

Abstract
We report that visual arrestin can regulate retinal release and late photoproduct formation in rhodopsin. Our experiments, which employ a fluorescently labeled arrestin and rhodopsin solubilized in detergent/phospholipid micelles, indicate that arrestin can trap a population of retinal in the binding pocket with an absorbance characteristic of Meta II with the retinal Schiff-base intact. Furthermore, arrestin can convert Metarhodopsin III (formed either by thermal decay or blue-light irradiation) to a Meta II-like absorbing species. Together, our results suggest arrestin may be able to play a more complex role in the rod cell besides simply quenching transducin activity. This possibility may help explain why arrestin deficiency leads to problems like stationary night blindness (Oguchi disease) and retinal degeneration.
Keywords: Arrestin, Rhodopsin, Retinal, Metarhodopsin III, Photo-regeneration
1. Introduction
For over twenty years arrestin has been known as the ultimate quencher of light-induced rhodopsin signaling (Kühn, Hall, & Wilden, 1984; Wilden, Hall, & Kühn, 1986). It does so by binding to the phosphorylated form of light-activated rhodopsin, thus blocking further interaction of rhodopsin with transducin (Burns & Baylor, 2001; Hurley, Spencer, & Niemi, 1998; Ridge, Abdulaev, Sousa, & Palczewski, 2003). In this paper, we present evidence obtained in vitro that suggests interaction of arrestin with rhodopsin can also regulate the photochemistry of rhodopsin, which could have important functions in vivo.
The molecular details of the rhodopsin photo-cycle have been well studied. Brieffy, activation of rhodopsin (λmax ~ 500 nm) begins with light-induced isomerization of the covalently attached chromophore 11-cis retinal to all-trans retinal (Hubbell, Altenbach, Hubbell, & Khorana, 2003; Wald, 1968). This event leads to the formation of two photoproducts, Meta I (λmax ~ 480 nm) and Meta II (λmax ~ 380 nm), which are in equilibrium (Parkes, Gibson, & Liebman, 1999; Schertler, 2005). Meta II is the active signaling form of the receptor that couples to transducin (Burns & Baylor, 2001; Hamm, 2001).
Light-activated rhodopsin can decay by different but parallel pathways. The retinal Schiff-base of Meta II is sensitive to cleavage, and thus Meta II eventually decays to opsin and free retinal (Bownds & Wald, 1965; Farrens & Khorana, 1995). Alternatively, retinal Schiff-base isomerization in Meta I forms the long-lived photoproduct Meta III (λmax ~ 470 nm) (Vogel et al., 2003; Vogel, Siebert, Zhang, Fan, & Sheves, 2004b). Up to half of the rhodopsin in the human retina may be converted to Meta III in bright-light conditions (Lewis, van Kuijk, Carruthers, & Kliger, 1997). Meta III may thus exist for retinal storage purposes (Heck et al., 2003a), although it also retains some signaling ability (Zimmermann, Ritter, Bartl, Hofmann, & Heck, 2004).
Other wavelengths of light can cause further photo-conversions of rhodopsin (Hubbard & Kropf, 1958). For example, blue light (<420 nm) can convert Meta II into other photoproducts, including Meta III (Ritter, Zimmermann, Heck, Hofmann, & Bartl, 2004) and P500 (λmax ~ 500 nm) (Arnis & Hofmann, 1995). P500 appears to be similar to dark-state rhodopsin in its inactivity (Cone, 1967; Huddleston & Williams, 1977), although it might actually bind an all-trans retinal (Bartl, Ritter, & Hofmann, 2001).
In the current study, we address how arrestin can affect the formation and decay of these photoproducts. Understanding these processes is important for understanding the visual photo-cycle, especially since arrestin is likely to encounter these photoproducts in the light-exposed rod cell. We are exploring these questions using new in vitro approaches, which include a fluorescently labeled arrestin mutant, as well as a functional solubilized system in which to monitor arrestin–rhodopsin interactions (Sommer, Smith, & Farrens, 2005; Sommer, Smith, & Farrens, 2006). Here, we summarize this recent work, which shows that arrestin can stabilize Meta II, both by inhibiting retinal release and Meta III formation, and by interacting with Meta III and converting it to a Meta II-like species. We also present new data, which shows that irradiation of arrestin-bound Meta II can result in a P500-like product that no longer binds arrestin.
Finally, based on a review of the literature (see Section 4) as well as our results, we present a hypothesis that arrestin may have a biological role to limit retinal release in the photo-bleached retina, which could help protect the cell from long-term oxidative damage (Sparrow et al., 2003). In addition, we speculate that arrestin may regulate the photo-product Meta III, and this interaction may be necessary for proper dark adaptation (Lamb & Pugh, 2004).
2. Materials and methods
2.1. Materials
Except where noted below, all reagents were obtained from Sigma (St. Louis, MO). Frozen bovine retinas were obtained from Lawson and Lawson, Inc. (Lincoln, NE), and 11-cis retinal was a generous gift from Rosalie Crouch (Medical University of South Carolina and National Eye Institute). Monobromobimane was purchased from Molecular Probes (Eugene, OR), and spectroscopic-grade buffers were from USB Corporation (Cleveland, OH). Acrylamide/bisacrylamine solution (37:5:1) was purchased from Bio-Rad, and Concavalin A Sepharose was obtained from Amersham (Piscataway, NJ). N-dodecyl-β-d-maltopyranoside (DM) was from Anatrace (Maumee, OH), and asolectin was purchased from Fluka (Buchs, Switzerland). Asolectin, an inexpensive mixture of soybean phospholipids, is often used to reconstitute purified rhodopsin (Mansoor, Palczewski, & Farrens, 2006; Niu, Kim, & Khorana, 2002). It consists of roughly equal parts of phosphatidylcholine, phosphatidylethanolamine, and phosphatidylinositol (product information as provided from manufacturer). To prepare asolectin/DM mixed micelles, a stock of 1% asolectin and 1% DM was made by dispersing the powdered asolectin into solution with a syringe and needle. The solution was clarified by centrifugation (100,000g, 20 min) before use.
2.2. Purification of native rhodopsin and recombinant arrestin
Rod outer segment (ROS) membranes containing native rhodopsin, and highly phosphorylated ROS, were prepared from bovine retinas. Phosphorylated rhodopsin (Rho-P) was purified from ROS using ConA and solubilized in DM as described previously (Sommer et al., 2005, 2006), and purified samples had a 280/500 nm absorbance ratio of 1.6. Mutant arrestin constructs W194F and I72C/W194F were created from the bovine visual arrestin cDNA (a gift from V.V. Gurevich) cloned in the pET15b vector (Invitrogen). All constructs were verified by DNA sequencing, arrestin was expressed and purified from Escherichia coli BL21(DE3) cells, and arrestin samples were subsequently labeled with monobromobimane as described (Sommer et al., 2006). Recombinant arrestin I72C/W194F labeled at ~92% efficiency; recombinant arrestin W194F labeled at less than 2% efficiency. No free label contamination was detected in the labeled arrestin samples. The bimane-labeled arrestin mutant I72C/W194F is subsequently referred to as I72B.
2.3. Fluorescence spectroscopy
All steady-state and time-resolved fluorescence measurements were made using a Photon Technologies steady-state spectrophotometer with a single excitation source (DeltaRam) and two emission detectors (T format). Excitation slits were set <0.25 nm to avoid bleaching of rhodopsin, and emission slits were set at 15 nm band pass. For emission scans, the sample was excited at 380 nm, and emission was measured from 400 to 600 nm using 2 nm increments (0.25 s integration per point). For simultaneous monitoring of retinal and arrestin release, the sample was excited for 1 s at 295 nm, followed by 1 s at 380 nm. Emission was detected at 330 and 456 nm, and the shutter was closed for 8 s between measurements. In this way, retinal release was monitored as an increase in opsin tryptophan fluorescence (λex: 295 nm, λem: 330 nm), and arrestin binding and release were monitored as changes in bimane fluorescence (λex: 380 nm, λem: 456 nm). Samples were photoactivated using a 150-W fiber optic light source (>495 nm). Background fluorescence was subtracted from all data, and steady-state scans were smoothed using the program Felix (Sommer et al., 2005, 2006).
2.4. UV–visible absorbance spectroscopy
All UV–visible absorption spectra were recorded with a Shimadzu UV-1601 spectrophotometer (bandwidth of 2 nm) (Sommer et al., 2006). For the photodecay experiments, the absorbance of 1 μM Rho-P (120 μl) was recorded in the dark after base-lining the spectrophotometer with the appropriate buffer. The sample was photoactivated (>495 nm) for 20 s, and spectra were subsequently recorded every 90 s for 120 min. For blue-light experiments, the sample was illuminated by a Machine Vision Strobe light source filtered through a 400 ± 20 nm band pass filter for 20 s. The presence of Schiff-base was ascertained by the addition of 5 μl of 0.8 N H2SO4 to the sample.
2.5. NaBH4 reduction and V8 proteolysis of rhodopsin
The fluorescent n-retinylidene opsin species (λex: 340 nm, λem: 480 nm) results from reduction of the Schiff-base in rhodopsin with NaBH4 (Bownds & Wald, 1965; Farrens & Khorana, 1995). Samples of 3 μM Rho-P, with or without 6 μM arrestin W194F, solubilized in 0.02% DM or 0.02% DM and 0.02% asolectin (20 μl) were photo-activated (>495 nm, 20 s, 20 °C), incubated in the dark for 120 min, and then 5 μl of 1% NaBH4 was added to each sample. After 10 min, 15 μl of 1 M sodium phosphate (pH 7.0) was added, and each sample was split into two 20 μl aliquots. Five microliters of 4.8 μM V8 protease was added to half the samples, and proteolysis occurred for 30 min at 20 °C. To assess the amount of Schiff-base present in Meta II rhodopsin immediately after activation, 1% NaBH4 was added to Rho-P (0.02% DM) in the dark, followed by photo-activation at 4 °C. Samples were subjected to 15% Tris–tricine–SDS–PAGE, and gels were soaked in 30% methanol before visualization. Bands were visualized using excitation from a short wave UV source (Alpha-Innotech FluorChem 5500 gel-doc) and a CCD camera (535 ± 50 nm cut-off filter; 10 min exposure). AlphaEase FC software was used to quantify the fluorescence of the bands.
2.6. Arrestin functional “pull-down” assay
Arrestin’s ability to bind Rho-P in native membranes (ROS-P) was performed as described previously with some modifications (Sommer et al., 2005). Briefly, a single sample of sonicated ROS-P (15 μM) was mixed with the fluorescently labeled arrestin mutant I72B (3 μM) in the dark (120 μl, 20 mM Hepes, 150 mM NaCl, pH 7.4, 20 °C). 20 μl aliquots were removed and immediately placed on ice in the dark, after green light illumination (>495 nm, 60 s), after subsequent blue-light illumination (<420 nm, 60 s), after subsequent green light illumination (60 s), and after the addition of 10 mM hydroxylamine. The sample was illuminated from the top to prevent the plastic tube from filtering the light. The samples were then diluted 10-fold with ice-cold buffer, centrifuged at 100,000g for 10 min (4 °C), and the pellets were solubilized in loading buffer and subjected to SDS–PAGE (10%). The gel was visualized as described above using a gel-doc apparatus, and bands were quantified using the AlphaEase FC software.
3. Results
3.1. Fluorescent arrestin binding to rhodopsin in mixed micelles
Arrestin binding and release from light-activated phosphorylated rhodopsin (Rho*-P) in native ROS membranes can be monitored with fluorescently labeled arrestin mutants (Sommer et al., 2005). Specifically, bimane-labeled arrestin I72C (I72B) exhibits a blue-shift and increase in fluorescence upon binding Rho*-P. The blue-shift indicates the probe is relocated into a more hydrophobic domain (Mansoor, McHaourab, & Farrens, 1999). Thus, this fluorescence change is most likely due to a burying of the bimane probe in a protein-protein or a protein-lipid interface. Fig. 1 illustrates the location of the fluorescent probe on arrestin and the likely binding interfaces between rhodopsin and arrestin.
Fig. 1.
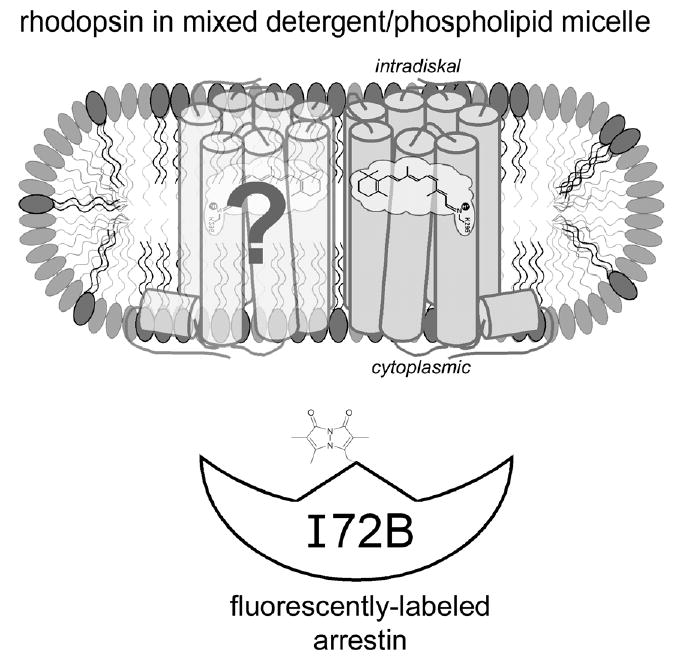
Cartoon representation of the experimental system used in this study. Possible arrangement of dark-state rhodopsin (gray) and a hypothetical dimer partner (light gray) (Liang et al., 2003) in a mixed detergent/phospholipid micelle. The intradiskal and cytoplasmic domains of detergent-solubilized rhodopsin are presumed to be exposed to solvent based on the dimensions of the DM micelle (Dupuy et al., 1997) and the facts that detergent-solubilized rhodopsin can be bound by both N- and C-terminal antibodies and ConA. Arrestin I72B (white) is a mutant labeled with the fluorescent probe monobromobimane at the approximate location shown (note—the fluorescent probe structure is not to scale with the protein model). The fluorescence of I72B increases upon binding to rhodopsin, and this fact was used to assay the interaction of these two proteins. The possible dimerization of rhodopsin and/or arrestin was not addressed in this study, and is merely illustrated here for speculative interest (see text for more details).
Using this approach, we recently made an unexpected discovery. We found that although arrestin binding to Rho*-P is inhibited when Rho-P is purified and solubilized in n-dodecyl-β-d-maltopyranoside (DM), arrestin binding can be restored with the addition of exogenous phospholipids in the form of asolectin (Fig. 2A) (Sommer et al., 2006). Furthermore, we found that acidic phospholipids (phosphatidylserine, phosphatidylinositol, and phosphatidic acid) specifically enable arrestin binding (Sommer et al., 2006). The high phosphatidylinositol content (~30%) of asolectin probably explains why it serves as a good enhancer of arrestin binding to DM-purified Rho-P.
Fig. 2.
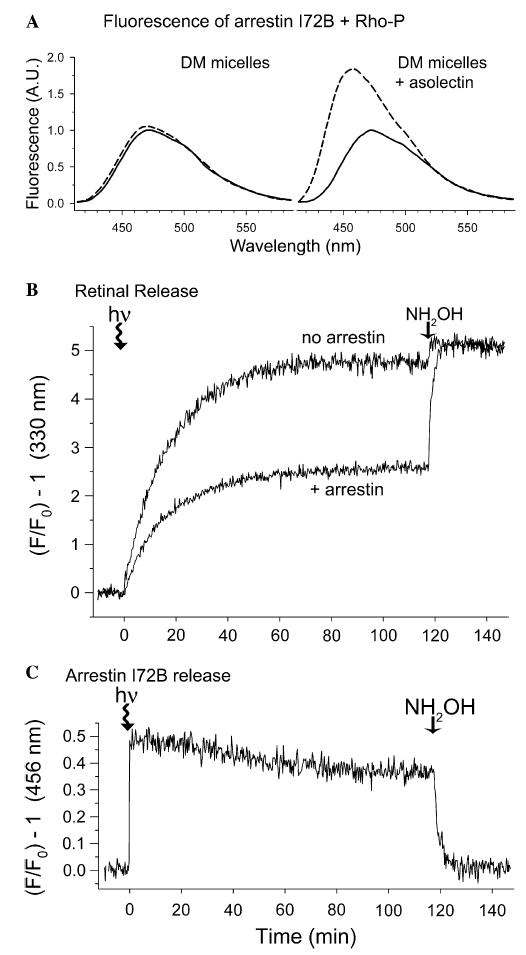
Arrestin can trap ~half of the retinal population in opsin. (A) Fluorescence of arrestin I72B (1 μM) in the presence of a twofold excess of purified Rho-P in DM-micelles (left panel) or DM/asolectin mixed micelles (right panel). Smoothed spectra show before (solid trace) and after (dashed trace) light-activation (λex = 380 nm, 20 °C). Data taken from: (Sommer et al., 2006). (B) Retinal release from Rho-P in mixed micelles measured as an increase in opsin tryptophan fluorescence (λex = 295 nm, λem = 330 nm). Photoactivation occurred at t = 0 min, and 10 mM hydroxylamine was added at t = 120 min. Samples contained 1 μM Rho-P (0.02% DM and 0.02% asolectin) with or without a twofold excess of arrestin I72B (20 mM Hepes, 150 mM NaCl, pH 7.4, 20 °C). (C) Arrestin binding and release measured during the same experiment as described in (B) (λex = 380 nm, λem = 456 nm). In parts (A–C) the data are plotted to show the relative increase in fluorescence, a value independent of instrumentation.
Importantly, the ability to monitor arrestin binding to Rho-P solubilized in mixed DM/asolectin micelles enables a soluble system in which to study the dynamics of the interaction of arrestin with the major photoproducts of rhodopsin and how this influences the release of the retinal chromophore from the binding pocket.1 Our results are described below.
3.2. Arrestin traps a population of retinal in mixed micelles
We measured retinal release from Rho*-P solubilized in mixed DM/asolectin micelles by monitoring the increase in native opsin tryptophan fluorescence that occurs when retinal is released from the binding pocket (Farrens & Khorana, 1995) (Fig. 2B). The data show that retinal was released from Rho*-P in mixed micelles with a t1/2 = 12.3 ± 0.8 min, which is ~1.6 times slower than in native ROS membranes (Sommer et al., 2006). The addition of hydroxylamine, a compound that cleaves the Schiff-base, resulted in a small (~5%) increase in fluorescence.
Strikingly, in the presence of a twofold excess of arrestin, the tryptophan fluorescence of Rho*-P plateaued at ~half that seen in the absence of arrestin, implying that arrestin traps ~half of the retinal population in the binding pocket (Heck et al., 2003a). The addition of hydroxylamine caused the fluorescence to increase to the same level as seen in the absence of arrestin. Interestingly, arrestin appeared to only affect the plateau of tryptophan fluorescence and not the rate at which the fluorescence increased (t1/2 = 13.2 ± 0.7 min).
The fluorescent arrestin I72B was used to monitor arrestin binding and release during the same experiment described above (Fig. 2C). Arrestin binding to Rho*-P in mixed micelles resulted in a ~50% increase in bimane fluorescence. The rate of arrestin release was significantly slower (t1/2 ~ 25 min) compared to retinal release, and the bimane fluorescence plateaued at ~70% of the starting state intensity.2 Hydroxylamine returned the fluorescence to the starting-state level.
Note that, at this time, we are not able to correlate the relative fluorescence change of I72B to the concentration of Rho*-P-bound arrestin, so we cannot quantify the ratio of bound arrestin to trapped retinal. Thus, we can only conclude that while some arrestin remained bound in the complex, ~ half of the retinal population was trapped.
3.3. Arrestin traps retinal as a Schiff-base adduct
We determined that the trapped retinal observed in Fig. 2B represents a Schiff-base linked form, by reducing the retinal Schiff-base with NaBH4, which results in a fluorescent n-retinylidene species (Bownds & Wald, 1965; Farrens & Khorana, 1995). Subsequent SDS–PAGE enabled the separation the n-retinylidene opsin from possible retinal–phospholipid adducts (Sommer et al., 2006). We also digested the reduced opsin with V8 protease to reveal the relative location of the covalently attached retinal on the protein. V8 cleaves opsin in the third cytoplasmic loop, resulting in two fragments. The larger fragment F1 (~27 kDa) contains transmembrane helices 1–5, and the smaller fragment F2 (~15 kDa) consists of transmembrane helices 6 and 7 (Farrens, Altenbach, Yang, Hubbell, & Khorana, 1996). Fragment F2 also contains Lys296, to which retinal is attached in dark-state and Meta II rhodopsin (Bownds, 1967).
When Rho-P in DM-micelles was reduced immediately after photo-activation (Meta II), retinal is clearly attached to F2 (Fig. 3B, lane 6). Two hours after photo-activation, ~25% of the original retinal population was attached to opsin (Fig. 3A, lane 7). The V8 proteolysis assay revealed some of this retinal is attached to both F1 and F2 of Rho-P (Fig. 3B, lane 7). This residual retinal may represent Meta III as well as retinal which attached to peripheral lysines on Rho*-P after release from the binding pocket (Fishkin, Jang, Itagaki, Sparrow, & Nakanishi, 2003). Note that the fluorescence of the V8 protease implies that some retinal may attach to this enzyme as well. In the presence of arrestin (Fig. 3B, lane 8), the amount of retinal attached to opsin in DM micelles was not significantly changed.
Fig. 3.
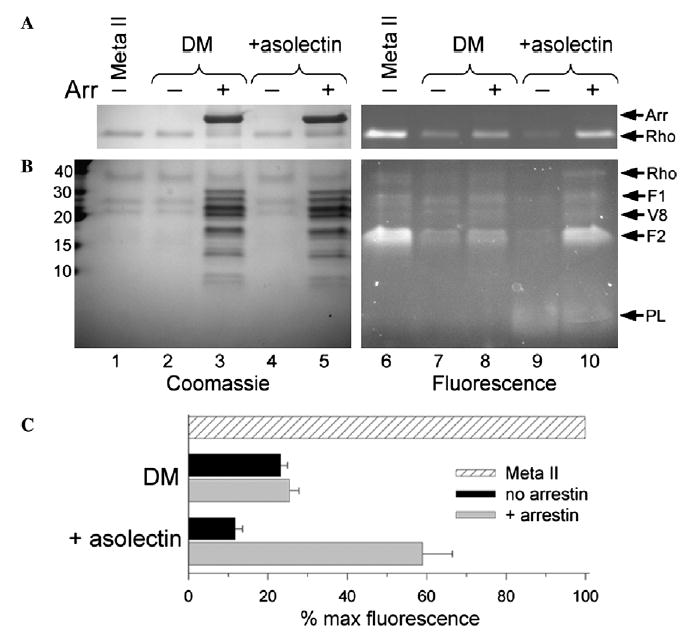
Arrestin traps retinal as a Schiff-base adduct on F2 of Rho*-P. (A) Reducing the retinal Schiff-base with NaBH4 produces a fluorescent Rho*-P that indicates the amount of retinal covalently attached to opsin. Briefly, Rho-P solubilized in 0.02% DM or 0.02% DM and 0.02% asolectin, without or with a twofold excess of arrestin, was photoactivated, allowed to decay at 20 °C in the dark, reduced after 120 min, and then subjected to SDS–PAGE. The total amount of Schiff-base linked retinal was assessed by the reduction of Meta II Rho immediately after bleach (lanes 1 and 6). (B) The same samples as described in (A) were subjected to V8 proteolysis. Molecular weight markers (kDa) are indicated on the left. In both (A) and (B), the Coomassiestain (left, lanes 1–5) and fluorescence (right, lanes 6–10) of each gel is shown, and arrows on the right identify the bands: arrestin (Arr), rhodopsin (Rho), Rho fragments resulting from V8 proteolysis (F1 and F2), the V8 protease (V8), and asolectin phospholipids (PL). (C) Plot of the average quantified fluorescence from four independent experiments as shown in (A). The fluorescence is expressed as a percent of the total “Meta II” fluorescence, and the error bars represent the standard error.
In DM/asolectin mixed micelles, only ~10% of retinal remained attached to opsin after photodecay (Fig. 3A, lane 9). Why is this value lower than in DM alone? A likely explanation is that the phosphatidylethanolamine in asolectin competes with lysine residues for retinal (Plack & Pritchard, 1969; Sparrow et al., 2003), and this conclusion is supported by the presence of a low molecular-weight fluorescent species (Fig. 3B, lanes 9 and 10). Significantly, arrestin caused ~60% of the retinal to remain attached to opsin, and most of this retinal (~80%) was attached to F2 (Fig. 3B, lane 10). Besides illustrating the phospholipid-dependence of retinal trapping by arrestin, these results confirm that arrestin acts to trap ~ half the retinal in opsin, as suggested from the plateau in rhodopsin tryptophan fluorescence (Fig. 2B). In addition, it appears that arrestin traps retinal on F2 of rhodopsin, possibly at Lys296.
3.4. Arrestin blocks Meta III formation and traps spectral Meta II
The photo-intermediates of Rho-P in DM/asolectin micelles were assayed using UV–visible absorption spectroscopy (Fig. 4A). Dark-state Rho-P exhibited a characteristic absorption maximum at 500 nm that shifted to 380 nm after photo-activation. Over time, there was a loss of absorbance at 380 nm and an increase in absorbance between 450 and 480 nm. This event can be explained as the conversion of Meta II (380 nm) to Meta III (470 nm) and the release of retinal from the binding pocket to form adducts with phospholipid head-groups (440–450 nm).
Fig. 4.
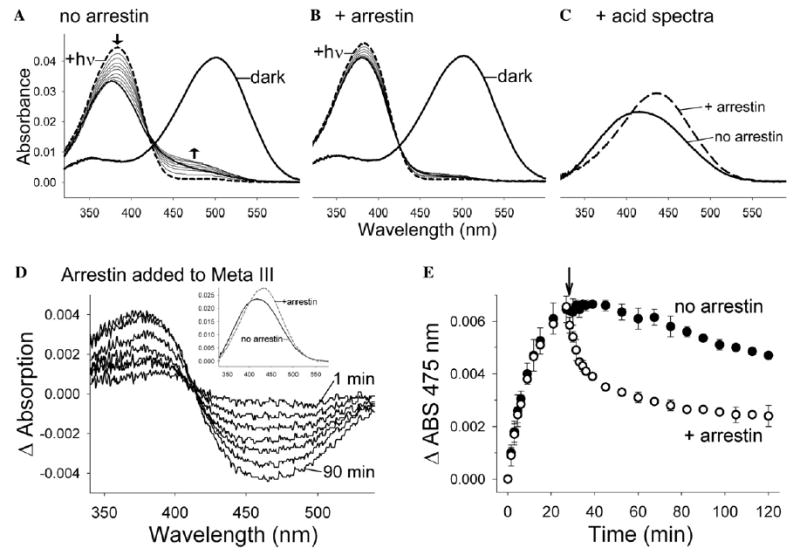
Arrestin stabilizes Meta II and converts Meta III to spectral Meta II. (A) The absorbance of Rho-P in DM/asolectin mixed micelles was observed in the dark (thick solid spectrum) and after photoactivation (dashed spectrum). Spectra were subsequently recorded every 3 min (thin solid spectra) for 120 min (the last spectrum is thick). The arrows indicate the loss of absorbance at 380 nm and the increase in absorbance at 440–480 nm over time. (B) The same experiment as described in (A) in the presence of a twofold excess of arrestin. (C) The amount of retinal Schiff-base remaining at the end of the experiments described in (A) and (B) was assessed by adding H2SO4 (protonated Schiff-bases absorb at 440 nm). (D) Difference spectra representing the conversion of ~470 nm absorbance to ~380 nm absorbance at 1, 2.5, 4, 8.5, 17.5, 50 and 90 min after the addition of arrestin (the first and last spectra are labeled). Arrestin was added to photo-decayed Rho*-P 27 min after photoactivation, and absorbance spectra were subsequently recorded. Difference spectra were calculated by subtracting a base line spectrum derived from a control experiment in which an equal volume of buffer was added at t = 27 min after photo-activation. (Inset) The converted species (380 nm) is most likely Meta II, since acidification at 90 min after the addition of arrestin yields more of the 440 nm absorbance (indicative of retinal Schiff-base) than the control where no arrestin was added. (E) Arrestin speeds the depletion of the 475 nm species over time. The arrow marks the time at which buffer (closed circles) or arrestin (open circles) was added. The data points represent the average from three independent experiments, and the error bars represent the standard error. In each experiment, 1 μM Rho-P in 20 mM Hepes, 150 mM NaCl, pH 7.4, 0.02% DM, 0.02% asolectin was used (20 °C), and in part (D) arrestin was added to a final concentration of 1.5 μM. The data shown in parts (D and E) was reported previously (Sommer et al., 2006).
However, when an excess of arrestin was present, the photodecay absorption spectra of Rho-P were dramatically different (Fig. 4B). There was a significant inhibition of the absorbance loss at 380 nm, and the increase of absorbance between 450 and 480 nm was inhibited. These data suggest that arrestin stabilizes rhodopsin in a Meta II-like state and inhibits retinal release and Meta III formation.
The presence of Schiff-base was assessed by acidification of the samples after photodecay in the experiments described above (Fig. 4C). Protonated Schiff-bases absorb at 440 nm and are indicative of retinal attached to opsin (as in the case of Meta II and Meta III) or linked to phospholipid. In the absence of arrestin, addition of acid to photo-decayed Rho*-P resulted in a broadened absorbance (λmax ~ 416 nm) that is typical for retinal adducts with asolectin phospholipids. In the presence of arrestin, acidification resulted in a 440 nm absorbance peak that verifies the NaBH4-reduction data (Fig. 3): arrestin traps retinal in a Schiff-base linked form.
3.5. Arrestin converts thermal Meta III to spectral Meta II
The above results clearly show that arrestin can prevent accumulation of Meta III. We next tested whether Meta III could interact with arrestin directly, as it can with transducin (Zimmermann et al., 2004). 3 The ability to measure this interaction in detergent allows clear observation of rhodopsin’s spectral intermediates free of scattering artifacts. With membranes, these experiments are complicated by the fact that arrestin binding to vesicles results in a significant increase in scattering (Schleicher, Kühn, & Hofmann, 1989).
Adding arrestin to Rho*-P 27 min after photoactivation (20 °C) resulted in a clear decrease at 470 nm (Meta III) and increase at 380 nm (Fig. 4D). This 380 nm species is characterized by an intact Schiff-base, and is thus likely Meta II (Fig. 4D, inset). Although some decay of Meta III in the absence of arrestin was observed, arrestin clearly speeds its depletion (Fig. 4E). Furthermore, in the absence of arrestin, no significant increase in absorbance at 380 nm was observed as Meta III decays, because Meta II was not stabilized (data not shown). Note that in the presence of arrestin, the absorbance at 475 nm did not return to the starting state level, probably because retinal-phospholipid adducts absorbing in that region were formed before the addition of arrestin.
3.6. Arrestin interacts with the blue-light photoproduct P470 but not with photoproduct P500
We next explored arrestin’s interaction with the photoproducts that result from blue-light irradiation of Meta II, which include species that absorb at ~470 and ~500 nm (called P470 and P500, respectively) (Arnis & Hofmann, 1995; Ritter et al., 2004). Although P470 has been shown to be identical to Meta III (Ritter et al., 2004), the identity of P500 remains unresolved. Long-believed to be photo-regenerated rhodopsin (Arnis & Hofmann, 1995), P500 may in fact bind all-trans retinal (Bartl et al., 2001). In addition, the relative proportions of P470 and P500 that form after irradiation of Meta II depend on the pH used and may differ between detergent- and membrane-bound rhodopsin (Arnis & Hofmann, 1995; Bartl et al., 2001; Ritter et al., 2004). It is important to note that in the preliminary experiments described below, we observed species that absorb at ~470 and ~500 nm, but we have not formally shown that these are the same P470 and P500 described above. In addition, we cannot rule out the possibility that P500 in our experiments is photo-regenerated dark-state rhodopsin.
The properties of blue-light photoproducts in mixed micelles are illustrated in Fig. 5A and B. When Meta II (Fig. 5A, curve ii) is illuminated with blue light (<420 nm), two absorbance peaks were formed (Fig. 5A, curve iii): unconverted Meta II (λmax ~ 380 nm) and the “P-products” (λmax ~ 480 nm). When hydroxylamine was added to these photoproducts (Fig. 5B, curve iv), absorbance peaks corresponding to retinaloxime (λmax ~ 365 nm) and P500 (λmax ~ 500 nm) were observed. Thus, while P470 is sensitive to hydroxylamine, P500 is thermally stable and resistant to hydroxylamine. When P500 was illuminated with green light in the presence of hydroxylamine, retinaloxime resulted (Fig. 5B, curve v), suggesting that green light converts P500 to a form that is hydroxylamine sensitive, like Meta II.
Fig. 5.
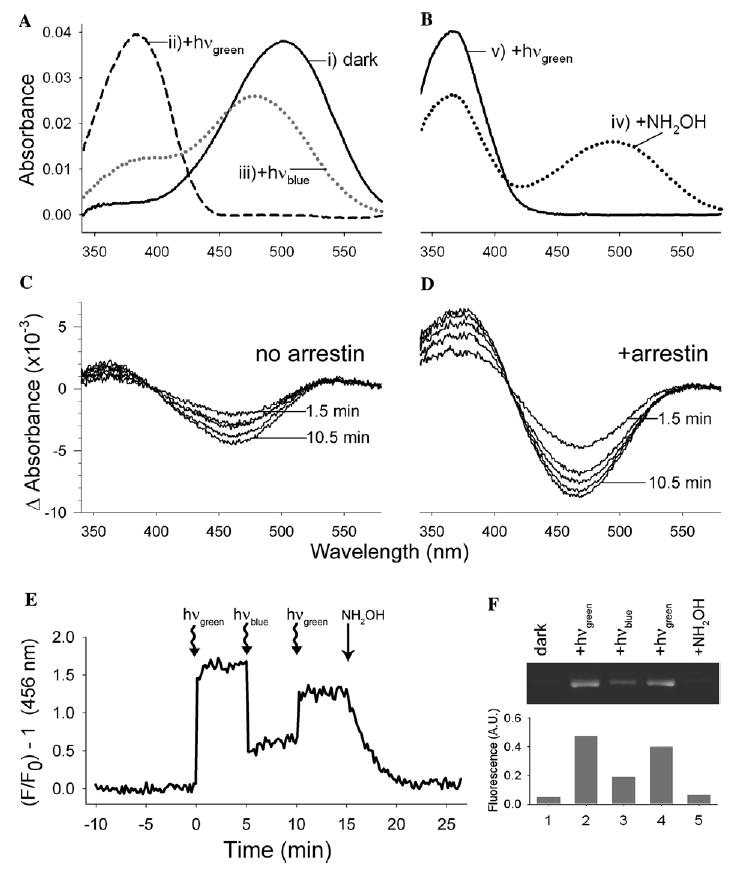
Arrestin interacts with the blue-light photoproduct P470 but not P500. (A) The absorbance spectra of Rho-P were recorded (i) in the dark state, (ii) after illumination with green light (>495 nm, 20 s), and (iii) after subsequent illumination with blue light (<420 nm, 20 s), which results in a mixtures of the products P470 (Meta III) and P500. (B) The same sample as described in (A), (iv) after the addition of 10 mM hydroxylamine, and (v) after subsequent illumination with green light (20 s). (C and D) Rho-P was illuminated with green light followed by blue light, as described in (A), and spectra were subsequently recorded at 1.5, 3, 4.5, 7.5, and 10.5 min after the addition of 2 μM arrestin (D) or and equal volume of buffer (C). Difference spectra were calculated by subtracting the first spectrum after blue-light irradiation from all subsequent spectra after the addition of arrestin or buffer. In each experiment, 1 μM Rho-P in 20 mM Hepes, 150 mM NaCl, pH 7.4, 0.02% DM, 0.02% asolectin was used (20 °C). (E) Fluorescence of arrestin I72B (1 μM) in the presence of a twofold excess of Rho-P (0.02% DM, 0.02% asolectin). The sample was illuminated at the indicated times with green or blue light (20 s), and 15 min after the initial illumination, 10 mM hydroxylamine was added. The sample was excited at 380 nm (20 °C). (F) The ability of ROS-P to “pull-down” arrestin in the dark (lane 1), after green-light illumination (lane 2), after subsequent blue-light illumination (lane 3), after subsequent green-light illumination (lane 4), and after the addition of 10 mM hydroxylamine (lane 5). The upper panel shows the fluorescence of the pulled-down arrestin resolved by SDS–PAGE, and in the lower panel, the fluorescence of the bands is plotted as a percent of the total arrestin present.
The population of “P-products” formed from the blue-light irradiation of Meta II showed a gradual loss of 470 nm absorbance and gain of 380 nm absorbance over time (Fig. 5C). We assume this is due to thermal decay of P470 to Meta II and/or opsin and free retinal. Since P470 is equivalent to Meta III (Ritter et al., 2004), we were curious if arrestin would also interact with P470. Thus, we added arrestin to a sample of blue-light irradiated Meta II and found that arrestin speeds the conversion of the ~470 nm species to a Meta II-like species (λmax ~ 380 nm) (Fig. 5D). Furthermore, the relative intensity of the 380 nm peak two hours after illumination was much greater in the presence of arrestin. Since Meta II is more absorptive than free retinal, this result suggests that arrestin can interact with P470 and convert it to Meta II, which it stabilizes. In the absence of arrestin, P470 appears to decay ultimately to opsin and free retinal.
Interestingly, the rates at which arrestin converts thermal Meta III (Fig. 4E, t1/2 ~ 10 min) and P470 (Fig. 5D, t1/2< 1.5 min) are significantly different, even though similar amounts of arrestin were used in these experiments (1.5 and 2 μM, respectively). We are unsure of the cause of this apparent discrepancy, but it is worth noting that the rhodopsin photoproduct population differed in these experiments. When arrestin was added to photo-decayed Rho*-P (Fig. 4E), a significant amount of Meta II was present (~25% of original population), which would have competed for arrestin binding to Meta III. This complicated situation may explain why the loss of Meta III seen in Fig. 4E is not described by a simple single-exponential rate (data not shown). In contrast, the blue light irradiation used in Fig. 5D resulted in a greatly depleted population of Meta II, which would have made much more arrestin available for interaction with P470.
Our data also suggest that arrestin does not interact with P500, since we find that a hydroxylamine-insensitive peak (λmax ~ 500 nm) remained, even after arrestin was added to blue-light irradiated Meta II (data not shown).4 We explored this possibility further by monitoring arrestin I72B binding after blue-light illumination of Meta II (Fig. 5E). Note that blue-light irradiation of arrestin-bound Meta II caused a dramatic drop in the fluorescence of arrestin, suggesting that blue light releases a significant fraction of arrestin. We could restore arrestin binding with subsequent green-light illumination (note the increase in fluorescence), and hydroxylamine-catalyzed decay of Meta II returned the fluorescence to the dark-state intensity.
Biochemical “pull-down” analysis using membrane-bound Rho*-P confirmed these results (Fig. 5F). Interestingly, the fluorescence profile in Fig. 5F closely mirrors that in Fig. 5E. Together, these results suggest arrestin cannot bind P500 since blue-light irradiation of arrestin-bound Meta II causes dissociation of arrestin. However, because arrestin can bind P470, some arrestin remains bound after blue-light irradiation. Finally, green light can convert P500 to a form which arrestin can bind.
4. Discussion
4.1. Overview of paper
We investigated arrestin binding to Rho*-P in mixed detergent (DM)/phospholipid micelles to assess how arrestin can regulate rhodopsin photochemistry, in a system unencumbered by the optical artifacts present in membrane-bound rhodopsin.
Our findings are summarized in Fig. 6. Briefly, we find that arrestin can stabilize ~half the population of retinal in a Schiff-base linked form with a Meta II-like absorbance (Figs. 2–4). In this stabilized form, retinal is likely attached to Lys296, although we cannot exclude the possibility that retinal may have migrated to another nearby site, perhaps near Helix VIII (Schädel et al., 2003).
Fig. 6.

A cartoon proposing how arrestin can interact with the major photoproducts of phosphorylated rhodopsin. Dark state rhodopsin (Rho) is converted by green light (>495 nm) to Meta I (MI), which is in equilibrium with Meta II (MII). The Meta I/Meta III transition is reversible (Chabre & Breton, 1979; Kolesnikov et al., 2003) and is thought to involve an isomerization of the retinal Schiff-base (Vogel et al., 2003; Vogel et al., 2004b). Meta II decays ultimately to opsin and all-trans retinal (atR), and Meta III slowly decays to opsin and atR as well (Zimmermann et al., 2004), although it is not known if this happens directly or through additional intermediates. Blue-light irradiation (<420 nm) of Meta II leads to two photoproducts, P470 (Meta III) and P500 (Arnis & Hofmann, 1995). Green light can convert P500 back to Meta II (Bartl et al., 2001). The conformation of the chromophore and the protonation state of the Schiff-base (blue proton) is illustrated for each photoproduct, including the isomerization around the Schiff base that occurs during Meta III formation (Vogel et al., 2003, 2004a, 2004b; Zimmermann et al., 2004). Although it has been shown that P500 most likely binds an all-trans retinal (Bartl et al., 2001), the exact nature of this photoproduct has not been determined. The selectivity of arrestin for the photoproducts is also illustrated: arrestin does not bind dark state Rho-P, P500, or opsin. Arrestin stabilizes Meta II at the expense of Meta I, yielding “extra Meta II” (Schleicher et al., 1989), and we have shown that arrestin will interact with Meta III/P470 and convert it to Meta II. Arrestin-bound Meta II is stabilized, and its decay to opsin and free retinal is inhibited. In the figure, the filled and empty arrows represent conversions that occur in the absence and presence of arrestin, respectively, and the T-like symbols indicate reactions or interactions which are blocked or inhibited. Note that the protein models are not to scale, and possible binding stoichiometries (does arrestin bind a dimer of rhodopsin?) are not considered. (For interpretation of the references to colors in this figure legend, the reader is referred to the web version of this paper.)
Furthermore, we find that arrestin clearly inhibits Meta III formation (Fig. 4B). Arrestin is known to favor Meta II at the expense of Meta I (Schleicher et al., 1989), and since Meta III evolves from Meta I (Vogel et al., 2004b), these facts may explain why we see Meta III inhibition in the presence of arrestin. However, we also find that arrestin can interact with thermal and blue-light-generated Meta III and convert it to a species with Meta II-like characteristics (Figs. 4 and 5D). The fast conversion of Meta III to Meta II may also explain the ability of arrestin to inhibit Meta III formation.
Finally, we find that arrestin does not interact with the blue-light photoproduct P500, although green light can convert P500 to a form to which arrestin can bind (Fig. 5). To the best of our knowledge, these results provide the first insights into the effect of blue-light on arrestin–rhodopsin interactions and suggest further investigations are warranted.
4.2. Caveats for this study
There are several caveats that must accompany the results reported here. Although the functional soluble system described here enables new insight into how arrestin regulates rhodopsin photochemistry, mixed detergent/lipid micelles obviously differ from the native ROS membrane in several ways. For example, solubilization in detergent is known to favor Meta II, perhaps by lowering the activation energy for the Meta I → Meta II transition (König, Welte, & Hofmann, 1989; Litman, Kalisky, & Ottolenghi, 1981). This may be why we do not observe the significant levels of Meta III observed in membranes (up to 40%) (Heck et al., 2003a). Furthermore, we have not formally shown that the Meta III and blue-light photoproducts we observe are the same as those reported in membranes.5
The presence of detergent may also disrupt possible secondary retinal-binding pockets and could thus affect the hypothesized channeling of retinal through the protein during release (Heck, Schädel, Maretzki, & Hofmann, 2003b; Schädel et al., 2003). The possible linkage between our results and the prior reports on retinal channeling is especially intriguing, considering the differences in retinal and arrestin release in micelles versus membranes. In native ROS membranes, arrestin slows retinal release, and arrestin and retinal release occur at the same time (Hofmann et al., 1992; Sommer et al., 2005). In contrast, only ~half of retinal is released and arrestin releases only very slowly in detergent/lipid micelles (Fig. 2 and (Sommer et al., 2006)). This phenomenon might suggest a coupling of retinal channeling and arrestin release. Interestingly, addition of detergent to rhodopsin-containing membrane extracts from Drosophila has also been reported to decouple arrestin release and the rates of regeneration in the invertebrate visual cycle (Kiselev & Subramaniam, 1996).
Another possible explanation for the “decoupling” of retinal and arrestin release in detergent is related to the phosphorylation status of our rhodopsin samples. Although our phosphorylation procedure yielded an average of ~6 phosphates per rhodopsin (Sommer et al., 2005), and thus makes a large population of nonphosphorylated rhodopsin unlikely, we cannot rule out the possibility that a population of rhodopsin exists that contains only one or two phosphates. Perhaps arrestin requires higher levels of phosphorylation (>2) in order to form a long-lived complex with rhodopsin in mixed micelles. We are currently investigating this possibility, by separating the different species of phosphorylated rhodopsin as described (Adamus, Arendt, Hargrave, Heyduk, & Palczewski, 1993; Aton, Litman, & Jackson, 1984).
Finally, the possible dimerization of rhodopsin might affect the dynamics of arrestin and retinal release (Liang et al., 2003; Mansoor et al., 2006). For example, our data is consistent with a scenario where arrestin(s) binds a dimer of rhodopsin, and retinal is released from only one dimer partner (Sommer et al., 2006). However, we cannot rule out the possibility that, in mixed micelles, rhodopsin may dimerize in a head-to-tail fashion, as observed in the crystal structure (Palczewski et al., 2000). We cannot predict what effect this manner of dimerization might have on arrestin binding.
Despite these unknowns, the preliminary results we present here represent valuable insight into how arrestin regulates retinal release and photoproduct formation from rhodopsin. In the following discussion, we speculate about the possible implications of our results in the visual photo-cycle.
4.3. Possible mechanisms of arrestin translocation
Recent studies have explored the mechanisms underlying the massive light-dependent translocation of arrestin, which appears important for light adaptation in the rod cell (Elias, Sezate, Cao, & McGinnis, 2004; Nair et al., 2004; Peterson et al., 2003; Strissel, Sokolov, Trieu, & Arshavsky, 2006). In the dark-adapted rod cell, arrestin is normally sequestered in the inner segment, where microtubules might serve as a “sink” for arrestin binding (Hanson, Francis, Vishnivetskiy, Klug, & Gurevich, 2006; Nair et al., 2004). Upon exposure to light, arrestin translocates from the inner segment to the outer segment, where it remains until the removal of light (Elias et al., 2004). This translocation has been proposed to be governed by mass action, where arrestin’s affinity for Rho*-P keeps it in the outer segment (Nair et al., 2005). However, other studies suggest that more complex mechanisms, including active transport, might be involved (Mendez, Lem, Simon, & Chen, 2003; Peterson et al., 2005; Strissel et al., 2006; Zhang et al., 2003). Remarkably, there may be enough arrestin in the rod to bind the entire pool of rhodopsin in a completely photo-bleached cell (Strissel et al., 2006). Since little regeneration of rhodopsin probably takes place under constant illumination (Palczewski et al., 1999), the persistence of arrestin in the outer segment suggests that it might be interacting with a long-lived photoproduct of rhodopsin. This possibility is supported by the results we present in this paper.
The fact that arrestin only enters the rod outer segment in bright light, when the rod cell becomes nonfunctional, suggests arrestin may do more than simply act as a classical signal terminator. It is more likely that p44, the short splice variant of arrestin, regulates rhodopsin signaling in the dim-light operational range of the rod cell (Langlois, Chen, Palczewski, Hurley, & Vuong, 1996). Consistent with this role, p44 is present in the outer segment even in the dark (Smith et al., 1994). The difference in dark-state localization between arrestin and p44 might be due to the high affinity of p44 for dark phosphorylated rhodopsin and opsin, which may constitute ~1% of the rhodopsin in the dark-adapted rod cell (Binder, O’Connor, Bownds, & Arshavsky, 1996; Pulvermüller et al., 1997; Schröder, Pulvermüller, & Hofmann, 2002). Although p44 has a similar affinity for dark Rho-P and Rho*-P, experiments using a fluorescently labeled p44 analogous to arrestin I72B suggest that p44 binds dark Rho-P differently than Rho*-P (Sommer, Smith, & Farrens, unpublished results). The localization of p44 to the ROS membrane may be a key determinate in its ability to quench Rho* activity quickly (illustrated in Fig. 7) (Langlois et al., 1996; Schröder et al., 2002). Since p44 is expressed at only ~10% the levels of arrestin, there is sufficient p44 to bind the limited number of Rho* produced by dim light (Palczewski et al., 1994). As described below, we postulate that an unappreciated role for full-length arrestin in the rod cell is a regulator of late photo-products and retinal release, rather than simply a quencher of Meta II signaling.
Fig. 7.
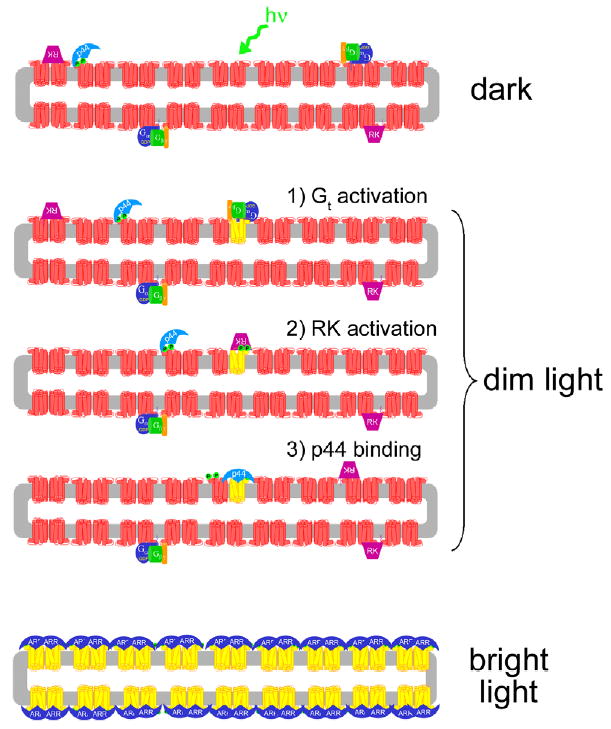
Model of light-dependent attenuation of Rho* by p44 and arrestin. In the dark-adapted rod disk (upper panel), p44 (light blue) is “tethered” to the membrane by its interaction with Rho-P (red), which comprises ~1% of the dark-state rhodopsin population (Binder et al., 1996). Transducin (Gt, blue, green, and orange subunits) and rhodopsin kinase (RK, purple) are also localized to the membrane by lipid membrane anchors (for simplicity, the interactions of RK with recoverin and Gβγ are ignored in this schematic). Upon exposure to dim light (middle panels), a single rhodopsin molecule (red) is converted to Meta II (yellow). (1) Gt binds Meta II and is activated. (2) Meta II is phosphorylated by RK. (3) p44 binds phosphorylated Meta II and blocks further Gt activation. It is not known exactly how p44 “hops” from Rho-P to Rho*-P, but rhodopsin dimerization may facilitate this process (Schröder et al., 2002). Furthermore, the rapid diffusion of rhodopsin monomeric units in the ROS membrane (Cone, 1972; Liebman & Entine, 1974) may allow p44-bound Rho-P to come into contact with Rho*-P, but this possibility is speculative. Note that different modes of p44 binding to Rho-P and Rho*-P are illustrated (see text for more details). Upon exposure to bright light (lowest panel), which results in complete rhodopsin bleaching, arrestin (dark blue) translocation from the inner segment might provide a pool of protein sufficient to bind up every photo-activated receptor (Strissel et al., 2006). In the figure, rhodopsin dimers and a binding stoichiometry of 2 arrestin to 2 Rho*-P are suggested. (For interpretation of the references to colors in this figure legend, the reader is referred to the web version of this paper.)
4.4. Possible physiological implications of arrestin’s interaction with metarhodopsin III
A form of stationary night blindness called Oguchi disease is a recessive genetic disorder caused by mutations in either rhodopsin kinase or arrestin. Patients with Oguchi disease require 5–7 h to dark-adapt, which is much longer than the normal 40 min (Dryja, 2000; Lamb & Pugh, 2004). This phenomenon suggests that arrestin activity is necessary for proper dark adaptation, which involves the removal of all metarhodopsin species from the rod cells and the regeneration of rhodopsin with 11-cis retinal. In bright light conditions, up to half of the rhodopsin in the human retina is estimated to be converted to Meta III (Lewis et al., 1997). Lamb and Pugh have hypothesized that Oguchi disease is caused by a build-up of Meta III, which decays much slower in the absence of arrestin and thus prevents regeneration (Lamb & Pugh, 2004).
Our results support this hypothesis. We find that phosphorylated Meta III can be bound by arrestin and stabilized as a Meta II-like species. In contrast to Meta III, arrestin-bound Meta II may be more accessible to the regeneration machinery, which begins with the reduction of all-trans retinal by retinol dehydrogenase (Hofmann et al., 1992). Furthermore, arrestin may serve to attenuate Meta III signaling by competing with transducin (Zimmermann et al., 2004).
4.5. Arrestin may help limit retinal release to prevent retinal degeneration
Arrestin knock-out mice, as well as some Oguchi disease patients, suffer light-dependent retinal degeneration, which may be due to excessive signaling (Chen, Simon, Matthes, Yasumura, & LaVail, 1999; Dryja, 2000; Hao et al., 2002). However, this phenomenon may not always be caused by excessive signaling, since bright light-induced retinal degeneration in arrestin knock-out mice is independent of transducin (Hao et al., 2002). Furthermore, bright light-induced retinal degeneration is characterized by an induction of the transcription factor AP-1, which suggests that oxidative stress might be the cause (Reme, 2005).
How might bright light trigger this pro-apototic redox-sensitive transcription factor? In contrast to other visual processes, the reduction of retinal to retinol occurs quite slowly (tens of minutes), which might be due to the low enzymatic activity of retinol dehydrogenase (Saari, Garwin, Van Hooser, & Palczewski, 1998), although other mechanisms might also be involved (Chen et al., 2005). Thus, in bright-light conditions, levels of free all-trans retinal might become quite high, which can lead to the formation of di-retinal conjugates with phosphatidylethanolamine (A2E). Light-dependent oxidation of A2E yields dangerous epoxides, which may be a causative agent in both age-related (AMD) and Stargardt’s macular degeneration (Radu, Mata, Bagla, & Travis, 2004; Sparrow et al., 2003; Wenzel, Grimm, Samardzija, & Reme, 2005). Note that animals lacking arrestin are susceptible to retinal damage from bright light, supporting the suggestion that arrestin serves a protective role in the rod cell. Consistent with this hypothesis, arrestin expression appears to be up-regulated when animals are exposed to light (Organisciak et al., 1991), as is our finding that arrestin can limit the release of retinal and trap it in stabilized Meta II.
A possible contradiction to this hypothesis is presented in an in vivo study utilizing arrestin knock-out mice (Palczewski et al., 1999). The authors found no significant difference in the retinoid levels in wild-type and arrestin knock-out mice, suggesting that arrestin does not play a critical role in regulating all-trans retinal release. However, these experiments used dark-adapted mice exposed to a bright flash of light. Under these conditions, arrestin would be mostly sequestered in the inner segment of the rod cell at the time of the flash, and this might explain why they observed no major difference between wild-type and arrestin knock-out mice. Arrestin’s effect might be more pronounced in mice which are exposed to constant illumination. Also, note that even a slight increase in the amount of free retinal caused by a lack of arrestin could, over time, lead to an increase in the levels of A2E and other damaging retinal byproducts.
It is interesting to note that when wild-type mice were exposed to constant illumination, levels of all-trans retinal remained elevated (at ~20% of total retinoids) while the levels of down-stream metabolic products (retinol) plateaued (Palczewski et al., 1999). This result might imply that arrestin stabilized a population of Meta II in the mouse retina. This possibility would be similar to the invertebrate system, where arrestin stabilizes Metarhodopsin and inhibits the release of retinal (Kiselev & Subramaniam, 1996, 1997). It is also interesting to consider that certain rhodopsin mutations, which constitutively activate the receptor and are implicated in Retinitis Pigmentosa, might lead to inappropriately long-lived arrestin–rhodopsin complexes and cytotoxic aggregates (Chuang, Vega, Jun, & Sung, 2004; Li, Franson, Gordon, Berson, & Dryja, 1995; Rim & Oprian, 1995). Finally, it has recently been suggested that retinol dehydrogenase activity may not be the sole factor that contributes to the relatively slow conversion of all-trans retinal to retinol in the rod cell (Chen et al., 2005). It is possible that the stabilization of Meta II by arrestin also contributes (Hofmann et al., 1992).
4.6. Possible roles of the blue-light photoproducts
In vitro studies suggest that rod cell exposure to full-spectrum light might generate P500 as well as Meta III (Arnis & Hofmann, 1995; Bartl et al., 2001). Although Meta III appears to have some activity, since it is bound by both transducin and arrestin, P500 seems to be similar to dark-state rhodopsin in its inactivity. In vivo investigations have shown that blue-light irradiation can deactivate a bleached rod cell (Cone, 1967; Grimm, Reme, Rol, & Williams, 2000; Huddleston & Williams, 1977), presumably by regenerating rhodopsin or isorhodopsin, which binds 9-cis retinal. Thus, the blue-light photoproduct absorbing at ~500 nm might represent another storage form of the protein, and as such it would make sense that it would not interact with arrestin. Since P500 can undergo light-dependent activation to a Meta II-like species, this photoproduct might serve to preserve some light-sensitive pigment in the rod cells after being exposed to high-bleaching conditions. This mechanism would allow the cell to maintain some sensitivity when transitioning from bright to dim light conditions before dark adaptation has occurred.
5. Summary
We described the following results for arrestin–rhodopsin interactions in a detergent-solubilized system: (1) Arrestin can inhibit some retinal release from rhodopsin. The retinal is trapped in a Schiff-base linked form, most likely at Lys296, with an absorbance characteristic of Meta II (380 nm). (2) Arrestin can interact with Meta III, formed either by thermal decay or blue-light irradiation, and convert it to a Meta II-like species. (3) Arrestin does not interact with the blue-light photoproduct P500, but green light can convert P500 to a form which arrestin can bind. Although these results may be unique for arrestin–rhodopsin interactions in a detergent-solubilized system, they do suggest the possibility that another role for arrestin in the rod cell might be to limit retinal release and regulate Meta III, which is consistent with recent physiological evidence supporting this hypothesis.
Acknowledgments
This work was supported in part by National Institutes of Health Grants DA018169 and EY015436 (to D.L.F.), a National Defense Science and Engineering Graduate Fellowship (to M.E.S.), and an N.L. Tartar Research Fellowship (to M.E.S.).
Footnotes
An earlier study also looked at arrestin–rhodopsin interactions using detergent-solubilized rhodopsin (Imamoto, Tamura, Kamikubo, & Kataoka, 2003). However, as far as we are aware, our study is the first to examine the effect of arrestin on retinal release and photoproduct formation using detergent-solubilized rhodopsin.
The plateau reported here is higher than in our previously published data (Sommer et al., 2006), even though both experiments used DM/ asolectin mixed micelles. We do not know the cause of this variation, but it may be due to the difference in background fluorescence due to the heterogenous nature of this phospholipid source. However, the retinal release as ascertained by opsin tryptophan fluorescence consistently plateaus at 50%.
Previously, it was shown that arrestin can convert 470 nm-absorbing “pseudo-photoproducts” of rhodopsin, which consist of nonspecific complexes of membrane-bound phosphorylated opsin and all-trans retinal, into species absorbing at 380 nm (Hofmann, Pulvermüller, Buczylko, Van Hooser, & Palczewski, 1992).
Note that blue-light illuminated arrestin-bound Meta II produced a similar distribution of absorbing species as in the absence of arrestin, suggesting that arrestin cannot inhibit the conversion of Meta II to the P-products (data not shown). In this regard, arrestin may be different than transducin, which can inhibit the formation of P500 (Arnis & Hofmann, 1995; Bartl et al., 2001).
This would require FTIR analysis of the protein as well as isolation and identification of the conformation of the retinal chromophore, as described (Bartl et al., 2001; Heck et al., 2003a; Vogel et al., 2003; Zimmermann et al., 2004).
References
- Adamus G, Arendt A, Hargrave PA, Heyduk T, Palczewski K. The kinetics of multiphosphorylation of rhodopsin. Archives of Biochemistry and Biophysics. 1993;304(2):443–447. doi: 10.1006/abbi.1993.1373. [DOI] [PubMed] [Google Scholar]
- Arnis S, Hofmann KP. Photoregeneration of bovine rhodopsin from its signaling state. Biochemistry. 1995;34(29):9333–9340. doi: 10.1021/bi00029a008. [DOI] [PubMed] [Google Scholar]
- Aton BR, Litman BJ, Jackson ML. Isolation and identification of the phosphorylated species of rhodopsin. Biochemistry. 1984;23(8):1737–1741. doi: 10.1021/bi00303a024. [DOI] [PubMed] [Google Scholar]
- Bartl FJ, Ritter E, Hofmann KP. Signaling states of rhodopsin: absorption of light in active metarhodopsin II generates an all-trans-retinal bound inactive state. Journal of Biological Chemistry. 2001;276(32):30161–30166. doi: 10.1074/jbc.M101506200. [DOI] [PubMed] [Google Scholar]
- Binder BM, O’Connor TM, Bownds MD, Arshavsky VY. Phosphorylation of non-bleached rhodopsin in intact retinas and living frogs. Journal of Biological Chemistry. 1996;271(33):19826–19830. doi: 10.1074/jbc.271.33.19826. [DOI] [PubMed] [Google Scholar]
- Bownds D. Site of attachment of retinal in rhodopsin. Nature. 1967;216(121):1178–1181. doi: 10.1038/2161178a0. [DOI] [PubMed] [Google Scholar]
- Bownds D, Wald G. Reaction of the rhodopsin chromophore with sodium borohydride. Nature. 1965;205:254–257. doi: 10.1038/205254a0. [DOI] [PubMed] [Google Scholar]
- Burns ME, Baylor DA. Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annual Review of Neuroscience. 2001;24:779–805. doi: 10.1146/annurev.neuro.24.1.779. [DOI] [PubMed] [Google Scholar]
- Chabre M, Breton J. The orientation of the chromophore of vertebrate rhodopsin in the “meta” intermediate states and the reversibility of the meta II-meta III transition. Vision Research. 1979;19(9):1005–1018. doi: 10.1016/0042-6989(79)90226-8. [DOI] [PubMed] [Google Scholar]
- Chen C, Tsina E, Cornwall MC, Crouch RK, Vijayaraghavan S, Koutalos Y. Reduction of all-trans retinal to all-trans retinol in the outer segments of frog and mouse rod photoreceptors. Biophysical Journal. 2005;88(3):2278–2287. doi: 10.1529/biophysj.104.054254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Simon MI, Matthes MT, Yasumura D, LaVail MM. Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness) Investigative Ophthalmology and Vision Science. 1999;40(12):2978–2982. [PubMed] [Google Scholar]
- Chuang JZ, Vega C, Jun W, Sung CH. Structural and functional impairment of endocytic pathways by Retinitis pigmentosa mutant rhodopsin–arrestin complexes. Journal of Clinical Investigation. 2004;114(1):131–140. doi: 10.1172/JCI21136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone RA. Early receptor potential: photoreversible charge displacement in rhodopsin. Science. 1967;155(766):1128–1131. doi: 10.1126/science.155.3766.1128. [DOI] [PubMed] [Google Scholar]
- Cone RA. Rotational diffusion of rhodopsin in the visual receptor membrane. Nature: New Biology. 1972;236(63):39–43. doi: 10.1038/newbio236039a0. [DOI] [PubMed] [Google Scholar]
- Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus, and other forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. American Journal of Ophthalmology. 2000;130(5):547–563. doi: 10.1016/s0002-9394(00)00737-6. [DOI] [PubMed] [Google Scholar]
- Dupuy C, Auvray X, Petipas C. Anomeric effects on the structure of micelles of alkyl maltosides in water. Langmuir. 1997;13(15):3965. [Google Scholar]
- Elias RV, Sezate SS, Cao W, McGinnis JF. Temporal kinetics of the light/dark translocation and compartmentation of arrestin and alpha-transducin in mouse photoreceptor cells. Molecular Vision. 2004;10:672–681. [PubMed] [Google Scholar]
- Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274(5288):768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- Farrens DL, Khorana HG. Structure and function in rhodopsin. Measurement of the rate of metarhodopsin II decay by fluorescence spectroscopy. Journal of Biological Chemistry. 1995;270(10):5073–5076. doi: 10.1074/jbc.270.10.5073. [DOI] [PubMed] [Google Scholar]
- Fishkin N, Jang YP, Itagaki Y, Sparrow JR, Nakanishi K. A2-rhodopsin: a new fluorophore isolated from photoreceptor outer segments. Organic and Biomolecular Chemistry. 2003;1(7):1101–1105. doi: 10.1039/b212213h. [DOI] [PubMed] [Google Scholar]
- Grimm C, Reme CE, Rol PO, Williams TP. Blue light’s effects on rhodopsin: photoreversal of bleaching in living rat eyes. Investigative Ophthalmology and Vision Science. 2000;41(12):3984–3990. [PubMed] [Google Scholar]
- Hamm HE. How activated receptors couple to G proteins. Proceeding of the National Academic Sciences of the United States of America. 2001;98(9):4819–4821. doi: 10.1073/pnas.011099798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Klug CS, Gurevich VV. Visual arrestin binding to microtubules involves a distinct conformational change. The Journal of Biological Chemistry. 2006 doi: 10.1074/jbc.M510738200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Wenzel A, Obin MS, Chen CK, Brill E, Krasnoperova NV, et al. Evidence for two apoptotic pathways in light-induced retinal degeneration. Nature Genetics. 2002;32(2):254–260. doi: 10.1038/ng984. [DOI] [PubMed] [Google Scholar]
- Heck M, Schädel SA, Maretzki D, Bartl FJ, Ritter E, Palczewski K, et al. Signaling states of rhodopsin. Formation of the storage form, metarhodopsin III, from active metarhodopsin II. Journal of Biological Chemistry. 2003a;278(5):3162–3169. doi: 10.1074/jbc.M209675200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck M, Schädel SA, Maretzki D, Hofmann KP. Secondary binding sites of retinoids in opsin: characterization and role in regeneration. Vision Research. 2003b;43(28):3003–3010. doi: 10.1016/j.visres.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Hofmann KP, Pulvermüller A, Buczylko J, Van Hooser P, Palczewski K. The role of arrestin and retinoids in the regeneration pathway of rhodopsin. Journal of Biological Chemistry. 1992;267(22):15701–15706. [PubMed] [Google Scholar]
- Hubbard R, Kropf A. The action of light of rhodopsin. Proceeding of the National Academic Sciences of the United States of America. 1958;44(2):130–139. doi: 10.1073/pnas.44.2.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell WL, Altenbach C, Hubbell CM, Khorana HG. Rhodopsin structure, dynamics, and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Advances in Protein Chemisty. 2003;63:243–290. doi: 10.1016/s0065-3233(03)63010-x. [DOI] [PubMed] [Google Scholar]
- Huddleston SK, Williams TP. Physiological activity of isorhodopsin in rat rods. Vision Research. 1977;17(6):711–714. doi: 10.1016/s0042-6989(77)80007-2. [DOI] [PubMed] [Google Scholar]
- Hurley JB, Spencer M, Niemi GA. Rhodopsin phosphorylation and its role in photoreceptor function. Vision Research. 1998;38(10):1341–1352. doi: 10.1016/s0042-6989(97)00459-8. [DOI] [PubMed] [Google Scholar]
- Imamoto Y, Tamura C, Kamikubo H, Kataoka M. Concentration-dependent tetramerization of bovine visual arrestin. Biophysical Journal. 2003;85(2):1186–1195. doi: 10.1016/S0006-3495(03)74554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselev A, Subramaniam S. Modulation of arrestin release in the light-driven regeneration of Rh1 Drosophila rhodopsin. Biochemistry. 1996;35(6):1848–1855. doi: 10.1021/bi951399k. [DOI] [PubMed] [Google Scholar]
- Kiselev A, Subramaniam S. Studies of Rh1 metarhodopsin stabilization in wild-type Drosophila and in mutants lacking one or both arrestins. Biochemistry. 1997;36(8):2188–2196. doi: 10.1021/bi9621268. [DOI] [PubMed] [Google Scholar]
- Kolesnikov AV, Golobokova EY, Govardovskii VI. The identity of metarhodopsin III. Visual Neuroscience. 2003;20(3):249–265. doi: 10.1017/s0952523803203047. [DOI] [PubMed] [Google Scholar]
- König B, Welte W, Hofmann KP. Photoactivation of rhodopsin and interaction with transducin in detergent micelles. Effect of ‘doping’ with steroid molecules. FEBS Letter. 1989;257(1):163–166. doi: 10.1016/0014-5793(89)81811-3. [DOI] [PubMed] [Google Scholar]
- Kühn H, Hall SW, Wilden U. Light-induced binding of 48-kDa protein to photoreceptor membranes is highly enhanced by phosphorylation of rhodopsin. FEBS Letter. 1984;176(2):473–478. doi: 10.1016/0014-5793(84)81221-1. [DOI] [PubMed] [Google Scholar]
- Lamb TD, Pugh EN., Jr Dark adaptation and the retinoid cycle of vision. Progress in Retinal and Eye Research. 2004;23(3):307–380. doi: 10.1016/j.preteyeres.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Langlois G, Chen CK, Palczewski K, Hurley JB, Vuong TM. Responses of the phototransduction cascade to dim light. Proceeding of the National Academic Sciences of the United States of America. 1996;93(10):4677–4682. doi: 10.1073/pnas.93.10.4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JW, van Kuijk FJ, Carruthers JA, Kliger DS. Metarhodopsin III formation and decay kinetics: comparison of bovine and human rhodopsin. Vision Research. 1997;37(1):1–8. doi: 10.1016/s0042-6989(96)00138-1. [DOI] [PubMed] [Google Scholar]
- Li T, Franson WK, Gordon JW, Berson EL, Dryja TP. Constitutive activation of phototransduction by K296E opsin is not a cause of photoreceptor degeneration. Proceeding of the National Academic Sciences of the United States of America. 1995;92(8):3551–3555. doi: 10.1073/pnas.92.8.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Fotiadis D, Filipek S, Saperstein DA, Palczewski K, Engel A. Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. Journal of Biological Chemistry. 2003;278(24):21655–21662. doi: 10.1074/jbc.M302536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebman PA, Entine G. Lateral diffusion of visual pigment in photorecptor disk membranes. Science. 1974;185(149):457–459. doi: 10.1126/science.185.4149.457. [DOI] [PubMed] [Google Scholar]
- Litman BJ, Kalisky O, Ottolenghi M. Rhodopsin-phospholipid interactions: dependence of rate of the meta I to meta II transition on the level of associated disk phospholipid. Biochemistry. 1981;20(3):631–634. doi: 10.1021/bi00506a028. [DOI] [PubMed] [Google Scholar]
- Mansoor SE, McHaourab HS, Farrens DL. Determination of protein secondary structure and solvent accessibility using site-directed fluorescence labeling. Studies of T4 lysozyme using the fluorescent probe monobromobimane. Biochemistry. 1999;38(49):16383–16393. doi: 10.1021/bi991331v. [DOI] [PubMed] [Google Scholar]
- Mansoor SE, Palczewski K, Farrens DL. Rhodopsin self-associates in asolectin liposomes. Proceeding of the National Academic Sciences of the United States of America. 2006;103(9):3060–3065. doi: 10.1073/pnas.0511010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez A, Lem J, Simon M, Chen J. Light-dependent translocation of arrestin in the absence of rhodopsin phosphorylation and transducin signaling. Journal of Neuroscience. 2003;23(8):3124–3129. doi: 10.1523/JNEUROSCI.23-08-03124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Kennedy MJ, Hurley JB, Gurevich VV, Slepak VZ. Direct binding of visual arrestin to microtubules determines the differential subcellular localization of its splice variants in rod photoreceptors. Journal of Biological Chemistry. 2004;279(39):41240–41248. doi: 10.1074/jbc.M406768200. [DOI] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, et al. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein–protein interactions. Neuron. 2005;46(4):555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu L, Kim JM, Khorana HG. Structure and function in rhodopsin: asymmetric reconstitution of rhodopsin in liposomes. Proceeding of the National Academic Sciences of the United States of America. 2002;99(21):13409–13412. doi: 10.1073/pnas.212518899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organisciak DT, Xie A, Wang HM, Jiang YL, Darrow RM, Donoso LA. Adaptive changes in visual cell transduction protein levels: effect of light. Experimental Eye Research. 1991;53(6):773–779. doi: 10.1016/0014-4835(91)90113-s. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Buczylko J, Ohguro H, Annan RS, Carr SA, Crabb JW, et al. Characterization of a truncated form of arrestin isolated from bovine rod outer segments. Protein Science. 1994;3 (2):314–324. doi: 10.1002/pro.5560030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Van Hooser JP, Garwin GG, Chen J, Liou GI, Saari JC. Kinetics of visual pigment regeneration in excised mouse eyes and in mice with a targeted disruption of the gene encoding interphotoreceptor retinoid-binding protein or arrestin. Biochemistry. 1999;38 (37):12012–12019. doi: 10.1021/bi990504d. [DOI] [PubMed] [Google Scholar]
- Parkes JH, Gibson SK, Liebman PA. Temperature and pH dependence of the metarhodopsin I–metarhodopsin II equilibrium and the binding of metarhodopsin II to G protein in rod disk membranes. Biochemistry. 1999;38(26):8598. doi: 10.1021/bi995078a. [DOI] [PubMed] [Google Scholar]
- Peterson JJ, Orisme W, Fellows J, McDowell JH, Shelamer CL, Dugger DR, et al. A role for cytoskeletal elements in the light-driven translocation of proteins in rod photoreceptors. Investigative Ophthalmology and Vision Science. 2005;46(11):3988–3998. doi: 10.1167/iovs.05-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JJ, Tam BM, Moritz OL, Shelamer CL, Dugger DR, McDowell JH, et al. Arrestin migrates in photoreceptors in response to light: a study of arrestin localization using an arrestin-GFP fusion protein in transgenic frogs. Experimental Eye Research. 2003;76(5):553–563. doi: 10.1016/s0014-4835(03)00032-0. [DOI] [PubMed] [Google Scholar]
- Plack PA, Pritchard DJ. Schiff bases formed from retinal and phosphatidylethanolamine, phosphatidylserine, ethanolamine or serine. Biochemical Journal. 1969;115(5):927–934. doi: 10.1042/bj1150927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulvermüller A, Maretzki D, Rudnicka-Nawrot M, Smith WC, Palczewski K, Hofmann KP. Functional differences in the interaction of arrestin and its splice variant, p44, with rhodopsin. Biochemistry. 1997;36(30):9253–9260. doi: 10.1021/bi970772g. [DOI] [PubMed] [Google Scholar]
- Radu RA, Mata NL, Bagla A, Travis GH. Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt’s macular degeneration. Proceeding of the National Academic Sciences of the United States of America. 2004;101(16):5928–5933. doi: 10.1073/pnas.0308302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reme CE. The dark side of light: rhodopsin and the silent death of vision the proctor lecture. Investigative Ophthalmology and Vision Science. 2005;46(8):2672–2682. doi: 10.1167/iovs.04-1095. [DOI] [PubMed] [Google Scholar]
- Ridge KD, Abdulaev NG, Sousa M, Palczewski K. Photo-transduction: crystal clear. Trends in Biochemical Sciences. 2003;28(9):479–487. doi: 10.1016/S0968-0004(03)00172-5. [DOI] [PubMed] [Google Scholar]
- Rim J, Oprian DD. Constitutive activation of opsin: interaction of mutants with rhodopsin kinase and arrestin. Biochemistry. 1995;34 (37):11938–11945. doi: 10.1021/bi00037a035. [DOI] [PubMed] [Google Scholar]
- Ritter E, Zimmermann K, Heck M, Hofmann KP, Bartl FJ. Transition of rhodopsin into the active metarhodopsin II state opens a new light-induced pathway linked to Schiff base isomerization. Journal of Biological Chemistry. 2004;279(46):48102–48111. doi: 10.1074/jbc.M406857200. [DOI] [PubMed] [Google Scholar]
- Saari JC, Garwin GG, Van Hooser JP, Palczewski K. Reduction of all-trans-retinal limits regeneration of visual pigment in mice. Vision Research. 1998;38(10):1325–1333. doi: 10.1016/s0042-6989(97)00198-3. [DOI] [PubMed] [Google Scholar]
- Schädel SA, Heck M, Maretzki D, Filipek S, Teller DC, Palczewski K, et al. Ligand channeling within a G-protein-coupled receptor. The entry and exit of retinals in native opsin. Journal of Biological Chemistry. 2003;278(27):24896–24903. doi: 10.1074/jbc.M302115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schertler GF. Structure of rhodopsin and the metarhodopsin I photointermediate. Current Opinion in Structural Biology. 2005;15(4):408–415. doi: 10.1016/j.sbi.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Schleicher A, Kühn H, Hofmann KP. Kinetics, binding constant, and activation energy of the 48-kDa protein–rhodopsin complex by extra-metarhodopsin II. Biochemistry. 1989;28(4):1770–1775. doi: 10.1021/bi00430a052. [DOI] [PubMed] [Google Scholar]
- Schröder K, Pulvermüller A, Hofmann KP. Arrestin and its splice variant Arr1-370A (p44). Mechanism and biological role of their interaction with rhodopsin. Journal of Biological Chemistry. 2002;277(46):43987–43996. doi: 10.1074/jbc.M206211200. [DOI] [PubMed] [Google Scholar]
- Smith WC, Milam AH, Dugger D, Arendt A, Hargrave PA, Palczewski K. A splice variant of arrestin. Molecular cloning and localization in bovine retina. Journal of Biological Chemistry. 1994;269(22):15407–15410. [PubMed] [Google Scholar]
- Sommer ME, Smith WC, Farrens DL. Dynamics of arrestin–rhodopsin interactions: arrestin and retinal release are directly linked events. Journal of Biological Chemistry. 2005;280(8):6861–6871. doi: 10.1074/jbc.M411341200. [DOI] [PubMed] [Google Scholar]
- Sommer ME, Smith WC, Farrens DL. Dynamics of arrestin–rhodopsin interactions: acidic phospholipids enable binding of arrestin to purified rhodopsin in detergent. Journal of Biological Chemistry. 2006;281(14):9407–9417. doi: 10.1074/jbc.M510037200. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Fishkin N, Zhou J, Cai B, Jang YP, Krane S, et al. A2E, a byproduct of the visual cycle. Vision Research. 2003;43(28):2983–2990. doi: 10.1016/s0042-6989(03)00475-9. [DOI] [PubMed] [Google Scholar]
- Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. Journal of Neuroscience. 2006;26(4):1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel R, Ludeke S, Radu I, Siebert F, Sheves M. Photoreactions of metarhodopsin III. Biochemistry. 2004a;43(31):10255–10264. doi: 10.1021/bi049182q. [DOI] [PubMed] [Google Scholar]
- Vogel R, Siebert F, Mathias G, Tavan P, Fan G, Sheves M. Deactivation of rhodopsin in the transition from the signaling state meta II to meta III involves a thermal isomerization of the retinal chromophore C[double bond]D. Biochemistry. 2003;42(33):9863–9874. doi: 10.1021/bi034684+. [DOI] [PubMed] [Google Scholar]
- Vogel R, Siebert F, Zhang XY, Fan G, Sheves M. Formation of Meta III during the decay of activated rhodopsin proceeds via Meta I and not via Meta II. Biochemistry. 2004b;43(29):9457–9466. doi: 10.1021/bi049337u. [DOI] [PubMed] [Google Scholar]
- Wald G. Molecular basis of visual excitation. Science. 1968;162(850):230–239. doi: 10.1126/science.162.3850.230. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Grimm C, Samardzija M, Reme CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Progress in Retinal and Eye Research. 2005;24 (2):275–306. doi: 10.1016/j.preteyeres.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Wilden U, Hall SW, Kühn H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proceeding of the National Academic Sciences of the United States of America. 1986;83(5):1174–1178. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Huang W, Zhu X, Craft CM, Baehr W, Chen CK. Light-dependent redistribution of visual arrestins and transducin subunits in mice with defective phototransduction. Molecular Vision. 2003;9:231–237. [PubMed] [Google Scholar]
- Zimmermann K, Ritter E, Bartl FJ, Hofmann KP, Heck M. Interaction with transducin depletes metarhodopsin III: a regulated retinal storage in visual signal transduction? Journal of Biological Chemistry. 2004;279(46):48112–48119. doi: 10.1074/jbc.M406856200. [DOI] [PubMed] [Google Scholar]