstrating that TAP-1 acts at the level of Raf recruitment to Ras.
The identity of the specific hydrophobic ligand for TAP-1 remains controversial. TAP-1 was originally identified by its ability to catalyze the cholesterol biosynthetic pathway in liver microsomes and can bind squalene in vitro (Tchen and Bloch, 1957). However, C. elegans is unable to synthesize cholesterol, discounting this as a function of CGR-1 in vivo. TAP-1 was also identified through its ability to bind α-tocopherol in vitro, leading to the suggestion that it may function as a α-tocopherol transporter (Kempna et al, 2003). Interestingly, α-tocopherol and its analogues negatively regulate oncogenic Ras signaling (Donapaty et al, 2006; Ni et al, 2005). These findings raise the interesting possibility that TAP-1 may mediate the effects of α-tocopherol on Ras signaling. Our results indicate that α-tocopherol and TAP-1 may interact in HeLa cells, but this interaction was not observed in two other cell lines.
In C. elegans, loss-of-function mutations of cgr-1 reduce Ras-mediated signaling, indicating that CGR-1 positively regulates Ras signaling (Goldstein et al, 2006). Here we show that endogenous TAP-1 negatively regulates Ras signaling in specific cell types. CGR-1 and TAP-1 may function in both positive and negative manners depending upon the cellular context. This is analogous to the regulation of the ETS domain transcription factor LIN-1 in C. elegans, which acts as both a positive and negative regulator of the vulval cell fate (Tiensuu et al, 2005; Leight et al, 2005). Elk-1, a mammalian homologue of LIN-1, was originally identified as a positive regulator of transcription, but also possesses negative regulatory functions, much like LIN-1 (Yang and Sharrocks, 2004). Alternatively, the functions of TAP-1 and CGR-1 may have diverged during evolution so that CGR-1 is now primarily a positive regulator of signaling whereas TAP-1 is primarily a negative regulator of signaling.
TAP-1 regulation of Ras signaling
Exactly how TAP-1 affects Raf activation remains to be fully defined. TAP-1 may influence Raf activity either prior to, or following, the binding of Raf to Ras, by influencing either the formation or the dissociation of the signaling complex. TAP-1 may physically impinge upon the interaction of Raf and Ras in a manner analogous to SUR-8 (Li et al, 2000), although we have been unable to detect any physical interactions between TAP-1 and Ras or Raf-1. Alternatively, TAP-1 may act upon Raf prior to cellular activation, either regulating the ability of Raf to translocate to the membrane or by priming Raf within the cytosol to make it more or less able to bind to Ras. Finally, TAP-1 may have lipid exchange activity as proposed for other CRAL/TRIO domain proteins (Phillips et al, 1999). In this case TAP-1 may regulate the lipid environment at the cell membrane, either increasing or decreasing the concentration of specific hydrophobic molecules, and these changes may influence the interaction of Ras and Raf. We have demonstrated that the biological activities of TAP-1 and Raf activation are interlinked, and favor a mechanism where these molecular components constitute a critical aspect of Ras biology. The biological function of TAP-1 has previously remained undefined. We have now discovered a role for TAP-1 in vivo and identified a major new aspect of Raf activation.
Acknowledgments
We are grateful to members of the Kornfeld Lab for advice and comments about the manuscript. The research was supported by grants to K.K. from the National Institutes of Health (5R01 GM06859804 and 5R01 CA08427107). K.K. was a Scholar of the Leukemia and Lymphoma Society and is a Senior Scholar of the Ellison Medical Foundation.
Abbreviations
- RBD
Ras Binding Domain
- GOLD
GOLgi Dynamics
- CRAL/TRIO
cellular retinaldehyde and TRIO domain
- TAP
tocopherol associated protein
- CGR-1
CRAL/TRIO and GOLD domain suppressor of activated ras
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anantharaman V, Aravind L. The GOLD domain, a novel protein module involved in Golgi function and secretion. Genome Biol. 2002;3:research0023. doi: 10.1186/gb-2002-3-5-research0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol. 2000;20:2475–87. doi: 10.1128/mcb.20.7.2475-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman J, Shu H, Van Vactor D. The guanine nucleotide exchange factor trio mediates axonal development in the Drosophila embryo. Neuron. 2000;26(1):93–106. doi: 10.1016/s0896-6273(00)81141-1. [DOI] [PubMed] [Google Scholar]
- Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–9. [PubMed] [Google Scholar]
- Brtva TR, Drugan JK, Ghosh S, Terrell RS, Campbell-Burk S, Bell RM, Der CJ. Two distinct Raf domains mediate interaction with Ras. J Biol Chem. 1995;270:9809–12. doi: 10.1074/jbc.270.17.9809. [DOI] [PubMed] [Google Scholar]
- Chong H, Vikis HG, Guan KL. Mechanisms of regulating the Raf kinase family. Cell Signal. 2003;15:463–9. doi: 10.1016/s0898-6568(02)00139-0. [DOI] [PubMed] [Google Scholar]
- Clark GJ, Cox AD, Graham SM, Der CJ. Biological assays for Ras transformation. Methods Enzymol. 1995;255:395–412. doi: 10.1016/s0076-6879(95)55042-9. [DOI] [PubMed] [Google Scholar]
- Cutler RE, Jr, Stephens RM, Saracino MR, Morrison DK. Autoregulation of the Raf-1 serine/threonine kinase. Proc Natl Acad Sci U S A. 1998;95:9214–9. doi: 10.1073/pnas.95.16.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donapaty S, Louis S, Horvath E, Kun J, Sebti SM, Malafa MP. RRR-alpha-tocopherol succinate down-regulates oncogenic Ras signaling. Mol Cancer Ther. 2006;5:309–16. doi: 10.1158/1535-7163.MCT-05-0330. [DOI] [PubMed] [Google Scholar]
- Douziech M, Sahmi M, Laberge G, Therrien M. A KSR/CNK complex mediated by HYP, a novel SAM domain-containing protein, regulates RAS-dependent RAF activation in Drosophila. Genes Dev. 2006;20:807–19. doi: 10.1101/gad.1390406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian JR, Vojtek AB, Cooper JA, Morrison DK. A single amino acid change in Raf-1 inhibits Ras binding and alters Raf-1 function. Proc Natl Acad Sci U S A. 1994;91:5982–6. doi: 10.1073/pnas.91.13.5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilhooly EM, Rose DP. The association between a mutated ras gene and cyclooxygenase-2 expression in human breast cancer cell lines. Int J Oncol. 1999;15:267–70. [PubMed] [Google Scholar]
- Goldstein JL, Glossip D, Nayak S, Kornfeld K. The CRAL/TRIO and GOLD domain protein CGR-1 promotes induction of vulval cell fates in Caenorhabditis elegans and interacts genetically with the Ras signaling pathway. Genetics. 2006;172:929–42. doi: 10.1534/genetics.104.035550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald IS. Development of the vulva. In: Riddle DL, Blumenthal T, Meyer BJ, Preiss JR, editors. C elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 519–541. [PubMed] [Google Scholar]
- Kempna P, Zingg JM, Ricciarelli R, Hierl M, Saxena S, Azzi A. Cloning of novel human SEC14p-like proteins: ligand binding and functional properties. Free Radic Biol Med. 2003;34:1458–72. doi: 10.1016/s0891-5849(03)00173-4. [DOI] [PubMed] [Google Scholar]
- Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–37. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- Kornfeld K. Vulval development in Caenorhabditis elegans. Trends Genet. 1997;13:55–61. doi: 10.1016/s0168-9525(97)01005-6. [DOI] [PubMed] [Google Scholar]
- Kornfeld K, Hom DB, Horvitz HR. The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell. 1995;83:903–13. doi: 10.1016/0092-8674(95)90206-6. [DOI] [PubMed] [Google Scholar]
- Leight ER, Glossip D, Kornfeld K. Sumoylation of LIN-1 promotes transcriptional repression and inhibition of vulval cell fates. Development. 2005;132:1047–56. doi: 10.1242/dev.01664. [DOI] [PubMed] [Google Scholar]
- Lemercier C, Verdel A, Galloo B, Curtet S, Brocard MP, Khochbin S. mHDA1/HDAC5 histone deacetylase interacts with and represses MEF2A transcriptional activity. J Biol Chem. 2000;275:15594–9. doi: 10.1074/jbc.M908437199. [DOI] [PubMed] [Google Scholar]
- Li W, Han M, Guan KL. The leucine-rich repeat protein SUR-8 enhances MAP kinase activation and forms a complex with. Ras and Raf Genes Dev. 2000;14:895–900. [PMC free article] [PubMed] [Google Scholar]
- Moghal N, Sternberg PW. The epidermal growth factor system in Caenorhabditis elegans. Exp Cell Res. 2003;284:150–9. doi: 10.1016/s0014-4827(02)00097-6. [DOI] [PubMed] [Google Scholar]
- Ni J, Wen X, Yao J, Hong-Chang C, Yin Y, Zhang M, Xie S, Chen M, Simons B, Chang P, di Sant'Agnese A, Messin EM, Yeh S. Tocopherol-associated protein suppresses prostate cancer cell growth by inhibition of the phosphoinositide 3-kinase pathway. Cancer research. 2005;65(21):9807–16. doi: 10.1158/0008-5472.CAN-05-1334. [DOI] [PubMed] [Google Scholar]
- Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol. 2003;13:1356–64. doi: 10.1016/s0960-9822(03)00535-9. [DOI] [PubMed] [Google Scholar]
- Panagabko C, Morley S, Hernandez M, Cassolato P, Gordon H, Parsons R, Manor D, Atkinson J. Ligand specificity in the CRAL-TRIO protein family. Biochemistry. 2003;42:6467–74. doi: 10.1021/bi034086v. [DOI] [PubMed] [Google Scholar]
- Phillips SE, Sha B, Topalof L, Xie Z, Alb JG, Klenchin VA, Swigart P, Cockcroft S, Martin TF, Luo M, Bankaitis VA. Yeast Sec14p deficient in phosphatidylinositol transfer activity is functional in vivo. Mol Cell. 1999;4:187–97. doi: 10.1016/s1097-2765(00)80366-4. [DOI] [PubMed] [Google Scholar]
- Sha B, Phillips SE, Bankaitis VA, Luo M. Crystal structure of the Saccharomyces cerevisiae phosphatidylinositol-transfer protein. Nature. 1998;391:506–10. doi: 10.1038/35179. [DOI] [PubMed] [Google Scholar]
- Shibata N, Arita M, Misaki Y, Dohmae N, Takio K, Ono T, Inoue K, Arai H. Supernatant protein factor, which stimulates the conversion of squalene to lanosterol, is a cytosolic squalene transfer protein and enhances cholesterol biosynthesis. Proc Natl Acad Sci U S A. 2001;98:2244–9. doi: 10.1073/pnas.041620398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker A, Baumann U. Supernatant protein factor in complex with RRR-alpha-tocopherylquinone: a link between oxidized Vitamin E and cholesterol biosynthesis. J Mol Biol. 2003;332:759–65. doi: 10.1016/s0022-2836(03)00924-0. [DOI] [PubMed] [Google Scholar]
- Stocker A, Tomizaki T, Schulze-Briese C, Baumann U. Crystal structure of the human supernatant protein factor. Structure. 2002;10:1533–40. doi: 10.1016/s0969-2126(02)00884-5. [DOI] [PubMed] [Google Scholar]
- Tchen TT, Bloch K. On the conversion of squalene to lanosterol in vitro. J Biol Chem. 1957;226:921–30. [PubMed] [Google Scholar]
- Tiensuu T, Larsen MK, Vernersson E, Tuck S. lin-1 has both positive and negative functions in specifying multiple cell fates induced by Ras/MAP kinase signaling in C. elegans. Dev Biol. 2005;286:338–51. doi: 10.1016/j.ydbio.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Ulku AS, Der CJ. Ras signaling, deregulation of gene expression and oncogenesis. Cancer Treat Res. 2003;115:189–208. [PubMed] [Google Scholar]
- Valenzuela DM, Groffen J. Four human carcinoma cell lines with novel mutations in position 12 of c-K-ras oncogene. Nucleic Acids Res. 1986;14:843–52. doi: 10.1093/nar/14.2.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–85. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- Yang SH, Sharrocks AD. SUMO promotes HDAC-mediated transcriptional repression. Mol Cell. 2004;13:611–7. doi: 10.1016/s1097-2765(04)00060-7. [DOI] [PubMed] [Google Scholar]
- Zimmer S, Stocker A, Sarbolouki MN, Spycher SE, Sassoon J, Azzi A. A novel human tocopherol-associated protein: cloning, in vitro expression, and characterization. J Biol Chem. 2000;275:25672–80. doi: 10.1074/jbc.M000851200. [DOI] [PubMed] [Google Scholar]