

Abstract
The manganese transport regulator (MntR) from Bacillus subtilis binds cognate DNA sequences in response to elevated manganese concentrations. MntR functions as a homodimer that binds two manganese ions per subunit. Metal binding takes place at the interface of the two domains that comprise each MntR subunit: an N-terminal DNA-binding domain and a C-terminal dimerization domain. In order to elucidate the link between metal binding and activation, a crystallographic study of MntR in its metal-free state has been undertaken. Here we describe the structures of the native protein and a selenomethionine-containing variant, solved to 2.8 Å. The two structures contain five crystallographically unique subunits of MntR, providing diverse views of the metal-free protein. In apo-MntR, as in the manganese complex, the dimer is formed by dyad-related C-terminal domains that provide a conserved structural core. Similarly, each DNA-binding domain largely retains the folded conformation found in metal bound forms of MntR. However, compared to metal-activated MntR, the DNA-binding domains move substantially with respect to the dimer interface in apo-MntR. Overlays of multiple apo-MntR structures indicate that there is a greater range of positioning allowed between N and C-terminal domains in the metal-free state and that the DNA-binding domains of the dimer are farther apart than in the activated complex. To further investigate the conformation of the DNA-binding domain of apo-MntR, a site-directed spin labeling experiment was performed on a mutant of MntR containing cysteine at residue 6. Consistent with the crystallographic results, EPR spectra of the spin-labeled mutant indicate that tertiary structure is conserved in the presence or absence of bound metals, though slightly greater flexibility is present in inactive forms of MntR.
Keywords: X-ray structure, transcriptional regulator, metal homeostasis, allostery
The manganese transport regulator (MntR) controls manganese homeostasis in Bacillus subtilis.1 When cellular levels of manganese are high, MntR represses the transcription of genes encoding manganese uptake transporters by binding to cognate operator sequences.1–3 Manganese is an essential nutrient in bacteria and plays an important role in cellular defense against reactive oxygen species. In several pathogenic species of bacteria, the regulation of manganese uptake has been linked to virulence.4 Notably, Bacillus anthracis, the causative agent of anthrax, possesses a close homolog of MntR,5 and mutants of B. anthracis lacking proteins involved in manganese uptake have weakened virulence.6 Furthermore, MntR is a member of the DtxR/IdeR family of metalloregulators, named for the diphtheria toxin repressor (DtxR) of Corynebacterium diphtheriae and the iron-dependent regulator (IdeR) of Mycobacterium tuberculosis, both of which play key roles in the virulence of the organisms from which they have been isolated.7–10 Because of the importance of these regulatory proteins in bacterial physiology and virulence, the mechanism of their action is of interest. In the past several years, the structural changes that relate to the activation of DtxR have received considerable attention,11–16 but little is known about how its distantly related structural homolog, MntR, is activated by manganese binding.
Structures of MntR have been solved in the presence of Mn2+ or Cd2+, another strongly activating metal ion.17,18 MntR is a functional homodimer of 142-residue subunits, each composed of two domains. The 71-residue N-terminal DNA-binding domain consists of three α-helices and two strands of antiparallel β-sheet (Figure 1), the latter forming the flexible “wing” of a winged helix-turn-helix (HTH) motif that interacts with DNA.17,19 The C-terminal domain contains four α-helices and forms the dimerization interface with its dyad-related mate. The N and C-terminal domains are connected by a long linker helix (α4; residues 64–86) that extends from the wing to the dimer interface. Two manganese ions bind at the interface of the two domains in a single subunit, forming a binuclear complex that involves residues from the DNA-binding domain (Asp8 and Glu11), the dimerization domain (Glu99, Glu102, and His103) and from the linker helix (His 77; Figure 1). The geometry of the binuclear complex with manganese appears to be sensitive to crystal packing forces. However, multiple lines of evidence suggest that the physiological complex places two Mn2+ at a separation of 4.4 Å, bridged by Glu99 and Glu102.5,17,18 Cadmium binds in a similar geometry to the two sites, which have been identified as A and C (Figure 1). Interestingly, the structure of MntR complexed with Zn2+, a weakly activating metal, reveals only a single bound metal ion, in the A site.3,18
Figure 1.
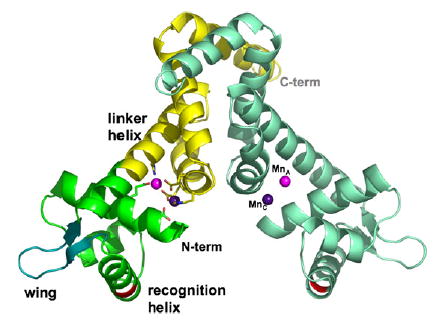
The structure of the MntR•Mn2+ complex (PDB ID 2F5F). Two subunits form a homodimer. The left subunit is colored to show the C-terminal dimerization domain in yellow and the N-terminal DNA-binding domain in green. The N and C termini are also labeled. The flexible “wing” is shown in turquoise. The linker helix, α4, and recognition helix of the helix-turn-helix motif are identified. The Mn2+ ions occupying the A and C sites of both subunits are colored light and dark purple, respectively. The backbone positions of Lys41 residues are shown in red. The residues that comprise the metal-binding site of the left subunit are shown in stick representation. Figures are drawn with PyMOL.36
Although structures of complexes of MntR with strongly and weakly activating metals have been obtained, there is little variation between these forms. The chief difference involves helix α1, which includes residues 3–20 in the Mn2+ and Cd2+ complexes. In the zinc complex of MntR, the helix unwinds at its N terminus so that α1 is shortened to include only residues 5–20. While this structural difference hints at greater flexibility in the MntR•Zn2+ complex than in the MntR•Mn2+ or MntR•Cd2+ complexes, there are no other substantial changes in backbone conformation, perhaps due to restraints imposed by crystal packing. As evidence that the crystalline environment can influence the conformation of MntR, a comparison of structures of the MntR•Mn2+ complex from monoclinic and hexagonal crystal forms shows that the DNA-binding domains can move somewhat independently of the dimeric core formed by dyad-related C-terminal domains, via slight flexibility in the linker helix, α4.18 This conformational shift repositions the metal-binding residues in the N-terminal domain (Asp8 and Glu11) by as much as 1.5 Å, and the recognition helices of the HTH motif can move by roughly 3 Å with respect to each other in the MntR dimer.
In addition to the conformational plasticity observed in the MntR•Mn2+ complex, there is presumably a unique set of structural features associated with the metal-free form of MntR that renders it inactive and distinct from the metal-bound form. A spectroscopic investigation into the activation of MntR revealed that structural reorganization takes place upon metal binding, but it remains unclear whether the reorganization originated from a stably folded conformation or from a molten globule-like state.20 Thus, there is no explicit description of the tertiary and/or quaternary changes that take place upon activation of MntR by metal binding. To further investigate the mechanism of activation, we have determined the structure of MntR in its metal-free state and investigated the conformation of the N-terminal domain using site-directed spin labeling.21,22 The results presented here are consistent with a disorder to order transition in the tertiary structure of MntR that orients independent, folded domains in positions to promote high affinity interactions with DNA.
The structure of Apo-MntR
The crystal structures of native apo-MntR (apo-MntRnat) and the selenomethionine-containing deri-vative (apo-MntRSeMet) have been solved to 2.8 Å resolution. Apo-MntRSeMet crystallized in the orthorhombic space group P212121 and the structure was solved using the anomalous signal from five of the 12 selenium atoms from the two dimers in the asymmetric unit (Table 1). Dimer 1 is composed of subunits A and B, and dimer 2 is composed of subunits C and D. The refined model contains 545 of the 564 residues present. Residues 3–136 of subunit A, and residues 2–138 of subunits C and D could be positioned in electron density. In subunit B, the flexible “wing” of the DNA-binding motif (residues 54–58) is absent from the model, which otherwise includes residues 3–134. The refined model possesses good stereochemistry, except that the backbone conformation of two residues, Tyr57 of subunit D and Tyr57 of subunit A, lie in unfavorable regions of the Ramachandran plot (Table 1). This residue was observed to adopt a strained conformation in the manganese-bound form of MntR as well.17
Table 1.
Data and refinement statistics
| apo-MntRSeMet |
|||||
|---|---|---|---|---|---|
| Anomalous data separated
|
Merged
|
||||
| Se peak | Se edge | Se remote | Se peak | apo-MntRnat | |
| Data collection | |||||
| Space group | P212121 | P3121 | |||
| Unit cell dimensions (Å) | a=55.89, b =65.03, c=186.97 | a=b=42.0, c=149.1 | |||
| Beam line | ALS 8.2.1 | ALS 8.3.1 | |||
| Wavelength (Å) | 0.9793 | 0.9796 | 0.9641 | 0.9793 | 1.1159 |
| Resolution rangea (Å) | 64.55–3.00 (3.16–3.00) | 64.55–3.00 (3.16–3.00) | 65.09–3.30 (3.48–3.30) | 64.55–2.80 (2.95–2.80) | 74.54–2.80 (2.95–2.80) |
| Observations | 57,416 | 55,571 | 29,614 | 65,335 | 54,743 |
| Unique reflections | 24,633 | 25,373 | 9810 | 16,290 | 4162 |
| Completeness (%) | 92.8 (84.7) | 90.2 (81.5) | 78.4 (56.8) | 93.6 (79.5) | 99.9 (99.9) |
| I/σ(I) | 7.8 (3.3) | 7.1 (2.7) | 7.7 (3.7) | 6.3 (2.0) | 8.8 (2.0) |
| Rmerge (%)b | 8.2 (22.4) | 9.0 (27.0) | 8.9 (19.1) | 10.6 (38.3) | 4.1 (35.0) |
| Wilson B factor (Å2) | 55 | 104 | |||
| Phasing | |||||
| Sites in asuc | 5 | ||||
| Resolution range | 15.0–3.8 | ||||
| Figure of merit | 0.46 | ||||
| Refinement | |||||
| Resolution (Å) | 20.0–2.80 | 20.0–2.80 | |||
| Rcryst/Rfree (%)d | 21.8/27.6 | 27.4/34.2 | |||
| Subunits per asue | 4 | 1 | |||
| Number of atoms | |||||
| Protein | 4423 | 1103 | |||
| Solvent | 22 | 1 | |||
| Solute | 60 | 0 | |||
| RMSD bonds (Å) | 0.008 | 0.009 | |||
| RMSD angles (°) | 1.20 | 1.09 | |||
| Ramachandrand | |||||
| Most favored (%) | 88.8 | 88.5 | |||
| Additionally allowed (%) | 10.8 | 11.5 | |||
| Generously allowed (%) | 0.2 | 0 | |||
| Disallowed (%) | 0.2 | 0 | |||
Apo-MntRnat was purified as described.17 Apo-MntRSeMet was expressed in Escherichia coli cells grown in the presence of selenomethionine according to standard methods,29 and purified in the same manner as the native protein. Crystals of apo-MntRSeMet were obtained by the hanging-drop vapor diffusion method. Drops of protein solution (10 mg/ml) were mixed with an equal volume of reservoir solution (15% (w/v) PEG 4000, 0.2 M ammonium sulfate, 5 mM cobalt(II) chloride) and allowed to equilibrate. Crystals of apo-MntRnat were obtained similarly, except that PEG 400 substituted for PEG 4000. Suitable crystals were transferred to a solution of the mother liquor with 10% (v/v) glycerol added as a cryo-protectant and flash-cooled in liquid nitrogen prior to data collection under a flowing stream of cold nitrogen (100 K). Data were processed using MOSFLM30 and scaled using SCALA from the CCP4 program suite.31 The structure of apo-MntRSeMet was determined using phases obtained by multiwavelength anomalous dispersion as implemented in Solve.32 Phases were extended to 2.8 Å using density modification as implemented in CNS.23 Model building was performed in O33 and subsequent refinement was performed using CNS software.23 The structure of apo-MntRnat was determined using molecular replacement, using a subunit of the apo-MntRSeMet structure as a search model. Virtually identical rotation and translation functions were obtained using EPMR24 and CNS. Following rigid body refinement and simulated annealing in CNS, further refinement in Refmac5 from the CCP4 program suite implementing TLS refinement27 permitted model rebuilding using COOT software.34 Despite the presence of Co2+ in the crystallization buffers, no evidence of bound cobalt was found in electron density maps or anomalous difference Fourier maps.
Numbers in parentheses reflect the highest resolution shell.
Rmerge = ∑h∑j|Ih,j−<Ih>|/∑j∑h|Ih,j|, where Ih,j is the jth observation of reflection h.
Asymmetric unit.
Rcryst = ∑h||Fo|−|Fc||/∑h|Fo|, where Fo and Fc are the observed and calculated structure factors for reflection h. Rfree is calculated similarly for 10% of the data not used in refinement.
Determined using Procheck software.35
Native apo-MntR crystallized in the trigonal space group P3121. A molecular replacement solution was obtained using subunit D from the selenomethionine structure as a search model. The model was trimmed to include residues 4–53 and 60–130, which omits wing residues 54–59, and yielded the same solution using CNS23 and EPMR.24 A single subunit occupies the asymmetric unit, and the 2-fold axis of the dimer coincident is the crystallographic 2-fold axis. The initial R-factor obtained after rigid body refinement was 41%, and attempts to improve the quality of the model by simulated annealing and rebuilding yielded only small improvements in the model statistics. Careful analysis of the diffraction data with Xtriage showed no evidence of merohedral twinning,25 and no evidence of or anisotropy in the data or pseudo-symmetry was found. Molecular replacement solutions in lower symmetry space groups also failed to improve refinement statistics. Ultimately, the implementation of TLS refinement in Refmac526,27 led to the greatest improvement in the model, though the Rcryst and Rfree values remain high (27.3% and 34.2%). Electron density maps permit continuous tracing of the backbone from residues 3–135 of the single subunit in the asymmetric unit, but density is often weak for side-chains. It may be that static or dynamic disorder within the crystals is limiting efforts to accurately model native apo-MntR in the trigonal crystal form. Consistent with this explanation, the Wilson B factor of 104 Å2 suggests a mobile structure (Table 1). Nevertheless, the apo-MntRnat structure provides an additional, useful observation of the backbone conformation of MntR in its metal-free form.
The structure of MntR observed in metal-bound forms is largely retained in its metal-free state. Five crystallographically independent subunits constituting three dimers of apo-MntR are present in the two crystal structures. In all instances, the quaternary interactions are largely conserved in that the C-terminal domain of each subunit maintains a constant conformation, contributing to a largely unvarying dimer interface (Figure 2(a)). As a result the subunits of apo-MntR superimpose closely with metal complexes of MntR in the C-terminal regions, with an RMSD in the positions of the Cα atoms of less than 0.6 Å (Figure 2(a)). These observations are consistent with the previous solution studies showing that MntR retains dimeric structure in the presence and absence of activating metal ions.3,5
Figure 2.
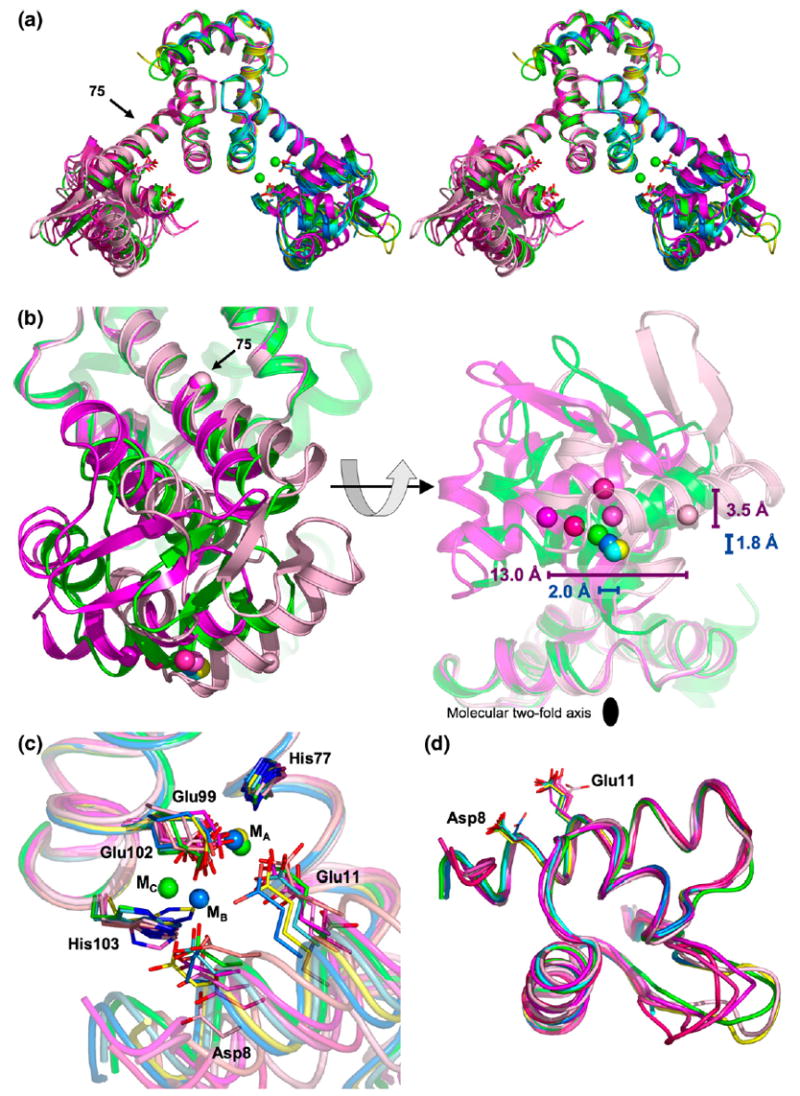
Comparison of MntR-metal complexes with apo-MntR. (a) Stereoview of an overlay of MntR-metal complexes and apo-MntR. The full dimers of apo-MntRnat (dark magenta) and the AC conformation of MntR•Mn2+ from the hexagonal crystal form (in green) are shown as ribbon diagrams. Other MntR-metal subunits are shown on the right, and the other apo-MntR subunits on the left. The ribbon cartoons of apo-MntR are colored in varying shades of magenta, from light (the A subunit of apo-MntRSeMet) to dark (apo-MntRnat). The AB and AC conformations of the MntR•Mn2+ complex from monoclinic crystal forms are light and dark blue, respectively. The MntR•Zn2+ complex is colored yellow. Metal-binding residues Asp8 and Glu11 are shown, with carbon atoms colored according to the above scheme, oxygen atoms in red and nitrogen atoms colored blue. The Mn2+ ions in the AC conformation are shown in green on the right side. The position of the Cα atom of Tyr75 is denoted by a labeled arrow. (b) Two views of the DNA-binding domains from the MntR•Mn2+ complex with the A subunit of apo-MntRSeMet and apo-MntRnat (colored as in (a)), aligned by their C-terminal domains. The left image is obtained by rotating 90° about the molecular 2-fold axis of the MntR dimer from the orientation shown in (a). Other subunits are omitted for clarity. The right image is obtained by an additional rotation of 90° about the horizontal axis. In the left-hand image, the position of the hinge is denoted by spheres marking the position of the Cα atom of Tyr75. The positions of Cα atoms of Lys41 from all subunits of apo-MntR and the MntR-metal complexes shown in (a), are shown as spheres colored as above. In the right-hand image, the lateral displacements of the DNA-binding domains, as defined by the range of positions of the Cα atoms of Lys41 (colored spheres), are shown with horizontal bars. The magenta bar indicates the range found among apo-MntR subunits, and the blue bar by subunits of the MntR-metal complexes. The distances indicate the respective ranges of displacement. The range of positions obtained due to spreading of the DNA-binding domains is shown by vertical bars, with labels indicating the range of spreading. The molecular 2-fold axis is identified by a black oval. (c) Comparison of metal-binding regions of apo-MntR and MntR-metal complexes aligned by their C-terminal domains. Carbon and metal atoms are colored as in (a). (d) DNA-binding domains of apo-MntR and MntR-metal complexes aligned with one another. Metal-binding residues Asp8 and Glu11 are shown.
In comparison to the fixed structure of the C-terminal domains in the MntR dimer, the positions of the N-terminal, DNA-binding domains vary greatly, via slight unwinding of the linker helix, α4, between residues 72–75 (Figure 2(a) and (b)). This behavior is similar to that observed among the multiple reported structures of the MntR•Mn2+ complex,18 though it is far more dramatic in the metal-free state. There is no loss of hydrogen-bonding within α4, nor departure from φ and φ dihedral angles consistent with a helical conformation, yet even two turns away, the Cα atom of residue 64, at the N terminus of α4, varies in position by up to 5.5 Å. Without bound metal ions, the DNA-binding domain swings, like a pendulum, about a pivot point located near residue 75. As measured by the Hingefind routine,28 the DNA-binding domain of apo-MntR can experience a 36° deflection about this hinge position, whereas comparison of the MntR-metal complex structures reveals a maximum motion of only 4°.18
The wide range of positions adopted by the DNA-binding domain can be appreciated by monitoring the location of the Cα atom of Lys41, which is at the center of the recognition helix of the HTH motif. Notably, the dyad-related Lys41 residues in apo-MntR dimers are at least 3.4 Å farther apart than in the most-separated conformation of the MntR•Mn2+ complex from the hexagonal crystal form (Figure 2(b)).18 The increased distance between Lys41 residues in apo-MntR is both due to increased spreading of the DNA-binding domains away from the molecular 2-fold axis and through lateral displacement perpendicular to the direction of spreading (Figure 2(b)). The DNA-binding domains of the apo-MntR subunits are spread farther apart than in MntR-metal complexes and over a greater range as well: 3.5 Å versus 1.8 Å (Figure 2(b)). The lateral variation in position of DNA-binding domains in apo-MntR is even more dramatic. Encompassing the relatively modest 2 Å range observed between MntR-metal complexes, the Cα atom of Lys41 may be laterally displaced by up to 13 Å between apo-MntR subunits (Figure 2(b)). The variation in the positioning of the DNA-binding domain is also reflected in the positions of metal-binding residues Asp8 and Glu11, which vary by nearly 5 Å (Figure 2(c)). These results suggest that bound metals both alter and substantially restrain the position of the DNA-binding domains.
Despite its variable location with respect to the dimerization domain, the DNA-binding domain of apo-MntR retains the tertiary fold seen in metal-activated forms of MntR.17,18 An overlay of the DNA-binding domains of each of the five subunits of apo-MntR with the same domain from the MntR•Mn2+ can be made with a RMSD in Cα atom positions of between 0.32 Å and 0.67 Å in pairwise comparisons (excluding the wing residues, 52–60; Figure 2(c)). The absence of bound metal ions does not significantly affect the folded structure of the DNA-binding domain. There are, however, two flexible regions within the DNA-binding domain that adopt multiple conformations in the structures presented here. The position of the wing motif, when visible in electron density, varies by up to 8.0 Å (measured at the Cα atom of Tyr57; Figure 2(d)). Mobility of the wing region has previously been suggested by weak electron density in maps from the MntR-metal complexes and is apparently unrelated to metal activation.17,18 A second, more intriguing variation occurs at the N terminus of helix α1, where a conformational change at residue Ser5 leads to the unwinding and loss of residues Thr3 and Pro4 from the helix, displacing the Cα atom of Thr2 by as much as 17.0 Å (Figures 2(d) and 3(a) and (b)). The unwound conformation is present in all four subunits of apo-MntRSeMet and has previously been observed in the MntR•Zn2+ complex. In the latter case, unwinding could be a sign of partial disordering of the N-terminal domain, resulting from the absence of bound metal in the C site.18 In all binuclear metal complexes of MntR, helix α1 includes residues 3 and 4. However, the correlation of α1 unwinding to metal binding is complicated by the fully wound helix in apo-MntRnat. Based on these conflicting observations from the two crystal forms of apo-MntR, it is impossible to say whether addition of residues 3 and 4 to the N terminus of α1 is dependent upon metal binding or is simply a result of environmental conditions associated with crystallization.
Figure 3.
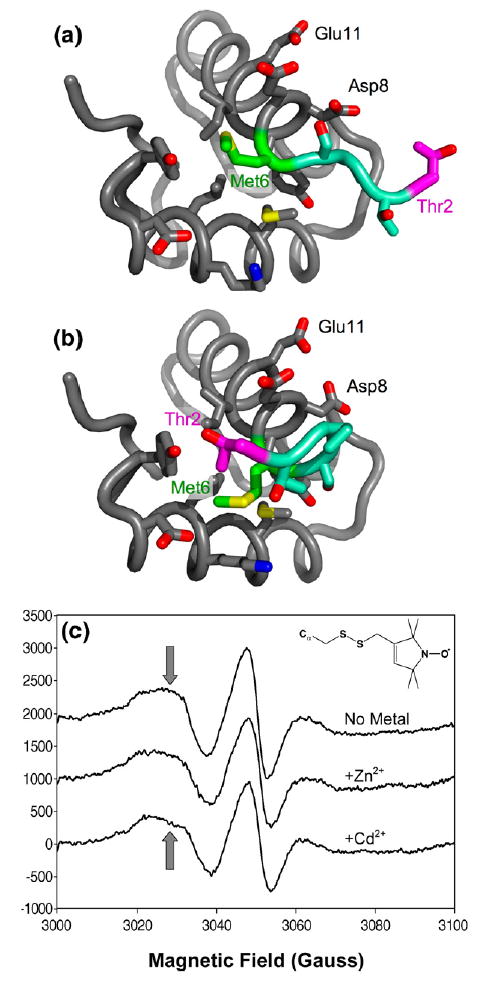
Site-directed spin-labeling at residue 6. (a) Structure of the DNA-binding domain of apo-MntR. The backbone is shown in this cartoon as a flexible tube. The side-chains of residues within 5 Å of residue 6 are shown. Carbon atoms are colored gray, except for those in Met6 (green), Thr2 (magenta), and those in residues 2–5, which are colored aqua. Oxygen atoms are red, nitrogen atoms are blue and sulfur is colored yellow. (b) Structure of the DNA-binding domain of metal-activated MntR colored as in (a). (c) EPR spectra of spin-labeled MntR:M6C without metal, or in the presence of excess Cd2+ and Zn2+. Inset shows the structure of a spin-label modified cysteinyl residue. Arrows indicate the perturbation of the down-field resonance that accompanies addition of metal ions. MntR:M6C was prepared by site-directed mutagenesis of the native mntR gene in plasmid pHB75061,17 using the QuickChange mutagenesis kit (Stratagene). The mutated plasmid was transformed into BL21(DE3) cells. Expression and purification proceeded as per wild-type MntR,17 except that the protein was maintained in 1 mM DTT throughout the purification and dialysis steps. Prior to labeling, 100 μM MntR:M6C (concentration of subunits) was dialyzed against deoxygenated buffer L (25 mM Mes, 500 mM NaCl, pH 6.0). The methanethiosulfonate spin label (MTSSL; Toronto Research) was added as a 50 mM stock solution in acetonitrile to a 1.5 molar excess over MntR:M6C and the reaction mixture was incubated at 4 °C for over 4 h with stirring. The solution was passed through a Sephadex G-25 desalting column (1 cm×15 cm) pre-equilibrated with buffer L and fractions containing protein were pooled and concentrated to 100 μM for spectral measurement. Spectra were recorded at room temperature on a Varian E-104 instrument fitted with a loop-gap resonator and the EWWIN 5.22 data acquisition package (Scientific Software Services, Plymouth, MI). Measurements were carried out at 9.3 GHz microwave frequency using 2-mW incident power, a modulation amplitude of 2 gauss, and a 100 gauss sweep. Metal ions were added from 0.5 M stock solutions of the chloride salt to final concentrations of 600 μM.
Nevertheless, these structures indicate that metal binding activates MntR by orienting the DNA-binding domains with respect to the fixed dimerization interface and with respect to each other. In the absence of activating metals, the DNA-binding domains adopt a range of positions that presumably place them too far apart in an MntR dimer to successfully interact with cognate DNA sequences. Furthermore, there is no evidence to suggest that the internal structure of the DNA-binding domain is substantially changed by the presence or absence of metal ions.
Site-directed spin labeling as a probe of conformation
To determine if the conserved fold of the DNA- binding domain in structures of apo-MntRnat and apo-MntRSeMet is merely the product of crystal-packing forces, a site-directed spin labeling study was undertaken to investigate the conformational behavior of MntR in solution. The mobility of a nitroxide spin label is reflected in its elecron paramagnetic resonance (EPR) spectrum, and when it is covalently attached to a protein, the label thus reports of the dynamics of that region of the protein.21,22 Methionine 6 was mutated to cysteine (MntR:M6C) in order to permit covalent labeling of MntR with a nitroxide spin label (inset, Figure 3(c)). Methionine at position 6 occupies a partially solvent-exposed position that receives additional shielding from Thr2 when residues 3–5 adopt a helical conformation at the N terminus of helix α1, and additionally forms numerous tertiary contacts (Figure 3(a) and (b)). Spin-labeling at residue 6 allows investigation of both the global fold of the N-terminal domain, which is constant among the crystal structures of MntR obtained to date, and the local conformation at the N terminus of α1, which is more variable.
The results of the spin-labeling experiment support the hypothesis that the DNA-binding domain retains substantial tertiary structure in the absence of bound metals, but increased flexibility at the N terminus of α1 may exist in apo-MntR, and to a lesser extent in the MntR•Zn2+ complex. Only subtle differences are visible in the EPR spectra of spin-labeled MntR:M6C taken without metal, or with either Cd2+ and Zn2+ present in excess (Figure 3(c)). The line width of the central resonance does not vary significantly in the three spectra, and the value of the inverse line width, 0.091 Hz−1, is consistent with a spin label in a protein environment where mobility is restricted by tertiary contacts.21,22 Slight perturbation of the downfield, M−1, resonance away from a Gaussian shape can be seen upon addition of Zn2+ and is more pronounced for Cd2+. These changes are consistent with a slight decrease in conformational distribution of the spin label, as would be expected if the N-terminal residues fold over residue 6 as they adopt a helical conformation, but overall, the change takes place in the context of a largely ordered protein environment.
The activation of MntR
The results presented here provide substantial insight into the structure of apo-MntR. The two domains of MntR retain stable folds, while the DNA-binding domain is able to adopt a variety of orientations with respect to the dimerization domain. The only notable variation in local structure is the unwinding of the N terminus of helix α1 in apo-MntR, a feature previously observed in the structure of MntR•Zn2+.18 EPR spectra from the site-directed spin-labeling study confirm that the DNA-binding domain remains folded in the absence of bound metal ion, and suggest that the changes observed at the N terminus of α1 in crystal structures take place in solution as well. Previous studies left open the possibility that metal binding might induce significant structural rearrangement from a molten globule state,20 but such a transition is clearly inconsistent with the structural and spectroscopic results presented here.
In general terms, the activation of the MntR dimer appears to proceed by the organization of two independently folded DNA-binding domains into positions appropriate for productive interaction with cognate DNA sequences. More specifically, the activation of MntR may be proposed to proceed through three stages. (1) In the absence of metal ions, MntR possesses substantial conformational entropy due to the motion of folded but structurally independent DNA-binding domains. Entropic costs associated with adopting a fixed, active conformation might be responsible for the low affinity of apo-MntR for cognate DNA sequences.3,18,20 However, it may also be that the active conformation is inaccessible to apo-MntR. Comparison of the positions of the DNA binding domains of metal-activated MntR to apo-MntR demonstrates that the recognition helix in the HTH motif adopts a small range of positions that does not overlap with any of the positions observed among the five apo-MntR subunits. However, these structures cannot be expected to represent all possible conformations allowed to MntR, so it may yet be that apo-MntR can sample the active conformation, but does not stably assume it. (2) Metal binding likely proceeds first through occupancy of the A site, which slightly increases the affinity of MntR for DNA, as seen with zinc.3 The argument for initial binding to the A site draws on two observations. First, all MntR-metal complexes possess occupancy at the A site, while there are at least two possible binding sites for the second metal (B or C). Second, sequential binding has been indicated by solution studies employing calorimetry and EPR.5,17,18 MntR with an occupied A site likely has an altered conformational repertoire that is somewhat more restricted than in apo-MntR. The MntR•Zn2+ complex is capable of adopting a conformation similar to other MntR-metal complexes (Figure 2), and occupancy of this site does put some constraints on the position of residue Glu11 from the DNA-binding domain (Figure 2(c)). However, the affinity of MntR•Zn2+ for DNA is 100-fold lower than the MntR•Mn2+ complex. It may be that the entropic costs of adopting a single conformation appropriate to high-affinity DNA binding continue to be prohibitive. (3) Following productive binding of an activating metal, such as Mn2+ or Cd2+, to the A site, bridging ligands in the metal-binding regions become organized, allowing a second metal to bind to the B or, more likely, the C site.18 Metal binding to either of these sites engages Asp8, a second metal-binding residue in the DNA-binding domain, further restricting conformational flexibility of the domain. At this point, MntR is capable of binding DNA with high affinity.
These findings provide an interesting comparison to the metal-ion activation of DtxR, the namesake and best-studied member of the family of metalloregulators to which MntR belongs. In an early crystallographic study, it was observed that although apo-DtxR and the metal-activated form of DtxR crystallize isomorphously, conformational changes are discernable between the two forms.11 As with MntR, the dimerization domains of apo-DtxR and metal-activated DtxR form a core of immobile residues, but display variable positioning of the DNA-binding domains. However, solution studies describe a more complex set of behaviors. Apo-DtxR exists as a monomer in dilute solutions12,13 and only forms a stable dimer after both metals have bound, though the sequence of metal ion binding is in dispute.14,15 In the absence of bound metals, the first 124 residues of DtxR, spanning the DNA-binding and dimerization domain, experience significant flexibility, perhaps sampling multiple conformations.16 Upon metal binding, dimers form and the structure becomes ordered. From the results of solution studies, it has been argued that the N-terminal 124 residues comprise a single domain that becomes ordered only in the dimeric, metal-activated form of DtxR.16
In contrast, the results presented here indicate that the N-terminal and C-terminal domains of apo-MntR form discrete, folded units that can move independently of each other. Perhaps these domains are only “loosely folded”, as in apo-DtxR and as indicated by ANS binding to the metal-free form of MntR.16,20 Nevertheless it appears that the folded form of each domain in MntR is favored even in the absence of bound metal. Metal binding to MntR serves principally to organize the DNA-binding domains with respect to the quaternary interface formed by dyad-related C-terminal domains. This organization places the recognition helices of the HTH motifs in closer proximity and presumably in a functional conformation. The favorable enthalpy of manganese binding18 is thus used to reduce the conformational entropy of the apo-MntR dimer. The weaker binding of the mononuclear MntR•Zn2+ complex to DNA is likely related to a partial loss of restraints between domains, suggesting a mechanism for the selective activation of MntR by Mn2+ and Cd2+ over other competing metal ions.1–3,18,20 Ongoing studies will further explore the conformational and structural changes associated with the activation of MntR to its high-affinity DNA-binding state upon metal binding.
Protein Data Bank accession codes
Crystallographic coordinates and structure factor amplitudes have been deposited in the RCSB Protein Data Bank with accession codes 2HYF (apo-MntRSeMet) and 2HYG (apo-MntRnat).
Acknowledgments
This work was supported in part by NIH Grant GM069644 (A. G.) and NIH Grant GM059323 (J. D. H.). We thank Dr Peter Zwart for his assistance with Xtriage software and Dr Wendy Breyer for many helpful discussions. Portions of this research were carried out at the Advanced Light Source national user facility operated by Lawrence Berkeley National Laboratory on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The Berkeley Center for Structural Biology is supported in part by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
Abbreviations used
- DtxR
diphtheria toxin repressor
- EPR
electron paramagnetic resonance
- HTH
helix-turn-helix
- IdeR
iron-dependent regulator
- MntR
manganese transport regulator
- RMSD
root mean square deviation
Footnotes
Edited by J. Doudna
References
- 1.Que Q, Helmann JD. Manganese homeostasis in Bacillus subtilis is regulated by MntR, a bifunctional regulator related to the diphtheria toxin repressor family of proteins. Mol Microbiol. 2000;35:1454–1468. doi: 10.1046/j.1365-2958.2000.01811.x. [DOI] [PubMed] [Google Scholar]
- 2.Guedon E, Helmann JD. Origins of metal ion selectivity in the DtxR/MntR family of metallor-egulators. Mol Microbiol. 2003;48:495–506. doi: 10.1046/j.1365-2958.2003.03445.x. [DOI] [PubMed] [Google Scholar]
- 3.Lieser SA, Davis TC, Helmann JD, Cohen SM. DNA-binding and oligomerization studies of the manganese(II) metalloregulatory protein MntR from Bacillus subtilis. Biochemistry. 2003;42:12634–12642. doi: 10.1021/bi0350248. [DOI] [PubMed] [Google Scholar]
- 4.Kehres DG, Maguire ME. Emerging Themes in manganese transport, biochemistry and pathogenesis in bacteria. FEMS Microbiol Rev. 2003;27:263–290. doi: 10.1016/S0168-6445(03)00052-4. [DOI] [PubMed] [Google Scholar]
- 5.Sen KI, Sienkiewicz A, Love JF, vanderSpek JC, Fajer PG, Logan TM. Mn(II) binding by the Anthracis repressor from Bacillus anthracis. Biochemistry. 2006;45:4295–4303. doi: 10.1021/bi052288g. [DOI] [PubMed] [Google Scholar]
- 6.Gat O, Mendelson I, Chitlaru T, Ariel N, Altboum Z, Levy H, et al. The solute-binding component of a putative Mn(II) ABC transporter (MntA) is a novel Bacillus anthracis virulence determinant. Mol Microbiol. 2005;58:533–551. doi: 10.1111/j.1365-2958.2005.04848.x. [DOI] [PubMed] [Google Scholar]
- 7.Boyd J, Oza MN, Murphy JR. Molecular cloning and DNA sequence analysis of a diphtheria tox iron-dependent regulatory element (dtxR) from Corynebacterium diphtheriae. Proc Natl Acad Sci USA. 1990;87:5968–5972. doi: 10.1073/pnas.87.15.5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schiering N, Tao X, Zeng H, Murphy JR, Petsko GA, Ringe D. Structures of the apo- and the metal ion-activated forms of the diphtheria tox repressor from Corynebacterium diphtheriae. Proc Natl Acad Sci USA. 1995;1995:9843–9850. doi: 10.1073/pnas.92.21.9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feese MD, Ingason BP, Goranson-Siekierke J, Holmes RK, Hol WGJ. Crystal Structure of the iron-dependent regulator from Mycobacterium tuberculosis at 2.0 Å resolution reveals the Src homology domain 3-like fold and metal binding function of the third domain. J Biol Chem. 2001;276:5959–5966. doi: 10.1074/jbc.M007531200. [DOI] [PubMed] [Google Scholar]
- 10.Pennella MA, Giedroc DP. Structural determinants of metal selectivity in prokaryotic metal-responsive transcriptional regulators. Biometals. 2005;18:413–428. doi: 10.1007/s10534-005-3716-8. [DOI] [PubMed] [Google Scholar]
- 11.Pohl E, Holmes RK, Hol WGJ. Motion of the DNA-binding domain with respect to the core of the Diphtheria toxin repressor (DtxR) revealed in the crystal structures of apo- and holo-DtxR. J Biol Chem. 1998;273:22420–22427. doi: 10.1074/jbc.273.35.22420. [DOI] [PubMed] [Google Scholar]
- 12.Tao X, Teng HY, Murphy JR. Transition-metal ion activation of DNA-binding by the diphtheria Tox repressor requires the formation of stable homodimers. Proc Natl Acad Sci USA. 1995;92:6803–6807. doi: 10.1073/pnas.92.15.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spiering MM, Ringe D, Murphy JR, Marletta MA. Metal stoichiometry and functional studies of the diphtheria toxin repressor. Proc Natl Acad Sci USA. 2003;100:3808–3813. doi: 10.1073/pnas.0737977100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rangachari V, Marin V, Bienkiewicz EA, Semavina M, Guerrero L, Love JF, et al. Sequence of ligand binding and structure change in the diphtheria toxin repressor upon activation by divalent transition metals. Biochemistry. 2005;44:5672–5682. doi: 10.1021/bi047825w. [DOI] [PubMed] [Google Scholar]
- 15.D'Aquino JA, Tetenbaum-Novatt J, White A, Berkovitch F, Ringe D. Mechanism of metal ion activation of the diphtheria toxin repressor DtxR. Proc Natl Acad Sci USA. 2005;102:18408–18413. doi: 10.1073/pnas.0500908102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Twigg PD, Parthasarathy G, Guerrero L, Logan TM, Caspar DLD. Disordered to ordered folding in the regulation of diphtheria toxin repressor activity. Proc Natl Acad Sci USA. 2001;98:11259–11264. doi: 10.1073/pnas.191354798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glasfeld A, Guedon E, Helmann JD, Brennan RG. Structure of the manganese-bound transporter of Bacillus subtilis. Nature Struct Biol. 2003;10:562–657. doi: 10.1038/nsb951. [DOI] [PubMed] [Google Scholar]
- 18.Kliegman JI, Griner SL, Helmann JD, Brennan RG, Glasfeld A. Structural basis for the metal-selective activation of the manganese transport regulator of Bacillus subtilis. Biochemistry. 2006;45:3493–3505. doi: 10.1021/bi0524215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brennan RG. The winged-helix DNA-binding motif: another helix-turn-helix takeoff. Cell. 1993;74:773–776. doi: 10.1016/0092-8674(93)90456-z. [DOI] [PubMed] [Google Scholar]
- 20.Golynskiy MV, Davis TC, Helmann JD, Cohen SM. Metal-induced structural organization and stabilization of the metalloregulatory protein MntR. Biochemistry. 2005;44:3380–3389. doi: 10.1021/bi0480741. [DOI] [PubMed] [Google Scholar]
- 21.Farrens DL. Site-directed spin-labeling (SDSL) studies of the G-protein couple receptor Rhodopsin. In: Strader C, Sibley D, Wess J, editors. Receptor Biochemistry and Methodology. Vol. 3. Wiley-Liss, Inc; Wilmington, DE: 1999. pp. 289–314. Structure-Function Analysis of G-protein -Coupled Receptors. [Google Scholar]
- 22.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nature Struct Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 23.Brünger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallog sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 24.Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallog sect D. 1999;55:484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- 25.Zwart PH, Grosse-Kunstleve RW, Adams PD. Characterization of X-ray data sets. CCP4 newsletter. 1999;42 [Google Scholar]
- 26.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallog sect D. 1997;54:1285–1294. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 27.Winn M, Isupov M, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallog sect D. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 28.Wriggers W, Schulten K. Protein domain movements: detection of rigid domains and visualization of hinges in comparisons of atomic coordinates. Proteins: Struct Funct Genet. 1997;29:1–14. [PubMed] [Google Scholar]
- 29.Doublié S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–529. [PubMed] [Google Scholar]
- 30.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992;26 [Google Scholar]
- 31.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallog sect D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallog sect D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones TA, Zhou J, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallog sect A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallog sect D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallog. 1993;26:283–291. [Google Scholar]
- 36.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]