

Abstract
We previously reported that the cancer drug clinical candidate tipifarnib kills the causative agent of Chagas disease, Trypanosoma cruzi, by blocking ergosterol biosynthesis at the level of inhibition of lanosterol 14α-demethylase. Tipifarnib is an inhibitor of human protein farnesyltransferase. We synthesized tipifarnib analogs that no longer bind to protein farnesyltransferase and display increased potency for killing parasites. This was achieved in a structure-guided fashion by changing the substituents attached to the phenyl group at the 4-position of the quinoline ring of tipifarnib and by replacing the amino group by OMe. Several compounds that kill Trypanosoma cruzi at sub-nanomolar concentrations and are devoid of protein farnesyltransferase inhibition were discovered. The compounds are shown to be advantageous over other lanosterol 14α-demethylase inhibitors in that they show only modest potency for inhibition of human cytochrome P450 (3A4). Since tipifarnib displays high oral bioavailability and acceptable pharmacokinetic properties, the newly discovered tipifarnib analogs are ideal leads for the development of drugs to treat Chagas disease.
Keywords: cytochrome P450, Chagas disease, drug discovery, sterol biosynthesis, structure-based drug design
Introduction
Chagas disease is caused by infection with the protozoan parasite, Trypanosoma cruzi (T. cruzi). When not spread by poor hygiene, infection is the result of exposure to the blood or the feces of triatomine bugs, which live in the mud in substandard housing in an area ranging from the southern part of South America to the southern part of the United States. Once infected, individuals usually become a life-long host to the parasite because no existing therapy is completely effective in the chronic stage of the disease. Eight to eleven million people in Latin America are infected with T cruzi, and 30% of those can be expected to develop complications ranging from mild to terminal1. There is no effective treatment for Chagas disease at this time. The two drugs which have been used are the nitrofuran, nifurtimox, and the nitroimidazole, benznidazole. The toxicity of these drugs is linked to their mode of action, and treatment is unavoidably accompanied by multiple undesirable side effects. Of the more than 1,200 new drugs discovered in the period from 1975 to 1997 only 4 were the result of a directed R&D effort by the pharmaceutical industry to treat a human tropical disease, and 1 of those 4 (nifurtimox) is no longer in widespread use2.
One of the guiding philosophies in our collaborative research effort for anti-parasite drug discovery is the concept that we can “piggy back” on the large research efforts underway in the pharmaceutical industry3. There is extensive medicinal chemistry and pharmacology data on inhibitors of protein farnesyltransferase since they were developed as anti-cancer agents4. Our thinking at the time was that we might find a human protein farnesyltransferase inhibitor that also had high affinity for the T. cruzi ortholog. Thus we previously investigated known protein farnesyltransferase inhibitors against a number of parasites including T. cruzi, and one compound that was highly potent at killing T. cruzi amastigotes (the clinically relevant parasite life cycle stage that grows in mammalian host cells) in an in vitro assay5 is tipifarnib (compound 1, figure 1)6. This compound is a candidate anti-cancer drug being developed by Johnson and Johnson Pharmaceuticals7. Our subsequent studies showed, surprisingly, that compound 1 kills T. cruzi not by blocking protein farnesylation but by disrupting sterol biosynthesis by inhibition of lanosterol 14α-demethylase (Tc-L14DM)6. T. cruzi amastigotes use ergosterol as a critical component of their membranes and cannot use host cell derived cholesterol. Since 1 shows a high degree of oral bioavailability, has ideal pharmacokinetic properties and is well tolerated in humans7, we considered 1 as an outstanding lead for the development of anti-Chagas disease drugs.
Figure 1.

Our goal shifted to designing analogs of 1 that no longer bind to human protein farnesyltransferase and remain as highly potent inhibitors of Tc-L14DM and as cytotoxic agents for T. cruzi amastigotes. To this end we used the X-ray structure of 1 bound to mammalian protein farnesyltransferase8 and a homology model of Tc-L14DM with 1 docked into the active site6 to develop novel tipifarnib analogs with the desired properties. Our model is shown in Figure 2 with bound 1 shown as a double-radii van der Waals surface. In this depiction, if the active site residues are in van der Waals contact with 1, the stick representation of these residues approximately sit on the doubled van der Waals surface of the inhibitor. It can be seen from Figure 1 that 1 fills the active site of Tc-L4DM well with the imidazole nitrogen of 1 coordinated to the heme-iron. Our modeling led to compound 2 (figure 1)9 which is essentially devoid of mammalian protein farnesyltransferase inhibition (IC50 > 5,000 nM versus ~1 nM for 1) and is about 10-fold more potent at killing T. cruzi amastigotes than 1 (EC50 = 0.6 nM versus 4 nM for 1). Modeling suggests that the extra methyl group on the 3-chlorophenyl ring of 2 clashes with the wall of the active site of protein farnesyltransferase but is tolerated in the active site of Tc-L14M. Also, the amino group of 1 is involved in a hydrogen-bonding network in the active site of protein farnesyltransferase but not in the active site of Tc-L14M. Although 2 is one of the most potent anti-T. cruzi compounds reported to date, subsequent studies showed that there is hindered rotation about the bond connecting the 3-chlorophenyl group and the quinolone ring presumably because of a clash between the ortho methyl group and the vinylic proton of the quinolone ring. Thus 2 is a mixture of rotamers with the ortho methyl group either above or below the plane of the quinolone ring. This is evident by the 1H-NMR spectrum of 2, which shows a doubling of resonance peaks (see Supplemental Material). Furthermore, the doubling does not collapse even when the temperature is raised to 65 °C (not shown) indicating that the rotamers are very stable at physiological temperature. Compound 2 also contains a chiral center so it is a mixture of 4 stable isomers, and this greatly diminishes its potential as a drug candidate. This is because the different isomers can be expected to have different affinities for Tc-L14DM, and it is possible that only 1 of the 4 isomers would be biologically active. Also, the pharmacokinetic and toxicity profiles of all 4 compounds would have to be separately investigated to go forward with drug candidate selection. We thus went on to design new analogs that either lack a 2-substitutent on the 3-chlorophenyl ring or have a C2-symmetric ring in order to avoid rotamer formation. In this study we reported the result of these two approaches. We also carried out additional structure-activity studies of these tipifarnib analogs.
Figure 2. Stereo diagram of tipifarnib (compound 1) docked into the active site of Tc-L14DM.

The 3-chlorophenyl group is shown in orange with its Cl in magenta, the 4-chlorophenyl group is shown in light pink with its Cl in magenta, and the rest of the inhibitor is shown in red. The heme is in yellow with the heme-iron in cyan. The active site protein residues are shown as sticks. The inhibitor is displayed as its doubled-radii van der Waals surface.
Chemistry
Scheme 1 shows the route used to synthesize some of the compounds reported in this study. Commercially available 5-bromoisatoic anhydride is converted to Weinreb amide 3, which reacts with a variety of phenyl lithiums (formed in situ from the phenyl halide using 2 eq. of n-BuLi, 1 eq. needed to deprotonate the NH) to give ketone 4. The order of addition of reagents is important to avoid lithium-bromide exchange between n-BuLi and the aryl-Br bond present in 3. Acetylation of the amino group followed by intramolecular ring closure using base gives quinolone 5. Conversion of 5 to the set of compounds 10 (Scheme 1) was carried out as described in our earlier study9.
Scheme 1. Synthesis of tipifarnib analogs from 5-bromoisatoic anhydride.

a) CH3ONHCH3 HCl, pyridine, CH2Cl2; b) R1PhBr (2eq.), n-BuLi (2eq.), THF; c) Ac2O, toluene, reflux; d) t-BuOK, 1,2-dimethoxyethane; e) i) BF4OMe3, CH2Cl2 ii) NaOH; f) i.) n-BuLi, THF, −78 °C ii. N(Me)Imidazole-CO-PhenylR2; g) 6N HCl, THF, reflux; h) CH3I, NaOH, BTEAC, THF; i) SOCl2, neat, 12 hrs j) NH3 (or CH3NH2), THF, rt; k) tosic acid 1eq + cat., MeOH, reflux.
We were also interested in analogs in which R1 is a 2-halide or a 2,6-dihalide. The use of 2-chlorophenyl lithium is problematic because of benzyne formation. We formed 2,6-difluorophenyl lithium but found that it reacted slugglishly with Weinreb amide 3 and also formed benzyne in the process. Thus we developed Scheme 2 for the preparation of the additional analogs. Commercially available halogenated cinnamic acid 11 was converted to the acid chloride and then reacted with 4-bromoaniline to give amide 12. Intramolecular Friedel-Crafts alkylation proceeded smoothly with concentrated H2SO4 to give lactam 13. Conversion to the quinolone was accomplished by oxidation of 13 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone to give 14. The latter was converted to the desired tipifarnib analogs by using steps from Scheme 1.
Scheme 2. Synthesis of tipifarnib analogs with R1 = ortho halogens.

a) SOCl2, (neat), reflux, 6 hours then 4-bromoaniline, DIEA (1.5 eq), CH2Cl2, 0 °C; b) H2SO4 (conc.), 105 °C; c) DDQ, dioxane, reflux.
Scheme 3 shows two routes to make the methanones needed for step f in Scheme 1.
Scheme 3. Synthesis of substituted 5-benzoyl-N1-alkyl-imidazoles.

Route 1: a) SOCl2, neat; b) CH3ONHCH3, pyridine, CH2Cl2; c) N-alkyl-imidazole, i) n-BuLi, THF −78°C, ii) Et3SiCl, −78 °C, iii) n-BuLi, THF, −78 °C. Route 2: a) TrCl, Et3N, CH3CN; b) i) Mg, I2, ether, RC6H4Br, room temperatureto reflux ii) MnO2, dioxane, reflux; c) MeOTf, CH2Cl2.
Results and Discussion
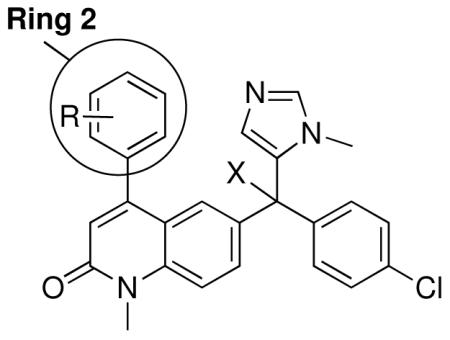
We synthesized a variety of analogs of 1 and tested them for their ability to kill T. cruzi amastigotes growing in mammalian host cells (3T3 cells). As noted above, adding a Me group at the 2-position of the 3-chlorophenyl ring of 1 and replacing the NH2 with OMe led to 2, which is devoid of protein farnesyltransferase inhibition and is ~10-fold more potent at killing parasites compared to 1. In an attempt to make analogs that do not exist as a pair of stable rotamers (see above), we synthesized analogs of 2 that have changes to the 3-chloro-2-methyl phenyl ring. Values of EC50 for killing T. cruzi amastigotes are listed in Table 1. Compound 3 lacks the ortho methyl group but retains the NH2 to OMe substitution. The compound is 100-fold less potent on protein farnesyltransferase compared to 1 and retains low nanomolar potency on T. cruzi. This result shows that the ortho methyl group and the OMe group of 2 act together to reduce the binding to protein farnesyltransferase. We also made compound 4, which like 2 has the ortho methyl group and the NH2-to-OMe change, but lacks the 3-chloro group. The properties are very similar to those of 2 showing that the 3-chloro group is not required for potent inhibition of T. cruzi growth and that the ortho methyl group is important for reducing binding to protein farnesyltransferase. However, compound 4 still exists as a pair of stable rotamers so it does not constitute the solution that we require. The same picture emerges with compound 5, which has an ortho CF3 instead of Me.
Table 1.
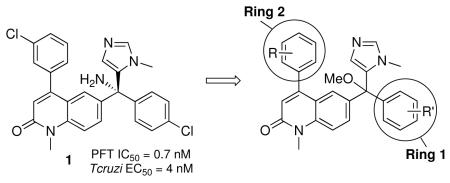 Tipifarnib analogs with ring 1 and ring 2 modifications.
Tipifarnib analogs with ring 1 and ring 2 modifications.
| Compound | Ring 2 | Ring 1 | X | Rat PFT IC50 | Tcruzi EC50 |
|---|---|---|---|---|---|
| 1 |
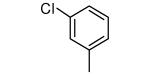
|
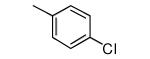
|
−NH2 | 0.7 | 4 |
| 2 |
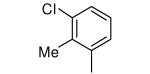
|
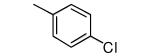
|
−OMe | >5000 | 0.6 |
| 3 |
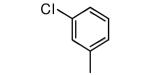
|
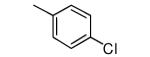
|
−OMe | 65 | 3.1 |
| 4 |
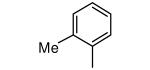
|
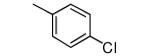
|
−OMe | 3,260 | 0.7 |
| 5 |
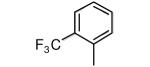
|
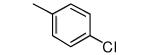
|
−OMe | >5,000 | 0.8 |
| 6 |
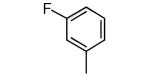
|
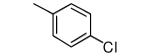
|
−OMe | 343 | 1.1 |
| 7 |
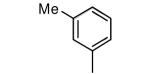
|
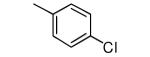
|
−OMe | 85 | 1.2 |
| 8 |
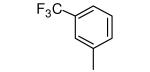
|
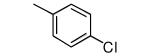
|
−OMe | 1,320 | 12 |
| 9 |
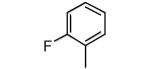
|
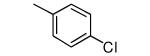
|
−OMe | 1,870 | 0.8 |
| 10 |
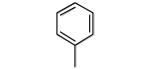
|
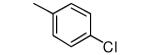
|
−OMe | 329.8 | 0.8 |
| 11 |
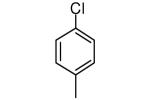
|
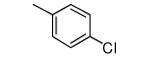
|
−OMe | 6035.0 | 0.82 |
| 12 |
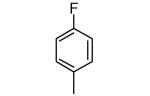
|
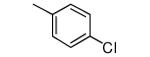
|
−OMe | 991 | 0.5 |
| 13 |
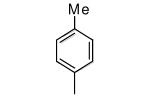
|
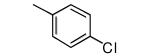
|
−OMe | 410 | 2.0 |
| 14 |
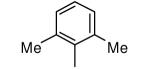
|
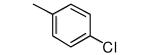
|
−OMe | > 10,000 | 1.8 |
| 15 |
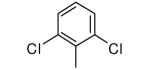
|
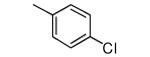
|
−OMe | >10,000 | 3.21 |
| 16 |
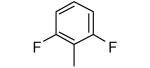
|
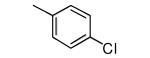
|
−OMe | >10,000 | 0.31 |
| 17 |
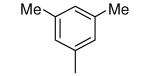
|
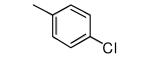
|
−OMe | 470 | 1.4 |
| 18 |
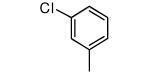
|
|
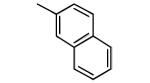
−OMe | 2,300 | 2.2 |
| posaconazole | not determined |
0.3 |
We made compounds that lack the ortho methyl group (and thus the rotamer problem) but contain replacements for 3-chloro group hoping to reduce binding to protein farnesyltransferase while retaining potency on T. cruzi. Replacing Cl with F, Me or CF3 reduced affinity to protein farnesyltransferase by ~100- to 2,000-fold while retaining good potency on T. cruzi (see 6-8, Table 1). This result shows that the Cl substituent gives an optimal fit in the active site of protein farnesyltransferase. This is apparent from the X-ray structure of 1 bound to protein farnesyltransferase.
Next we made compound 9, which contains an ortho F instead of the ortho Me hoping that the smaller F would not sterically interfere with the vinylic hydrogen. Compound 9 is similar to 2 in that it binds much more weakly to protein farnesyltransferase compared to 1 and is superior to 1 in its ability to kill T. cruzi. However, 9 was found to exist as stable rotamers that are distinguished in the 1H-NMR spectrum (not shown). The NMR peaks coalesce when the temperature is raised to 75 °C (not shown); however, the rotamers persist at physiological temperature.
We made compound 10, which lacks an ortho substituent (so no rotamers are possible) and also lacks the 3-chloro group, which is important for binding to protein farnesyltransferase. This compound ranks among the most potent of our compounds against T. cruzi and displays an intermediate loss in affinity for protein farnesyltransferase (~500-fold).
Compounds 11-13 lack ortho substituents and contain a single non-hydrogen substituent at the para position. All 3 compounds show good potency for killing T. cruzi and a substantial loss of binding to protein farnesyltransferase, with 11 showing the largest loss.
We also reasoned that addition of a non-hydrogen group to the ortho position is acceptable if both ortho positions are substituted since the two rotamers are chemically identical in this case. Compound 14 contains a 2,6-dimethylphenyl ring and like the 2-methylphenyl-containing compound 2, it is devoid of protein farnesyltransferase inhibition activity and displays good potency for killing parasites. Compounds 15 and 16 display similar properties as 14. The 2,6-difluorophenyl compound 16 is the most potent in the series against T. cruzi with an EC50 of 0.3 nM. This compound is as potent as posaconazole against T. cruzi in our in vitro amastigote killing assay. These are the most potent anti-T. cruzi compounds that we are aware of.
Compound 17 contains a 3,5-dimethyphenyl group. It is highly potent against parasites and displays a ~600-fold reduction in affinity for protein farnesyltransferase compared to 1. In our previous study we showed that the tipifarnib analog with a β-naphthyl replacing the 4-chlorophenyl group of 1 displayed ~20-fold less affinity to protein farnesyltransferase compared to 1 and displayed an EC50 against T. cruzi of 45 nM. Given the lowering of EC50 and the loss of protein farnesyltransferase affinity when the amino group of 1 is replaced with OMe, we prepared compound 18. Indeed this compound now is a very weak inhibitor of protein farnesyltransferase, and the EC50 for killing T. cruzi drops to 2 nM.
Table 2 shows the effect of a wider variation in substitution of the amino group of 1. Replacing NH2 with OH produces only a slight reduction in protein farnesyltransferase affinity. Presumably the OH group can still engage in the active site hydrogen bond network. Replacement of the NH2 group with OMe, OEt, OPr, and NHMe has a much more dramatic effect. Modeling shows that the NH2 group does not contact Tc-L14DM residues. It is quite likely that the improved potency for killing T. cruzi when NH2 is replaced with OMe is the result of better penetration of the more hydrophobic compound across host cell and parasite membranes. This was our motivation for preparing OPr and NHEt analogs. However, OMe remains as the best group in terms of EC50 values. We attempted to prepare the analog containing N(Me)2, but it was unstable presumably by breaking down in an sn-1 type reaction.
Table 2.
 Tipifarnib analogs with X group modifications
Tipifarnib analogs with X group modifications
| Compound | Ring 2 | Ring 1 | X | Rat PFT IC50 | Tcruzi EC50 |
|---|---|---|---|---|---|
| 1 |
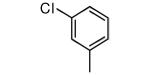
|
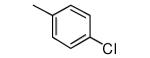
|
NH2 | 0.7 | 4 |
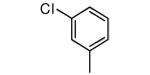
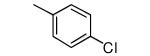
| 19 |
|
|
OH | 8 | 17 |
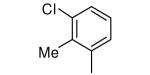
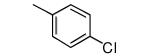
| 2 |
|
|
−OMe | >5000 | 0.6 |
| 20 |
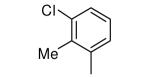
|
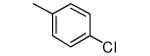
|
−OH | 796 | 112 |
| 21 |
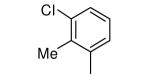
|
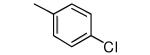
|
−OEt | >10,000 | 27 |
| 22 |
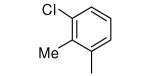
|
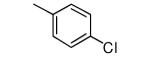
|
−OPr | >5000 | 69 |
| 23 |
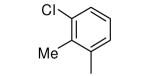
|
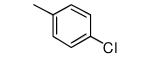
|
−NHMe | 957 | 5 |
Table 3 shows data for compounds in which the imidazole N-Me is replaced with N-Et. Compound 26 is as potent against T. cruzi as the N-Me comparator, compound 3, but is superior to 3 in that it binds ~1,500-fold weaker to protein farnesyltransferase than 1. Again compounds with the OMe group are superior against T. cruzi. Compound 26 also lacks rotamers (no ortho substituent).
Table 3.
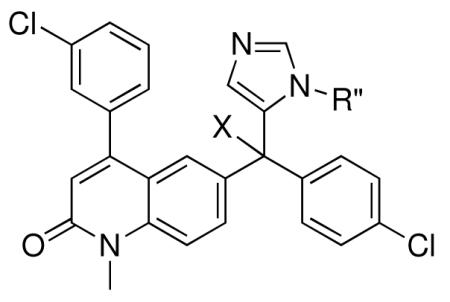 Tipifarnib analogs with imidazole and X group modifications.
Tipifarnib analogs with imidazole and X group modifications.
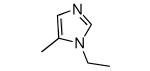
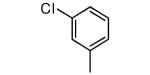
| Compound | Ring 2 | Imidazole | X | Rat PFT IC50 | Tcruzi EC50 |
|---|---|---|---|---|---|
| 1 |
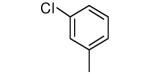
|
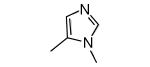
|
−NH2 | 0.7 | 4 |
| 24 |
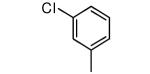
|
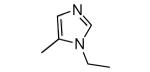
|
−NH2 | 28 | 118 |
| 25 |
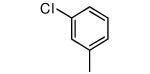
|
|
−NHMe | 66 | 100 |
| 26 |
|
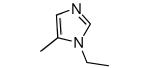
|
−OMe | 970 | 3 |
| 27 |

|
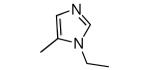
|
−OH | 30 | 228 |
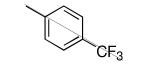
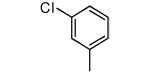
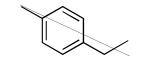
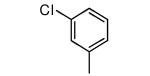
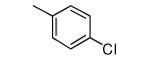
We made additional modifications to 1 to further test the validity of our homology model (Figure 2), and the results are summarized in Table 4. Modeling shows that the Cl of the 4-chlorophenyl group of 1 fills the active site well. It can be seen that replacement of this Cl with Me and CF3 are tolerated, but the larger groups Et and iPr lead to less potent compounds against T. cruzi. Modeling also suggests that the Cl of the 3-chlorophenyl group fills the active site of Tc-L14DM well (Figure 2). Replacement of this Cl with larger groups, Ph and Bn, lead to a dramatic reduction in potency against T. cruzi parasites.
Table 4.
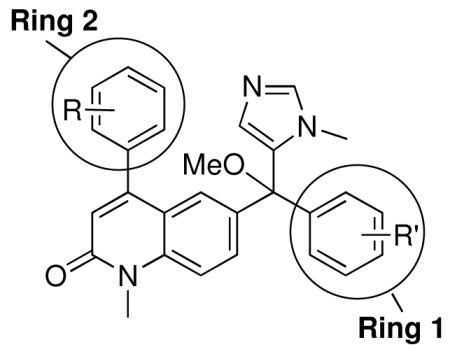 Tipifarnib analogs with additional ring 1 and ring 2 modifications.
Tipifarnib analogs with additional ring 1 and ring 2 modifications.
| Compound | Ring 2 | Ring 1 | X | Tcruzi EC50 |
|---|---|---|---|---|
| 2 |
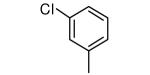
|
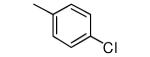
|
−OMe | 3 |
| 28 |
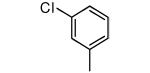
|
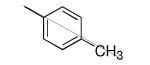
|
−OMe | 3 |
| 29 |
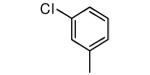
|
|
−OMe | 5 |
| 30 |
|
|
−OMe | 10 |
| 31 |
|

|
−OMe | 33 |
| 32 |

|
|
−OMe | 320 |
| 33 |
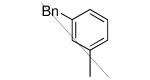
|
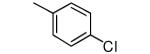
|
−OMe | 83 |
We tested our most potent anti-T. cruzi compound, 16, for binding to recombinant Tc-L14DM in vitro. As shown in Figure 3, when 1.4 μM Tc-L14DM is titrated with 0.2-2 μM 16, the visible absorption spectrum of the heme Soret band undergoes a spectral shift (as seen in the difference spectrum, Figure 3). The spectral features strongly suggest that the imidazole nitrogen of 16 coordinates directly to the heme iron. When the difference spectrum is quantified as a function of the equivalents of 16 added to the enzyme, it is found that the change in the spectrum ceases when 1 enzyme equivalent of inhibitor is added (not shown). This shows that the equilibrium dissociation constant for the enzyme-inhibitor complex, Kd, is much less the enzyme concentration of 1.4 μM. From this result we can say that 16 binds tightly to Tc-L14DM, but we cannot determine the value of Kd precisely. It is for this reason that we prefer to rank our tipifarnib analogs based on values of EC50 for killing T. cruzi amastigotes (Tables 1-4). EC50 is the most important parameter for our drug discovery effort since the compound not only has to bind to the target enzyme but it also has to cross the host cell and parasite membranes.
Figure 3. Difference Spectra for the binding of 16 to recombinant Tc-14DM.

Compound 16 was added at 200 nM – 2 uM with a Tc-14DM concentration of 1.4 μM.
As these tipifarnib analogs contain an imidazole ring, there is some concern that they may be potent inhibitors of hepatic cytochrome P450s. We tested some of our best compounds in the series for inhibition of the major drug metabolizing enzyme, cytochrome P450 CYP3A4 isoform, using an in vitro assay. The well established CYP3A4 inhibitor ketoconazole was a highly potent inhibitor with an IC50 measured at 11 nM (Table 5). We also tested posaconazole since it is being considered for clinical testing as an anti-Chagas disease drug. This compound was the next most potent inhibitor (IC50 = 82 nM). Tipifarnib was previously reported to be a weak inhibitor (>10,000 nM) of P450 enzymes,10 and we also observed relatively weak inhibition against CYP3A4 (IC50 = 4,060 nM). The tipifarnib analogs designed in these studies had either ~ micromolar level IC50 values (JK-27, JK-36, and SM-2) or somewhat more potent activity as observed with JK-35, and CHN-3 (Table 5). It is encouraging that the CYP3A4 inhibition by some of the tipifarnib analogs is considerably less than for the azole drugs. In future studies, it will be necessary to profile the compounds against additional liver microsomal enzymes to more fully establish their potential to cause drug-drug interactions.
Table 5.
In vitro inhibition of cytochrome P450 CYP3A4 isoform
| Compound | IC50 (nM) | SEM (nM) |
|---|---|---|
| Tipifarnib | 4,060 | 338.5 |
| 10 | 2,345 | 847.5 |
| 11 | 923 | 212 |
| 12 | 1628 | 594 |
| 16 | 275 | 57 |
| Posaconazole | 82 | 13 |
| Ketoconazole | 11 | 0.5 |
Conclusions
In this continued study of analogs of the cancer drug clinical candidate tipifarnib, 1, as inhibitors of Tc-L14DM and as anti-T. cruzi agents, we have developed 20 new compounds that display values of EC50 for killing T. cruzi of < 10 nM and 7 new compounds that display values of EC50 < 1 nM. Many of these compounds either fail to bind to mammalian protein farnesyltransferase or bind with > 100-fold reduced affinity. In this study we have solved the rotamer problem that we encountered in our earlier study of tipifarnib analogs containing an ortho substituted phenyl group. The availability of a large set of highly potent anti-T. cruzi compounds sets the stage for the next steps of drug development which include pharmacokinetic, efficacy and toxicology studies.
Experimental
Biological and modeling studies
Assays of rat protein farnesyltransferase to obtain the IC50 for inhibitors and of T. cruzi amastigote growth to obtain EC50 values for the inhibitors were carried out as described9. Binding of inhibitors to recombinant Tc-L14DM was carried out by difference optical spectroscopy as described11. Molecular modeling studies were carried out as described6. EC50 values reported in the tables have standard errors of less than 50% based on duplicate or triplicate independent determinations.
Inhibition of recombinant human CYP3A4 enzyme was determined using a commercial kit (CYP3A4/BFC High Throughput Inhibitor Screening Kit, GenTest, Inc.) following the manufacturers instructions. The compounds were tested in serial dilutions in duplicate with the IC50 values calculated by non-linear regression analysis using the Prism software (GraphPad Software, Inc.). The assay was performed twice, and average IC50 values are shown. Ketoconazole was the positive control and gave results within the expected range provided by the manufacturer.
Chemistry
Starting materials were purchased from Aldrich, Acros, Alfa-Aesar, EMD, Fisher, Lancaster, Mallinckrodt, TCI-America, or VWR and used without further purification, unless otherwise specified. N-methylimidazole (M50834) was purchased from Sigma-Aldrich and distilled at reduced pressure (10mm Hg, bp: 67-69 °C) after being stirred over sodium at room temperature overnight. Solvents were purified using a J.C. Meyer type Solvent Dispensing System utilizing Al2O3 and/or Copper cartridges, depending on the particular solvent. n-BuLi was titrated with diphenylacetic acid prior to use. Nitrogen gas used in reactions requiring an inert atmosphere was house-supplied and run through Dri-Rite dessicant. Glassware for distillation and water sensitive reactions was flame dried under vacuum or dried in an oven. Silica was EMD Silica Gel 60, 40-63 μm (11567-1). TLC plates were aluminum backed EMD Silica Gel 60 F254 (5554/7). 1H-NMR spectra were recorded on a Bruker Avance AV300. ESI-MS were recorded on a Bruker Esquire Ion Trap Mass Spectrometer. Purification of all final compounds was by reverse phase HPLC utilizing an octadecylsilane stationary phase and a water-to-methanol gradient with 0.08% v/v Trifluoroacetic Acid, (TFA). HPLC was carried out on a Varian Pro-Star fitted with a Waters YMC Pack-ODS-A column (2 × 10 cm) running at 12 mL/min using a detector excitation wavelength of 254 nm. All target compounds possessed a purity of ≥ 95% as verified by HPLC.
Methanone synthesis (route 1)
4-Chloro-N-methoxy-N-methylbenzamide
20.0 g (0.128 mols) 4-chlorobenzoic acid (156.57 g/mol) was placed in a 500 mL round bottomed flask. 120 mL Thionyl chloride was added, and the mixture was refluxed overnight. Thionyl chloride was removed under reduced pressure to produce a red oil. Anhydrous toluene was added and then removed under reduced pressure two times. The crude product was dissolved in 200 mL anhydrous CH2Cl2. 13.73 g (0.140 mols) N,O-dimethylhydroxylamine•HCl (97.54 g/mol) was added. 52 mL (0.640 mols) anhydrous pyridine was added over a period of 10 minutes. The reaction was stirred under nitrogen at room temperature overnight. Volatiles were removed under reduced pressure. The solid was partitioned between CHCl3 and water. The organic phase was washed with brine, then collected and dried over anhydrous magnesium sulfate. The solvents were removed to produce a red colored oil which was purified on silica with 5% MeOH/CH2Cl2 as eluent. 22.9 g of product was produced, yield 90%. 1H NMR (300 MHz, CDCl3, δ): 7.66 (m, 2H), 7.38 (m, 2H), 3.54 (s, 3H, −OMe), 3.36 (s, 3H, −NMe) ESI-MS m/z 200.4 (M + H+)+ MW: 199.63 g/mol.
(4-Chlorophenyl)(1-methyl-1H-imidazol-5-yl)methanone
A flame dried 125 mL round bottom flask was charged with a stir bar, and over-pressurized with dry nitrogen gas and 3.0 mL (37.6 mmols) freshly distilled N-methylimidazole (82.11 g/mol, 1.035 g/mL). The flask was sealed with a new rubber septum and 30 mL anhydrous THF was added. The solution was stirred for about 10 minutes, and then the temperature was lowered to −78 °C and stirred for an additional 10 minutes. 16.2 mL (41.3mmols) Freshly titrated n-BuLi (2.5 M in hexanes) was added dropwise through the septum over a period of 10 minutes under an overpressure of dry nitrogen. A slight color change to pale yellow was observed. The reaction mixture was allowed to stir at this temperature for 45 minutes, then 6.3 mL (37.6 mmols) 99% chlorotriethylsilane (Et3SiCl) was added dropwise over 5 minutes. The reaction was allowed to stir for 1 hour at −78 °C, at which point 15.0 mL (37.6 mmols) freshly titrated n-BuLi (2.5 M in hexanes) was added dropwise through the septum over a period of 10 minutes and allowed to stir at −78 °C for an additional 45 minutes. A separate flask was flame dried and charged with 5.0 g (25.1 mmols) 4-chloro-N-methoxy-N-methylbenzamide (199.63 g/mol) and sealed with a rubber septum. 15 mL Anhydrous THF was added and the mixture was stirred at room temperature for 10 minutes, then at the appropriate time, transferred via cannula to the flask containing the in situ generated C-2 triethylsilyl protected N-methylimidazole at a slow rate to maintain low temperature as indicated by the slow sublimation of CO2. The mixture was left to stir overnight and became a deep red color. The reaction was quenched by the addition of 1 M HCl until the pH of the aqueous phase was no longer basic as indicated by litmus paper, then allowed to stir for one hour. The pH of the aqueous phase was adjusted to above 8 with 1.5 M NaOH, and the mixture was partitioned between CHCl3 and water. The organic phase was washed with brine and dried with anhydrous MgSO4. Solvents were removed under reduced pressure to produce a reddish solid. Product was purified by re-crystallization from CH2Cl2 to produce 4.24 g of fluffy golden crystals, 76.6% yield. 1H NMR (300 MHz, CDCl3, δ): 7.83 (d, J = 8.4 Hz, 2H), 7.68 (s, 1H), 7.59 (s, 1H), 7.50 (d, J = 8.4 Hz, 2H), 4.03 (s, 1H) ESI-MS m/z 221.4 (M + H+)+ MW: 220.65 g/mol.
(4-Chlorophenyl)(1-ethyl-1H-imidazol-5-yl)methanone
The compound was prepared in a manner analogous to (4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methanone. 1H NMR (300 MHz, CDCl3, δ): 7.81 (m, 2H), 7.75 (s, 1H), 7.58 (s, 1H), 7.50 (m, 2H), 4.03 (s, 1H) ESI-MS m/z 221.4 (M + H+)+ MW: 234.68 g/mol.
Methanone synthesis (route 2)
1-Triphenylmethyl-4-imidazole Carboxaldehyde
To a 1 L three–necked round-bottomed flask with an addition funnel was added 3H-imidazole-4-carbaldehyde (12 g, 0.125 mol, Aldirch), tritylchloride (38.3 g, 0.137 mol) and acetonitrile (400 mL). The mixture was stirred atroom temperatureto give a slurry. Triethylamine (30 mL, 0.125 mol) was added dropwise over 20 min. After the addition was complete, the reaction mixture was stirred atroom temperaturefor 20 h. Hexane (40 mL) and water (400 mL) were added. The slurry was stirred for 30 min and filtered. The cake was washed with water (3 × 100 mL) and dried in oven at 50°C for 20 h to give 2 as a white solid. (39.8 g, 94%). 1H NMR (CDCl3, δ) 9.81 (s, 1H), 7.54 (s, 1H), 7.46 (s, 1H), 7.20-7.29 (m, 10H), 7.09-7.04 (m, 5H). MS m/z 339.4 (M + H)+ MW: 338.4.
p-Tolyl-(1-trityl-1-H-imidazol-4-yl)- methanone
An oven dried 250 mL round bottom flask fitted with reflux condenser was sealed with a rubber septum under argon atmosphere and charged with magnesium turnings (1.12 g, 46.7 mmol) and a pinch of iodine. Anhydrous ether (50 mL) was added and stirred for 5 min at room temperature. To this mixture p-bromo toluene (5 g, 29.2 mmol) was added dropwise in anhydrous ether (10 mL) slowly, at a rate to allow the ether to barely reflux. The mixture was then refluxed for 30 min and cooled to 0 °C. 1-Triphenylmethyl-4-imidazole carboxaldehyde (7.9 g 23.3 mmol) was dissolved in anhydrous ether (20mL) and added dropwise with continuous stirring. The reaction mixture was stirred for 3 h at room temperature and then refluxed for 1 h. The reaction mixture was cooled and poured into 50 mL of ice cold 1 M HCl. The phases were separated and the aqueous phase was extracted with ether (2×50 mL). The combined ether layers were washed with saturated NaHCO3 solution and the resulting ether layer kept in an ice cold bath for 3 h and then filtered. The resulting white solid (9 g) was transferred to a 200 mL round bottomed flask fitted with a reflux condenser. The solid was suspended in 1,4-dioxane (50 mL) and stirred for 5 min and then MnO2 (9 g, 104 mmol) was added. The resulting mixture was refluxed for 3 h and cooled to room temperature and diluted with CH2Cl2 (50 mL) then filtered through a celite pad. Solvent was removed under reduced pressure to produce 8.45 g of product as a crispy solid, yield 94.4%. 1H NMR (300 MHz, CDCl3, δ) 8.12 (d, J = 8.1 Hz, 2H), 7.69 (s, 1H), 7.54 (s, 1H), 7.43-7.34 (m, 10H), 7.25 (d, J = 8.1 Hz, 2H), 7.20-7.12 (m, 5H), 2.55 (s, 3H) ESI-MS m/z 429.2 (M+H)+ MW: 428.56 g/mol.
(3-Methyl-3H-imidazol-4-yl)-p-tolyl-methanone
A flask was charged with 5.0 g of the previous compound (11.6 mmol) under nitrogen atmosphere which was suspended in 40 mL anhydrous CH2Cl2 and stirred rapidly for 10 min. A solution of methyl trifluoromethane sulfonate (1.95 mL, 17.5 mmol) in anhydrous CH2Cl2 (10 mL) was added dropwise over 10 min and the reaction mixture was stirred at room temperature for 20 h. The volume of the reaction was reduced to about 1/3 of its original volume and to this was added 30 mL of hexane. The slurry was stirred for 30 min and filtered. The cake was washed with hexane and dissolved in 30 ml of 1:2 water acetone mixture and stirred for 3 h. The reaction mixture was filtered to remove trityl alcohol and the cake was washed with water (2×10 mL). The filtrate was concentrated under reduced pressure to remove acetone and the resulting slurry was filtered again to remove traces of trityl alcohol and the cake washed with water (10 mL). The filtrate was washed with saturated NaHCO3 and extracted with CH2Cl2 (2×40 mL). Combined organic layers were dried with MgSO4. Solvent was removed under reduced pressure to produce 2.09 g of product as a pulpy white solid, yield 89.6% 1H NMR (300 MHz, CDCl3, δ) 7.78 (d, J = 8.1 Hz, 2H), 7.64 (s, 1H), 7.59 (s, 1H), 7.31 (d, J = 8.1 Hz, 2H), 4.02 (s, 3H), 2.45(s, 3H) ESI-MS m/z 201.1 (M+H)+ MW: 200.24 g/mol.
(3-Methyl-3H-imidazol-4-yl)-(4-trifluoromethyl-phenyl)-methanone
The compound was prepared in a manner analogous to the above methanone. 1H NMR (300 MHz, CDCl3, δ) 7.95 (d, J = 8.7 Hz, 2H), 7.78 (d, J = 8.7Hz, 2H), 7.69 (s, 1H), 7.58 (s, 1H), 4.03 (s, 3H), ESI-MS m/z 255.4 (M+H)+ MW: 254.21 g/mol.
(4-ethyl-phenyl)-(3-Methyl-3H-imidazol-4-yl)-methanone
The compound was prepared in a manner analogous to the above methanone. 1H NMR (300 MHz, CDCl3, δ) 7.80 (d, J = 9.0 Hz, 2H), 7.64 (s, 1H), 7.59 (s, 1H), 7.32 (d, J = 9.0Hz, 2H), 4.01 (s, 3H), 2.75 (q, J = 3.3 Hz, 1.8 Hz, 2H), 1.28 (t, J = 7.2 Hz, 3H) ESI-MS m/z 215.1 (M+H)+ MW: 214.26 g/mol.
(4-Isopropyl-phenyl)-(3-Methyl-3H-imidazol-4-yl)-methanone
The compound was prepared in a manner analogous to the above methanone. 1H NMR (300 MHz, CDCl3, δ) 7.82 (d, J = 8.6 Hz, 2H), 7.65 (s, 1H), 7.61 (s, 1H), 7.36 (d, J = 8.6 Hz, 2H), 4.02 (s, 3H), 3.0 (m, 1H), 1.33 (d, J = 6.9 Hz, 6H) ESI-MS m/z 229.1 (M+H)+ MW: 228.29 g/mol.
2-Amino-5-bromo-N-methoxy-N-methylbenzamide
A 2 L flask was charged with 25.0 g (103.3 mmols) 5-bromoisatoic anhydride (242.03 g/mol) and 15.1 g (155 mmols) N,O-dimethylhydroxylamine hydrochloride (97.54 g/mol). The solids were suspended in 500 mL anhydrous CH2Cl2 and stirred rapidly. 37.5 mL Pyridine (465 mmol) was added slowly, and the mixture was allowed to stir until all solids had dissolved, about 36 hours. The crude mixture was partitioned between CHCl3 and water. The organic phase was washed with brine and dried with anhydrous MgSO4. Solvents were removed under reduced pressure to produce a gold oil which crystallized upon standing. Product was triturated with hexanes, filtered then used without further purification. 23.9 g of a lightly colored crystalline product was produced, 89% yield. 1H NMR (300 MHz, CDCl3, δ): 7.52 (d, J = 2.4 Hz, 1H), 7.28 (dd, J = 2.4Hz, 8.1Hz 1H), 6.61 (d, J = 8.7Hz, 1H), 4.69 (s (broad), 2H), 3.61 (s, 3H), 3.36 (s, 3H) ESI-MS m/z 283.1 (M + Na+)+ MW: 259.10 g/mol.
(2-Amino-5-bromophenyl)(3-chloro-2-methylphenyl)methanone
A 250 mL flask was flame dried, and charged with a stirbar and a rubber septum. 2-Bromo-6-chlorotoluene (6 mL, 45.9 mmols) was added to the flask and dissolved in 60 mL of anhydrous THF under an atmosphere of dry nitrogen. The solution was stirred for about 5 minutes and then cooled to −78 °C then stirred for about 10 minutes. 18 mL (45.9 mmols, 1 eq.) n-BuLi (2.5 M in hexanes) was added dropwise at a rate such that temperature of the reaction remained close to −78 °C, as indicated by the slow sublimation of CO2. The solution was allowed to stir for 20 minutes. 5.95 g (22.95 mmols, 0.5 eq) 2-amino-5-bromo-N-methoxy-N-methylbenzamide was added to a separate flask and dissolved in 60 mL anhydrous THF. After stirring for about 5 min, this solution was added drop wise by canula to the flask containing the in situ generated aryl lithiium. The solution was allowed to stir at this temperature for 2 hours at which time the cooling bath was removed allowing the flask to rise to room temperature. 50 mL 1 M aqueous HCl was added and the bi-phasic mixture was allowed to stir for 30 minutes. The mixture was partitioned between CHCl3 and water. The organic phase was washed three times with saturated, aqueous NaHCO3, then separated and dried with MgSO4. Solvents were removed under reduced pressure to produce an orange-brown oil. This was purified on silica with 20% EtOAc/hexanes to produce 6.3 g of yellow crystalline product, yield 85%. 1H NMR (300 MHz, CDCl3, δ): 7.47 (d, J = 7.8Hz, 1H), 7.36 (dd, J = 8.7Hz, 2.1Hz, 1H), 7.25 (d, J = 2.1Hz 1H), 7.21 (m, 2H), 7.11 (d, J = 7.5Hz 1H), 6.64 (d, J = 8.7Hz, 1H), 6.49 (s (broad), 2H), 2.27 (s, 3H) ESI-MS m/z 324.3 (M + H+)+ MW: 324.60g/mol.
6-Bromo-4-(3-chloro-2-methylphenyl)quinolin-2(1H)-one
A 250 mL flask was fitted with a stir-bar and 11.9 g of (2-amino-5-bromophenyl)(3-chloro-2-methylphenyl)methanone was added. A water condenser was attached, then 50 mL anhydrous toluene and 24.3 mL (257 mmols, 7 eq.) acetic anhydride were added. The solution was heated to reflux for 6 hours then volatiles were removed under reduced pressure. Approximately 50 mL of toluene was added and removed at reduced pressure two times. The crude product was set aside. A separate flask was fitted with a stir-bar and a septum and loaded with 24.7 g (220 mmols, 6 eq.) of 95% t-BuOK powder. This was suspended in 120 mL 1,2-dimethoxyethane and stirred for about 10 minutes. The temperature was lowered to 0 °C. The crude product set aside previously was dissolved in 45 mL 1,2-dimethoxyethane then transferred dropwise by cannula to the flask containing the t-BuOK suspension. The color of the solution changed to yellow, and the mixture was allowed to stir under an inert atmosphere overnight. At this point the volaties were removed under reduced pressure, and the resulting paste was suspended in ~300 mL of water. The solid was collected by filtration and used in the next step without purification. 8.4 g (24.10 mmols) of product was produced as a fluffy, white solid, yield 66%. 1H NMR (300 MHz, CD3OD, δ): 7.62 (dd, J = 8.7Hz, 1.8Hz, 1H), 7.53 (d, J = 7.8Hz, 1H), 7.39 (d, J = 8.7Hz, 1H), 7.30 (m, 1H), 7.21 (d, J = 1.8Hz, 1H), 7.11 (d, J = 7.2Hz, 1H), 6.62 (s, 1H), 2.17 (s, 3H) ESI-MS m/z 348.3 (M + H+)+ MW: 348.62 g/mol.
6-Bromo-4-(3-chloro-2-methylphenyl)-2-methoxyquinoline
A flame dried 25 mL flask was fitted with a stir-bar and charged with 500 mg (1.43 mmols) of 6-bromo-4-(3-chloro-2-methylphenyl)quinolin-2(1H)-one and 423 mg (2.86 mmols, 2 eq.) BF4OMe3 then sealed with a septum. The solids were suspended in 5.5 mL of anhydrous CH2Cl2 and stirred for 20 hours at which time 5.5 mL of 1 M aqueous NaOH was added. The mixture was stirred for about 3 hours, then partitioned between CHCl3 and water. The organic was washed three times with brine, then dried with MgSO4. Solvents were removed under reduced pressure to produce a clear yellow oil which crystallized upon standing. This was purified on silica with 50:50 CH2Cl2:hexane to produce 329 mg of product as a flaky yellow-green solid, yield 63%. 1H NMR (300 MHz, CDCl3, δ): 7.79 (d, J = 9.0 Hz, 1H), 7.69 (dd, J = 8.7Hz, 2.3Hz, 1H), 7.50 (dd, J = 7.8Hz, 1.2Hz, 1H), 7.40 (d, J = 2.1 Hz, 1H), 7.26 (m, 1H), 7.09 (dd, J = 7.5Hz, 1.2Hz, 1H), 6.77 (s, 1H), 4.10 (s, 3H), 2.08 (s, 3H) ESI-MS m/z 362.3 (M + H+)+ MW: 362.65 g/mol.
4-(3-Chloro-2-methylphenyl)-6-((4-chlorophenyl)(hydroxy)(1-methyl-1H-imidazol-5-yl)methyl)-2-methoxyquinoline
A flame dried flask was charged with a stir bar, 4.2 g (11.58 mmols) of 6-bromo-4-(3-chloro-2-methylphenyl)-2-methoxyquinoline and sealed with a rubber septum. 20 mL of anhydrous THF was added and the solution stirred at room temperature for 10 minutes. The temperature was lowered to −78 °C, and stirred for another 10 minutes. 5.0 mL (1.1 eq. 6.369 mmols) 2.5 M n-BuLi was added dropwise accompanied by a color shift to dark yellow-orange. This was allowed to stir for 20 minutes. A separate flask was flame dried and charged with 2.8 g (4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methanone (1.1 eq., 6.369 mmols). This was dissolved in 55 mL THF and added to the flask containing quinoline in three increments by syringe, rapidly drop wise over 10 minutes. The reaction was stirred overnight and allowed to warm slowly to room temperature, and the color shifted to gold. The reaction was quenched by addition of 1 volume of a saturated aqueous solution of NH4Cl. This was partitioned between 1 M NH4OH and CHCl3. The organic phase was collected and solvents were removed under reduced pressure to produce a foamy, white semi-solid. This was purified on silica with a mobile phase consisting of MeOH/CH2Cl2 1:10 v/v to produce 3.52 g (6.91 mmols) of product, 60% yield. TLC (CH2Cl2:MeOH 9:1 v/v): Rf = 0.45; 1H NMR (300 MHz, CDCl3, δ): 7.86 (m, 1H), 7.59 (m, 1H), 7.43 (m, 1H), 7.20 (m, 6H), 7.01 (m, 2H), 6.79 (s, 1H), 6.17 (m, 1H), 4.12 (s, 3H), 3.30 (m, 3H), 1.90 (m, 3H) ESI-MS m/z 504.3 (M + H+)+ MW: 504.41 g/mol.
4-(3-Chloro-2-methylphenyl)-6-((4-chlorophenyl)(hydroxy)(1-methyl-1H-imidazol-5-yl)methyl)quinolin-2(1H)-one
3.5 g (6.91 mmols) of the previous compound was dissolved in 15 mL THF. 30 mL 6 N HCl (25 eq) was added dropwise. The flask was fitted with a water-cooled condenser and the mixture was stirred at 60 °C for 5 hours. Most of the THF was removed by a stream of nitrogen and the heterogenous mixture that remained was made basic with excess aqueous 1 M NH4OH then partitioned between water and CHCl3. The organic was dried with MgSO4, and solvents were removed at reduced pressure. Product was purified on silica with a mobile phase consisting of MeOH/CH2Cl2 1:10 v/v to produce 2.1 g (4.28 mmols) of product as a white solid, 62% yield. TLC (CH2Cl2:MeOH 9:1 v/v): Rf = 0.30; 1H NMR (300 MHz, CD3OD, δ): 7.68 (m, 1H), 7.59 (m, 1H), 7.47 (m, 2H), 7.28 (m, 3H), 7.12 (m, 3H), 6.73 (m, 1H), 6.51 (m, 1H), 6.1 (m, 1H), 3.41 (m, 3H), 1.96 (m, 3H) ESI-MS m/z 490.4 (M + H+)+ MW: 490.38 g/mol.
4-(3-Chloro-2-methylphenyl)-6-((4-chlorophenyl)(hydroxy)(1-methyl-1H-imidazol-5-yl)methyl)-1-methylquinolin-2(1H)-one
2.1 g (4.28 mmols) of the previous compound was added to a 100 mL flask and dissolved in 30 mL THF. 487 mg (0.5 eq., 2.14 mmols) benzyltriethylammonium chloride was added as a phase transfer catalyst. 25.5 mL 40% wt. NaOH (120 eq., 17.1 g) was added and the mixture allowed to stir for approximately 10 minutes. 375 μL (1.4 eq., 6 mmols) CH3I was added and the mixture was allowed to stir overnight. The THF was removed at reduced pressure and the product was partitioned between CHCl3 and 1M NH4OH. The product was purified on silica with a mobile phase consisting of MeOH/CH2Cl2 1:10 v/v to produce 2.0 g (3.97 mmols) of product as a colorless semi-solid, 59% yield. TLC (CH2Cl2:MeOH 9:1 v/v): Rf = 0.45; 1H NMR (300 MHz, CDCl3, δ): 7.80 (m, 1H), 7.72 (m, 1H), 7.63 (m, 1H), 7.49 (m, 1H), 7.29 (m, 3H), 7.12 (m, 3H), 6.78 (m, 1H), 6.58 (m, 1H), 6.14 (m, 1H), 3.85 (s, 3H), 3.41 (m, 3H), 1.95 (m, 3H) ESI-MS m/z 504.3 (M + H+)+ MW: 504.41 g/mol.
4-(3-chloro-2-methylphenyl)-6-((4-chlorophenyl)(ethoxy)(1-methyl-1H-imidazol-5-yl)methyl)-1-methylquinolin-2(1H)-one (21)
10.0 mg (0.02 mmols) of the previous compound was dissolved in 10 mL of EtOH and approximately 15 mg tosic acid was added. The reaction was heated to reflux for 48 hours. TLC analysis indicated only one major product and complete conversion of starting material. Solvents were removed under reduced pressure to produce a colorless, oily semi-solid. Product was purified by HPLC using a water-methanol gradient with 0.08% v/v trifluoroacetic acid. 0-5 minutes 20% MeOH, 5-25 minutes 20-65% MeOH, 25-30 minutes 65-100% MeOH. Product elutes at 27.9 minutes. 4.0 mg (0.0062 mmols) produced as a mono-TFA salt. Yield 31%. TLC (CH2Cl2: MeOH 9:1 v/v): Rf = 0.55; 1H NMR (300 MHz, CDCl3, δ): 8.98 (s, 1H), 7.85 (m, 1H), 7.75 (m, 1H), 7.57 (m, 1H), 7.52 (m, 1H), 7.35 (m, 5H), 7.17 (m, 1H), 7.12 (m, 1H), 6.63 (m, 1H), 3.82 (m, 3H), 3.52 (m, 3H) 2.05 (s, 3H), 1.13 (m, 3H) ESI-MS m/z 533.6 (M + H+)+ MW: 532.46 g/mol. Mono-TFA salt FW: 646.45 g/mol.
4-(3-chloro-2-methylphenyl)-6-((4-chlorophenyl)(propoxy)(1-methyl-1H-imidazol-5-yl)methyl)-1-methylquinolin-2(1H)-one (22)
10.0 mg (0.02 mmols) 4-(3-Chloro-2-methylphenyl)-6-((4-chlorophenyl)(hydroxy)(1-methyl-1H-imidazol-5-yl)methyl)-1-methylquinolin-2(1H)-one was dissolved in 10 mL of PrOH and approximately 15 mg tosic acid was added. The reaction was heated to reflux for 48 hours. TLC analysis indicated only one major product and complete conversion of starting material. Solvents were removed under reduced pressure to produce a colorless, oily semi-solid. Product was purified by HPLC using a water-methanol gradient with 0.08% v/v trifluoroacetic acid. 0-5 minutes 20% MeOH, 5-25 minutes 20-65% MeOH, 25-30 minutes 65-100% MeOH. Product elutes at 28.6 minutes. 3.9 mg (0.0059 mmols) produced as a mono-TFA salt. Yield 30%. TLC (CH2Cl2: MeOH 9:1 v/v): Rf = 0.55; 1H NMR (300 MHz, CDCl3, δ): 8.97 (s, 1H), 7.76 (ddd, J = 9.0, 2.4 Hz, 1H), 7.73 (d, J = 9.0 Hz, 1H), 7.54 (m, 2H), 7.32 (m, 5H), 7.19 (dd, J = 2.4 Hz, 1H), 7.15 (m, 1H), 6.59 (m, 1H), 3.78 (s, 3H), 3.47 (m, 3H) 2.02 (m, 3H), 1.50 (m, 2H), 0.86 (m, 3H) ESI-MS m/z 546.6 (M + H+)+ MW: 546.49 g/mol. Mono-TFA salt FW: 660.51 g/mol.
4-(3-chloro-2-methylphenyl)-6-((4-chlorophenyl)(methylamino)(1-methyl-1H-imidazol-5-yl)methyl)-1-methylquinolin-2(1H)-one (23)
10.0 mg (0.02 mmols) 4-(3-Chloro-2-methylphenyl)-6-((4-chlorophenyl)(hydroxy)(1-methyl-1H-imidazol-5-yl)methyl)-1-methylquinolin-2(1H)-one was dissolved in 5 mL of SOCl2. The flask was covered with aluminum foil and the reaction mixture was stirred overnight. SOCl2 was removed at reduced pressure to produce a whitish solid. 10 mL THF was added and stirring with the flask open, anhydrous methylamine was bubbled through the solution for a period of 30 minutes. Then the flask was sealed and stirred overnight under an overpressure of methylamine. Solvents were removed at reduced pressure, and the crude mixture was dissolved in 1 mL of MeOH and centrifuged. The supernatant was collected and injected onto HPLC. Product was purified using a water-methanol gradient with 0.08% v/v trifluoroacetic acid. 0-5 minutes 20% MeOH, 5-25 minutes 20-65% MeOH, 25-30 minutes 65-100% MeOH. Product elutes at 24.4 minutes. 1.4 mg (0.0018 mmols) produced as a bis-TFA salt. Yield 9%. TLC (CH2Cl2: MeOH 9:1 v/v): Rf = 0.55; 1H NMR (300 MHz, CDCl3, δ): 8.86 (m, 1H), 7.82 (m, 2H), 7.54 (m, 1H), 7.41-7.27 (m, 6H), 7.15 (m, 1H), 7.00 (m, 1H), 6.64 (s, 1H), 3.86 (m, 3H), 3.57 (m, 3H) 2.16 (m, 3H), 2.01 (m, 3H) ESI-MS m/z 517.5 (M + H+)+ MW: 517.45 g/mol. Bis-TFA salt FW: 745.5 g/mol.
4-(3-Chloro-2-methylphenyl)-6-((4-chlorophenyl)(hydroxy)(1-methyl-1H-imidazol-5-yl)methyl)-1-methylquinolin-2(1H)-one (20)
The title compound was injected on HPLC using a water-methanol gradient with 0.08% v/v trifluoroacetic acid. 0-5 minutes 20% MeOH, 5-25 minutes 20-65% MeOH, 25-30 minutes 65-100% MeOH. Product elutes at 23.9 minutes. Produced as a mono-TFA salt. TLC (CH2Cl2: MeOH 9:1 v/v): Rf = 0.55; 1H NMR (300 MHz, CD3OD, δ): 8.94 (m, 1H), 7.86 (dd, J = 9.0, 2.1 Hz, 1H), 7.79 (m, 2H), 7.51 (m, 1H), 7.37 (m, 2H), 7.31-7.18 (m, 3H), 7.09 (m, 1H), 6.85-6.76 (m, 2H), 6.62 (m, 1H), 3.85 (s, 3H), 3.65 (m, 3H), 1.96 (m, 3H) ESI-MS m/z 504.6 (M + H+)+ MW: 504.41 g/mol. Mono-TFA salt FW: 618.43 g/mol.
6-Bromo-N-(E)-2,6-difluorocinnamoylanilide
15.0 g (81.5 mmols) (E)-2,6-difluorocinnamic acid was dissolved in 25 mL SOCl2 and heated to reflux and stirred overnight. Thionyl chloride was removed at reduced pressure, then anhydrous toluene was added and removed at reduced pressure two times to produce a flaky white solid. Crude product was triturated and transferred to a separate flame-dried flask, which was placed under vacuum overnight. 16.1 g (79.5 mmols) was produced and used without further purification. Yield, 97%. 6.50 g (37.8 mmols) p-bromoaniline was placed in a separate 500 mL round bottomed flask and dissolved in 100 mL anhydrous CH2Cl2. 13.2 mL (75.5 mmols, 2eq.) diisopropylethylamine was added and the solution was allowed to stir for several minutes, at which time the temperature was lowered to 0 °C. 11.5 g (57.0 mmols, 1.5 eq.) crude cinnamoyl chloride from above was dissolved in approximately 40 mL CH2Cl2 and added rapidly, dropwise. This was allowed to stir overnight. The color became dark green. The crude mixture was partitioned between CH2Cl2 and water. The organic phase was collected and dried with MgSO4, then solvents were removed under reduced pressure to produce a solid which was re-crystallized from CHCl3 to produce 9.72 g (28.74 mmols) of long yellowish needles of product for a yield of 76%. 1H NMR (300 MHz, CDCl3, δ): 7.87 (d, J = 15.9 Hz, 1H), 7.54 (m, 2H), 7.46 (m, 2H), 7.32 (m, 1H), 6.96 (m, 2H), 6.85 (d, J = 15.9 Hz, 1H) MW: 338.15 g/mol.
6-Bromo-4-(2,6-difluorophenyl)-2-oxotetrahydroquinoline
4.35 g (12.9 mmols) of the previous compound was added to a 250 mL round bottomed flask, which was fitted with a reflux condenser and a stir-bar. 45 mL concentrated H2SO4 was added and reaction was heated to 105 °C for four hours, at which time the reaction was quenched by pouring mixture into ice water. A white precipitate formed and was collected by filtration to produce 3.42 g (10.1 mmols) of crude product, yield 79%. 1H NMR (300 MHz, CD3OD, δ): 7.45 (m, 1H), 7.36 (dd, J = 8.4 Hz, 1.2 Hz, 1H), 7.09 (m, 2H), 7.07 (d, J = 8.4 Hz, 1H), 6.94 (s, 1H), 3.00 (dd, J = 16.5 Hz, 12 Hz, 1H), 2.79 (dd, J = 16.5 Hz, 6.6 Hz, 1H) MW: 338.15 g/mol.
6-Bromo-4-(2,6-difluorophenyl)quinolin-2(1H)-one
A mixture of the previous compound (1.0 g, 2.95 mmols) and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (4.03 g, 17.75 mmols) in dioxane (50 mL) was stirred under reflux for 48 hours and concentrated in vacuo. The residue was dissolved in potassium carbonate solution (2.5%, 50 mL) and extracted with CH2Cl2-MeOH (20:1, 2×50 mL). The combined dried (MgSO4) organic extracts were evaporated, and the residue was purified with flash chromatography over silica to produce 678 mg (2.02 mmols) of pure product, 68.4% yield. 1H NMR (300 MHz, CD3OD, δ): 7.73 (dd, J = 9 Hz, 2.1 Hz, 1H), 7.65 (m, 1H), 7.40 (d, J = 8.7 Hz, 1H), 7.25 (m, 3H), 6.69 (s, 1H) MW: 336.13 g/mol.
6-Bromo-4-(2,6-difluorophenyl)-2-methoxyquinoline
A 50 mL round bottomed flask was charged with 360 mg (1.07 mmols) of the previous compound. 15 mL CH2Cl2 was added to the flask follwed by 316 mg (2.14 mmols, 2 eq.) BF4OMe3. The flask was sealed with a septum and stirred overnight with a slight overpressure of N2 vented through a bubbler. The following day, several mL 1.5 M NaOH was added and then allowed to stir for about an hour, at which time the mixture was partitioned between CHCl3 and H2O. The organic phase was collected, then washed with brine and dried with anhydrous MgSO4. Solvents were removed under vacuum. Crude material was adsorbed onto silica, then purified using a mobile phase of hexanes:CH2Cl2; 1:1 (v/v) to produce 189 mg (0.54 mmols) of product, 50% yield. TLC (hexanes:CH2Cl2 1:1 v/v): Rf = 0.50; 1H NMR (300 MHz, CDCl3, δ): 7.82 (d, 7.36 (dd, J = 9 Hz, 1H), 1H), 7.73 (dd, J = 8.9 Hz, 2.1 Hz, 1H), 7.55 (d, J = 1.8 Hz, 1H), 7.50 (m, 1H), 7.28 (s, 1H), 7.11 (m, 2H), 6.95 (s, 1H), 4.13 (s, 3H) MW: 350.16 g/mol.
4-(2,6-Difluorophenyl)-6-((4-chlorophenyl)(hydroxy)(1-methyl-1H-imidazol-5-yl)methyl)-2-methoxyquinoline
A 25 mL round bottomed flask was flame-dried, fitted with a magnetic stir-bar and charged with 100 mg of the previous compound. 5 mL of THF was added and mixture was stirred for 10 minutes, at which time the temperature was lowered to −78 °C and stirred for an additional 10 minutes. 125 μL (1.05 eq. 0.3124 mmols) 2.5 M n-BuLi was added dropwise accompanied by a color shift to dark yellow-orange. This was allowed to stir for approximately 5 minutes after the sublimation of CO2 subsided as indicated by the evolution of gas from the dry-ice bath. A separate flask was flame dried and charged with 60 mg 13 (0.9 eq., 0.2678 mmols). This was dissolved in 55 mL THF and added to the flask containing quinoline in three increments, rapidly dropwise over 10 minutes. Color shifted steadily to yellow-gold after stirring overnight and warming slowly to room temperature. The reaction was quenched by addition of 1 volume equivalent of a saturated aqueous solution of NH4Cl. The bi-phasic mixture was partitioned between 1 M NH4OH and CHCl3. Organic phase was collected and solvents were removed under reduced pressure to produce a foamy, white semi-solid. This was purified on silica with a mobile phase consisting of MeOH/CH2Cl2 1:10 v/v to produce 71.5 mg (0.1443 mmols) of product, 49% yield. TLC (CH2Cl2:MeOH 9:1 v/v): Rf = 0.45; 1H NMR (300 MHz, CDCl3, δ): 7.91 (d, 1H), 7.64 (dd, 1H), 7.41 (m, 1H), 7.23 (m, 4H), 7.14 (m, 1H), 6.98 (m, 3H), 4.13 (s, 3H), 3.50 (s, 3H), 3.35 (s, 3H) ESI-MS m/z 492.5 (M + H+)+ MW: 491.92 g/mol
4-(2,6-Difluorophenyl)-6-((4-chlorophenyl)(hydroxy)(1-methyl-1H-imidazol-5-yl)methyl)-quinolin-2(1H)-one
71.5 mg (0.1453 mmols) of the previous compound was loaded into a 25 mL round bottomed flask and dissolved in 5 mL THF. 0.61 mL (3.63 mmols, 25 eq.) 6N aqueous HCl was added and mixture was refluxed for 4 hours at which time reaction was neutralized with a 1M NH4OH in water. The mixture was partitioned between 1M NH4OH and CHCl3. The aqueous was extracted 3 times with CHCl3, and the organic fractions were combined and dried with anhydrous MgSO4. Solvents were removed under vacuum. Crude product was purified on silica with a mobile phase consisting of MeOH/CH2Cl2 1:10 v/v to produce 71.5 mg (0.1443 mmols) of product, 78% yield. TLC (CH2Cl2:MeOH 9:1 v/v): Rf = 0.35; 1H NMR (300 MHz, CDCl3, δ): 7.91 (dd, J = 8.4 Hz, 1.8 Hz, 1H), 7.64 (dd, 1H), 7.41 (m, 1H), 7.23 (m, 4H), 7.14 (m, 1H), 6.98 (m, 3H), 4.13 (s, 3H), 3.50 (s, 3H), 3.35 (s, 3H) MW: 477.89 g/mol.
4-(2,6-Difluorophenyl)-6-((4-chlorophenyl)(hydroxy)(1-methyl-1H-imidazol-5-yl)methyl)-1-methylquinolin-2(1H)-one
54 mg (0.11 mmols) of the previous compound was added to a 25 mL flask and dissolved in 5 mL THF. 13 mg (0.5 eq., 0.057 mmols) benzyltriethylammonium chloride was added as a phase transfer catalyst. 0.82 mL 40% wt. NaOH (120 eq., 540 mg) was added and allowed to stir for approximately 10 minutes. 10 μL (1.4 eq., 0.16 mmols) CH3I was added and the mixture was allowed to stir overnight. The THF was removed at reduced pressure and the residue was partitioned between CHCl3 and 1M NH4OH. The product was purified on silica with a mobile phase consisting of MeOH/CH2Cl2 1:10 v/v to produce 27 mg (3.97 mmols) of product as a colorless semi-solid, 48% yield. TLC (CH2Cl2:MeOH 9:1 v/v): Rf = 0.45; 1H NMR (300 MHz, CDCl3, δ): 7.77 (dd, 1H), 7.73 (d, 1H), 7.64 (s, 1H), 7.55 (m, 1H), 7.31 (m, 2H), 7.18 (m, 2H), 7.11 (m, 2H), 6.97 (d, 1H), 6.73 (s, 1H), 3.85 (s, 3H), 3.42 (m, 3H) MW: 491.92 g/mol.
4-(2,6-Difluorophenyl)-6-((4-chlorophenyl)(methoxy)(1-methyl-1H-imidazol-5-yl)methyl)-1-methylquinolin-2(1H)-one (16)
13 mg (0.027 mmols) of the previous compound was dissolved in 10 mL of MeOH and approximately 7 mg tosic acid was added. The reaction was heated to reflux for 48 hours. TLC analysis indicated only one major product and complete conversion of starting material. Solvents were removed under reduced pressure to produce a colorless, oily semi-solid. Product was purified by HPLC using a water-methanol gradient with 0.08% v/v trifluoroacetic acid. 30-100% over 20 minutes followed by 10 minutes at 100%. Product elutes at 12.2 minutes. 6.7 mg (0.0893 mmols) produced as a mono-TFA salt. Yield 59%. TLC (CH2Cl2: MeOH 9:1 v/v): Rf = 0.55; 1H NMR (300 MHz, CD3OD, δ): 9.02 (s, 1H), 7.85 (dd, J = 9.0, 2.1 Hz, 1H), 7.76 (d, J = 9.0 Hz, 1H), 7.63 (m, 1H), 7.58 (d, J = 1.5 Hz, 1H), 7.38 (m, 4H), 7.20 (m, 3H), 6.78 (s, 1H), 6.73 (s, 1H), 3.83 (s, 3H), 3.55 (s, 3H), 3.21 (s, 3H) ESI-MS m/z 506.5 (M + H+)+ MW: 505.94 g/mol. Mono-TFA salt FW: 619.97 g/mol.
Supplementary Material
Acknowledgements
This work was supported by a grant from the National Institutes of Health (AI070218) and from the Drugs for Neglected Diseases initiative (DNDi).
Abbreviations
- Tc-14DM
lanosterol 14α-demethylase from T. cruzi
- T cruzi
Trypanosoma Cruzi
- IC50
concentration of inhibitor resulting in 50% enzyme inhibition
- EC50
concentration of inhibitor resulting in 50% parasite growth inhibition
- ESI-MS
electrospray ionization mass spectrometry.
Footnotes
Supporting Information Available: Additional synthesis information and NMR are provied. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Division of Parasitic Diseases CfDC Chagas Disease. http://www.cdc.gov/chagas/
- 2.Pecoul B, Chirac P, Trouiller P, Pinel J. Access to Essential Drugs in Poor Countries. A Lost Battle? Journal of the American Medical Association. 1999;281:361–367. doi: 10.1001/jama.281.4.361. [DOI] [PubMed] [Google Scholar]
- 3.Gelb MH, Hol WG. Drugs to Combat Tropical Protozoan Parasites. Science. 2002;297:343–344. doi: 10.1126/science.1073126. [DOI] [PubMed] [Google Scholar]
- 4.Kohl NE, Mosser SD, deSolms J, Giuliani EA, Pompliano DL, Graham SL, et al. Selective Inhibition of ras-Dependent Transformation by a Farnesyl Transferase Inhibitor. Science. 1993;260:1934–1937. doi: 10.1126/science.8316833. [DOI] [PubMed] [Google Scholar]
- 5.Buckner FS, Verlinde CLMJ, La Flamme AC, Van Voorhis WC. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing beta-galactosidase. Antimicrobial Agents and Chemotherapy. 1996;40:2592–2597. doi: 10.1128/aac.40.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hucke O, Gelb MH, Verlinde CLMJ, Buckner FS. The Protein Farnesyltransferase Inhibitor Tipifarnib as a New Lead for the Development of Drugs against Chagas Disease. Journal of Medicinal Chemistry. 2005;48:5415–8. doi: 10.1021/jm050441z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karp JE, Kaufmann SH, Adjei AA, Lancet JE, Wright JJ, End DW. Current status of clinical trials of farnesyltransferase inhibitors. 2001. pp. 470–476. [DOI] [PubMed]
- 8.Reid TS, Beese LS. Crystal structures of the anticancer clinical candidates R115777 (Tipifarnib) and BMS-214662 complexed with protein farnesyltransferase suggests a mechanism of FTI selectivity. Biochemistry. 2004;43:6877–6884. doi: 10.1021/bi049723b. [DOI] [PubMed] [Google Scholar]
- 9.Kraus JM, Verlinde CLMJ, Karimi M, Lepesheva GI, Gelb MH, Buckner FS. Rational modification of a candidate cancer drug for use against Chagas disease. J. Med. Chem. 2008;52:1639–1647. doi: 10.1021/jm801313t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Venet M, End D, Angibaud P. Farnesyl protein transferase inhibitor ZARNESTRA R115. Curr. Top. Med. Chem. 2003:1095–1102. doi: 10.2174/1568026033452050. [DOI] [PubMed] [Google Scholar]
- 11.Suryadevara PK, Olepus S, Lockman JW, Ohkanda J, Karimi M, Verlinde CLMJ, Kraus JM, Schoepe J, Van Voorhis WC, Hamilton AD, Buckner FS, Gelb MH. J. Med. Chem. 2009;52:3703–3715. doi: 10.1021/jm900030h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.