

Abstract
Novel phage-displayed random linear dodecapeptide (X12) and cysteine-constrained decapeptide (CX10C) libraries constructed in fusion to the amino-terminus of P99 β-lactamase molecules were used for identifying β-lactamase-linked cancer cell-specific ligands. The size and quality of both libraries were comparable to the standards of other reported phage display systems. Using the single-round panning method based on phage DNA recovery, we identified severalβ-lactamase fusion peptides that specifically bind to live human breast cancer MDA-MB-361 cells. The β-lactamase fusion to the peptides helped in conducting the enzyme activity-based clone normalization and cell-binding screening in a very time- and cost-efficient manner. The methods were suitable for 96-well readout as well as microscopic imaging. The success of the biopanning was indicated by the presence of ~40% cancer cell-specific clones among recovered phages. One of the binding clones appeared multiple times. The cancer cell-binding fusion peptides also shared several significant motifs. This opens a new way of preparing and selecting phage display libraries. The cancer cell-specific β-lactamase-linked affinity reagents selected from these libraries can be used for any application that requires a reporter for tracking the ligand molecules. Furthermore, these affinity reagents have also a potential for their direct use in the targeted enzyme prodrug therapy of cancer.
Keywords: Phage display, β-lactamase, peptide fusion libraries, cancer cell-specific ligands, targeted enzyme prodrug therapy, enzyme reporter
1. Introduction
The major problem in the treatment of cancer is the lack of a therapy that specifically attacks tumor cells. Developing medications that can target cancer cells without harming the rest of the body is one of the most important challenges for cancer researchers today. The field of targeted therapy is providing an entirely new generation of anticancer drugs that are highly promising (Slamon et al., 2001; Hurwitz et al., 2004; Mass et al., 2005; Dancey & Chen, 2006). The addition of targeted agents such as transtuzumab (Herceptin, Genentech/Roche) and bevacizumab (Avastin, Genentech/Roche) to a standard chemotherapy regimen has shown improved survival in patients with breast cancer (Slamon et al., 2001) and with colorectal cancer (Hurwitz et al., 2004), respectively. It is believed that an ability to focus on cancer cells makes targeted therapy both more effective and less likely to cause side effects. A variety of strategies are being evaluated to achieve this goal (Allen, 2002; Crown & Pegram, 2003; Neri & Bicknell, 2005; Pegram et al., 2005). Targeted therapies are generally based on identifying and exploiting specific differences between cancer cells and normal cells. One of the successful strategies requires developing the ligand molecules that specifically bind to cancer cells. These cancer-specific ligands may have therapeutic effects of their own and also can be used for targeted delivery of cytotoxic drugs, genes, or enzymes at the cancer site (Fuchs & Bachran, 2009).
Delivering an enzyme to cancer cells with the help of cancer-specific ligands is an important step in the targeted enzyme prodrug therapy of cancer, which is based on the concept that a systemically administered non-toxic prodrug can be converted locally to high concentrations of a cytotoxic drug by an enzyme predominantly present at the tumor site (Bagshawe, 2009). The development of molecules that bind to cancer cells and not to normal cells is challenging as most cancer cells share many common features with the normal host cells from which they are derived. There are now unprecedented opportunities for increased throughput to develop novel molecules that target cancer. Phage display is one example of a high throughput method of generating massive libraries of molecules that have different structural properties (Smith, 1985; Irving, Pan, & Scott, 2001; Hoogenboom, 2002; Van Beijnum et al., 2002). Phage display libraries have been found very useful in generating ligands against a large variety of targets, including tumor-associated antigens (Begent et al., 1996; Szardenings et al., 1997; Desai et al., 1998; Shukla & Krag, 2005b; Shukla & Krag, 2005a). Products derived from phage display technology have now been approved by the FDA for human use and are available as commercial agents (Shukla & Krag, 2006). In the past few years we have been developing strategies to develop tumor-targeting β-lactamase molecules β-lactam hydrolase, E.C. # 3.5.2.6), an enzyme candidate ideal for prodrug therapy (Kuhn et al., 2004). Our efforts were focused on engineering catalytically active Enterobacter cloacae P99 cephalosporinase (P99 β-lactamase) molecules with a built-in target binding site, using phage display technology (Shukla et al., 2007; Shukla & Krag, 2009). In these studies, we used a P99 β-lactamase scaffold to construct the libraries by inserting the random loops in the enzyme molecules in such a way that they are on the outer surface available for target contact, and, most importantly, that such alterations do not cause misfolding and the loss of enzyme activity. These restrictions pose very serious limitations to the size and type of the insertions. We have presently completed the work on the randomization of 5 selected loops of P99 β-lactamase molecule and none of them could accept cysteine-constrained peptides without a dramatic loss of the enzyme-active clones (Shukla & Krag, 2009).
In order to deal with these problems, we have developed novel phage display random peptide libraries in fusion to the N-terminal end of P99 β-lactamase. β-Lactamase fusion to the selected ligands from this library serves not only as a potent enzyme candidate for their use in prodrug therapy but also as an excellent reporter for their biological studies. In the published protocols, the selected cancer-specific ligands from phage display selections are usually conjugated with a reporter for their cell-binding and other studies and later to a selected enzyme for the prodrug therapy. These processes involve peptide synthesis and chemical or biological modifications of the ligands that are costly and time-consuming and may also result in a ligand losing the binding to its target. The present study has shown a successful selection of phage-displayed β-lactamase-random peptide (linear and cysteine-constrained) fusion libraries on live cancer cells for identifying cancer-cell specific ligands.
2. Materials and methods
2.1. Cell culture
Human breast cancer cell line MDA-MB-361, human non-cancer breast epithelial cell line MCF 10A, and mouse fibroblast cell line 3T3 were obtained from American Type Culture Collection (ATCC; Manassas, Virginia). MDA-MB-361 cells were cultured in Leibovitz's L-15 Medium (ATCC) containing 20% fetal bovine serum. MCF 10A were propagated in Clonetics® mammary epithelial cell growth medium (MEGM®, Lonza group Ltd., Basel, Switzerland) containing 52 μg/ml BPE, 0.5 μg/ml hydrocortisone, 10 ng/ml hEGF, 5 μg/ml insulin, and 100 ng/ml cholera toxin (Calbiochem, Gibbstown, New Jersey). 3T3 cells were grown in Dulbecco's Modified Eagle's Medium (ATCC) containing 10% bovine calf serum. The cells were cultured in a humid atmosphere with 5% CO2 at 37°C.
2.2. β-Lactamase-random peptide fusion libraries
A schematic presentation of the vector design that was used for constructing the libraries is given in Figure 1. The presence of chloramphenicol acetyl transferase and P99 β-lactamase help in the selection of the vector-transformed bacterial host against chloramphenicol and cefotaxime, respectively. Random peptide libraries were attached to the N-terminal end of P99 β-lactamase with the help of the (Gly)3-Ser linker. The FLAG (Asp-Tyr-Lys-(Asp)4-Lys) and hexahistidine tags were cloned at the C-terminal end of the P99 β-lactamase with the Gly-(Ala)2 linkers between them. The vector has an amber stop at the C-terminal end of 6xHis tag for producing the peptide-β-lactamase-FLAG-6xHis fusion protein in a non-suppressor bacterial host.
Figure 1.

(A) Schematic presentation of phagemid vector design. The vector expressed peptide random libraries at the amino-terminus of P99 β-lactamase. This vector in suppressor host TG1 E. coli, with the help of KM13 helper phage, generated phage particles displaying β-lactamase-random peptide as an N-terminal fusion protein to phage coat pIII. In non-suppressor strain Top10 E. coli, which recognizes the amber stop (tag), the same vector expressed free β-lactamase protein with a random peptide library at the N-terminal end and FLAG and 6xHis tags at the C-terminal end. (B) This magnified view of the encircled portion of the vector shows the cloned insert piece at AgeI/SpeI digested site. The linear dodecapeptide (X12) and cysteine-constrained decapeptide (CX10C) libraries were created between the β-lactamase (BLA) and pIII signal peptide (PSP). The random peptides were linked to the N-terminal end of the β-lactamase molecule with (Gly)3-Ser linker.
Two random peptide libraries, the linear dodecapeptide (X12) and the cysteine-constrained decapeptide (CX10C), were constructed in fusion to P99 β-lactamase molecules. The gel-purified oligonucleotides for this study were provided by Biosynthesis (Lewisville, Texas). The half-site cloning method (Cwirla et al., 1990) was employed for constructing the β-lactamase fusion libraries using the nnk-scheme of randomization. The randomization was achieved in 2 steps in order to avoid library clones without an insert. In the first step, a set of annealed 5ƍ-phosphorylated oligonucleotides containing stop-codons were cloned at the place of intended randomization. In the second step, the vector containing stop-codon insert was purified, digested with SpeI and AgeI, and cloned with the set of 5′-phosphorylated oligonucleotides with the random region (forward strand, 5′ctagtcgttcctttctattctcactctgct(nnk)12ggtggaggttcgaca3′; complementary, 5′agcagagtgagaatagaaaggaa cga3′ and 5′ccggtgtcgaacctccacc3′) for the linear dodecapeptide (X12) library. The oligonucleotides for the cysteine-constrained (CX10C) library were the same as above, except the forward strand had tgt(nnk)10tgc at the place of (nnk)12. Thirty transformations were performed with the TG1 bacterial cells (≥1×1010 transformations/μg; Stratagene, La Jolla, California) using an electroporator (Bio-Rad Labs, Hercules, California). The β-lactamase-active clones were selected on LB/Agar bio-dishes (BD Biosciences, Acton, Massachusetts) containing 10 μg/ml chloramphenicol and 0.1 μg/ml cefotaxime. All the cefotaxime-selected clones had random peptide expression ‘in-frame’ because the libraries were cloned at the amino-terminus of β-lactamase. The phage libraries were produced following the super-infection with KM13 helper phage (Kristensen & Winter, 1998).
2.3. Biopanning of the fusion libraries on the live human breast cancer cells
Human breast cancer MDA-MB-361 cells and human non-cancer epithelial MCF 10A cells were plated in a 35-mm CellBIND® dish and T25 flask (Corning Incorporated, Corning, New York) and allowed to grow to about 80% confluency. The original medium was changed to Dulbecco's Modification of Eagle's Medium (DMEM; ATCC) containing BSA (1 mg/ml; Sigma Chemical Co., St. Louis, Missouri) and the cells were incubated for 2 hours at 37°C in a humidified, 5% CO2 atmosphere. The mixture (1:1) of phage particles from both linear and cysteine-constrained libraries in DMEM containing BSA and mammalian protease inhibitor cocktail (20 μl/ml; Sigma Chemical Co.) was subtracted for plastic binding clones by incubation in a CellBIND® flask. The plastic-subtracted libraries were added to the MCF 10A cells and incubated for 2 hours at room temperature for the negative subtraction of the clones binding to normal breast epithelial cells. The subtracted libraries were incubated with live MDA-MB-361 cells for 2 hours at room temperature. The cells were washed 5 times with DMEM containing BSA and protease inhibitors for a total time of 1 hour. The cells were gently scraped and transferred into a tube. The tube was centrifuged and the cells were suspended in 50 μl sample buffer (10 mM Tris-HCl, 0.5 mM EDTA, pH 8.2) containing random hexamer primers and heated at 95°C for 3 min. The supernatant collected by centrifugation was mixed with 50 μl buffered dNTPs and 2 μl φ 29 DNA polymerase enzyme (TempliPhi kit; GE Healthcare, Little Chalfont, UK) and incubated overnight at 30°C. The next day, the enzyme was inactivated by heating the tubes at 60°C for 10 minutes. The amplified DNA was subjected to BstAPI digestion, and T4 DNA ligation, and the phagemids recovered from this process were used for the electrotransformation of non-suppressor TOP10 cells (Invitrogen) (Shukla et al., 2007). The isolated colonies on LB/agar plate containing chloramphenicol and cefotaxime were randomly selected, amplified, and processed for preparing bacterial lysates as described earlier (Shukla et al., 2007; Shukla & Krag, 2009). The lysates of selected clones were purified for the β-lactamase-peptide fusion proteins using nickel-sepharose columns (GE Health Care, Bio-sciences AB, Uppsala, Sweden).
2.4. Anti-FLAG ELISA of recombinant β-lactamase-peptide fusion protein
Different dilutions of the β-lactamase preparations (100 μl) were added to the 96-well Ni-NTA microplates (HisSorb™; Qiagen, Valencia, California) and incubated overnight in cold. The wells were washed 4 times with PBST and blocked with 1% casein solution (Thermo Fisher Scientific, Rockford, Illinois) for 1 hour at room temperature. After washing the wells 2 times with PBS, 100 μl FLAG antibody-HRP conjugate (Sigma Chemical Co., St. Louis, Missouri), diluted 1:20000, was added and incubated for 1 hour at room temperature. After washing 4 times with PBST and 2 times with PBS, the luminescence was developed with SuperSignal® ELISA kit (Thermo Fisher Scientific, Rockford, Illinois) for horse radish peroxidase. The luminescence was read using GloRunner™ microplate luminometer (Turner BioSystems, Sunnyvale, California).
2.5. β-Lactamase enzyme activity assays
The β-lactamase enzyme activities were assayed in different preparations by colorimetric and fluorometric methods, using nitrocefin (Oxoid, Basingstoke, UK) and soluble fluorocillin green (Invitrogen, Carlsbad, California) as substrates, respectively, as described earlier (Shukla et al., 2007; Shukla & Krag, 2009). Briefly, the assay was conducted in 96-well microplates by mixing 20 μl of β-lactamase preparation with 100 μl solution of either nitrocefin (100 μg/ml PBS containing 0.125% n-octyl-β-D-glucopyranoside) or fluorocillin green (10 μg/ml PBS). The changes in the optical densities (490 nm) or fluorescence intensities (~495Ex/525 Em) were measured at 2-minute intervals for 10 minutes using Synergy-HT microplate reader (Biotek Instruments, Winooski, Vermont). The activity was calculated from the data falling within the linear range. While the colorimetric method was employed for the normalization of different β-lactamase preparations, the fluorometric assay was the method of choice for the clone binding studies. The enzyme stability of the β-lactamase-peptide fusion proteins was tested by assaying the β-lactamase activities of different clones following 4 hours incubation at 37°C.
2.6. Clone screening for their binding to cancer cells
Thirty thousand MDA-MB-361, MCF 10A, or 3T3 cells were plated in 96-well CellBIND® culture plates (Corning) and incubated overnight at 37°C in a humidified, 5% CO2 atmosphere. The next day, original medium was changed to Dulbecco's Modification of Eagle's Medium (DMEM; ATCC) containing BSA (1 mg/ml; Sigma Chemical Co., St. Louis, Missouri) and the cells were incubated for 2 hours at 37°C. The β-lactamase-peptide fusion samples selected for screening were normalized by diluting in DMEM with BSA and protease inhibitors. One-hundred μl solutions of β-lactamase-peptide fusion proteins were added to the wells and the plate was incubated at room temperature for 2 hours. The medium was removed and the wells were washed 3 times with DMEM and 2 times with Dulbecco's PBS containing Ca2+ and Mg2+ (Mediatech, Inc., Herndon, Virginia). Using soluble fluorocillin substrate (Invitrogen), the clone binding was assessed fluorometrically (Ex. 495/Em. 525 nm) by assaying the residual β-lactamase enzyme activity associated with the cells. MCF 10A and 3T3 cells served as negative cell-controls in this screening. A pool of β-lactamase-peptide fusions from the unselected libraries and the wild-type β-lactamase molecules (without a peptide fusion) were run in parallel as negative β-lactamase controls.
The clones that showed positive binding to live MDA-MB-361 cells were also tested for their binding to fixed cells. The above protocol was adopted for these studies in which the cells were fixed with 0.5% buffered-paraformaldehyde, washed and blocked with 1% Hammerstein grade casein (Thermo Scientific, Rockford, Illinois) for 1 hour before their incubation with purified β-lactamase-peptide clones. In a separate set of experiments, the binding of positive clones was further confirmed by microscopic studies. The MDA-MB-361 cancer cells and non-cancer breast epithelial MCF 10A cells were exposed to the purified β-lactamase-peptide fusion protein preparations and after washing the residual β-lactamase were detected by using a precipitating fluorocillin green substrate (Invitrogen). This β-lactamase substrate has been developed specifically for microscopy as its fluorescent enzyme product is insoluble and gets precipitated. The cells were observed under a Nikon TE2000-U inverted fluorescence microscope (Nikon Corp., Kanagawa, Japan) using 360±20 nm excitation, 535±20 nm emission, and 400 nm dichroic filters.
2.7. DNA sequencing, library quality analysis, and consensus sequence motif determination
The DNA of the vectors after each cloning and that of the inserts of MDA-MB-361 cell-binding clones were sequenced using appropriate primers. The plasmids were purified using QIAprep® miniprep columns (Valencia, California). The cycle sequencing reactions were performed using BigDye® Terminator version 3.1 kit (Applied Biosystems, Foster City, California). The DNA sequencing was carried out by the Vermont Cancer Center DNA Analysis Facility at the University of Vermont using an automated ABI Prism® 3130xl Genetic Analyzer. The quality assessment of linear and cysteine-constrained libraries was done by analyzing the overall and positional diversities of amino acids in the random region of both libraries using receptor ligands contacts (RELIC) program (Mandava et al., 2004). The translated amino acids of N-terminal fusion peptides in MDA-MB-361 cell-binding β-lactamase clones were searched for significant motifs using the IBM Sequence Pattern Discovery Tool software program based on the TEIRESIAS algorithm (Rigoutsos & Floratos, 1998).
3. Results
The linear dodecapeptide and cysteine-constrained decapeptide libraries had ~5×107 and ~6×107 primary β-lactamase-active clones, respectively. DNA sequencing of the analyzed clones showed no β-lactamase molecule without a fusion peptide. The observed frequencies of most of the amino acids in both libraries were close to their calculated frequencies. The results also showed high amino acid diversities at different positions in the random region. A low diversity was observed at position 1 of both libraries, indicating a preference for certain amino acids at this position. These results indicate that the overall and positional diversity of different amino acids in the random region of both libraries was fairly good. Randomly selected library clones exhibited strong β-lactamase enzyme activities in their purified phage particles, pIII-conjugated and pIII-independent β-lactamase-peptide fusion protein preparations. There were no remarkable differences in the ratios of enzyme activity to β-lactamase protein levels in different clones and their expression yield and stability were comparable to β-lactamase enzyme without peptide fusion (data not presented).
3.1. Binding of the selected clones to cancer cells
The binding results of randomly selected clones to different cell lines are presented in Figure 2. Twenty-three of the 58 screened clones (~40%) showed significantly higher binding to the live human breast cancer MDA-MB-361 cells against which they were panned, in comparison to the live non-cancer human breast epithelial MCF 10A cells and unrelated 3T3 cells. The mouse fibroblast 3T3 cells were used as an irrelevant negative cell line. The cancer cell-binding of these clones was also significantly higher as compared to the β-lactamase-peptide fusion proteins from a pool of unselected library clones and to the wild-type β-lactamase, which were used as negative clone controls. The clones which were found to bind live MDA-MB-361 cells showed significantly higher binding to the paraformaldehyde-fixed cancer cells as well. Furthermore, these clones did not show binding to casein blocker or well plastics (data not presented). Using another β-lactamase substrate that forms green fluorescent precipitate following the enzymatic action, the bindings of positively screened clones were confirmed by microscopic studies. These experiments showed higher binding of these clones to breast cancer MBA-MB-361 cells in comparison to non-cancer breast epithelial MCF 10A cells. The results of the imaging experiments are represented by the photomicrographs (Figure 3) from the cell-binding studies of clones 2415, 1982, 1987, 2436 and 1996.
Figure 2.

Clone screening of their binding to live human breast cancer cells. Randomly selected 58 clones following panning were screened for their binding to live MDA-MB-361 cells. The human non-cancer MCF 10A epithelial cells and mouse fibroblast 3T3 cell-line were used as negative control cells. The screening was based on assaying the cell-bound β-lactamase activities. The clones expressing wild-type β-lactamase (Wt-BLA; without fusion peptide) and unselected library were used as negative clone controls. The bars represent β-lactamase activity as change in relative fluorescence unit (RFU)/min ± SE. *p≤0.05, significance of difference in the binding of clones to MDA-MB-361 cells in comparison to MCF 10A or 3T3 cells were determined using Student's T-test.
Figure 3.

Fluorescence imaging of human breast cancer cells for their binding to the positive clones identified from primary screening. The precipitating green fluorescence was produced by the β-lactamase activity associated with the cells. Figure shows high binding of the selected representative clones 2415 (a,b), 1982 (c,d), 1987 (e,f), 2436 (g,h), 1996 (i,j) in human breast cancer MDA-MB-361 cells in comparison to human non-cancer breast MCF 10A cells. The pool of unselected library clones (k,l), which was used as a negative clone control, did not show any significant binding to both cell lines.
3.2. Identity, frequency, and consensus sequence motifs of cancer cell-binding clones
The amino acid sequences at the amino-terminus of β-lactamase and their frequency of occurrence among the MDA-MB-361 cancer cell-binding clones are presented in Table 1. There were 15 unique sequences in the 23 cancer cell-binding fusion peptides; 7 of them were cyclic peptides. The fusion peptide sequence GLWWHGAGISLI appeared 9 times.
Table 1.
Amino acid sequences of dodecamer linear (X12) and cysteine-constrained decamer (CX10C) peptides at the amino-terminus of β-lactamase clones that bind to human breast cancer MDA-MB-361 cells.
| Clone numbers |
Amino acid sequences |
Clone numbers |
Amino acid sequences |
|---|---|---|---|
| 2416, 2417, 2422, 2426, 2430, 2432, 2433, 2435, 2436 |
GLWWHGAGISLI | 1998 | GSAVCVWSPRHY |
| 2000 | CGGMCRFLVGCC | ||
| 1982 | HCCFTGMVTRLV | 2001 | CEIFRSGVGMRC |
| 1984 | CGKGWVSLVGSC | 2002 | CREMGYLNWLAC |
| 1987 | CCALVGGGSVYC | 2005 | WGLQALVLSKKG |
| 1995 | AGGVCSFIFRVS | 2008 | CGFWGVWARWGC |
| 1996 | SLLAGSWACGVF | 2415 | CAVLWPILGVVC |
| 1997 | WLLDSFSALPKC | 2431 | LFACSVRCTPLW |
Amino acids are represented by their single-letter codes.
The alignments of the fusion peptide sequences from the cancer cell-binding clones revealed several significant consensus sequence motifs (Table 2). The motifs [V/L]X[A/G]XSXXC, LVG, [A/G][V/M]C, GXW[A/G], G[V/M]XXR, and WX[V/L/I]X[A/G] are shared by 3 peptides. The consensus sequences GG[V/M]CXF[I/L], [V/L]X[A/G]XW[A/G]C, GXW[A/G]XW[A/G] and [A/G]X[A/G]VCXXXXR are examples of some of the other significant motifs shared by these β-lactamase fusion peptides.
Table 2.
Consensus sequence motifs among the β-lactamase fusion peptides that were found to specifically bind human breast cancer MDA-MB-361 cells.
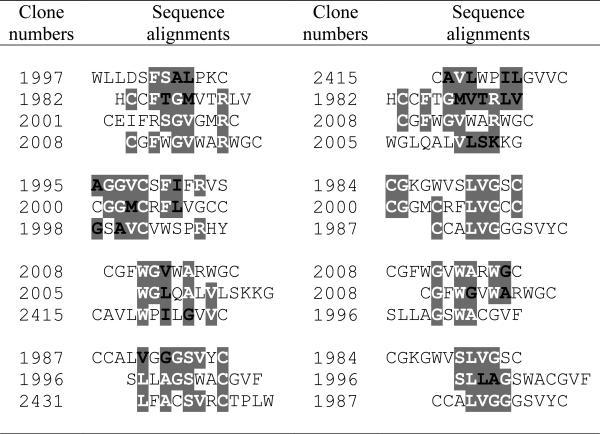
|
Amino acids sharing consensus motifs are written in bold and highlighted; white letters= exactly the same, black letters=conservative substitution. Amino acid sequences are represented by their single-letter codes.
4. Discussion
The identification of cancer target-specific ligands is an important step in developing various targeted therapies of cancer. Phage display technology has been recently utilized in a very effective way for developing ligands against a variety of targets, including cancer-specific antigens (Begent et al., 1996; Szardenings et al., 1997; Desai et al., 1998; Shukla & Krag, 2005b; Shukla & Krag, 2005a; Shukla & Krag, 2006). The ligands selected from a typical phage display process require a chemical or biological conjugation with reporter molecules, such as enzymes, biotin, fluorescent molecules, or radionuclides for their binding and kinetics studies, tumor imaging, and for developing targeted cancer therapeutics. For the last few years we have focused on constructing and selecting phage display libraries for identifying cancer cell-specific ligands that do not require their conjugation with a reporter and that can be used directly in a variety of applications. Using a phage display approach, we made an effort to engineer bifunctional β-lactamase molecules, which retain catalytic activity and have a random peptide library embedded within their scaffold (Shukla et al., 2007; Shukla & Krag, 2009). The construction of these libraries requires modifications in the structure of β-lactamase molecules without incurring a loss in their enzyme activity. This severely restricts the length and the type of random peptides that can be inserted into different β-lactamase loops. In our experience, the insertion of only small linear random peptides of 7 to 8 amino acids into 2 of the 5 tested P99 β-lactamase loops could successfully generate the size and quality of catalytically-active β-lactamase libraries that were suitable for the phage-display selection. The insertions of cysteine-constrained peptides at these sites were simply incompatible with the P99 β-lactamase protein folding required for its optimal enzyme activity. The present fusion libraries are a practical solution to these problems.
In the present investigation, we used novel phage display β-lactamase-random peptide fusion libraries for identifying β-lactamase-linked cancer cell-specific ligands. In these libraries, the random peptides are not embedded within the enzyme molecule scaffold but are in fusion to the N-terminal end of P99 β-lactamase molecules through a linker. The size and quality of both linear and cysteine-constrained random peptide libraries constructed in fusion to P99 β-lactamase enzyme molecules were comparable to the standards of other reported phage display systems (Rodi, Soares, & Makowski, 2002). The size of the libraries can be further improved by modifying the vector design and increasing the efficiency of bacterial transformation. The expression yields, specific activity, and stability of β-lactamase in fusion to peptides were not affected. It may be attributed to the fact that, unlike our previously reported β-lactamase-scaffold libraries (Shukla et al., 2007; Shukla & Krag, 2009), the random peptides in the present libraries are not embedded within the enzyme molecules. Using the single-round panning method based on phage DNA recovery, we identified several β-lactamase fusion peptides that specifically bind to live human breast cancer MDA-MB-361 cells. The success of biopanning was indicated by the presence of ~40% cancer cell-specific clones among recovered phages, with one of the binding clones appearing multiple times. The success rate of positive hits in the present study with β-lactamase fusion libraries is comparable to those usually reported from conventional phage display libraries (Shukla & Krag, 2005a; Shukla & Krag, 2005b). The successful single-round panning involving a phage recovery from the amplified DNA has been reported earlier also (Tanaka et al., 2002; Naik et al., 2004; Shukla et al., 2007). The selected clones did not show binding to human non-cancer breast epithelial cells (MCF 10A), an unrelated mouse fibroblast (3T3) cell line, or non-specific proteins. Furthermore, these cancer cell-binding fusion peptides also shared several significant motifs, which show their selectivity and possible binding to some common cancer cell targets. Although we used single-round panning method with phage DNA recovery protocol in the present study, the vector designs of these β-lactamase-peptide fusion libraries are also compatible with the conventional multiple rounds of phage display selection protocols based on the phage recovery by bacterial host infection.
The presence of the β-lactamase fusion molecule with each peptide enabled us to conduct the enzyme activity-based clone normalization and cell-binding screening in a very time- and cost-efficient manner. Unlike phage-ELISA of a typical phage display screening protocol, the β-lactamase enzyme-based screening did not require antibodies, secondary reagents, or several lengthy incubations and washing steps. β-Lactamase is an excellent reporter with several available chromogenic and fluorogenic substrates (O'Callaghan et al., 1972; Jones et al., 1982; Gao et al., 2003; Xing, Khanamiryan, & Rao, 2005). In the present investigation, a highly sensitive fluorometric β-lactamase assay that had a dynamic range of over 3 orders of magnitude was used for primary cell-binding screening. The results were confirmed by microscopic studies using another β-lactamase substrate that forms fluorescent product precipitate. The selected β-lactamase-peptide ligands in the present study can be used for any application that requires a reporter for tracking the molecules. β-Lactamase-peptide fusion proteins can be readily produced in bulk and purified (Cartwright & Waley, 1984). This eliminates the need of peptide synthesis and their chemical conjugation with a reporter for their applications. Since the selected β-lactamase-peptide ligands from the present libraries are derived from the screening of final peptide-reporter products, no questions are raised about whether a phage-independent synthetic peptide will not bind to the target or whether its chemical conjugation to a reporter will interfere with the binding.
β-Lactamase enzyme has also been used for antibody-directed enzyme prodrug therapy (ADEPT) in experimental animals (Meyer et al., 1993; Vrudhula et al., 1993; Kerr et al., 1995; Svensson et al., 1995; Vrudhula, Svensson, & Senter, 1995). In this type of targeted therapy, the β-lactamase enzyme protein is chemically (Meyer et al., 1993; Svensson et al., 1995) or biologically (Rodrigues et al., 1995; McDonagh et al., 2003; Cortez-Retamozo et al., 2004; Alderson et al., 2006) conjugated to a cancer-specific ligand (usually an antibody) for delivering the enzyme to the tumor site. The presence of β-lactamase enzyme predominantly at the tumor site amplifies availability of the active drug concentrations locally following a cephalosporin prodrug infusion. The β-lactamase-induced hydrolytic cleavage of the β-lactam ring in the prodrugs triggers a secondary reaction that releases the anticancer drug at the C-3′ position of the cephalosporin nucleus. Cephalosporin prodrugs of mechanistically diverse anticancer agents such as doxorubicin (Svensson et al., 1995), taxol (Vrudhula et al., 2003), platinum complexes (Hanessian & Wang, 1993), phenylenediamine mustard (Kerr et al., 1995), and vinblastine (Meyer et al., 1993) have been successfully used with β-lactamase in experimental studies.
The cancer cell-specific ligands selected from β-lactamase-random peptide fusion libraries can be used directly for the targeted enzyme prodrug therapy. Furthermore, the smaller size of these cancer-specific β-lactamase-peptide ligands will facilitate better tumor delivery and avoid the issues related to the large size of antibodies, which include a slow conjugate clearance from blood and normal tissues and limited delivery in the poorly vascularized tumors (Martin et al., 1997; Denny & Wilson, 1998; Senter & Springer, 2001). P99 β-lactamase, being a bacterial protein, is likely to be immunogenic in humans. However, the P99 β-lactamase-specific CD4+ T-cell epitopes have been identified and their removal in the variants showed significantly lower immunogenicity in mice (Harding et al., 2005). Using an appropriate substrate, the cancer-specific β-lactamase-peptide ligands can also be used for in vivo tumor imaging (Yao, So, & Rao, 2007).
In conclusion, the present study has shown that good quality phage-displayed β-lactamase-random peptide (linear and cysteine-constrained) fusion libraries can be constructed and selected against cancer cells. This opens a new way of preparing and selecting phage display libraries. The presence of β-lactamase fusion not only helps in accelerating the clone screening process but can also be useful for tracking the ligand molecule in a variety of biological studies. The cancer-specific β-lactamase-linked affinity reagents selected from such libraries have a potential for direct use in targeted enzyme prodrug therapy.
5. Acknowledgements
This work was supported by the National Institutes of Health [RO1CA112091] and the SD Ireland Cancer Research Fund.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
6. References
- Alderson RF, Toki BE, Roberge M, Geng W, Basler J, Chin R, Liu A, Ueda R, Hodges D, Escandon E, Chen T, Kanavarioti T, Babe L, Senter PD, Fox JA, Schellenberger V. Characterization of a CC49-based single-chain fragment-beta-lactamase fusion protein for antibody-directed enzyme prodrug therapy (ADEPT) Bioconjug Chem. 2006;17:410–418. doi: 10.1021/bc0503521. [DOI] [PubMed] [Google Scholar]
- Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750–763. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- Bagshawe KD. Targeting: the ADEPT story so far. Curr Drug Targets. 2009;10:152–157. doi: 10.2174/138945009787354520. [DOI] [PubMed] [Google Scholar]
- Begent RH, Verhaar MJ, Chester KA, Casey JL, Green AJ, Napier MP, Hope-Stone LD, Cushen N, Keep PA, Johnson CJ, Hawkins RE, Hilson AJ, Robson L. Clinical evidence of efficient tumor targeting based on single-chain Fv antibody selected from a combinatorial library. Nat Med. 1996;2:979–984. doi: 10.1038/nm0996-979. [DOI] [PubMed] [Google Scholar]
- Cartwright SJ, Waley SG. Purification of beta-lactamases by affinity chromatography on phenylboronic acid-agarose. Biochem J. 1984;221:505–512. doi: 10.1042/bj2210505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez-Retamozo V, Backmann N, Senter PD, Wernery U, De Baetselier P, Muyldermans S, Revets H. Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 2004;64:2853–2857. doi: 10.1158/0008-5472.can-03-3935. [DOI] [PubMed] [Google Scholar]
- Crown J, Pegram M. Platinum-taxane combinations in metastatic breast cancer: an evolving role in the era of molecularly targeted therapy. Breast Cancer Res Treat. 2003;79(Suppl 1):S11–18. doi: 10.1023/a:1024373306493. [DOI] [PubMed] [Google Scholar]
- Cwirla SE, Peters EA, Barrett RW, Dower WJ. Peptides on phage: a vast library of peptides for identifying ligands. Proc Natl Acad Sci U S A. 1990;87:6378–6382. doi: 10.1073/pnas.87.16.6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancey JE, Chen HX. Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat Rev Drug Discov. 2006;5:649–659. doi: 10.1038/nrd2089. [DOI] [PubMed] [Google Scholar]
- Denny WA, Wilson WR. The design of selectively-activated anticancer prodrugs for use in antibody-directed and gene-directed enzyme-prodrug therapies. J Pharm Pharmacol. 1998;50:387–394. doi: 10.1111/j.2042-7158.1998.tb06878.x. [DOI] [PubMed] [Google Scholar]
- Desai SA, Wang X, Noronha EJ, Kageshita T, Ferrone S. Characterization of human antihigh molecular weight-melanoma-associated antigen single-chain Fv fragments isolated from a phage display antibody library. Cancer Res. 1998;58:2417–2425. [PubMed] [Google Scholar]
- Fuchs H, Bachran C. Targeted tumor therapies at a glance. Curr Drug Targets. 2009;10:89–93. doi: 10.2174/138945009787354557. [DOI] [PubMed] [Google Scholar]
- Gao W, Xing B, Tsien RY, Rao J. Novel fluorogenic substrates for imaging beta-lactamase gene expression. J Am Chem Soc. 2003;125:11146–11147. doi: 10.1021/ja036126o. [DOI] [PubMed] [Google Scholar]
- Hanessian S, Wang J. Design and synthesis of a cephalosporin-carboplatinum prodrug activable by a E-lactamase. Can. J. Chem. 1993;71:896–906. [Google Scholar]
- Harding FA, Liu AD, Stickler M, Razo OJ, Chin R, Faravashi N, Viola W, Graycar T, Yeung VP, Aehle W, Meijer D, Wong S, Rashid MH, Valdes AM, Schellenberger V. A beta-lactamase with reduced immunogenicity for the targeted delivery of chemotherapeutics using antibody-directed enzyme prodrug therapy. Mol Cancer Ther. 2005;4:1791–1800. doi: 10.1158/1535-7163.MCT-05-0189. [DOI] [PubMed] [Google Scholar]
- Hoogenboom HR. Overview of antibody phage-display technology and its applications. Methods Mol Biol. 2002;178:1–37. doi: 10.1385/1-59259-240-6:001. [DOI] [PubMed] [Google Scholar]
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- Irving MB, Pan O, Scott JK. Random-peptide libraries and antigen-fragment libraries for epitope mapping and the development of vaccines and diagnostics. Curr Opin Chem Biol. 2001;5:314–324. doi: 10.1016/S1367-5931(00)00208-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RN, Wilson HW, Novick WJ, Jr., Barry AL, Thornsberry C. In vitro evaluation of CENTA, a new beta-lactamase-susceptible chromogenic cephalosporin reagent. J Clin Microbiol. 1982;15:954–958. doi: 10.1128/jcm.15.5.954-958.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr DE, Schreiber GJ, Vrudhula VM, Svensson HP, Hellstrom I, Hellstrom KE, Senter PD. Regressions and cures of melanoma xenografts following treatment with monoclonal antibody beta-lactamase conjugates in combination with anticancer prodrugs. Cancer Res. 1995;55:3558–3563. [PubMed] [Google Scholar]
- Kristensen P, Winter G. Proteolytic selection for protein folding using filamentous bacteriophages. Fold Des. 1998;3:321–328. doi: 10.1016/S1359-0278(98)00044-3. [DOI] [PubMed] [Google Scholar]
- Kuhn D, Coates C, Daniel K, Chen D, Bhuiyan M, Kazi A, Turos E, Dou QP. Beta-lactams and their potential use as novel anticancer chemotherapeutics drugs. Front Biosci. 2004;9:2605–2617. doi: 10.2741/1420. [DOI] [PubMed] [Google Scholar]
- Mandava S, Makowski L, Devarapalli S, Uzubell J, Rodi DJ. RELIC--a bioinformatics server for combinatorial peptide analysis and identification of protein-ligand interaction sites. Proteomics. 2004;4:1439–1460. doi: 10.1002/pmic.200300680. [DOI] [PubMed] [Google Scholar]
- Martin J, Stribbling SM, Poon GK, Begent RH, Napier M, Sharma SK, Springer CJ. Antibody-directed enzyme prodrug therapy: pharmacokinetics and plasma levels of prodrug and drug in a phase I clinical trial. Cancer Chemother Pharmacol. 1997;40:189–201. doi: 10.1007/s002800050646. [DOI] [PubMed] [Google Scholar]
- Mass RD, Press MF, Anderson S, Cobleigh MA, Vogel CL, Dybdal N, Leiberman G, Slamon DJ. Evaluation of clinical outcomes according to HER2 detection by fluorescence in situ hybridization in women with metastatic breast cancer treated with trastuzumab. Clin Breast Cancer. 2005;6:240–246. doi: 10.3816/CBC.2005.n.026. [DOI] [PubMed] [Google Scholar]
- McDonagh CF, Beam KS, Wu GJ, Chen JH, Chace DF, Senter PD, Francisco JA. Improved yield and stability of L49-sFv-beta-lactamase, a single-chain antibody fusion protein for anticancer prodrug activation, by protein engineering. Bioconjug Chem. 2003;14:860–869. doi: 10.1021/bc0340316. [DOI] [PubMed] [Google Scholar]
- Meyer DL, Jungheim LN, Law KL, Mikolajczyk SD, Shepherd TA, Mackensen DG, Briggs SL, Starling JJ. Site-specific prodrug activation by antibody-beta-lactamase conjugates: regression and long-term growth inhibition of human colon carcinoma xenograft models. Cancer Res. 1993;53:3956–3963. [PubMed] [Google Scholar]
- Naik RR, Jones SE, Murray CJ, McAuliffe JC, Vaia RA, Stone MO. Peptide-templates for nanoparticle synthesis derived from PCR-driven phage display. Adv. Func. Mater. 2004;14:25–30. [Google Scholar]
- Neri D, Bicknell R. Tumour vascular targeting. Nat Rev Cancer. 2005;5:436–446. doi: 10.1038/nrc1627. [DOI] [PubMed] [Google Scholar]
- O'Callaghan CH, Morris A, Kirby SM, Shingler AH. Novel method for detection of beta-lactamases by using a chromogenic cephalosporin substrate. Antimicrob Agents Chemother. 1972;1:283–288. doi: 10.1128/aac.1.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram MD, Pietras R, Bajamonde A, Klein P, Fyfe G. Targeted therapy: wave of the future. J Clin Oncol. 2005;23:1776–1781. doi: 10.1200/JCO.2005.11.029. [DOI] [PubMed] [Google Scholar]
- Rigoutsos I, Floratos A. Combinatorial pattern discovery in biological sequences: The TEIRESIAS algorithm. Bioinformatics. 1998;14:55–67. doi: 10.1093/bioinformatics/14.1.55. [DOI] [PubMed] [Google Scholar]
- Rodi DJ, Soares AS, Makowski L. Quantitative assessment of peptide sequence diversity in M13 combinatorial peptide phage display libraries. J Mol Biol. 2002;322:1039–1052. doi: 10.1016/s0022-2836(02)00844-6. [DOI] [PubMed] [Google Scholar]
- Rodrigues ML, Presta LG, Kotts CE, Wirth C, Mordenti J, Osaka G, Wong WL, Nuijens A, Blackburn B, Carter P. Development of a humanized disulfide-stabilized anti-p185HER2 Fv-beta-lactamase fusion protein for activation of a cephalosporin doxorubicin prodrug. Cancer Res. 1995;55:63–70. [PubMed] [Google Scholar]
- Senter PD, Springer CJ. Selective activation of anticancer prodrugs by monoclonal antibody-enzyme conjugates. Adv Drug Deliv Rev. 2001;53:247–264. doi: 10.1016/s0169-409x(01)00206-x. [DOI] [PubMed] [Google Scholar]
- Shukla GS, Krag DN. Selection of tumor-targeting agents on freshly excised human breast tumors using a phage display library. Oncol Rep. 2005a;13:757–764. PMID: 15756454. [PubMed] [Google Scholar]
- Shukla GS, Krag DN. Phage display selection for cell-specific ligands: development of a screening procedure suitable for small tumor specimens. J Drug Target. 2005b;13:7–18. doi: 10.1080/10611860400020464. PMID: 15848950. [DOI] [PubMed] [Google Scholar]
- Shukla GS, Krag DN. Selective delivery of therapeutic agents for the diagnosis and treatment of cancer. Expert Opin Biol Ther. 2006;6:39–54. doi: 10.1517/14712598.6.1.39. [DOI] [PubMed] [Google Scholar]
- Shukla GS, Krag DN. Developing bifunctional beta-lactamase molecules with built-in target-recognizing module for prodrug therapy: identification of Enterobacter Cloacae P99 cephalosporinase loops suitable for randomization and phage-display selection. J Mol Recognit. 2009;22:425–436. doi: 10.1002/jmr.957. PMID: 19437416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla GS, Murray CJ, Estabrook M, Shen GP, Schellenberger V, Krag DN. Towards a ligand targeted enzyme prodrug therapy: single round panning of a beta-lactamase scaffold library on human cancer cells. Int J Cancer. 2007;120:2233–2242. doi: 10.1002/ijc.22138. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- Svensson HP, Vrudhula VM, Emswiler JE, MacMaster JF, Cosand WL, Senter PD, Wallace PM. In vitro and in vivo activities of a doxorubicin prodrug in combination with monoclonal antibody beta-lactamase conjugates. Cancer Res. 1995;55:2357–2365. [PubMed] [Google Scholar]
- Szardenings M, Tornroth S, Mutulis F, Muceniece R, Keinanen K, Kuusinen A, Wikberg JE. Phage display selection on whole cells yields a peptide specific for melanocortin receptor 1. J Biol Chem. 1997;272:27943–27948. doi: 10.1074/jbc.272.44.27943. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Ito T, Furuta M, Eguchi C, Toda H, Wakabayashi-Takai E, Kaneko K. In situ phage screening. A method for identification of subnanogram tissue components in situ. J Biol Chem. 2002;277:30382–30387. doi: 10.1074/jbc.M203547200. [DOI] [PubMed] [Google Scholar]
- Van Beijnum JR, Moerkerk PT, Gerbers AJ, De Bruine AP, Arends JW, Hoogenboom HR, Hufton SE. Target validation for genomics using peptide-specific phage antibodies: a study of five gene products overexpressed in colorectal cancer. Int J Cancer. 2002;101:118–127. doi: 10.1002/ijc.10584. [DOI] [PubMed] [Google Scholar]
- Vrudhula VM, Svensson HP, Senter PD. Cephalosporin derivatives of doxorubicin as prodrugs for activation by monoclonal antibody-beta-lactamase conjugates. J Med Chem. 1995;38:1380–1385. doi: 10.1021/jm00008a016. [DOI] [PubMed] [Google Scholar]
- Vrudhula VM, Svensson HP, Kennedy KA, Senter PD, Wallace PM. Antitumor activities of a cephalosporin prodrug in combination with monoclonal antibody-beta-lactamase conjugates. Bioconjug Chem. 1993;4:334–340. doi: 10.1021/bc00023a005. [DOI] [PubMed] [Google Scholar]
- Vrudhula VM, Kerr DE, Siemers NO, Dubowchik GM, Senter PD. Cephalosporin prodrugs of paclitaxel for immunologically specific activation by L-49-sFv-beta-lactamase fusion protein. Bioorg Med Chem Lett. 2003;13:539–542. doi: 10.1016/s0960-894x(02)00935-6. [DOI] [PubMed] [Google Scholar]
- Xing B, Khanamiryan A, Rao J. Cell-permeable near-infrared fluorogenic substrates for imaging beta-lactamase activity. J Am Chem Soc. 2005;127:4158–4159. doi: 10.1021/ja042829+. [DOI] [PubMed] [Google Scholar]
- Yao H, So MK, Rao J. A bioluminogenic substrate for in vivo imaging of beta-lactamase activity. Angew Chem Int Ed Engl. 2007;46:7031–7034. doi: 10.1002/anie.200701931. [DOI] [PubMed] [Google Scholar]