

Abstract
We use the Glazier-Graner-Hogeweg model to simulate three-dimensional (3D), single-phenotype, avascular tumors growing in an homogeneous tissue matrix (TM) supplying a single limiting nutrient. We study the effects of two parameters on tumor morphology: a diffusion-limitation parameter defined as the ratio of the tumor-substrate consumption rate to the substrate-transport rate, and the tumor-TM surface tension. This initial model omits necrosis and oxidative/hypoxic metabolism effects, which can further influence tumor morphology, but our simplified model still shows significant parameter dependencies. The diffusion-limitation parameter determines whether the growing solid tumor develops a smooth (noninvasive) or fingered (invasive) interface, as in our earlier two-dimensional (2D) simulations. The sensitivity of 3D tumor morphology to tumor-TM surface tension increases with the size of the diffusion-limitation parameter, as in 2D. The 3D results are unexpectedly close to those in 2D. Our results therefore may justify using simpler 2D simulations of tumor growth, instead of more realistic but more computationally expensive 3D simulations. While geometrical artifacts mean that 2D sections of connected 3D tumors may be disconnected, the morphologies of 3D simulated tumors nevertheless correlate with the morphologies of their 2D sections, especially for low-surface-tension tumors, allowing the use of 2D sections to partially reconstruct medically-important 3D-tumor structures.
Introduction
In [1] we studied the effects of nutrient limitation and surface tension in a simplified two-dimensional (2D) model of tumor invasion using the Glazier-Graner-Hogeweg (GGH) (also known as the Cellular Potts Model) [2]–[4], as implemented in the CompuCell3D (CC3D) modeling environment [5]–[10]. In 2D, the selection of smooth-interface (noninvasive) vs. fingered (invasive) growth depends on the tumor's substrate-consumption rate per unit substrate-transport rate, the diffusion-limitation parameter
 , while the detailed morphology also depends on the tumor-tissue-matrix (TM) surface tension
, while the detailed morphology also depends on the tumor-tissue-matrix (TM) surface tension
 . Lack of competition for nutrients promotes spherical, noninvasive tumors. Low concentrations of nutrients in the environment which cause tumor-cell competition, or cells with a very high substrate-consumption rate generate a fingering instability and irregular, invasive tumors. Our results agree with in vitro experiments showing that tumors branch into the surrounding tissues if the nutrient supply is too small [11], [12], and with other tumor-model predictions [13]–[18].
. Lack of competition for nutrients promotes spherical, noninvasive tumors. Low concentrations of nutrients in the environment which cause tumor-cell competition, or cells with a very high substrate-consumption rate generate a fingering instability and irregular, invasive tumors. Our results agree with in vitro experiments showing that tumors branch into the surrounding tissues if the nutrient supply is too small [11], [12], and with other tumor-model predictions [13]–[18].
Our three-dimensional (3D) model extension of our 2D model [1], includes growing, spatially-extended tumor cells, surrounding TM represented as a nondiffusing field secreting nutrients, a diffusing field representing matrix-degrading enzymes (MDEs) that degrade TM, and a diffusing nutrient field (substrate) which governs the rate of tumor-cell growth. As in our 2D model we assume that all tumor cells are identical in their capacities and responses, and that the specific growth rate of tumor cells increases linearly with the local concentration of a single limiting substrate, with no concentration threshold for tumor cells to grow.
For a detailed discussion of tumor initiation and progression, see [19] and [20]. Growth of avascular tumor spheroids depends on diffusion of nutrients and waste products, usually limiting the spheroid's maximum size, as discussed in [1]. Tumors can be benign (noninvasive) or malignant (invasive). Most tumors are initially noninvasive and gradually become more invasive as they reduce their cell-cell adhesiveness and (often) increase their cell-extracellular-matrix (ECM) adhesiveness [21]. Tumor cells can secrete MDEs that degrade the ECM which maintains the integrity of normal tissues [22]–[24] and modifies the distribution in the ECM of molecules to which cells adhere, e.g., fibronectin, increasing effective cell motility [22], [25]–[30]. Intrinsic cell motility can also increase during tumor progression [31]–[33].
Hypoxia (a shortage of oxygen) [34] activates transcription of the met proto-oncogene [12], which increases levels of the Met tyrosine kinase, a receptor for hepatocyte growth factor (HGF) [35]–[37]. HGF is a scatter factor [38] that coordinates a number of specific cytokines [39] which weaken cell-cell contacts and increase cell motility [40], [41]. Thus hypoxia indirectly enhances HGF-induced cell motility [12], [42]. Hypoxia can also induce epithelial-mesenchymal transitions (EMT) [43] through Notch signaling [44], one of the initial steps in metastasis, transforming nonmotile, epithelial cells into migratory and invasive cells, e.g., through down-regulation of E-cadherin [45], [46].
Mathematical models of tumor growth [47] range from simple fitting of experimental data on the growth kinetics of tumor spheroids using various growth laws [48] to more complex simulations of tumor-induced angiogenesis and capillary network formation [47], [49]–[52], and tumor spreading at early [53] and later invasive stages [18], [54]–[58]. Continuum and solid-mechanics models [14]–[17], [59]–[62] consider physical forces among cells and TM, capturing tumor structure at the tissue level, but do not describe the tumor's cellular and subcellular properties, making mechanisms such as cell-cell adhesion difficult to include. Point-cell models (cellular automata) allow stochastic descriptions at cellular [63]–[67] and subcellular levels [68], [69] but neglect the shapes of cells. Hybrid multi-cell models combine discrete representations of individual tumor cells with continuum representations of diffusible chemicals [70]–[74] and either discrete or continuum models of the surrounding tissue. For comprehensive reviews of mathematical models of tumor growth see [75], [76] and references therein.
Three recent models of tumor growth have analyzed tumor-growth morphologies in a two-dimensional parameter phase space. The model of [15] analyzed nonlinear tumor morphological response to two nondimensional parameters representing the balance of cell death to birth and tissue mechanics (proliferation-induced pressure), the model of [16] analyzed fragmenting and compact morphologies in the nutrient-adhesion phase space, and the model of [17] analyzed the fragmenting, fingering, and compact morphologies in the nutrient-mechanics phase space.
In our recent 2D GGH simulations of growing avascular tumors [1] we found that smaller tumor-TM surface tension speeds diffusion of tumor cells and that simulated substrate-deprived tumor morphologies are more sensitive to variations in tumor-TM surface tension. These results agree with the observation that hypoxia enhances the sensitivity of tumor cell motility to scatter factors, and suggest an experimentally-testable hypothesis that HGF decreases tumor-TM surface tension, which we could measure, e.g., using compression apparatus [77]–[80]. They also agree with a recent study of the analogy between branching instabilities in a growing tumor and instabilities in a drop of water impacting a solid surface, which suggested promoting tumor cell-tumor cell adhesion as a clinical strategy in oncological therapies [81].
As in [1], we do not model explicitly the haptotactic repulsion and pressure on the tumor cells from the surrounding normal tissue. Thus our simulated tumor cells move freely, which is realistic only for environments that do not constrain tumor-cell motility, such as idealized gliomas or those growing in mechanically unconfined areas, e.g. in vitro [61], [82]. However, the tumor-TM surface tension creates an effective hydrostatic pressure on the tumor. While not identical to a tumor growing in an elastic or viscoelastic tissue, increasing the surface tension reproduces many of the effects of increasing the rigidity of an explicitly-modeled external tissue.
Necrosis can be essential to instability mechanisms at later stages of tumor growth, destabilizing the shape of the tumor [17]. While diffusional instabilities lead to fingering morphologies, the connecting portions of the fingers experience necrosis due to prolonged hypoxia and anoxia, leading to a disconnection of the fingers and a fragmented morphology. Necrosis becomes biologically significant when hypoxia of a layer of cells surrounding the necrotic core of the tumor triggers a cascade of signals mediated by hypoxia-inducible factor-1 (HIF-1) [83] and vascular endothelial growth factor (VEGF) [84], which initiate angiogenesis, i.e. tumor vascularization, by inducing growth and extension of nearby blood vessels. Since we focus on the role of cell-cell adhesion and competition for nutrients at the tumor-TM interface where the tumor cells are alive and proliferating, as in [1], we simulate single-cell-type avascular tumors without angiogenesis, omitting necrotic and quiescent cells, which are absent at the tumor-TM interface during early stages of fingering. While necrosis certainly has a profound effect on the late-stage morphology of fingered tumors, its primary effect on the instabilities we are studying is to reduce the competition for substrate, thus changing the  values for the onset of different instabilities. Since the degree of shift depends on details of the necrotic mechanism, we feel that studying the effects of nutrient limitation separately from the effects necrosis is clearer. We will combine the effects in a later paper. We do not model quiescence explicitly because the substrate concentration in the central regions of our simulated tumors is nearly zero, so the cells there barely grow (see Mathematical Structure of the Tumor Model), effectively behaving like quiescent cells.
values for the onset of different instabilities. Since the degree of shift depends on details of the necrotic mechanism, we feel that studying the effects of nutrient limitation separately from the effects necrosis is clearer. We will combine the effects in a later paper. We do not model quiescence explicitly because the substrate concentration in the central regions of our simulated tumors is nearly zero, so the cells there barely grow (see Mathematical Structure of the Tumor Model), effectively behaving like quiescent cells.
In this paper we extend our 2D model of tumor-interface instabilities to more realistic 3D tumors. We find that our results for our 3D simulations agree with our 2D results, which is surprising because certain relationships, such as mutually penetrating connected structures, cannot exist in 2D. Such structures form in real tumors during neoangiogenesis [85], during which tumors recruit blood vessels from the surrounding vascular network to supply nutrients and remove waste. We need to understand the physics of instabilities in growing avascular 3D tumors before we proceed to investigate how 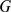 and
and  affect vascular tumors undergoing neoangiogenesis, extending the recent GGH simulations in [52].
affect vascular tumors undergoing neoangiogenesis, extending the recent GGH simulations in [52].
We aim to answer two questions: 1) what causes front instabilities and invasiveness in 3D avascular tumors? and 2) can we reconstruct medically-important 3D tumor structures from 2D sections? We hypothesize that hypoxia and surface tension will have similar effects on tumor morphology in 2D and 3D simulations (which is not obvious a priori). We show that the diffusion-limitation parameter  determines whether the 3D tumor has a uniform or fingered margin, while the tumor-TM surface tension
determines whether the 3D tumor has a uniform or fingered margin, while the tumor-TM surface tension  affects the detailed tumor morphology, as in 2D. We construct a
affects the detailed tumor morphology, as in 2D. We construct a 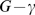 phase diagram showing the effects of these parameters on simulated 3D avascular tumors and their 2D sections. Although geometrical artifacts mean that the 2D sections of many connected 3D simulated tumors are disconnected, the morphologies of 3D simulated tumors nevertheless correlate strongly with the morphologies of their 2D sections, especially at later stages, allowing the use of 2D sections to partially reconstruct 3D tumor structures.
phase diagram showing the effects of these parameters on simulated 3D avascular tumors and their 2D sections. Although geometrical artifacts mean that the 2D sections of many connected 3D simulated tumors are disconnected, the morphologies of 3D simulated tumors nevertheless correlate strongly with the morphologies of their 2D sections, especially at later stages, allowing the use of 2D sections to partially reconstruct 3D tumor structures.
Results
We can describe tumor morphologies in terms of their sphericity [86],
| (1) |
where 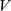 is the volume of an object and
is the volume of an object and 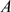 is its surface area. Sphericity is a computationally convenient measure of how round an object is for 3D images. We study the time dependence of the sphericity of simulated tumors as a function of
is its surface area. Sphericity is a computationally convenient measure of how round an object is for 3D images. We study the time dependence of the sphericity of simulated tumors as a function of  and
and  . As in [1], we call structures with pronounced orientational order dendritic, and structures without apparent orientational order seaweed-like or diffusion-limited-aggregation-like (DLA-like). We also study how changes in
. As in [1], we call structures with pronounced orientational order dendritic, and structures without apparent orientational order seaweed-like or diffusion-limited-aggregation-like (DLA-like). We also study how changes in  and
and  affect the time at which the middle 2D section of the simulated tumor reaches the boundary of the simulation domain, and the conditions under which the simulated tumor stops proliferating before reaching this boundary. Because 2D sections of real tumors are more convenient to analyze medically, we also measure the circularity
[1],
affect the time at which the middle 2D section of the simulated tumor reaches the boundary of the simulation domain, and the conditions under which the simulated tumor stops proliferating before reaching this boundary. Because 2D sections of real tumors are more convenient to analyze medically, we also measure the circularity
[1],
| (2) |
where 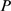 is the perimeter of an object, in our case, of the middle 2D sections of the simulated tumors, and compare it with the sphericity for the corresponding 3D images. If the sphericity of a 3D simulated tumor corresponds to the circularity of its 2D section, analysis of 2D sections of real tumors may allow us to reconstruct their 3D morphology, helping to determine whether they are invasive or not, and possibly predicting the effectiveness of antiangiogenic therapies.
is the perimeter of an object, in our case, of the middle 2D sections of the simulated tumors, and compare it with the sphericity for the corresponding 3D images. If the sphericity of a 3D simulated tumor corresponds to the circularity of its 2D section, analysis of 2D sections of real tumors may allow us to reconstruct their 3D morphology, helping to determine whether they are invasive or not, and possibly predicting the effectiveness of antiangiogenic therapies.
Small  (
(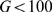 ) corresponds to a growth-limited regime
[1], [87]. The substrate penetrates most of the tumor and reaches most cells, so the simulated tumor remains spherical [1]. Larger
) corresponds to a growth-limited regime
[1], [87]. The substrate penetrates most of the tumor and reaches most cells, so the simulated tumor remains spherical [1]. Larger  slows the growth of the tumor (since the local substrate concentration decreases in the presence of tumor cells), and diffusing substrate penetrates fewer cell diameters past the surface layer of the tumor. Initially, substrate is present throughout the tumor, which grows in all directions. As the tumor grows, its cells consume substrate and the substrate concentration develops a gradient, the concentration increasing in the radial direction away from the tumor centroid. Locally, cells near the tumor surface in protruding regions experience higher concentrations of substrate and hence grow faster than others. These cells proliferate more quickly, while the cells in the narrow valleys, where the interface between the tumor and TM lags significantly behind the furthest local radial position of the tumor, experience low concentrations of substrate and slow their growth. Figure 1 shows the time evolution of a simulated tumor with
slows the growth of the tumor (since the local substrate concentration decreases in the presence of tumor cells), and diffusing substrate penetrates fewer cell diameters past the surface layer of the tumor. Initially, substrate is present throughout the tumor, which grows in all directions. As the tumor grows, its cells consume substrate and the substrate concentration develops a gradient, the concentration increasing in the radial direction away from the tumor centroid. Locally, cells near the tumor surface in protruding regions experience higher concentrations of substrate and hence grow faster than others. These cells proliferate more quickly, while the cells in the narrow valleys, where the interface between the tumor and TM lags significantly behind the furthest local radial position of the tumor, experience low concentrations of substrate and slow their growth. Figure 1 shows the time evolution of a simulated tumor with  (which is near the value
(which is near the value 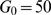 corresponding to the parameters used in [70]), for
corresponding to the parameters used in [70]), for 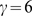 (a),
(a), 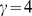 (b),
(b), 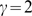 (c), and
(c), and 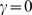 (d). The simulated tumors are dense, fast-growing and initially spherical. As they grow, their tumor-TM interfaces become slightly irregular (grooved) (the lower
(d). The simulated tumors are dense, fast-growing and initially spherical. As they grow, their tumor-TM interfaces become slightly irregular (grooved) (the lower  , the rougher the surface of the developing tumor) but remain compact.
, the rougher the surface of the developing tumor) but remain compact.
Figure 1. Simulated growing tumors with 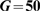 .
.

(a) 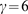 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially spherical; it then becomes slightly irregular. Fourth row: 3D visualization of the same simulation. (b)
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially spherical; it then becomes slightly irregular. Fourth row: 3D visualization of the same simulation. (b) 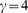 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially spherical; it then becomes grooved. Fourth row: 3D visualization of the same simulation. (c)
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially spherical; it then becomes grooved. Fourth row: 3D visualization of the same simulation. (c) 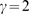 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially spherical; it then becomes grooved. Fourth row: 3D visualization of the same simulation. (d)
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially spherical; it then becomes grooved. Fourth row: 3D visualization of the same simulation. (d) 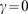 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially spherical; it then becomes grooved with a rough surface. Fourth row: 3D visualization of the same simulation. The simulation time is indicated in days beneath each column, where 1 day = 400 MCS.
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially spherical; it then becomes grooved with a rough surface. Fourth row: 3D visualization of the same simulation. The simulation time is indicated in days beneath each column, where 1 day = 400 MCS.
Large  corresponds to a transport-limited or diffusion-limited regime
[1], [87]. The substrate penetrates only a short distance into the tumor, so the tumor grows more slowly than for smaller values of
corresponds to a transport-limited or diffusion-limited regime
[1], [87]. The substrate penetrates only a short distance into the tumor, so the tumor grows more slowly than for smaller values of  . Figure 2 shows the time evolution of simulated tumors with
. Figure 2 shows the time evolution of simulated tumors with 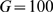 , for
, for 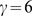 (a),
(a), 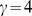 (b),
(b), 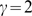 (c), and
(c), and 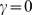 (d). For high TM-tumor surface tensions, the simulated tumors are compact and dendritic (with anisotropic branches), while for low TM-tumor surface tensions, the simulated tumors are DLA-like (with isotropic branches), as in our previous 2D simulations [1]. The effect of surface tension on morphology is more dramatic for larger
(d). For high TM-tumor surface tensions, the simulated tumors are compact and dendritic (with anisotropic branches), while for low TM-tumor surface tensions, the simulated tumors are DLA-like (with isotropic branches), as in our previous 2D simulations [1]. The effect of surface tension on morphology is more dramatic for larger  , again as in 2D [1]. Figure 3 shows the time evolution of simulated tumors with
, again as in 2D [1]. Figure 3 shows the time evolution of simulated tumors with 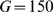 , for
, for 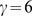 (a),
(a), 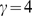 (b),
(b), 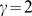 (c), and
(c), and 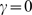 (d).
(d).
Figure 2. Simulated growing tumors with 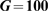 .
.

(a) 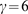 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially compact; it then becomes dendritic. The disconnected parts in the last image connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (b)
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially compact; it then becomes dendritic. The disconnected parts in the last image connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (b) 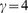 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially compact; it then becomes dendritic. The disconnected parts in the last two images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (c)
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially compact; it then becomes dendritic. The disconnected parts in the last two images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (c) 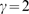 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially compact with a rough surface; it then becomes seaweed-like. The disconnected parts in the last two images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (d)
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially compact with a rough surface; it then becomes seaweed-like. The disconnected parts in the last two images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (d) 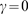 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is seaweed-like with a rough surface. The disconnected parts in the images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. The simulation time is indicated in days beneath each column, where 1 day = 400 MCS.
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is seaweed-like with a rough surface. The disconnected parts in the images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. The simulation time is indicated in days beneath each column, where 1 day = 400 MCS.
Figure 3. Simulated growing tumors with 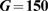 .
.

(a) 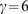 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially compact; it then becomes dendritic. The disconnected parts in the last two images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (b)
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is initially compact; it then becomes dendritic. The disconnected parts in the last two images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (b) 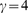 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor becomes dendritic. The disconnected parts in the last two images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (c)
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor becomes dendritic. The disconnected parts in the last two images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (c) 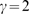 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor has a form intermediate between dendrite and seaweed. The disconnected parts in the images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (d)
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor has a form intermediate between dendrite and seaweed. The disconnected parts in the images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (d) 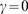 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is seaweed-like. The disconnected parts in the images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. The simulation time is indicated in days beneath each column, where 1 day = 400 MCS.
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor is seaweed-like. The disconnected parts in the images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. The simulation time is indicated in days beneath each column, where 1 day = 400 MCS.
At 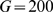 for a high surface tension,
for a high surface tension, 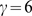 , the tumor occupies a region with a high concentration of MDE that has degraded all the TM Field. TM far from the tumor still produces substrate, but the substrate is essentially exhausted at the tumor surface. The availability of nutrients is so limited that cell proliferation nearly stops; the simulated tumor does not reach the boundary of the simulation domain (Figure 4 (a)). For a lower surface tension,
, the tumor occupies a region with a high concentration of MDE that has degraded all the TM Field. TM far from the tumor still produces substrate, but the substrate is essentially exhausted at the tumor surface. The availability of nutrients is so limited that cell proliferation nearly stops; the simulated tumor does not reach the boundary of the simulation domain (Figure 4 (a)). For a lower surface tension, 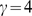 , the simulated tumor does reach the boundary of the simulation domain but so many cells stop dividing that some branches of the dendritic tumor stop growing (Figure 4 (b)). Figures 4 (c) and 4 (d) show the time evolution of simulated tumors for
, the simulated tumor does reach the boundary of the simulation domain but so many cells stop dividing that some branches of the dendritic tumor stop growing (Figure 4 (b)). Figures 4 (c) and 4 (d) show the time evolution of simulated tumors for 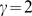 and
and 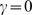 , respectively. For such low surface tensions, DLA-like structures form.
, respectively. For such low surface tensions, DLA-like structures form.
Figure 4. Simulated growing tumors with 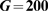 .
.

(a) 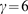 . 2D sections of a 3D simulation along the XY plane. The developing tumor remains compact and ceases proliferating. We do not show 2D sections along the XZ and YZ planes because they are essentially indistinguishable from those along the XY plane. We do not show 3D visualization of the same simulation because it does not provide any new information about the simulated tumor. (b)
. 2D sections of a 3D simulation along the XY plane. The developing tumor remains compact and ceases proliferating. We do not show 2D sections along the XZ and YZ planes because they are essentially indistinguishable from those along the XY plane. We do not show 3D visualization of the same simulation because it does not provide any new information about the simulated tumor. (b) 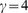 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor forms a truncated dendrite. The disconnected parts in the last image connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (c)
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor forms a truncated dendrite. The disconnected parts in the last image connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (c) 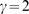 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor has a form intermediate between dendrite and seaweed, with thinner fingers. The disconnected parts in the images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (d)
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor has a form intermediate between dendrite and seaweed, with thinner fingers. The disconnected parts in the images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. (d) 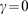 . 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor forms a seaweed. The disconnected parts in the images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. The simulation time is indicated in days beneath each column, where 1 day = 400 MCS.
. 2D sections of a 3D simulation along the XY (first row), XZ (second row) and YZ plane (third row). The developing tumor forms a seaweed. The disconnected parts in the images connect to the backbone of the tumor out of the section plate. Fourth row: 3D visualization of the same simulation. The simulation time is indicated in days beneath each column, where 1 day = 400 MCS.
Figure 1 shows that, for small  , the substrate reaches most cells, which grow in all directions, no valleys form and the tumor-TM interface remains smooth. As we increase
, the substrate reaches most cells, which grow in all directions, no valleys form and the tumor-TM interface remains smooth. As we increase  , the substrate penetrates less deeply into the tumor. Cells near the tumor surface experience higher concentrations of substrate and proliferate more quickly, producing fingers, while the space between fingers fills slowly or not at all with new cells, as Figures 2–
4 show. Thus the competition for substrate between tumor cells results in a fingering instability
[15], [88]–[90] which generates a fingered tumor morphology [1]. Table 1 shows the mean circularity
, the substrate penetrates less deeply into the tumor. Cells near the tumor surface experience higher concentrations of substrate and proliferate more quickly, producing fingers, while the space between fingers fills slowly or not at all with new cells, as Figures 2–
4 show. Thus the competition for substrate between tumor cells results in a fingering instability
[15], [88]–[90] which generates a fingered tumor morphology [1]. Table 1 shows the mean circularity 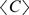 , defined as the average of the circularities of 2D sections of 3D tumors taken at the midplanes XY, XZ and YZ, for different values of
, defined as the average of the circularities of 2D sections of 3D tumors taken at the midplanes XY, XZ and YZ, for different values of  and
and  at the moment when the tumor reaches the boundaries of the simulation domain (
at the moment when the tumor reaches the boundaries of the simulation domain ( 6 mm). Figure 5 summarizes the morphologies and shows the sphericity
6 mm). Figure 5 summarizes the morphologies and shows the sphericity 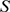 of 3D tumors for different values of
of 3D tumors for different values of  and
and  at the moment when the tumor reaches the boundaries of the simulation domain (
at the moment when the tumor reaches the boundaries of the simulation domain ( 6 mm). Although the 2D sections of many simulated tumors are disconnected, the underlying 3D tumors are connected (except for
6 mm). Although the 2D sections of many simulated tumors are disconnected, the underlying 3D tumors are connected (except for 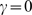 where a few cells migrate out of the backbone of the tumor). Thus the disconnectedness of tumors simulated in 2D results from the underlying physics of growth and diffusion and provides fundamental information about the growth dynamics, while the disconnectedness of 2D sections of connected 3D simulated tumors is a geometrical artifact and does not provide much information beyond indicating a rough tumor surface.
where a few cells migrate out of the backbone of the tumor). Thus the disconnectedness of tumors simulated in 2D results from the underlying physics of growth and diffusion and provides fundamental information about the growth dynamics, while the disconnectedness of 2D sections of connected 3D simulated tumors is a geometrical artifact and does not provide much information beyond indicating a rough tumor surface.
Table 1. The dependence on 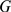 and
and  of the mean circularity
of the mean circularity 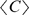 of 2D sections of the simulated 3D tumors, observed when the tumor reaches the boundaries of the simulation domain (
of 2D sections of the simulated 3D tumors, observed when the tumor reaches the boundaries of the simulation domain ( 6 mm).
6 mm).
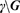
|
50 | 100 | 150 | 200 |

|
0.74 0.02 0.02 |
0.37 0.03 0.03 |
0.35 0.03 0.03 |
|

|
0.69 0.02 0.02 |
0.31 0.02 0.02 |
0.29 0.07 0.07 |
0.51 0.04 0.04 |

|
0.62 0.03 0.03 |
0.25 0.04 0.04 |
0.22 0.01 0.01 |
0.21 0.02 0.02 |

|
0.54 0.02 0.02 |
0.23 0.03 0.03 |
0.16 0.01 0.01 |
0.15 0.02 0.02 |
The space for 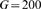 and
and 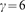 is blank because the corresponding tumor never grows to this size.
is blank because the corresponding tumor never grows to this size.
Figure 5. Morphologies of 3D tumors visualized in 3D and sphericity 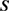 as a function of
as a function of  and
and 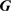 , observed when the simulated tumor reaches the boundaries of the simulation domain (
, observed when the simulated tumor reaches the boundaries of the simulation domain ( 6 mm).
6 mm).

The standard deviation for  is less than 0.02. The panel for
is less than 0.02. The panel for  and
and  is blank because the corresponding tumor never grows to this size.
is blank because the corresponding tumor never grows to this size.
Sphericity increases with surface tension  and decreases with increasing
and decreases with increasing  (the structure for
(the structure for  and
and 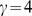 deviates from this general behavior because its growth is truncated). These dependencies of
deviates from this general behavior because its growth is truncated). These dependencies of 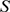 are consistent with our observation that competition for substrate favors branching instabilities while surface tension stabilizes the tumor-TM interface, as in 2D [1]. Table 2 shows how the sphericity of simulated tumors with 1000 Generalized Cells (which represent
are consistent with our observation that competition for substrate favors branching instabilities while surface tension stabilizes the tumor-TM interface, as in 2D [1]. Table 2 shows how the sphericity of simulated tumors with 1000 Generalized Cells (which represent 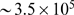 real tumor cells) depends on
real tumor cells) depends on  and
and  . Figure 6 shows how the sphericity
. Figure 6 shows how the sphericity 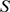 of simulated tumors, when they reach the boundaries of the simulation domain (a) or contain 1000 Generalized Cells (b), depends on surface tension
of simulated tumors, when they reach the boundaries of the simulation domain (a) or contain 1000 Generalized Cells (b), depends on surface tension  for different
for different  . This dependence, more monotonic for a fixed number of cells than for a fixed size, suggests that the mass of the tumor, which is proportional to the number of tumor cells, is a more accurate description of the tumor stage than its size.
. This dependence, more monotonic for a fixed number of cells than for a fixed size, suggests that the mass of the tumor, which is proportional to the number of tumor cells, is a more accurate description of the tumor stage than its size.
Table 2. The dependence on 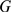 and
and  of the sphericity
of the sphericity 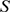 of the simulated tumors with 1000 Generalized Cells.
of the simulated tumors with 1000 Generalized Cells.
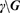
|
50 | 100 | 150 | 200 |

|
0.75 | 0.68 | 0.50 | |

|
0.72 | 0.61 | 0.45 | 0.39 |

|
0.66 | 0.47 | 0.33 | 0.28 |

|
0.58 | 0.32 | 0.22 | 0.18 |
The space for 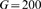 and
and 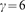 is blank because the corresponding tumor ceases to grow before reaching 1000 Generalized Cells.
is blank because the corresponding tumor ceases to grow before reaching 1000 Generalized Cells.
Figure 6. Sphericity 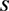 of simulated tumors as a function of
of simulated tumors as a function of  for different
for different 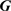 .
.

(a) When they reach the boundary of the simulation domain, (b) with 1000 Generalized Cells.
Table 1 and Figure 5 show that the sphericity 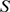 of 3D simulated tumors and the mean circularity
of 3D simulated tumors and the mean circularity 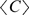 of their 2D sections differ by 0.05 or less, except for
of their 2D sections differ by 0.05 or less, except for 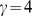 and
and  (truncated growth) and for
(truncated growth) and for  and
and  . The sphericity
. The sphericity 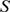 is smaller than the mean circularity
is smaller than the mean circularity 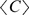 , except for
, except for 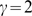 and
and 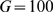 . Our results indicate a close relationship between the numerical values of
. Our results indicate a close relationship between the numerical values of 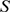 and
and 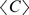 for a given tumor and provide a simple method for partial reconstruction of 3D tumor structure from 2D sections: the sphericity of a real 3D tumor of a size on the order of a millimeter approximately equals the mean circularity of the 2D sections taken at the midplanes XY, XZ and YZ through the 3D tumor. This partial reconstruction is the first step in determining the 3D tumor's morphology, and thus potentially has medical value in predicting its behavior.
for a given tumor and provide a simple method for partial reconstruction of 3D tumor structure from 2D sections: the sphericity of a real 3D tumor of a size on the order of a millimeter approximately equals the mean circularity of the 2D sections taken at the midplanes XY, XZ and YZ through the 3D tumor. This partial reconstruction is the first step in determining the 3D tumor's morphology, and thus potentially has medical value in predicting its behavior.
Figures 7 and 8 show that the sphericity 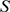 of 3D simulated tumors and the mean circularity
of 3D simulated tumors and the mean circularity 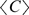 of their 2D sections both decrease in time
of their 2D sections both decrease in time  for all
for all  and
and  .
.  varies less with
varies less with  than
than  does. The
does. The  curves for
curves for 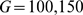 at
at 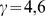 , and for
, and for 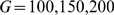 at
at 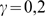 lie close to each other, indicating that
lie close to each other, indicating that  does not strongly affect how the sphericity of simulated low-surface-tension tumors changes in time. The
does not strongly affect how the sphericity of simulated low-surface-tension tumors changes in time. The 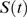 curve for
curve for 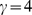 and
and 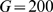 deviates from the curves for the other
deviates from the curves for the other 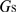 s because tumor growth is truncated. We do not show the
s because tumor growth is truncated. We do not show the 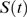 curve for
curve for 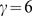 and
and 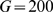 because the simulated tumor does not grow. The
because the simulated tumor does not grow. The 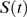 curves are more concave (have larger slopes) for lower surface tensions. The absolute value of the difference between
curves are more concave (have larger slopes) for lower surface tensions. The absolute value of the difference between 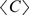 and
and 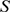 for almost all simulated tumors initially increases in time and then decreases, as Figure 9 shows (except for
for almost all simulated tumors initially increases in time and then decreases, as Figure 9 shows (except for  , where it is not monotonic and remains small). Also,
, where it is not monotonic and remains small). Also, 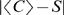 decreases with
decreases with  . Therefore, reconstructing 3D tumor structure from 2D sections is more accurate for avascular tumors either at very early or at later stages, and is more accurate for low-surface-tension tumors which grow faster and thus are medically relevant.
. Therefore, reconstructing 3D tumor structure from 2D sections is more accurate for avascular tumors either at very early or at later stages, and is more accurate for low-surface-tension tumors which grow faster and thus are medically relevant.
Figure 7. Mean circularity 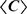 as a function of time for 2D sections of 3D simulations of tumor growth.
as a function of time for 2D sections of 3D simulations of tumor growth.

(a) 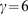 , (b)
, (b) 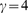 , (c)
, (c) 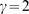 , and (d)
, and (d) 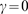 .
.
Figure 8. Sphericity 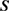 as a function of time for 3D simulations of tumor growth.
as a function of time for 3D simulations of tumor growth.

(a) 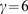 , (b)
, (b) 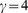 , (c)
, (c) 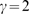 , and (d)
, and (d) 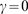 .
.
Figure 9.
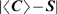 as a function of time for 3D simulations of tumor growth.
as a function of time for 3D simulations of tumor growth.

(a) 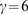 , (b)
, (b) 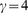 , (c)
, (c) 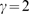 , and (d)
, and (d) 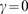 .
.
Table 3 shows how the time at which the simulated tumor reaches the boundary of the simulation domain, which has a size of 6 mm, depends on  and
and  . The smaller the tumor-TM surface tension,
. The smaller the tumor-TM surface tension,  , the faster the growth of the tumor, because for large
, the faster the growth of the tumor, because for large  , tumor cells must diffuse to find substrate to maintain their growth rather than having substrate diffuse to reach them. The smaller
, tumor cells must diffuse to find substrate to maintain their growth rather than having substrate diffuse to reach them. The smaller  , the larger the diffusion coefficient of the tumor cells [91]. Small surface tension enhances spreading of the tumor cells, so the tumor grows continuously. This result agrees with experiments showing that less adhesive tumors grow faster [43]. Table 4 shows how the time at which the simulated tumor grows to 1000 cells depends on
, the larger the diffusion coefficient of the tumor cells [91]. Small surface tension enhances spreading of the tumor cells, so the tumor grows continuously. This result agrees with experiments showing that less adhesive tumors grow faster [43]. Table 4 shows how the time at which the simulated tumor grows to 1000 cells depends on  and
and  .
.
Table 3. The dependence on 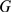 and
and  of the time (in days) at which the simulated tumor reaches the boundary of the simulation domain.
of the time (in days) at which the simulated tumor reaches the boundary of the simulation domain.
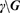
|
50 | 100 | 150 | 200 |

|
18 | 44 | 100 |

|

|
16 | 40 | 68 | 100 |

|
14 | 32 | 40 | 64 |

|
12 | 20 | 28 | 32 |
Table 4. The dependence on 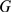 and
and  of the time (in days) at which the simulated tumor grows to 1000 Generalized Cells.
of the time (in days) at which the simulated tumor grows to 1000 Generalized Cells.
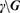
|
50 | 100 | 150 | 200 |

|
2 | 5 | 14 |

|

|
2 | 5 | 11 | 35 |

|
2 | 5 | 8 | 13 |

|
2 | 4 | 6 | 9 |
Almost all the 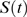 curves in Figure 8 have a quasi-Gaussian profile. Thus we can fit them to a function of form
curves in Figure 8 have a quasi-Gaussian profile. Thus we can fit them to a function of form 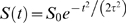 , where
, where 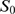 and
and  are constants. The characteristic time
are constants. The characteristic time
 then characterizes how fast the tumor develops fingers and thus is a useful way to characterize tumor morphology dynamics. Table 5 shows
then characterizes how fast the tumor develops fingers and thus is a useful way to characterize tumor morphology dynamics. Table 5 shows  as a function of
as a function of  and
and  . Sensitivity of simulated-tumor morphology dynamics to changes in
. Sensitivity of simulated-tumor morphology dynamics to changes in  decreases with increasing
decreases with increasing  in the growth-limited regime, then increases with increasing
in the growth-limited regime, then increases with increasing  in the diffusion-limited regime, in agreement with experiments showing that hypoxia enhances the sensitivity of diffusion-limited tumors to scatter factors which increase cells' motility [12], [42].
in the diffusion-limited regime, in agreement with experiments showing that hypoxia enhances the sensitivity of diffusion-limited tumors to scatter factors which increase cells' motility [12], [42].
Table 5. The dependence of  (in days) on
(in days) on 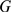 and
and  .
.
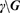
|
50 | 100 | 150 | 200 |

|
52 | 35 | 79 |

|

|
37 | 29 | 47 | 101 |

|
23 | 21 | 27 | 46 |

|
15 | 14 | 18 | 21 |
Discussion
We have shown that whether a simulated growing 3D tumor develops a smooth or fingered interface depends primarily on  , in agreement with in vitro experimental observations in [12], where tumor spheroids embedded in a 3D collagen matrix in hypoxic conditions developed a branched tubular structure, while in normoxic conditions they remained unbranched, confirming the previous results of [11] and recent studies employing different modeling approaches [17], [18], [92]. The transition from a smooth to fingered interface for GGH-simulated 3D avascular tumors occurs between
, in agreement with in vitro experimental observations in [12], where tumor spheroids embedded in a 3D collagen matrix in hypoxic conditions developed a branched tubular structure, while in normoxic conditions they remained unbranched, confirming the previous results of [11] and recent studies employing different modeling approaches [17], [18], [92]. The transition from a smooth to fingered interface for GGH-simulated 3D avascular tumors occurs between  and
and 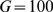 , while for GGH-simulated 2D avascular tumors it occurs between
, while for GGH-simulated 2D avascular tumors it occurs between  and
and 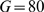 [1]. The transition regions overlap, which shows that the fingering instability is essentially dimension-independent and justifies using simpler 2D models of tumor growth instead of computationally expensive 3D models. Our GGH simulations of biofilm growth showed that changing the vertical dimension of the simulation domain,
[1]. The transition regions overlap, which shows that the fingering instability is essentially dimension-independent and justifies using simpler 2D models of tumor growth instead of computationally expensive 3D models. Our GGH simulations of biofilm growth showed that changing the vertical dimension of the simulation domain, 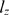 , greatly affected biofilm morphology, because
, greatly affected biofilm morphology, because  was proportional to
was proportional to 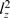 in those simulations. However, changing
in those simulations. However, changing 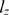 and keeping
and keeping  constant by changing, for instance, the background concentration of substrate did not significantly affect biofilm morphology [87]. Since the interaction between the growing tumor and the substrate does not depend on the boundaries of the simulation domain,
constant by changing, for instance, the background concentration of substrate did not significantly affect biofilm morphology [87]. Since the interaction between the growing tumor and the substrate does not depend on the boundaries of the simulation domain,  is a convenient parameter to define tumor-morphology regimes. In our simulations, we set the size of the cubic simulation domain
is a convenient parameter to define tumor-morphology regimes. In our simulations, we set the size of the cubic simulation domain 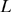 to the order of the typical size of avascular tumors, so
to the order of the typical size of avascular tumors, so  is an accurate, relative measure of how much the tumor cells compete for substrate.
is an accurate, relative measure of how much the tumor cells compete for substrate.
Figures 1–
5 show that while the tumor-TM surface tension does not affect the overall morphology significantly for low  , its effect grows for higher
, its effect grows for higher  . For low
. For low  , the tumor cells near the tumor-TM interface grow fast enough to find more substrate. For larger
, the tumor cells near the tumor-TM interface grow fast enough to find more substrate. For larger  , they grow more slowly, and in order to maintain their growth, they must migrate to reach substrate. The results of our 3D simulations agree with hypoxia's observed enhancement of the sensitivity of tumor cell motility to scatter factors, and support the hypothesis we suggested in [1] that HGF decreases tumor-TM surface tension.
, they grow more slowly, and in order to maintain their growth, they must migrate to reach substrate. The results of our 3D simulations agree with hypoxia's observed enhancement of the sensitivity of tumor cell motility to scatter factors, and support the hypothesis we suggested in [1] that HGF decreases tumor-TM surface tension.
2D simulated tumors [1] were partially disconnected for 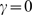 and for larger
and for larger  . Our 3D simulated tumors always remain connected (except again for
. Our 3D simulated tumors always remain connected (except again for 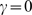 where a few cells migrate out of the backbone of the tumor), although their 2D sections appear disconnected in most cases. The ratio
where a few cells migrate out of the backbone of the tumor), although their 2D sections appear disconnected in most cases. The ratio 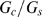 , where
, where 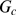 is the
is the  at which simulated tumors at high surface tensions cease to grow or produce truncated dendrites and
at which simulated tumors at high surface tensions cease to grow or produce truncated dendrites and 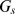 is the
is the  at which simulated tumors are roughly spherical, is about 5–7 in both 2D [1] and 3D. Thus the range in
at which simulated tumors are roughly spherical, is about 5–7 in both 2D [1] and 3D. Thus the range in  which spans all morphological regimes is very similar in 3D and 2D, i.e. 3D simulated tumors are as sensitive to competition for substrate as 2D tumors, which again justifies using simpler 2D models of tumor growth instead of more realistic, but computationally expensive 3D models.
which spans all morphological regimes is very similar in 3D and 2D, i.e. 3D simulated tumors are as sensitive to competition for substrate as 2D tumors, which again justifies using simpler 2D models of tumor growth instead of more realistic, but computationally expensive 3D models.
While the diffusion-limitation parameter  determines whether the tumor has a uniform or fingered margin, the tumor-TM surface tension
determines whether the tumor has a uniform or fingered margin, the tumor-TM surface tension  affects the detailed tumor morphology. These effects are also visible in 2D sections of simulated 3D tumors. Also as in 2D, the sensitivity of 3D tumor morphology to tumor-TM surface tension increases with
affects the detailed tumor morphology. These effects are also visible in 2D sections of simulated 3D tumors. Also as in 2D, the sensitivity of 3D tumor morphology to tumor-TM surface tension increases with  , causing a directional-solidification-like transition at high
, causing a directional-solidification-like transition at high  between dendritic structures, produced when the tumor-TM surface tension
between dendritic structures, produced when the tumor-TM surface tension  is high, and seaweed-like or DLA-like structures, produced when
is high, and seaweed-like or DLA-like structures, produced when  is low. Thus our 3D results support the idea, suggested in [12] and supported by our 2D simulations [1] and several other studies [16], [93], [94], that we need to therapeutically suppress cell motility and increase tumor cell-tumor cell adhesion when targeting tumor angiogenesis, in order to prevent the spread of tumor cells because of substrate deprivation.
is low. Thus our 3D results support the idea, suggested in [12] and supported by our 2D simulations [1] and several other studies [16], [93], [94], that we need to therapeutically suppress cell motility and increase tumor cell-tumor cell adhesion when targeting tumor angiogenesis, in order to prevent the spread of tumor cells because of substrate deprivation.
Using a cubic lattice for our GGH simulations may introduce artifacts related to anisotropic spatial evolution of the simulated tumor, which can influence high-rank tensor observables. Such artifacts are of limited significance in our simulations, but do appear to a limited extent in Figure 1 (a), and more prevalently in Figures 2 (a), 2 (b), 3 (a) and 3 (b). In general, anisotropy is significant for large values of tumor-TM surface tension (large  ), though the anisotropy does not appear to affect our general results. In these cases, we plan to cross-check our conclusions concerning orientational observables, such as dendritic orientation, with simulations on a higher-symmetry hexagonal lattice.
), though the anisotropy does not appear to affect our general results. In these cases, we plan to cross-check our conclusions concerning orientational observables, such as dendritic orientation, with simulations on a higher-symmetry hexagonal lattice.
As in [1], we coarse-grained tumor cells to speed our simulations. Thus our Generalized Cells represent tumor-cell clusters, describing the averaged behavior of many cells. Although our model discretizes tumors at approximately the same spatial resolution as continuum methods, it introduces surface energy and cell adhesion in a correct, physical manner. Moreover, the inclusion of cells as extended and deformable objects will allow us to study in the future the effects of elasticity on simulated tumor morphology.
In our study of the effects of  and
and  on tumor growth we did not include quiescence and necrosis explicitly. Because the substrate concentration in the central regions of our simulated tumors is nearly zero, we would expect the cells there to behave like quiescent cells (no growth). We repeated our simulations with quiescence for
on tumor growth we did not include quiescence and necrosis explicitly. Because the substrate concentration in the central regions of our simulated tumors is nearly zero, we would expect the cells there to behave like quiescent cells (no growth). We repeated our simulations with quiescence for  and
and 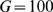 , both with
, both with 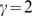 . To simulate quiescence, we impose a rule that a tumor cell stops growing, consuming substrate and producing MDE if the substrate concentration inside the cell drops below a threshold
. To simulate quiescence, we impose a rule that a tumor cell stops growing, consuming substrate and producing MDE if the substrate concentration inside the cell drops below a threshold 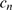 . Figures 10 (a) and 10 (b) show the morphologies of 2D sections of 3D tumors with quiescence for
. Figures 10 (a) and 10 (b) show the morphologies of 2D sections of 3D tumors with quiescence for  and
and 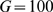 , respectively. These morphologies are slightly more compact than the corresponding structures in Figures 1 (c) and Figure 2 (c). The tumors reach the boundaries of the simulation domain (
, respectively. These morphologies are slightly more compact than the corresponding structures in Figures 1 (c) and Figure 2 (c). The tumors reach the boundaries of the simulation domain ( 6 mm) after 14 days (
6 mm) after 14 days ( ) and 26 days (
) and 26 days (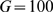 ). Their mean circularities at the moment when the tumors reach the boundaries of the simulation domain are slightly larger than without quiescence,
). Their mean circularities at the moment when the tumors reach the boundaries of the simulation domain are slightly larger than without quiescence, 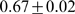 and
and 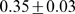 . This difference arises because quiescent cells do not grow and do not consume substrate, reducing the competition for substrate. The net effect is slightly faster growth (for
. This difference arises because quiescent cells do not grow and do not consume substrate, reducing the competition for substrate. The net effect is slightly faster growth (for 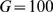 ) than without quiescence and morphologies corresponding to smaller values of
) than without quiescence and morphologies corresponding to smaller values of  .
.
Figure 10. Simulated growing tumors with quiescence or necrosis for 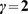 .
.

(a) 2D sections of 3D simulations with quiescence with 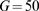 . (b) 2D sections of 3D simulations with quiescence with
. (b) 2D sections of 3D simulations with quiescence with 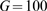 . Green - proliferating cells, blue - quiescent cells. (c) 2D sections of 3D simulations with necrosis with
. Green - proliferating cells, blue - quiescent cells. (c) 2D sections of 3D simulations with necrosis with 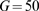 . (d) 2D sections of 3D simulations with necrosis with
. (d) 2D sections of 3D simulations with necrosis with 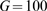 . Green - proliferating cells, red - necrotic cells.
. Green - proliferating cells, red - necrotic cells.
We also repeated our simulations with necrosis for  and
and 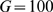 , both with
, both with 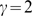 . To simulate necrosis, we impose a rule that a tumor cell stops consuming substrate and producing MDE and shrinks (grows with a negative growth rate
. To simulate necrosis, we impose a rule that a tumor cell stops consuming substrate and producing MDE and shrinks (grows with a negative growth rate 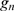 ) if the substrate concentration inside the cell drops below
) if the substrate concentration inside the cell drops below 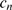 . Figures 10 (c) and 10 (d) show the morphologies of 2D sections of 3D tumors with necrosis for
. Figures 10 (c) and 10 (d) show the morphologies of 2D sections of 3D tumors with necrosis for  and
and 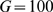 , respectively. These morphologies do not differ appreciably from the corresponding structures in Figure 1 (c) and Figure 2 (c). The tumors reach the boundaries of the simulation domain (
, respectively. These morphologies do not differ appreciably from the corresponding structures in Figure 1 (c) and Figure 2 (c). The tumors reach the boundaries of the simulation domain ( 6 mm) after 14 days (
6 mm) after 14 days ( ) and 28 days (
) and 28 days (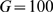 ). Their mean circularities at the moment when the tumors reach the boundaries of the simulation domain are slightly larger than without necrosis,
). Their mean circularities at the moment when the tumors reach the boundaries of the simulation domain are slightly larger than without necrosis,  and
and 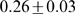 . This difference arises because necrotic cells do not grow and do not consume substrate, reducing the competition for substrate. The net effect is slightly faster growth (for
. This difference arises because necrotic cells do not grow and do not consume substrate, reducing the competition for substrate. The net effect is slightly faster growth (for 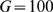 ) than without necrosis (but slower than with quiescence) and morphologies corresponding to slightly smaller values of
) than without necrosis (but slower than with quiescence) and morphologies corresponding to slightly smaller values of  . In addition, as we would expect, at late times, the tumors are more fragmented than without necrosis, which could affect their biomedically significant degree of invasiveness. However, at early times, tumor morphologies are essentially indistinguishable with and without necrosis. We will examine effects of quiescence and necrosis in more detail in a future paper.
. In addition, as we would expect, at late times, the tumors are more fragmented than without necrosis, which could affect their biomedically significant degree of invasiveness. However, at early times, tumor morphologies are essentially indistinguishable with and without necrosis. We will examine effects of quiescence and necrosis in more detail in a future paper.
While we recognize that most tumors are much more complex than our simple simulations, our goal was to understand the physics of the initial phase of the fingering instability as a function of the tumor-cell adhesivity and substrate consumption rate. We have shown that in 3D simulations, which are more expensive computationally, the physics of front instabilities and invasiveness in GGH-simulated tumors is the same as in 2D simulations (which was not obvious a priori). Therefore, our results justify using simpler 2D models of tumor growth instead of 3D models in some cases. We also found that the sphericity of 3D simulated tumors of a size on the order of a few mm (the typical size that avascular tumors reach) correlates strongly with the mean circularity of their 2D sections, especially for faster-growing, low-surface-tension tumors. Our results suggest that analyzing 2D sections of real avascular tumors at later stages should allow us to reconstruct the morphology of the underlying 3D tumors for use in tumor staging and guidance of therapeutic choices.
In future work, we will check for lattice artifacts and coarse-graining effects, and study the effects of necrosis and hypoxia-dependent growth rates on the morphological instability of growing tumors. We also plan to study the sensitivity of the tumor morphology to the other parameters in the simulations and the variability of morphology from replica to replica.
Methods
Mathematical Structure of the Tumor Model
We have discussed our model in detail in [1] and review it very briefly here. In our simplified solid, avascular tumor model, cells are spatially extended and deformable, move, adhere to each other, consume substrate, grow at a rate proportional to the local substrate concentration, divide when their volume doubles, and secrete MDE. Our choice of biological mechanisms follows our recent 2D model [1], which simplifies the HDC model of [70].
We use the GGH model [2]–[4] to represent tumor cells. As in [1], [70], we include three fields: a TM field representing a homogenized version of the cells and ECM of the normal tissue surrounding the tumor, an MDE field produced by tumor cells, which degrades the TM field, and a substrate field representing the concentration of a substrate limiting tumor-cell growth. The equations for these fields are the same as in [1]. MDE (denoted by  , which ranges between 0 and 1) degrades TM (denoted by
, which ranges between 0 and 1) degrades TM (denoted by  , which ranges between 0 and 1) according to [95], [96] (equation (1) in [1]):
, which ranges between 0 and 1) according to [95], [96] (equation (1) in [1]):
| (3) |
where  is a positive constant. Degradation does not consume MDE and MDE does not decay. However, the maximum MDE concentration is 1, which effectively imposes a decay in regions of high concentration. Tumor cells produce MDE at a constant rate
is a positive constant. Degradation does not consume MDE and MDE does not decay. However, the maximum MDE concentration is 1, which effectively imposes a decay in regions of high concentration. Tumor cells produce MDE at a constant rate  (the rate of MDE production per tumor cell); MDE then diffuses uniformly (equation (2) in [1]):
(the rate of MDE production per tumor cell); MDE then diffuses uniformly (equation (2) in [1]):
| (4) |
where 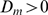 is the diffusion constant of MDE.
is the diffusion constant of MDE. 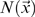 equals 1 inside tumor cells, otherwise it is 0. To model the transport of nutrient in surrounding normal tissue, TM produces substrate at a constant rate per unit density. We denote the substrate concentration
equals 1 inside tumor cells, otherwise it is 0. To model the transport of nutrient in surrounding normal tissue, TM produces substrate at a constant rate per unit density. We denote the substrate concentration  as a fraction of the maximum soluble concentration
as a fraction of the maximum soluble concentration 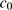 , so we require
, so we require 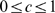 . The substrate diffuses and is consumed by the tumor cells at a constant rate per cell (equation (3) in [1]). Including saturation:
. The substrate diffuses and is consumed by the tumor cells at a constant rate per cell (equation (3) in [1]). Including saturation:
| (5) |
where 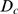 ,
, 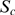 and
and 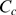 are positive constants representing respectively the substrate-diffusion constant, the substrate-production rate per unit TM and the substrate-consumption rate per tumor cell, and
are positive constants representing respectively the substrate-diffusion constant, the substrate-production rate per unit TM and the substrate-consumption rate per tumor cell, and  is the Heaviside step function. The substrate-consumption rate for the normalized substrate field is:
is the Heaviside step function. The substrate-consumption rate for the normalized substrate field is:
| (6) |
Initially 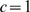 ,
,  and
and 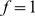 everywhere.
everywhere.
In [1] we showed that 2D-tumor morphology depended mainly on a single parameter, the nondimensional ratio of the maximum tumor-growth rate to the maximum substrate-transport rate: the Diffusion-Limitation Parameter (equation (8) in [1]):
| (7) |
where 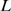 is the size of the simulation domain and
is the size of the simulation domain and 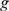 is the maximum specific growth rate (amount of new tumor produced per unit time per unit tumor). We vary
is the maximum specific growth rate (amount of new tumor produced per unit time per unit tumor). We vary  by varying
by varying 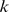 . Cells grow by increasing their volume
. Cells grow by increasing their volume 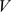 at a rate proportional to the local substrate concentration.
at a rate proportional to the local substrate concentration.
GGH Implementation of the Tumor Model
For a detailed description of GGH simulations, see [1] and [4]. For additional information on CC3D and open-source downloads of CC3D software for Windows, Mac OSX and Linux platforms, please visit: http://www.compucell3d.org/.
In our simulation, Generalized Cells are spatially-extended domains, which represent either tumor cells or non-tumor tissue and reside on a single 3D, square Cell Lattice [4], [97]. Generalized Cells carry state descriptors, e.g., cells' target volumes and volumes at which mitosis occurs. Fields are continuously-variable concentrations, each of which resides on its own lattice, here diffusing MDE and substrate, and nondiffusing TM, evolving according to equations (3), (4) and (5). The Effective Energy creates forces which determine a Generalized Cell's shape, motility, adhesion and response to extracellular signals [4].
We denote the unique index of a Generalized Cell by  , where the value at a Cell-Lattice site (voxel)
, where the value at a Cell-Lattice site (voxel) 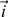 is
is  if this site lies in Generalized Cell
if this site lies in Generalized Cell  . Each Generalized Cell has an associated Generalized-Cell type
. Each Generalized Cell has an associated Generalized-Cell type
 . In our model, TM denotes tissue medium and t tumor cells.
. In our model, TM denotes tissue medium and t tumor cells.
The Effective Energy  in our tumor simulations includes three terms [1]–[4]:
in our tumor simulations includes three terms [1]–[4]:
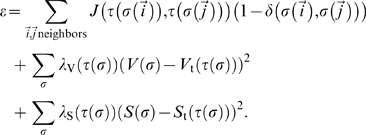 |
(8) |
The first term describes the surface adhesion between Generalized Cells in terms of Contact Energies
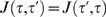 [1]–[4]. The units of
[1]–[4]. The units of 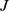 in 3D are energy/unit boundary surface area. We use a fourth-neighbor interaction range (32 neighbors for each voxel) to calculate the Contact Energies, which reduces Cell-Lattice anisotropy effects [98]. The second term constrains the Generalized Cells' volumes;
in 3D are energy/unit boundary surface area. We use a fourth-neighbor interaction range (32 neighbors for each voxel) to calculate the Contact Energies, which reduces Cell-Lattice anisotropy effects [98]. The second term constrains the Generalized Cells' volumes; 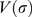 is the volume in Cell-Lattice sites of Generalized Cell
is the volume in Cell-Lattice sites of Generalized Cell  ,
, 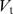 its Target Volume, and
its Target Volume, and 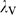 its inverse compressibility. The third term represents the elasticity of a cell membrane;
its inverse compressibility. The third term represents the elasticity of a cell membrane; 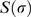 is the surface area of Generalized Cell
is the surface area of Generalized Cell  ,
, 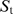 its Target Surface Area, and
its Target Surface Area, and 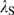 its inverse membrane elasticity. We model TM as one unconstrained Generalized Cell:
its inverse membrane elasticity. We model TM as one unconstrained Generalized Cell: 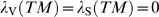 .
.
We define the tumor surface tension
 in terms of the Contact Energies
in terms of the Contact Energies 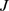 [2],[3]:
[2],[3]:
| (9) |
The surface tension controls the tendency of tumor cells to disperse or cluster.
To model cell motility, the Cell Lattice evolves through stochastic attempts by Generalized Cells to extend their boundaries into neighboring Cell-Lattice sites, slightly displacing the Generalized Cells which currently occupy those sites [2]–[4]. At each step, we randomly select a Cell-Lattice site 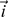 and attempt to change its index from
and attempt to change its index from 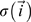 to the index
to the index 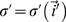 of a Cell-Lattice site
of a Cell-Lattice site 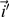 randomly chosen in its fourth-order neighborhood. If the difference in Effective Energy produced by the change
randomly chosen in its fourth-order neighborhood. If the difference in Effective Energy produced by the change 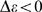 then we accept the change. If
then we accept the change. If 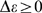 then we accept the change with a probability
then we accept the change with a probability 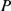 :
:
| (10) |
where 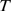 represents the cell's intrinsic cytoskeletally-driven mot ility. One Monte Carlo Step (MCS) corresponds to
represents the cell's intrinsic cytoskeletally-driven mot ility. One Monte Carlo Step (MCS) corresponds to  attempts, where
attempts, where  is the total number of Cell-Lattice sites.
is the total number of Cell-Lattice sites.
The dimension of our Cell Lattice in voxels is 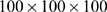 . In our simulations, each simulated tumor cell (Generalized Cell of type
. In our simulations, each simulated tumor cell (Generalized Cell of type  ) initially occupies a
) initially occupies a 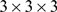 voxel cube. The initial 3D configuration of these simulations consists of 8 Generalized Cells of type
voxel cube. The initial 3D configuration of these simulations consists of 8 Generalized Cells of type  forming a cube in the middle of the simulation domain. We set the linear size of 1 voxel to 60
forming a cube in the middle of the simulation domain. We set the linear size of 1 voxel to 60 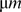 , which is on the order of the scale in [1], so the size of the simulation domain corresponds to 6 mm. Therefore the linear size of 1 simulated tumor cell is 180
, which is on the order of the scale in [1], so the size of the simulation domain corresponds to 6 mm. Therefore the linear size of 1 simulated tumor cell is 180 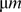 , which is about 7 times greater than the size of real tumor cells [70], [99], [100], so 1 simulated tumor cell represents
, which is about 7 times greater than the size of real tumor cells [70], [99], [100], so 1 simulated tumor cell represents 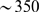 real tumor cells. Since the size of tumor cells is much smaller than the substrate-penetration and capillary lengths (see [1]), the cell size is not critical. Coarse-graining the cells greatly speeds our simulations. Since
real tumor cells. Since the size of tumor cells is much smaller than the substrate-penetration and capillary lengths (see [1]), the cell size is not critical. Coarse-graining the cells greatly speeds our simulations. Since  is dimensionless, its value is independent of our choice of length scale.
is dimensionless, its value is independent of our choice of length scale.
We simulate growth of tumor cells by increasing their 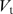 at a rate proportional to the local limiting-substrate concentration
at a rate proportional to the local limiting-substrate concentration  at the cell's center of mass [1], [52], [87], [101]:
at the cell's center of mass [1], [52], [87], [101]:
| (11) |
where 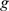 is a growth rate. For tumor cells, the initial
is a growth rate. For tumor cells, the initial 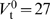 . When a tumor cell reaches the doubling volume
. When a tumor cell reaches the doubling volume
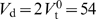 , it divides and splits along a random axis into two tumor cells (of the same phenotype) with Target Volumes
, it divides and splits along a random axis into two tumor cells (of the same phenotype) with Target Volumes 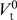 [97]. To prevent growing cells from changing shape, we adjust
[97]. To prevent growing cells from changing shape, we adjust 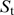 so that the nondimensional ratio
so that the nondimensional ratio 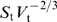 remains constant, i.e.
remains constant, i.e.
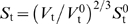 . We set
. We set 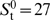 , which produces roughly spherical cells. Since we do not set a substrate-concentration threshold for cells to grow, all cells can proliferate. However, the substrate concentration in the inner part of the growing tumor spheroid is nearly zero. Thus only tumor cells near the surface of the spheroid grow appreciably, while cells in the middle of the spheroid effectively do not grow during the duration of the simulation.
, which produces roughly spherical cells. Since we do not set a substrate-concentration threshold for cells to grow, all cells can proliferate. However, the substrate concentration in the inner part of the growing tumor spheroid is nearly zero. Thus only tumor cells near the surface of the spheroid grow appreciably, while cells in the middle of the spheroid effectively do not grow during the duration of the simulation.
As in [1], we define three Fields: 1) diffusible substrate  , 2) diffusing MDE
, 2) diffusing MDE  , 3) nondiffusing TM
, 3) nondiffusing TM 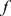 . These Fields can have nonzero values at each point simultaneously and co-occupy space with cells.
. These Fields can have nonzero values at each point simultaneously and co-occupy space with cells.
Tumor cells produce MDE at a constant rate at the cell's center of mass. Tumor cells absorb substrate at their respective centers of mass. Substrate and MDE diffuse uniformly on their Field lattices using a forward-Euler algorithm. If 1 voxel corresponds to  meters and 1 MCS to
meters and 1 MCS to 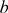 seconds then, for example,
seconds then, for example, 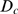 (in
(in 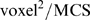 ) relates to the physical diffusion constant of substrate
) relates to the physical diffusion constant of substrate 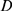 (in
(in 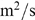 ) via:
) via: 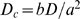 . In our simulations, we use no-flux boundary conditions at all Field edges.
. In our simulations, we use no-flux boundary conditions at all Field edges.
Parameter Values
To the best of our knowledge, no one has measured the adhesion parameters for a tumor cell line, although measurement should be possible [77]–[80]. Because the Contact Energy between two Cell-Lattice sites that belong to the same Generalized Cell is defined to be zero, we set all  positive to prevent Generalized Cells from dissociating. We also require
positive to prevent Generalized Cells from dissociating. We also require 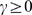 to keep the tumor cells from floating off into the TM spontaneously. Following our previous paper, our simulations use
to keep the tumor cells from floating off into the TM spontaneously. Following our previous paper, our simulations use 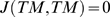 and
and 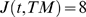 , and vary
, and vary 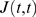 from 4, for which
from 4, for which 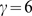 , to 16, for which
, to 16, for which 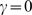 . For
. For 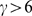 , the simulated morphologies do not differ much from those for
, the simulated morphologies do not differ much from those for 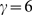 . We set
. We set 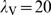 and
and 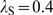 , which prevents Generalized Cells from nonbiological disappearing or freezing.
, which prevents Generalized Cells from nonbiological disappearing or freezing.
As in our earlier paper, we take 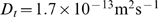 and
and 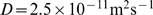 . For
. For 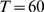 , the diffusion constant for our simulated tumor cells is about
, the diffusion constant for our simulated tumor cells is about 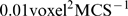 , as in our previous 2D simulations, so 400 MCS corresponds to approximately 1 day and
, as in our previous 2D simulations, so 400 MCS corresponds to approximately 1 day and 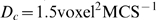 .
.
The remaining parameters come from [1]: 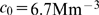 ,
, 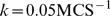 (which we denote
(which we denote 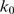 ),
), 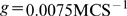 ,
, 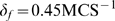 ,
, 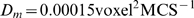 ,
, 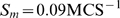 , and
, and 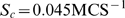 . We define the production and consumption parameters per Generalized Cell, so they are the same for both 2D and 3D. We also set
. We define the production and consumption parameters per Generalized Cell, so they are the same for both 2D and 3D. We also set 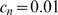 and
and 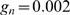 . Equation (7) gives, for
. Equation (7) gives, for 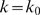 ,
, 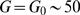 . Since MDE diffusion is very slow and TM degradation by MDE is strong,
. Since MDE diffusion is very slow and TM degradation by MDE is strong,  at all voxels occupied by tumor cells and
at all voxels occupied by tumor cells and 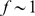 at all voxels occupied by TM.
at all voxels occupied by TM.
We vary 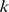 from 0.05 to 0.2, corresponding approximately to varying
from 0.05 to 0.2, corresponding approximately to varying  from 50 to 200, which covers the complete range of possible simulated-tumor morphologies. Smaller values of
from 50 to 200, which covers the complete range of possible simulated-tumor morphologies. Smaller values of  produce the same patterns as
produce the same patterns as  and values of
and values of  greatly slow or even halt tumor-cell proliferation. Table 6 lists our model parameters.
greatly slow or even halt tumor-cell proliferation. Table 6 lists our model parameters.
Table 6. Parameter values in our 3D simulations of tumor growth.
| 1 MCS |
 day day |
| 1 voxel | 60 
|
Diffusion-limitation parameter 
|
50–200 |
Tumor-TM surface tension 
|
0–6 |
| Tumor-TM Contact Energy | 8 |
Tumor-cell motility 
|
60 |
Tumor-cell doubling volume 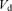
|
54 |
Tumor inverse compressibility 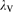
|
20 |
Tumor inverse membrane elasticity 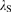
|
0.4 |
Tumor-cell shape parameter 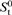
|
27 |
Substrate diffusion constant 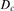
|
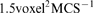
|
TM degradation rate 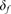
|
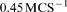
|
MDE diffusion constant 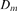
|
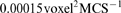
|
MDE production rate 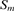
|
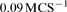
|
Substrate production rate 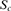
|
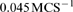
|
Tumor-cell growth rate 
|
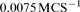
|
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported in part by National Institutes of Health, National Institute of General Medical Sciences grants R01 GM76692 and R01 GM077138, the Biocomplexity Institute at Indiana University and the Indiana University Faculty Research Support Program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Popławski NJ, Agero U, Gens JS, Swat M, Glazier JA, et al. Front instabilities and invasiveness of simulated avascular tumors. Bull Math Biol. 2009;71:1189. doi: 10.1007/s11538-009-9399-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Graner F, Glazier JA. Simulation of biological cell sorting using a two-dimensional extended Potts model. Phys Rev Lett. 1992;69:2013. doi: 10.1103/PhysRevLett.69.2013. [DOI] [PubMed] [Google Scholar]
- 3.Glazier JA, Graner F. Simulation of the differential adhesion driven rearrangement of biological cells. Phys Rev E. 1993;47:2128. doi: 10.1103/physreve.47.2128. [DOI] [PubMed] [Google Scholar]
- 4.Glazier JA, Balter A, Popławski NJ. Anderson ARA, Chaplain MAJ, Rejniak KA, editors. Magnetization to morphogenesis: a brief history of the Glazier-Graner-Hogeweg model. Single-Cell-Based Models in Biology and Medicine, Birkhäuser-Verlag. 2007. 79
- 5.Chaturvedi R, Izaguirre JA, Huang C, Cickovski T, Virtue P, et al. Multi-model simulations of chicken limb morphogenesis. Lect Notes Comput Sci. 2003;2659:39. [Google Scholar]
- 6.Izaguirre JA, Chaturvedi R, Huang C, Cickovski T, Coffland J, et al. CompuCell, a multi-model framework for simulation of morphogenesis. Bioinformatics. 2004;20:1129. doi: 10.1093/bioinformatics/bth050. [DOI] [PubMed] [Google Scholar]
- 7.Chaturvedi R, Huang C, Izaguirre JA, Newman SA, Glazier JA, et al. A hybrid discrete-continuum model for 3-D skeletogenesis of the vertebrate limb. Lect Notes Comp Sci. 2004;3305:543. [Google Scholar]
- 8.Chaturvedi R, Huang C, Kazmierczak B, Schneider T, Izaguirre JA, et al. On multiscale approaches to three-dimensional modeling of morphogenesis. J Roy Soc Interf. 2005;2:237. doi: 10.1098/rsif.2005.0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cickovski TM, Huang C, Chaturvedi R, Glimm T, Hentschel HGE, et al. A framework for three-dimensional simulation of morphogenesis. IEEE/ACM Trans Comp Biol Bioinf. 2005;2:1. doi: 10.1109/TCBB.2005.46. [DOI] [PubMed] [Google Scholar]
- 10.Swat MH, Hester SD, Balter AI, Heiland RW, Zaitlen BL, et al. Maly IV, editor. Multicell simulations of development and disease using the CompuCell3D simulation environment. Systems Biology, Humana. 2009. 361. [DOI] [PMC free article] [PubMed]
- 11.Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, et al. The contribution of VHL substrate binding and HIF1-α to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002;1:247. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]
- 12.Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, et al. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 13.Alarcon T, Byrne HM, Maini PK. A cellular automaton model for tumour growth in inhomogeneous environment. J Theor Biol. 2003;225:257. doi: 10.1016/s0022-5193(03)00244-3. [DOI] [PubMed] [Google Scholar]
- 14.Frieboes HB, Zheng X, Sun C, Tromberg B, Gatenby R, et al. An integrated computational/experimental model of tumor invasion. Cancer Res. 2006;66:1597. doi: 10.1158/0008-5472.CAN-05-3166. [DOI] [PubMed] [Google Scholar]
- 15.Cristini V, Lowengrub J, Nie Q. Nonlinear simulation of tumor growth. J Math Biol. 2003;46:191. doi: 10.1007/s00285-002-0174-6. [DOI] [PubMed] [Google Scholar]
- 16.Cristini V, Frieboes HB, Gatenby R, Caserta S, Ferrari M, et al. Morphologic instability and cancer invasion. Clin Cancer Res. 2005;11:6772. doi: 10.1158/1078-0432.CCR-05-0852. [DOI] [PubMed] [Google Scholar]
- 17.Macklin P, Lowengrub J. Nonlinear simulation of the effect of microenvironment on tumor growth. J Theor Biol. 2007;245:677. doi: 10.1016/j.jtbi.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Cristini V, Li X, Lowengrub JS, Wise SM. Nonlinear simulations of solid tumor growth using a mixture model: invasion and branching. J Math Biol. 2009;58:723. doi: 10.1007/s00285-008-0215-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinberg RA. The Biology of Cancer. Garland.: 2006. [Google Scholar]
- 20.Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 21.Huang S, Ingber DE. The structural and mechanical complexity of cell-growth control. Nat Cell Biol. 1999;1:E131. doi: 10.1038/13043. [DOI] [PubMed] [Google Scholar]
- 22.Lawrence JA, Steeg PS. Mechanisms of tumour invasion and metastasis. World J Urol. 1996;14:124. doi: 10.1007/BF00186891. [DOI] [PubMed] [Google Scholar]
- 23.Liotta LA, Rao CN, Barsky SH. Tumour invasion and the extracellular matrix. Lab Invest. 1983;49:636. [PubMed] [Google Scholar]
- 24.Stetler-Stevenson WG, Aznavoorian S, Liotta LA. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu Rev Cell Biol. 1993;9:541. doi: 10.1146/annurev.cb.09.110193.002545. [DOI] [PubMed] [Google Scholar]
- 25.Carter SB. Principles of cell motility: the direction of cell movement and cancer invasion. Nature. 1965;208:1183. doi: 10.1038/2081183a0. [DOI] [PubMed] [Google Scholar]
- 26.Quigley JP, Lacovara J, Cramer EB. The directed migration of B-16 melanoma-cells in response to a haptotactic chemotactic gradient of fibronectin. J Cell Biol. 1983;97:A450. [Google Scholar]
- 27.Lacovara J, Cramer EB, Quigley JP. Fibronectin enhancement of directed migration of B16 melanoma cells. Cancer Res. 1984;44:1657. [PubMed] [Google Scholar]
- 28.McCarthy JB, Furcht LT. Laminin and fibronectin promote the directed migration of B16 melanoma cells in vitro. J Cell Biol. 1984;98:1474. doi: 10.1083/jcb.98.4.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klominek J, Robert KH, Sundqvist KG. Chemotaxis and haptotaxis of human malignant mesothelioma cells: effects of fibronectin, laminin, type iv collagen, and an autocrine motility factor-like substance. Cancer Res. 1993;53:4376. [PubMed] [Google Scholar]
- 30.Debruyne PR, Bruyneel EA, Karaguni I-M, Li X, Flatau G, et al. Bile acids stimulate invasion and haptotaxis in human corectal cancer cells through activation of multiple oncogenic signalling pathways. Oncogene. 2002;21:6740. doi: 10.1038/sj.onc.1205729. [DOI] [PubMed] [Google Scholar]
- 31.Hynes RO. Integrins: versatility, modulation, and signalling in cell adhesion. Cell. 1992;69:11. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 32.Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 33.Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractability, and signalling. Annu Rev Cell Dev Biol. 1996;12:463. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- 34.Harris AL. Hypoxia - a key regulatory factor in tumor growth. Nat Rev Cancer. 2002;2:38. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura T, Teramoto H, Ichihara A. Purification and characterization of a growth factor from rat platelets for mature parenchymal hepatocytes in primary cultures. Proc Natl Acad Sci USA. 1986;83:6489. doi: 10.1073/pnas.83.17.6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamura T, Nishizawa T, Hagiya M, Seki T, Shimonishi M, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 37.Rubin JS, Bottaro DP, Aaronson SA. Hepatocyte growth factor/scatter factor and its receptor, the c-met proto-oncogene product. Biochim Biophys Acta. 1993;1155:357. doi: 10.1016/0304-419x(93)90015-5. [DOI] [PubMed] [Google Scholar]
- 38.Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nature Rev Cancer. 2002;4:289. doi: 10.1038/nrc779. [DOI] [PubMed] [Google Scholar]
- 39.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 40.Stoker N, Gherardi E, Perryman M, Grey J. Scatter factor is a fibroblast-derived modulator of epithelial cell motility. Nature. 1987;327:239. doi: 10.1038/327239a0. [DOI] [PubMed] [Google Scholar]
- 41.Gherardi E, Gray J, Stoker M, Perryman M, Furlong R. Purification of scatter factor, a fibroblast-derived basic protein that modulates epithelial interactions and movement. Proc Natl Acad Sci USA. 1989;86:5844. doi: 10.1073/pnas.86.15.5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Condeelis J, Singer RH, Segall JE. The great escape: When cancer cells hijack the genes for chemotaxis and motility. Annu Rev Cell Dev Biol. 2005;21:695. doi: 10.1146/annurev.cellbio.21.122303.120306. [DOI] [PubMed] [Google Scholar]
- 43.Christofori G. New signals from the invasive front. Nature. 2006;441:444. doi: 10.1038/nature04872. [DOI] [PubMed] [Google Scholar]
- 44.Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci USA. 2008;105:6392. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 46.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J Cell Biol. 2006;172:973. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chaplain MAJ. Avascular growth, angiogenesis and vascular growth in solid tumours: the mathematical modelling of the stages of tumour development. Math Comput Model. 1996;23:47. [Google Scholar]
- 48.Wheldon TE. Mathematical models in experimental and clinical oncology. In: Ingram D, Bloch RF, editors. Mathematical Methods in Medicine. London: J. Wiley and Sons; 1986. 1 [Google Scholar]
- 49.Anderson ARA, Chaplain MAJ. Continuous and discrete mathematical models of tumourinduced angiogenesis angiogenesis. Bull Math Biol. 1998;60:857. doi: 10.1006/bulm.1998.0042. [DOI] [PubMed] [Google Scholar]
- 50.Plank MJ, Sleeman BD. Lattice and non-lattice models of tumour angiogenesis. Bull Math Biol. 2004;66:1785. doi: 10.1016/j.bulm.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Owen MR, Alarcon T, Maini PK, Byrne HM. Angiogenesis and vascular remodelling in normal and cancerous tissues. J Math Biol. 2009;58:689. doi: 10.1007/s00285-008-0213-z. [DOI] [PubMed] [Google Scholar]
- 52.Shirinifard A, Gens JS, Zaitlen BL, Popławski NJ, Swat M, et al. 3D multi-cell simulation of tumor growth and angiogenesis. PLoS ONE. 2009;4:e7190. doi: 10.1371/journal.pone.0007190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sherratt JA, Nowak MA. Oncogenes, anti-oncogenes and the immune response to cancer: a mathematical model. Proc R Soc Lond B. 1992;248:261. doi: 10.1098/rspb.1992.0071. [DOI] [PubMed] [Google Scholar]
- 54.Orme ME, Chaplain MAJ. A mathematical model of vascular tumour growth and invasion. Math Comput Model. 1996;23:43. [Google Scholar]
- 55.Perumpanani AJ, Sherratt JA, Norbury J, Byrne HM. Biological inferences from a mathematical model of malignant invasion. Invasion Metastasis. 1996;16:209. [PubMed] [Google Scholar]
- 56.Anderson ARA, Chaplain MAJ, Newman EL, Steele RJC, Thompson AM. Mathematical modelling of tumour invasion and metastasis. J Theor Med. 2000;2:129. [Google Scholar]
- 57.Byrne HM, Chaplain MAJ, Pettet GJ, McElwain DLS. A mathematical model of trophoblast invasion. J Theor Med. 1999;1:275. [Google Scholar]
- 58.Zheng X, Wise SM, Cristini V. Nonlinear simulation of tumor necrosis, neo-vascularization and tissue invasion via an adaptive finite-element/level-set method. Bull Math Biol. 2005;67:211. doi: 10.1016/j.bulm.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 59.Chaplain MAJ, Sleeman BD. Modelling the growth of solid tumours and incorporating a method for their classification using nonlinear elasticity theory. J Math Biol. 1993;31:431. doi: 10.1007/BF00173886. [DOI] [PubMed] [Google Scholar]
- 60.Tracqui P. From passive diffusion to active cellular migration in mathematical models of tumour invasion. Acta Biotheor. 1995;43:443. doi: 10.1007/BF00713564. [DOI] [PubMed] [Google Scholar]
- 61.Frieboes HB, Lowengrub JS, Wise S, Zheng X, Macklin P, et al. Computer simulation of glioma growth and morphology. Neuroimage. 2007;37:S59. doi: 10.1016/j.neuroimage.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li XR, Cristini V, Nie Q, Lowengrub JS. Nonlinear three-dimensional simulation of solid tumor growth. Discr Cont Dyn Syst Ser B. 2007;7:581. [Google Scholar]
- 63.Kimmel M, Axelrod DE. Unequal cell division, growth regulation and colony size of mammalian cells: a mathematical model and analysis of experimental data. J Theor Biol. 1991;153:157. doi: 10.1016/s0022-5193(05)80420-5. [DOI] [PubMed] [Google Scholar]
- 64.Smolle J, Stettner H. Computer simulation of tumour cell invasion by a stochastic growth model. J Theor Biol. 1993;160:63. doi: 10.1006/jtbi.1993.1004. [DOI] [PubMed] [Google Scholar]
- 65.Qi A, Zheng X, Du C, An B. A cellular automaton model of cancerous growth. J Theor Biol. 1993;161:1. doi: 10.1006/jtbi.1993.1035. [DOI] [PubMed] [Google Scholar]
- 66.Kansal AR, Torquato S, Harsh GR, Chiocca EA, Deisboeck TS. Simulated brain tumor growth using a three-dimensional cellular automaton. J Theor Biol. 2000;203:367. doi: 10.1006/jtbi.2000.2000. [DOI] [PubMed] [Google Scholar]
- 67.Dormann S, Deutsch A. Modeling of self-organized avascular tumor growth with a hybrid cellular automaton. In Silico Biol. 2002;2:0035. [PubMed] [Google Scholar]
- 68.Düchting W. Tumor growth simulation. Comput Graph. 1990;14:505. [Google Scholar]
- 69.Düchting W, Ulmer W, Ginsberg T. Cancer: a challenge for control theory and computer modelling. Eur J Cancer. 1996;32A:1283. doi: 10.1016/0959-8049(96)00075-5. [DOI] [PubMed] [Google Scholar]
- 70.Anderson ARA. A hybrid mathematical model of solid tumour invasion: the importance of cell adhesion. Math Med Biol. 2005;22:163. doi: 10.1093/imammb/dqi005. [DOI] [PubMed] [Google Scholar]
- 71.Rejniak KA. A single-cell approach in modeling the dynamics of tumor microregions. Math Biosci Eng. 2005;2:643. doi: 10.3934/mbe.2005.2.643. [DOI] [PubMed] [Google Scholar]
- 72.Zhang L, Athale CA, Deisboeck TS. Development of a three-dimensional multiscale agent-based tumor model: simulating gene-protein interaction profiles, cell phenotypes and multicellular patterns in brain cancer. J Theor Biol. 2007;244:96. doi: 10.1016/j.jtbi.2006.06.034. [DOI] [PubMed] [Google Scholar]
- 73.Wang Z, Zhang L, Sagotsky J, Deisboeck TS. Simulating non-small cell lung cancer with a multiscale agent-based model. Theor Biol Med Model. 2007;4:50. doi: 10.1186/1742-4682-4-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Z, Birch CM, Sagotsky J, Deisboeck TS. Cross-scale, cross-pathway evaluation using an agent-based non-small cell lung cancer model. Bioinformatics. 2009;25:2389. doi: 10.1093/bioinformatics/btp416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sanga S, Frieboes HB, Zheng X, Gatenby R, Bearer EL, et al. Predictive oncology: a review of multidisciplinary, multiscale in silico modeling linking phenotype, morphology and growth. Neuroimage. 2007;37:S120. doi: 10.1016/j.neuroimage.2007.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Macklin P, McDougall S, Anderson AR, Chaplain MAJ, Cristini V, et al. Multiscale modelling and nonlinear simulation of vascular tumour growth. J Math Biol. 2009;58:765. doi: 10.1007/s00285-008-0216-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beysens DA, Forgacs G, Glazier JA. Embryonic tissues are viscoelastic materials. Can J Phys. 2000;78:243. [Google Scholar]
- 78.Foty RA, Forgacs G, Pfleger CM, Steinberg MS. Liquid properties of embryonic tissues: measurement of interfacial tensions. Phys Rev Lett. 1994;72:2298. doi: 10.1103/PhysRevLett.72.2298. [DOI] [PubMed] [Google Scholar]
- 79.Foty RA, Pfleger CM, Forgacs G, Steinberg MS. Surface tensions of embryonic tissues predict their mutual envelopment behavior. Development. 1996;122:1611. doi: 10.1242/dev.122.5.1611. [DOI] [PubMed] [Google Scholar]
- 80.Forgacs G, Foty RA, Shafrir Y, Steinberg MS. Viscoelastic properties of living embryonic tissues: a quantitative study. Biophys J. 1998;74:2227. doi: 10.1016/S0006-3495(98)77932-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guiot C, Delsanto PP, Deisboeck TS. Morphological instability and cancer invasion: a ‘splashing water drop’ analogy. Theor Biol Med Model. 2007;4:4. doi: 10.1186/1742-4682-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mariani L, Beaudry C, McDonough WS, Hoelzinger DB, Demuth T, et al. Glioma cell motility is associated with reduced transcription of proapoptotic and proliferation genes: a cDNA microarray analysis. J Neuro-Oncol. 2001;53:161. doi: 10.1023/a:1012253317934. [DOI] [PubMed] [Google Scholar]
- 83.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 85.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nature Med. 1995;1:21. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 86.Wadell H. Volume, shape and roundness of quartz particles. J Geol. 1935;43:250. [Google Scholar]
- 87.Popławski NJ, Shirinifard A, Swat M, Glazier JA. Simulation of single-species bacterial-biofilm growth using the Glazier-Graner-Hogeweg model and the CompuCell3D modeling environment. Math Biosci Eng. 2008;5:355. doi: 10.3934/mbe.2008.5.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moore MG, Juel A, Burgess JM, McCormick WD, Swinney HL. Fluctuations in viscous fingering. Phys Rev E. 2002;65:030601(R). doi: 10.1103/PhysRevE.65.030601. [DOI] [PubMed] [Google Scholar]
- 89.Miura T, Shiota K. Depletion of FGF acts as a lateral inhibitory factor in lung branching morphogenesis in vitro. Mech Dev. 2002;116:29. doi: 10.1016/s0925-4773(02)00132-6. [DOI] [PubMed] [Google Scholar]
- 90.Hartmann D, Miura T. Modelling in vitro lung branching morphogenesis during development. J Theor Biol. 2006;242:862. doi: 10.1016/j.jtbi.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 91.Turner S, Sherratt JA, Painter KJ, Savill NJ. From a discrete to a continuous model of biological cell movement. Phys Rev E. 2004;69:021910. doi: 10.1103/PhysRevE.69.021910. [DOI] [PubMed] [Google Scholar]
- 92.Wise SM, Lowengrub JS, Frieboes HB, Cristini V. Three-dimensional multispecies nonlinear tumor growth - I: Model and numerical method. J Theor Biol. 2008;253:524. doi: 10.1016/j.jtbi.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bello L, Lucini V, Carrabba G, Giussani C, Machluf M, et al. Simultaneous inhibition of glioma angiogenesis, cell proliferation, and invasion by a naturally occurring fragment of human metalloproteinase-2. Cancer Res. 2001;61:8730. [PubMed] [Google Scholar]
- 94.Deisboeck TS. Cancer: a profit-driven biosystem? Med Hypotheses. 2008;71:186. doi: 10.1016/j.mehy.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stetler-Stevenson WG, Hewitt R, Corcoran M. Matrix metallo-proteinases and tumour invasion: from correlation to causality to the clinic. Cancer Biol. 1996;7:147. doi: 10.1006/scbi.1996.0020. [DOI] [PubMed] [Google Scholar]
- 96.Chambers AF, Matrisian LM. Changing views of the role of matrix metalloproteinases in metastasis. J Natl Cancer Inst. 1997;89:1260. doi: 10.1093/jnci/89.17.1260. [DOI] [PubMed] [Google Scholar]
- 97.Balter A, Merks RMH, Popławski NJ, Swat M, Glazier JA. Anderson ARA, Chaplain MAJ, Rejniak KA, editors. The Glazier-Graner-Hogeweg model: extensions, future directions, and opportunities for further study. Single-Cell-Based Models in Biology and Medicine, Birkhäuser-Verlag. 2007. 151
- 98.Holm EA, Glazier JA, Srolovitz DJ, Grest GS. Effects of lattice anisotropy and temperature on domain growth in the two-dimensional Potts model. Phys Rev A. 1991;43:2662. doi: 10.1103/physreva.43.2662. [DOI] [PubMed] [Google Scholar]
- 99.Melicow MM. The three-steps to cancer: a new concept of carcinogenesis. J Theor Biol. 1982;94:471. doi: 10.1016/0022-5193(82)90322-8. [DOI] [PubMed] [Google Scholar]
- 100.Folkman J, Hochberg M. Self-regulation of growth in three dimensions. J Exp Med. 1973;138:745. doi: 10.1084/jem.138.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Popławski NJ, Swat M, Gens JS, Glazier JA. Adhesion between cells, diffusion of growth factors, and elasticity of the AER produce the paddle shape of the chick limb. Physica A. 2007;373:521. doi: 10.1016/j.physa.2006.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]