

Abstract
Kinesin superfamily motor proteins contain a structurally conserved loop near the ATP binding site, termed L5. The function of L5 is unknown, although several drug inhibitors of the mitotic kinesin Eg5 bind to L5. We used electron paramagnetic resonance spectroscopy (EPR) to investigate the function of L5 in Eg5. We site-specifically attached EPR probes to ADP, L5, and the neck linker element that docks along the enzymatic head to drive forward motility on microtubules (MTs). Nucleotide-dependent spectral mobility shifts occurred in all of these structural elements, suggesting that they undergo coupled conformational changes. These spectral shifts were altered by deletion of L5 or addition of S-trityl-l-cysteine (STLC), an allosteric inhibitor that binds to L5. In particular, EPR probes attached to the neck linker of MT-bound Eg5 shifted to a more immobilized component in the nucleotide-free state relative to the ADP-bound state, consistent with the neck linker docking upon ADP release. In contrast, after L5 deletion or STLC addition, EPR spectra were highly immobilized in all nucleotide states. We conclude that L5 undergoes a conformational change that enables Eg5 to bind to MTs in a pre-powerstroke state. Deletion or inhibition of L5 with the small-molecule inhibitor STLC blocks this pre-powerstroke state, forcing the Eg5 neck linker to dock regardless of the nucleotide state.
Introduction
Eg5 is a homotetrameric kinesin motor that is vital to mitotic spindle assembly. Small-molecule inhibitors of Eg5 cause mitotic spindle disruption and initiate apoptosis in dividing cells (1–3). Several of these inhibitors, including the drug ispinesib, which is being tested in clinical trials, and the compound S-trityl-l-cysteine (STLC), bind to the same high-affinity site on Eg5 (4,5). This site is a unique surface loop of unknown function called L5 (human Eg5 residues 116–133; Fig. 1 A).
Figure 1.
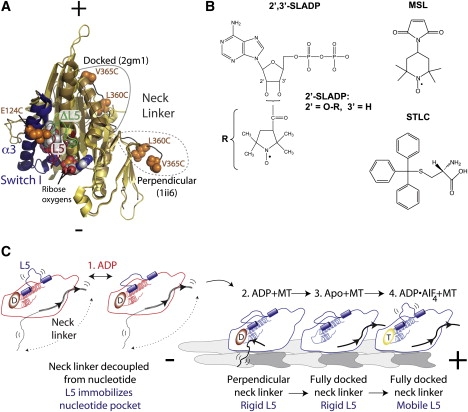
EPR probes, probe locations, and a model based on this study. (A) Structure of Eg5 head showing EPR probe locations. Alignment of PDB ID No. 2gm1 (34) and 1ii6 (15), rendered using PyMol Software (Schrodinger, LLC, New York, NY). ADP is shown in CPK format with ribose oxygens labeled, the α3 helix and Switch I elements are blue, and L5 is red except for the seven residues that were deleted in the ΔL5 constructs, which are shown in green. Added cysteines are indicated in orange CPK format. The two structures have distinct neck linker positions (circled in solid and dashed lines for 2gm1 and 1ii6, respectively), and the positions of the L360C and V365C mutations are shown in both structures. The 1ii6 neck linker, with the labeled cysteines, is in a perpendicular conformation and the 2gm1 neck linker is docked. (B) Structures of the EPR probes and STLC drug used. (C) Schematic diagram of conformational changes in Eg5-367. Eg5 heads are shown bound to the MT (blue) and free (red). The MT is gray, with plus and minus ends indicated. L5 and the neck linker elements are labeled, ADP is indicated as a red oval with D, and ATP is indicated as a yellow oval with T. State 1 shows two different conformations for Eg5 bound to ADP in solution. The neck linker position is not sensitive to the nucleotide state, whereas the L5 element is mobile in one of these conformations and rigid in the other. The rigid L5 conformation supports MT binding. Once ADP-bound Eg5 binds to the MT, its neck linker adopts the perpendicular conformation while L5 remains rigid (state 2). ADP release results in neck linker docking (state 3). Upon ATP binding, L5 returns to the more mobile conformation whereas the neck linker remains docked (state 4). Evidence for and implications of this model are discussed in the text.
In all kinesins, the α2 helix immediately follows the GXXGXGKS/T nucleotide-binding P-loop element. L5 is a structurally conserved loop found in all members of the kinesin superfamily that breaks α2 after five residues; α2 reforms after L5 and extends down the length of the kinesin head. L5 sequences vary among kinesin families in both amino acid content and length. Eg5 is a member of the kinesin-5 family, which have the longest L5 sequences of ∼15–20 residues (6).
L5 is a unique feature of kinesin motors and is not found in either myosin motors or G proteins. However, a structural comparison of kinesins, myosins, and G proteins reveals a potential role for L5. In myosin motors, the 25/50-kDa loop, which controls the rate of ADP release as well as entry into the latch state, is located 15 residues C-terminal to its GXXGXGKS/T P-loop. This location is 10 residues beyond the location of L5 in the kinesins (7,8). The α1 helix in G proteins, which corresponds structurally to α2 in kinesins, breaks at approximately the position of L5 and leads directly into Switch I, which controls the rate of GDP release (9). These comparisons suggest that L5 in kinesins may control conformational changes associated with ADP release. Surprisingly, though, replacement or deletion of several residues within L5 only modestly decreases ATPase activity in standard assays (10).
Despite the fact that L5 deletions have little apparent effect, kinetic data suggest a role for L5 in microtubule (MT)-stimulated ADP release. After it collides with the MT, the Eg5 head undergoes a slow kinetic isomerization (∼1 s−1) (11). The structural changes corresponding to this isomerization are unknown, but this step allows Eg5 to bind tightly to the MT and release ADP to proceed through its MT-stimulated ATPase cycle. Eg5 monomers and dimers alike must undergo the isomerization step before MT-stimulated ADP release occurs, suggesting that the isomerization involves a conformational shift in the Eg5 head, possibly in the neck linker and/or in L5 (11,12).
Small-molecule inhibitors that bind to L5 induce two notable changes in Eg5. First, they increase the ADP affinity of Eg5 heads, perhaps by interfering with the isomerization step described above (13). Second, the inhibitor monastrol has been shown to force the neck linker element (residues 354–367), which drives forward motility in kinesins, to dock along the enzymatic core of Eg5 in a post-powerstroke conformation (10). The combination of these two effects is perplexing because the neck linker of ADP-bound Eg5 in the absence of inhibitors is not fully docked, either in solution or on MTs (10,14). Rather, the neck linker of ADP-bound Eg5 is believed to be in a pre-powerstroke conformation that is roughly perpendicular to the axis of the Eg5 head on the MT (14,15). These data have led to the idea that monastrol and similar inhibitors allosterically force Eg5 into an off-pathway conformation having both high ADP affinity and a docked neck linker. However, it is unclear how a small molecule binding to L5 would induce this state.
Here we use EPR spectroscopy to investigate conformational changes of the nucleotide pocket, L5, and the neck linker of Eg5. Our data are consistent with a model in which L5 and the neck linker undergo a series of coupled conformational changes that integrate into the stepping mechanism of Eg5 (Fig. 1 C).
Materials and Methods
Cloning
Eg5-367 in the pRSET plasmid (Invitrogen, Carlsbad, CA) encodes the first 367 amino acids of Eg5 with a C-terminal 6X histidine tag. For cys-light Eg5, all native cysteines were replaced with alanine. Removal of native cysteines and introduction of cysteines into cys-light Eg5 for MSL labeling was performed via Quikchange site-directed mutagenesis (Stratagene, La Jolla, CA). Eg5-513 was cloned from the MegaMan human transcriptome library (Stratagene), and a C-terminal 6X-histidine tag was introduced by polymerase chain reaction. The ΔL5 mutants (lacking L5) were created using Exsite mutagenesis (Stratagene). All constructs were verified by DNA sequencing.
Expression and protein purification
All Eg5 constructs were grown and expressed in E. coli as described previously (16). Cells were resuspended in 20 mL of lysis buffer (10 mM Hepes, 2 mM MgCl2, 5 mM NaCl, 1 mM EGTA, 20 mM imidizole, 5 mM β-mercaptoethanol, leupeptin (1 μg/mL), pepstatin (1 μg/mL), aprotinin (1 μg/mL), phenylmethanesulfonyl fluoride (10 μg/mL), 5% sucrose, pH 6.8) and lysed by French press. Supernatants were batch bound in Ni-NTA resin (Qiagen, Valencia, CA) for 1 h, washed with lysis buffer containing 75 mM imidizole, and eluted with lysis buffer containing 350 mM imidizole. Peak fractions were dialyzed into lysis buffer with 1 mM DTT and 10 μM ATP, and then applied to a 5 mL HiTrap S-Sepharose column (GE Healthcare, Waukesha, WI) and eluted using a linear 5–500 mM NaCl gradient. Peak fractions were pooled and frozen with an additional 15% sucrose in liquid nitrogen.
Protein labeling for EPR spectroscopy
Purified Eg5 constructs were concentrated and exchanged into labeling buffer (20 mM HEPES, 2 mM Mg(OAc)2, 20 mM KOAc, 1 mM EGTA, 5% sucrose, pH 6.8, 4°C). The protein was labeled at the nucleotide-binding site by addition of 70 μM spin-labeled nucleotide analogs to 100 μM Eg5-367 in labeling buffer. Nucleotide-analog spin probes were synthesized according to a previously described protocol (17). The spin-labeled nucleotides 2′-SLATP, 3′-SLATP, and 2′,3′-SLATP all exchanged into the nucleotide pocket of Eg5-367 in a few minutes 2′,3′-SLAMPPNP exchanged in after an overnight incubation. The SLATP species are hydrolyzed by Eg5, resulting in Eg5•SLADP at the nucleotide site.
Covalent modification of single cysteine-containing Eg5 constructs was carried out by addition of 300 μM 4-maleimido-2,2,6,6-tetramethyl-1-piperidinyloxy (MSL; Sigma Aldrich, St. Louis, MO) and 100 μM protein to the labeling buffer. MSL labeling was allowed to proceed overnight at 4°C.
The activities of Eg5-367, Eg5-367 ΔL5, and MSL-lableled proteins were verified in a coupled-enzyme ATPase assay as described previously (16).
Sample preparation
Unreacted spin probe was removed using a P-30 spin column (Bio-Rad, Hercules, CA) equilibrated with labeling buffer as described previously (18). For experiments with the diphosphate•AlF4 species at the active site, 10 mM NaF and 2 mM AlCl3 were subsequently added. Nucleotide-free (apo) Eg5 was generated by incubating spin-labeled Eg5 with a solution of 2% apyrase (1 U/μL) for 15 min. The ADPase activity of apyrase was verified by thin-layer chromatography, which showed that all ATP was converted to AMP within 5 min. The ADP release rate of Eg5-367 is 0.05 s−1 (11); therefore, within the 15-min incubation period, essentially all Eg5 will be nucleotide-free. For EPR spectra taken under crystallization conditions, MSL-labeled Eg5 was exchanged into a buffer containing 18% polyethylene glycol (PEG)-3350, 100 mM PIPES, and 200 mM NaNO3 pH 6.8 (15). Experiments performed in the presence of STLC (Sigma Aldrich, St. Louis, MO) or Monastrol (Sigma Aldrich) used a 2:1 molar excess of inhibitor over spin-labeled Eg5 protein. Increasing the STLC concentration to a 4:1 molar excess did not change the EPR spectra obtained from these proteins.
For spectral accumulation of Eg5 samples in solution, the protein was loaded into a 25 μL quartz capillary. For the spectra of Eg5 bound to MTs, 20 μM spin-labeled protein and 60 μM paclitaxel-stabilized, polymerized MTs purified from porcine brain were added to the labeling buffer. For MSL-labeled protein, either 2 mM ADP or ADP•AlF4 was added to the labeling buffer. For the apo state, apyrase-treated protein was used. The MT-Eg5 solution was centrifuged at 100,000 × g for 15 min. The pellet was then scraped with a spatula onto a quartz flat cell, covered with a coverslip, sealed with vacuum grease, and placed in the EPR cavity as described previously (18).
EPR spectroscopy and data analysis
First-derivative, X-band EPR spectra were accumulated with a Bruker EMX spectrometer (Bruker Instruments, Billerica, MA) using a high-sensitivity microwave cavity. The instrument settings were as follows: microwave power = 25 mW, time constant = 164 ms, frequency = 9.83 GHz, and modulation = 0.1 mT at a frequency of 100 kHz. Spectral accumulation and temperature control were performed as described previously (19). All spectra were taken in 10-mT-wide sweeps.
Effective cone angles of mobility can be approximated using the order parameter S = (T‖′ − T0)/(T‖ − T0) as a measure of probe mobility. Here 2T‖′ is the observed splitting, 2T‖ is the splitting for an immobilized probe (7.16 mT for 2′,3′-SLADP), and 2T0 is the isotropic hyperfine splitting for freely tumbling SLADP in solution (3.23 mT for 2′,3′-SLADP). The cone angle is then given by cos θ = −0.5 ± 0.5∗(1 + 8S)1/2, where 2θ is the vertex angle of the cone of mobility. For additional details, see Griffith and Jost (20) and Alessi et al. (21), and Fig. S1 of the Supporting Material.
There is a small effect of polarity on the EPR spectra of nitroxide spin probes, leading to a small change in the broadening of the spectrum (maximally ∼0.1 mT) on going from the interior of a protein to an exposed, aqueous environment (22). The nitroxide probes used in these studies are attached to the ribose hydroxyls of the nucleotide or to solvent-exposed cysteine residues. Available structural data on Eg5 and other kinesin-family motors all indicate that the attached spin probe moieties would be solvent-exposed in both the open and closed conformations of the nucleotide pocket (15,23–26), making the above an overestimate of any polarity effects. However, still using this maximum value, the result would be to underestimate the change in the cone angle by 3–4°. This is too small an effect to alter any of our conclusions.
Deconvolutions to determine the ratios of MSL probes in the more mobile and more immobilized spectral components were performed using a Microsoft Excel-based least-squares fitting algorithm as detailed previously (27). For the mobile basis spectrum, we used the spectrum of MT•E124C-MSL•ADP•AlF4 or, alternatively, that of E124C-MSL•ADP in solution. For the immobilized basis spectrum, we used the spectrum of MT•V365C-MSL taken at 2°C. The basis spectra and sample deconvolutions are shown in Fig. S2. All deconvolutions produced χ2 values ≤ 2% using two basis spectral components.
Results and Discussion
Site-specific introduction of EPR probes into Eg5
To determine the effects of L5 on nucleotide-dependent conformational changes in Eg5, we performed EPR spectroscopy using spin probes located at several locations, either attached to the ribose oxygens of ADP or covalently attached to specific cysteines within a human monomeric Eg5 construct (Eg5-367; Fig. 1 A). To monitor conformational changes in the nucleotide pocket, we used the synthetic nucleotide spin probe 2′,3′-SLADP, as previously used for kinesin-1 and ncd (18,24) (Fig. 1 B).
To generate a triphosphate or transition state analog, we used ADP•AlF4, as described in Materials and Methods. We found that 2′,3′-SLADP•AlF4 yielded similar spectral components to 2′,3′-SLAMP-PNP. However, the ADP•AlF4 bound far more tightly and thus revealed much clearer spectra (data not shown).
To monitor conformational changes in L5 and the neck linker, we used site-directed mutagenesis to remove and replace all four native cysteines with alanines in Eg5-367. Single cysteines were then added back for covalent modification using MSL (Fig. 1 B, Materials and Methods). To conjugate probes to L5, we used an E124C mutant. For the neck linker, we used L360C and V365C mutants.
Changes caused by deleting residues 125–131 from L5 (referred to here as L5 deletion or Eg5-367 ΔL5) (28) or by adding the drug STLC were monitored at both the nucleotide pocket and the neck linker. Identical spectral shifts of Eg5-367 bound to 2′,3′-SLADP were obtained using the inhibitor monastrol (data not shown). However, STLC was used for subsequent experiments because it is a single isomer inhibitor (Fig. 1 B).
The maximal ATPase rate was 23.2 ± 0.67 s−1 for wild-type Eg5-367 and 17 ± 0.71 s−1 for the Eg5-367 ΔL5 mutant, which is consistent with previous results (28). To determine the activity of labeled Eg5-367, proteins were labeled with MSL as described in Materials and Methods. Double integration of EPR spectra verified that these proteins were fully labeled. The maximal MT-stimulated ATPase activities of labeled proteins were as follows: E124C-MSL, 18.4 ± 4.8; L360C-MSL, 17.7 ± 1.2; and V365C-MSL, 14.0 ± 1.7 ATP/head/s. Thus, MSL labeling appeared to have a relatively minor effect on ATPase activity, as reported previously for small fluorescent probes attached to monomeric Eg5 (14).
L5 causes an immobilization of 2′,3′-SLADP in the Eg5 nucleotide pocket that is blocked by STLC treatment
We tested whether L5 has an effect on the conformation of the nucleotide pocket of Eg5 in solution by comparing the EPR spectra of 2′,3′-SLADP bound to Eg5-367 and Eg5-367 ΔL5 (Fig. 2 A). Fig. 2 A shows our basic observation. The three sharp central spectral peaks in the spectra (denoted by arrows) result from unbound probe that is rapidly tumbling in solution. When 2′,3′-SLADP binds at the active site of Eg5, the adjacent protein surface spatially restricts the probe's motion, resulting in a broadening of the EPR spectral peaks. This is visualized as a greater splitting between the low-field peak and high-field dip components of the spectra (some of which are denoted by solid or dashed vertical lines). The more restricted the motion of the probe, the greater the low- to high-field splitting (Table 1). The spectrum of Eg5-367•2′,3′-SLADP (Fig. 2 A, black) has two components. There is a dominant low-field component (labeled [C1] in the inset) in equilibrium with another component seen as a low-field shoulder (labeled [C2] in the inset). The low- to high-field splittings of the [C1] and [C2] components are 4.69 and 6.12 mT, respectively. As a useful first approximation to relate the low- to high-field splitting in the magnetic field variable to the physical magnitude of the conformational change we are observing, EPR probe mobility can be modeled as motion in a cone of revolution (20,21). For the [C1] and [C2] components, these correspond to cones of mobility with vertex angles of 120.5° and 71.5°, respectively. We present our data on the magnitudes of the low- to high-field splittings both in mT and in terms of a cone angle through which the probe can move freely (Table 1). The same two spectral components were also visualized using the probes 2′-SLADP and 3′-SLADP (data not shown). Therefore, these two components are not due to the two enantiomer positions of the spin-label moiety on the ribose oxygens of 2′,3′-SLADP. Rather, we conclude that they reflect two different conformational states of the Eg5 protein that are detected by the spin probes.
Figure 2.
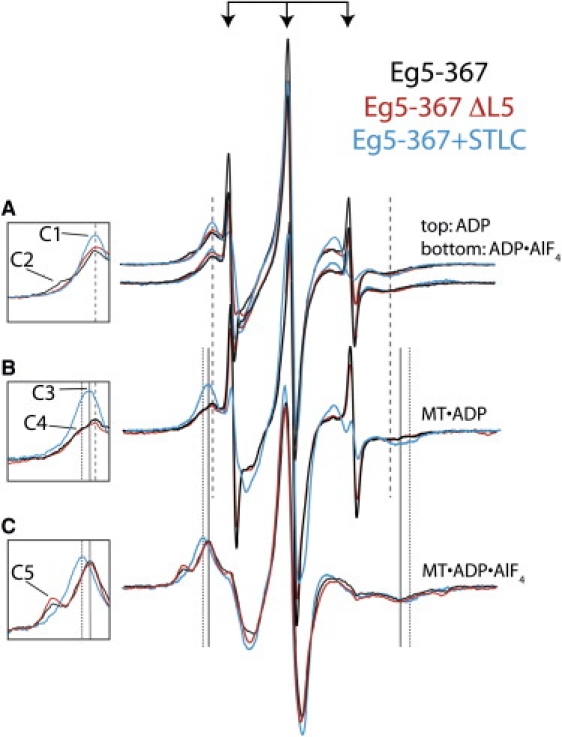
2′,3′-SLADP bound to Eg5-367 reveals mixed nucleotide pocket conformations in solution and on MTs. The innermost three sharp peaks of all spectra are from 2′,3′-SLADP that is free in solution (denoted by arrows). Insets show low-field spectral components, which are identified here by number ([C#]), as described below. (A) Top: Spectra of 2′,3′-SLADP bound to wild-type (black), ΔL5 (red), and STLC-treated Eg5-367 (blue) reveal a common inner component ([C1]) at the same magnetic field location. Bottom: Similar spectra of Eg5-367•2′,3′-SLADP•AlF4. Wild-type Eg5-367 spectra show a more immobilized component ([C2]) that is not present in STLC-treated and ΔL5 spectra. (B) When bound to MTs, 2′,3′-SLADP bound to Eg5-367 and Eg5-367 ΔL5 reveal the same inner component as in solution ([C1], coarse dashed line) as well as a broader component ([C4], fine dashed line). STLC-treated Eg5-367 displays an outward shift of a single spectral component (component [C3]). (C) 2′,3′-SLADP•AlF4 bound to Eg5-367 and Eg5-367 ΔL5 on MTs reveal identical two-component spectra. The inner component has the same low- to high-field splitting as STLC-treated MT•Eg5-367•2′,3′-SLADP ([C3], solid line). The outer component ([C5]) is from highly immobilized probes. The spectrum of STLC-treated 2′,3′-SLADP•AlF4 has a single component ([C4]) that is more immobilized than 2′,3′-SLADP.
Table 1.
Low-to-high field splittings and cone angles of mobility for 2′,3′-SLADP bound to Eg5
| Sample | Splittings (mT) | Cone angle (°) | ||||||
|---|---|---|---|---|---|---|---|---|
| 2′,3′-SLADP | 2′,3′-SLADP•AlF4 | 2′,3′-SLADP | 2′,3′-SLADP•AlF4 | |||||
| −MT | +MT | −MT | +MT | −MT | +MT | −MT | +MT | |
| Eg5-367 | 4.69, [C1] | 4.78, [C1] | 4.64, [C1] | 5.07, [C3] | 120.5 | 117.4 | 122.2 | 107.8 |
| 6.12, [C2] | 5.57 [C4] | 6.32, [C2] | 6.83, [C5] | 71.5 | 91.1 | 63.6 | 38.9 | |
| Eg5-367 ΔL5 | 4.69, [C1] | 4.74, [C1] | 4.71, [C1] | 5.14, [C3] | 120.5 | 118.8 | 119.8 | 105.5 |
| 5.52 [C4] | 6.88, [C5] | 92.8 | 35.8 | |||||
| Eg5-367 STLC | 4.69, [C1] | 4.97, [C3] | 4.75, [C1] | 5.44, [C4] | 120.5 | 111.1 | 118.4 | 95.5 |
Splittings in mT are on the left, and cone angles of probe mobility corresponding to those splittings in degrees are on the right. Bracketed numbers next to splittings indicate specific spectral components shown in the insets of Fig. 2. Standard errors of EPR splittings were ± 0.03 mT. Sample-to-sample variation in EPR splittings is on the order of 0.2 mT; therefore, splittings within 0.2 mT of each other are considered to be the same spectral component (denoted as [C1]–[C5] in Fig. 2 and in this table) (24).
Eg5-367 ΔL5 and Eg5-367 treated with STLC completely lacked the more immobilized spectral component ([C2], 71.5°). The more mobile component ([C1], cone angle 120.5°) remained unchanged (Fig. 2 A, red and blue spectra). This result suggests that L5 induces an immobilization of 2′,3′-SLADP bound to the nucleotide pocket that is not observed after STLC treatment or deletion of residues within L5. The spectral components of Eg5-367, Eg5-367 ΔL5, and STLC-treated Eg5-367 in solution with 2′,3′-SLADP•AlF4 at the active site were the same as those of bound 2′,3′-SLADP (Fig. 2 A, bottom).
Our data indicate that the spin probe samples at least two conformations of the Eg5 nucleotide site in solution, and at least one of these conformations is L5-dependent. However, the conformations detected by 2′,3′-SLADP are not nucleotide-dependent. These two L5-dependent conformations of the nucleotide pocket visualized by our EPR spectra may reflect several different conformations in a mixed population of Eg5-367•ADP or Eg5-367•ADP•AlF4. Kinetic studies on both Eg5 monomers and dimers have identified a conformational change of L5 upon nucleotide binding in solution (13,29). One of these two populations has high ADP affinity and low MT affinity, whereas the other has high MT affinity and low ADP affinity. The two visible components of 2′,3′-SLADP in solution may reflect these two populations of Eg5-367. To illustrate this idea, Fig. 1 C shows two conformations of Eg5-367•ADP in solution (state 1); the one with a more immobilized L5 element binds to the MT (state 2).
Spectra of MSL bound to the Eg5 neck linker do not depend on the nucleotide state in solution
To identify conformational changes of the Eg5 neck linker element in solution, MSL was covalently attached to residue 360 or 365 (L360C-MSL or V365C-MSL; positions shown in Fig. 1 A). Unreacted MSL was removed by column purification (see Materials and Methods). Thus, in contrast to the 2′,3′-SLADP spectra, the three central peaks in the MSL spectra result from highly mobile probes covalently bound to Eg5. The EPR spectra of probes at both locations had two spectral components that were not significantly affected by a change in nucleotide state (Fig. 3 A) or by STLC treatment. In addition, the spectra of V365C-MSL with and without STLC were identical at 25°C and 4°C (Fig. 3 B). This result indicates that STLC binding does not significantly alter the change in free energy between the available neck linker conformations corresponding to the two spectral components of V365C-MSL (30). Although it is possible that the Eg5 neck linker undergoes conformational changes that are not visualized by MSL covalently bound to Eg5 at these locations, we note that Eg5 neck linker docking is resolvable by EPR in the experiments described below. Thus, we conclude that the simplest explanation for these data is that neck linker docking of Eg5 in solution is not induced by the addition of either ADP•AlF4 or STLC. In our model, the neck linker conformation of Eg5 in solution is decoupled from its nucleotide state (shown in Fig. 1 C, state 1) and from STLC binding.
Figure 3.
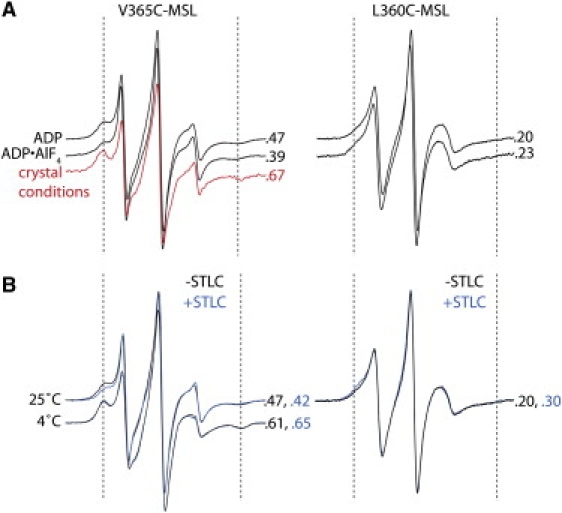
The neck linker position of Eg5 in solution does not depend on the nucleotide state or STLC treatment. More-immobilized spectral components are indicated with dashed lines, and the fraction of immobilized probes is indicated to the right of each spectrum. (A) Both V365C-MSL (left) and L360C-MSL (right) yield similar spectra in the presence of ADP and ADP•AlF4. V365C-MSL spectra shift to a more immobilized component in crystallization buffer conditions (red). The addition of PEG to the motor in solution is likely to induce this shift to the more-immobilized component. Of importance, the sample for this spectrum is not an actual crystal; rather, it is a sample of V365-MSL in a solution that generated crystals of Eg5 (15) (see Materials and Methods). (B) STLC treatment does not alter the spectrum of V365C-MSL in the presence of ADP at either 25°C or 4°C (left), indicating that the free-energy changes between available neck linker conformations are not altered by STLC treatment. Spectra of L360C-MSL in the presence of ADP at 25°C (right) were also identical in the presence and absence of STLC.
The above data stand in apparent contrast to a previous x-ray crystal structure (31) and to spectroscopic data showing that the allosteric inhibitor monastrol induces Eg5 neck linker docking in solution (10). To investigate this discrepancy, we took EPR spectra of V365C-MSL under altered buffer conditions. PEG has been shown to induce protein-protein interactions in a large number of systems (32), and is a component of some crystallization buffers used for Eg5 (15,31). The addition of crystallization buffer components containing PEG (15) (see Materials and Methods for details) increased the fraction of probe in the immobilized component by 20% (Fig. 3 A). This result demonstrates that the neck linker of V365C-MSL is able to dock in solution, but is not induced to do so by addition of STLC. The free-energy change between conformational states corresponding to the mobile and immobilized spectral components is extremely small (∼0.3 kJ/mol for V365C-MSL•ADP in solution), so it is quite possible that the neck linker conformation can be shifted by different buffer conditions or by a slightly different drug agent binding to Eg5. In support of this idea, a recent x-ray crystal structure of Eg5 bound to STLC showed different neck linker conformations for two Eg5 heads within the same crystal unit cell (25). Similarly, kinesin-1 can be induced to dock its neck linker by the mere addition of sulfate ions (27). We conclude that the neck linker of Eg5 in solution may be induced to dock by monastrol but is not induced to dock by STLC, due either to a difference between these two allosteric inhibitors or to differences in experimental conditions between past (10) and present experiments.
2′,3′-SLADP bound to the Eg5 nucleotide pocket is immobilized upon binding of Eg5 to MTs in the diphosphate state and further immobilized in the triphosphate state
EPR spectra of 2′,3′-SLADP bound to kinesin-1 or ncd are broadened upon binding of the MT and further broadened upon binding of AlF4 on the MT. This broadening has been attributed to a movement of Switch I toward the nucleotide pocket, referred to as closing of the nucleotide pocket (24). This interpretation is supported by a cryo-electron microscopy structure of MT-bound kinesin-1 (23). We observed a similar phenomenon in Eg5. EPR spectroscopy was performed on MT•Eg5-367• 2′,3′-SLATP. MT-Eg5 pellets were prepared as described in Materials and Methods, and elsewhere (18,24). The binding constant of Eg5•2′,3′-SLADP to MTs was 2.6 μM (determined via sedimentation) (11) and the concentration of MTs in the pellet was 700 μM. Thus, <1% of the Eg5 in the pellet was not associated with MTs. This is too small a value to affect our conclusions. The resulting spectrum has multiple components, as seen in the broad shoulder of the low-field peak and the broad high-field dip (Fig. 2 B, black spectrum). The innermost component has splitting identical to that of the inner, more mobile component of the spectrum of Eg5•2′,3′-SLADP in solution (component [C1], cone angle: 120.5°; Fig. 2, A and B, dashed vertical line). Upon MT binding, a significant fraction of the probes shift to a new, more immobilized component (component [C4]), with a corresponding cone angle of mobility of 91.1°. These data are consistent with a fraction of motors adopting a more closed conformation of the nucleotide pocket upon MT binding while in equilibrium with other motors that retain a more open conformation. Of interest, the L5-dependent outer component (component [C2]) of the spectrum of Eg5-367•2′,3′-SLADP in solution is lost upon MT binding, and the mobility of 2′,3′-SLADP in the nucleotide pocket of the MT•Eg5•2′,3′-SLADP state is not affected by deletion of L5 (Fig. 2 B, red versus black spectra). The spectrum of the MT•Eg5-367•2′,3′-SLADP•AlF4 state also has two components and is not affected by deletion of L5 (Fig. 2 C, red versus black spectra). The outermost spectral component (component [C5]; Fig. 2 C) is very highly immobilized (cone angle: 38.9°). These data are consistent with the idea that further closing of the Eg5 nucleotide pocket occurs in the triphosphate state relative to the diphosphate state when the motor is bound to MTs, as previously observed for kinesin and ncd (24).
STLC treatment alters but does not prevent MT-induced immobilization of an EPR probe in the Eg5 nucleotide pocket
STLC treatment of Eg5-367•2′,3′-SLADP resolves spectra into one clear component, both in solution and when Eg5 is bound to MTs. In solution, the splitting of this component is identical to the inner, more mobile component of the spectrum of 2′,3′-SLADP bound to untreated Eg5 (component [C1]; Fig. 2 A, blue spectrum). Several kinetic and spectroscopic studies have indicated that L5-binding drugs stabilize a high-ADP-affinity state, radically slowing ADP product release (10,13,26,33). The existence of a single spectral component for STLC-treated Eg5 is consistent with the idea that the conformational states reflected by the EPR spectrum in the presence of STLC together represent an open state of the nucleotide pocket.
The binding of STLC-treated Eg5 to MTs induces a shift in the spectrum of 2′,3′-SLADP to a more immobilized component (component [C3], cone angle: 111.1°; Fig. 2 B). This is the same mobility seen in the inner spectral component of MT•Eg5-367•2′,3′-SLADP•AlF4 (Fig. 2, B and C, solid vertical line). This result indicates that STLC treatment of MT•Eg5-367•2′,3′-SLADP may cause the nucleotide pocket to adopt an ATP-like conformation, consistent with kinetic data showing that drugs that bind to L5 appear to cause ATP resynthesis by Eg5 (13).
The single visible component in the spectrum of STLC-treated Eg5-367•2′,3′-SLADP•AlF4 is not distinguishable from component [C4] (cone angle: 95.5°). This component is more immobilized than component [C3] (cone angle: 111.1°; Fig. 2, B and C, blue spectra). The trend of a shift to a more immobilized component upon MT binding, and again upon 2′,3′-SLADP•AlF4 binding, is observed even in STLC-treated Eg5. Thus, although the EPR spectra suggest that STLC treatment alters the conformation of the nucleotide pocket of Eg5 in all nucleotide states, it does not prevent the MT- and ATP-induced probe immobilization observed for untreated Eg5, kinesin, and ncd (24). We conclude that STLC treatment of Eg5 does not block the closing of Switch I around the nucleotide. In contrast to L5 deletion, STLC treatment markedly alters the spectra of 2′,3′-SLADP or 2′,3′-SLADP•AlF4 bound to Eg5 on MTs. STLC may exert additional effects on the nucleotide pocket that are independent of L5. Consistent with this, a recent x-ray crystal structure indicates that STLC may possibly alter the Switch I/nucleotide state through contacts with the α3-helix or the core β-sheet of Eg5 (25).
MSL bound to L5 of Eg5 on MTs is moderately immobilized in the ADP and apo states, but is mobile in the triphosphate state
The above data indicate that L5 affects the conformation of the nucleotide pocket in solution. To further examine L5 function, we sought to identify conformational changes occurring in L5 as Eg5 binds to the MT, releases ADP, and then binds ATP (Fig. 1 C). Eg5-367 was labeled with MSL at a single cysteine at position 124 within L5 (E124C-MSL; Fig. 1 A). Spectra were collected from pellets of E124C-MSL•ADP in solution and MT•E124C-MSL in the presence of ADP, in the apo state, and in the presence of ADP•AlF4. The spectrum of E124C-MSL•ADP in solution shows a vast majority of covalently attached probes with high mobility (Fig. 4 A, three central peaks). For MT•E124C-MSL•ADP, 32% of probes are present in a more immobilized component (dashed line). A similar spectrum is observed in the MT-bound apo state, with 41% of probes in the immobilized component. When Eg5 is bound to MTs in the ADP•AlF4 state, the EPR spectrum again shows extremely mobile probes. We conclude that the EPR spectra reveal a reversible conformational transition of L5. Through this conformational transition, L5 may exert an influence over the conformation of Eg5, particularly in the ADP and apo states when bound to MTs. The conformation of L5 observed for MT•E124C-MSL in the ADP-bound and apo states is referred to throughout the rest of the text as “rigid” to differentiate the L5 conformational change from the separate, but correlated, closing of the nucleotide pocket detected for MT•Eg5-367•2′,3′-SLADP in the experiments described above.
Figure 4.
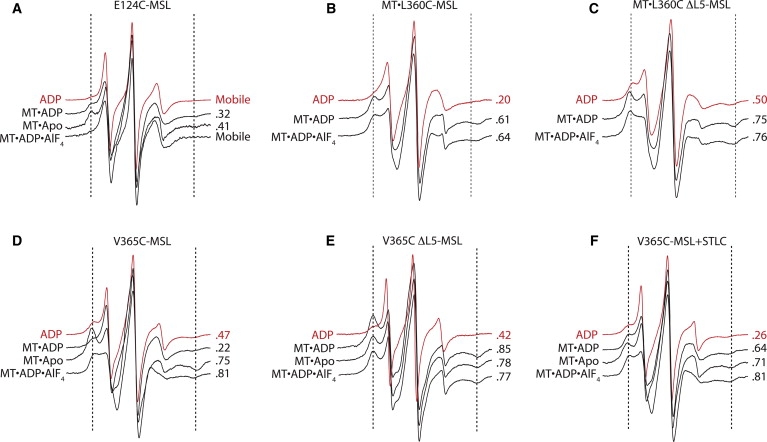
L5 and neck linker movement is coupled to the Eg5 nucleotide state on MTs. All proteins are labeled with the MSL probe at the site indicated. Nucleotide states are indicated to the left of each spectrum, and the fraction of immobilized probes is indicated to the right. Red spectra are in solution, black spectra are MT-bound. (A) E124C, (B) L360C, (C) L360C ΔL5, (D) V365C, (E) V365C ΔL5, and (F) V365C+200 μM STLC.
MSL bound to the neck linker of Eg5 on MTs is immobilized in the apo and triphosphate states
The Eg5 neck linker conformation has been shown to be tightly coupled to the nucleotide state when the motor is bound to MTs, transitioning between a conformation that is believed to be roughly perpendicular to the MT axis (15) and a conformation that is fully docked (14,34). We sought to detect these changes by EPR spectroscopy using L360C-MSL- and V365C-MSL-labeled Eg5. In the perpendicular neck linker conformation, L360 is tucked against the head and V365 is displaced away from it. In the docked neck linker conformation, both residues are tucked against the head (Fig. 1 A). All EPR spectra of both L360C-MSL and V365C-MSL bound to MTs displayed an immobilized component with a low- to high-field splitting of ∼6.4 mT, corresponding to a cone angle of ∼60°. The spectrum of MT•L360C-MSL•ADP has 61% of the probes in the more immobilized component (Fig. 4 B). In contrast, the spectrum of MT•V365C-MSL•ADP shows only 22% of probes in the more immobilized component (Fig. 4 D). The larger fraction of more immobilized probes bound to L360 relative to V365 is consistent with the Eg5 neck linker adopting the perpendicular conformation, or another conformation in which L360 is docked against the core of Eg5 whereas V365 is largely undocked (14,15) (depicted as state 2 in Fig. 1 C).
The spectrum of MT•V365C-MSL undergoes a dramatic shift to the more immobilized component in the apo state, with 75% of probes in the more immobilized component (Fig. 4 D). This is consistent with a transition from a perpendicular neck linker conformation in the ADP state to a fully docked conformation in the MT-bound apo state. Two pieces of existing data support this interpretation. First, this exact conformational transition of the Eg5 neck linker was observed by means of fluorescence spectroscopy (14). Second, the immobilization of neck linker-bound EPR probes was shown to correlate with neck linker docking in kinesin-1 (35). This transition is shown as state 3 in Fig. 1 C.
Very little additional immobilization occurs upon transition to the ADP•AlF4 state, for either MT•L360C-MSL (76% of probes in the more immobilized component) or MT•V365C-MSL (81%) (Fig. 4, B and D). According to the above interpretation, this result indicates that in the majority of Eg5 motors, the neck linkers are fully docked in the apo state and they remain docked in the triphosphate-analog state. Thus, the transition between the MT-bound apo state and the MT-bound ADP•AlF4 state induces a rigid-to-mobile conformational transition in L5, but does not affect the docked neck linker conformation. This is depicted as state 4 in Fig. 1 C.
MSL probes attached to the neck linker of MT-bound Eg5-367 ΔL5 or STLC-treated Eg5-367 are highly immobilized regardless of nucleotide state
To establish whether there is a direct structural link between L5 function and Eg5 neck linker conformation when bound to MTs, EPR spectra were collected from L360C-MSL (Fig. 4 C) and V365C-MSL (Fig. 4 E) using the Eg5-367 ΔL5 mutant. Spectra of MT-bound Eg5 with probes at both locations show a large fraction of probes in the more-immobilized component in all nucleotide states. MT•V365C-MSL ΔL5•ADP has 85% of probes in the more immobilized component, versus 22% for untreated V365C-MSL with L5 intact. If our structural interpretation of these EPR spectral data is correct, neck linker docking is heavily favored for Eg5-367 ΔL5 in all nucleotide states when bound to MTs.
Our spectra of STLC-treated MT•Eg5-367•2′,3′-SLADP are consistent with the idea that STLC induces an ATP-like state of the Eg5 nucleotide pocket when bound to MTs (33). To determine whether STLC induces an ATP-like, fully docked state of the Eg5 neck linker when bound to MTs, we collected EPR spectra from MT pellets of STLC-treated V365C-MSL. Similar to the case with L5 deletion, in the presence of STLC a large fraction of probes was in the more-immobilized component in all nucleotide states (Fig. 4 F). In particular, STLC-treated MT•V365C-MSL•ADP had 64% of probes in the immobilized component versus 22% in the absence of STLC.
Although the effects of L5 deletion in STLC treatment that we observed using 2′,3′- SLADP in the nucleotide pocket are distinct, L5 deletion and STLC treatment also have a similar effect on the neck linker. This functional defect prevents Eg5 from establishing its pre-powerstroke state by binding to MTs with its neck linker in the perpendicular conformation. Instead, the neck linker of Eg5-367 ΔL5 or Eg5-367 treated with STLC is fully docked regardless of the nucleotide.
Conclusions
This work demonstrates that L5 in the kinesin family member Eg5 undergoes conformational changes that are coupled to the nucleotide state and neck linker movement. L5 causes a significant reduction in the mobility of a spin-labeled ADP analog in the Eg5 nucleotide pocket. L5 itself undergoes a nucleotide-dependent conformational change when Eg5 is bound to MTs. Based on this role of L5 in Eg5's mechanochemical mechanism, we suggest that L5 in some kinesin motors may be analogous to the 25/50 kDa loop of the myosin motors. L5 and the 25/50 kDa loop are in structurally similar locations on kinesin and myosin motors, and both elements fine-tune the initial ADP release event that leads to MT or actin activation.
L5 deletion alters the series of conformational changes that take place in the Eg5 neck linker. In agreement with previous results (14), we found that Eg5 transitions from a pre-powerstroke neck linker conformation that is perpendicular to the head, to a post-powerstroke, docked neck linker conformation upon ADP release. Deletion of L5 blocks the pre-powerstroke conformation and forces Eg5 to bind to MTs with its neck linker docked, regardless of the nucleotide state. Together, these data suggest that L5, the nucleotide pocket, and the neck linker undergo a series of coupled conformational changes that integrate into the stepping mechanism of Eg5. High-resolution structural data will be necessary to reveal how these elements are coupled to elicit the conformational states and conformational changes that have been identified here.
Supporting Material
Two figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00344-9.
Supporting Material
Acknowledgments
The authors thank members of the Rice laboratory for assistance, Susan Gilbert for the Eg5-367 construct, Park Packing for tubulin materials, Chuck Sindelar for his spectral deconvolution routine, and Chris Felix (Medical College of Wisconsin, Milwaukee, WI) for providing the EPR facilities.
This work was supported by National Institutes of Health grants GM072656 (S.R. and A.L.), AR042895 (R.C. and N.N.), and GM077067 (E.P. and N.N.).
References
- 1.Mayer T.U., Kapoor T.M., Mitchison T.J. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- 2.Kapoor T.M., Mayer T.U., Mitchison T.J. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J. Cell Biol. 2000;150:975–988. doi: 10.1083/jcb.150.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skoufias D.A., DeBonis S., Kozielski F. S-trityl-L-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. J. Biol. Chem. 2006;281:17559–17569. doi: 10.1074/jbc.M511735200. [DOI] [PubMed] [Google Scholar]
- 4.Lad L., Luo L., Sakowicz R. Mechanism of inhibition of human KSP by ispinesib. Biochemistry. 2008;47:3576–3585. doi: 10.1021/bi702061g. [DOI] [PubMed] [Google Scholar]
- 5.Brier S., Lemaire D., Kozielski F. Identification of the protein binding region of S-trityl-L-cysteine, a new potent inhibitor of the mitotic kinesin Eg5. Biochemistry. 2004;43:13072–13082. doi: 10.1021/bi049264e. [DOI] [PubMed] [Google Scholar]
- 6.Dagenbach E.M., Endow S.A. A new kinesin tree. J. Cell Sci. 2004;117:3–7. doi: 10.1242/jcs.00875. [DOI] [PubMed] [Google Scholar]
- 7.Goodson H.V., Warrick H.M., Spudich J.A. Specialized conservation of surface loops of myosin: evidence that loops are involved in determining functional characteristics. J. Mol. Biol. 1999;287:173–185. doi: 10.1006/jmbi.1999.2565. [DOI] [PubMed] [Google Scholar]
- 8.Sweeney H.L., Rosenfeld S.S., Sellers J.R. Kinetic tuning of myosin via a flexible loop adjacent to the nucleotide binding pocket. J. Biol. Chem. 1998;273:6262–6270. doi: 10.1074/jbc.273.11.6262. [DOI] [PubMed] [Google Scholar]
- 9.Siderovski D.P., Willard F.S. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int. J. Biol. Sci. 2005;1:51–66. doi: 10.7150/ijbs.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maliga Z., Xing J., Rosenfeld S.S. A pathway of structural changes produced by monastrol binding to Eg5. J. Biol. Chem. 2006;281:7977–7982. doi: 10.1074/jbc.M511955200. [DOI] [PubMed] [Google Scholar]
- 11.Cochran J.C., Krzysiak T.C., Gilbert S.P. Pathway of ATP hydrolysis by monomeric kinesin Eg5. Biochemistry. 2006;45:12334–12344. doi: 10.1021/bi0608562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cochran J.C., Sontag C.A., Gilbert S.P. Mechanistic analysis of the mitotic kinesin Eg5. J. Biol. Chem. 2004;279:38861–38870. doi: 10.1074/jbc.M404203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cochran J.C., Gilbert S.P. ATPase mechanism of Eg5 in the absence of microtubules: insight into microtubule activation and allosteric inhibition by monastrol. Biochemistry. 2005;44:16633–16648. doi: 10.1021/bi051724w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenfeld S.S., Xing J., King P.H. Docking and rolling, a model of how the mitotic motor Eg5 works. J. Biol. Chem. 2005;280:35684–35695. doi: 10.1074/jbc.M506561200. [DOI] [PubMed] [Google Scholar]
- 15.Turner J., Anderson R., Sakowicz R. Crystal structure of the mitotic spindle kinesin Eg5 reveals a novel conformation of the neck-linker. J. Biol. Chem. 2001;276:25496–25502. doi: 10.1074/jbc.M100395200. [DOI] [PubMed] [Google Scholar]
- 16.Larson A.G., Landahl E.C., Rice S.E. Mechanism of cooperative behaviour in systems of slow and fast molecular motors. Phys. Chem. Chem. Phys. 2009;11:4890–4898. doi: 10.1039/b900968j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crowder M.S., Cooke R. Orientation of spin-labeled nucleotides bound to myosin in glycerinated muscle fibers. Biophys. J. 1987;51:323–333. doi: 10.1016/S0006-3495(87)83338-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong Y.L., Dietrich K.A., Rice S.E. The kinesin-1 tail conformationally restricts the nucleotide pocket. Biophys. J. 2009;96:2799–2807. doi: 10.1016/j.bpj.2008.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naber N., Purcell T.J., Cooke R. Dynamics of the nucleotide pocket of myosin measured by spin-labeled nucleotides. Biophys. J. 2007;92:172–184. doi: 10.1529/biophysj.106.090035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffith O.H., Jost P.C. Academic Press; New York: 1976. Lipid Spin Labels in Biological Membranes. [Google Scholar]
- 21.Alessi D.R., Corrie J.E., Trentham D.R. Synthesis and properties of a conformationally restricted spin-labeled analog of ATP and its interaction with myosin and skeletal muscle. Biochemistry. 1992;31:8043–8054. doi: 10.1021/bi00149a039. [DOI] [PubMed] [Google Scholar]
- 22.Owenius R., Osterlund M., Carlsson U. Spin and fluorescent probing of the binding interface between tissue factor and factor VIIa at multiple sites. Biophys. J. 2001;81:2357–2369. doi: 10.1016/S0006-3495(01)75882-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sindelar C.V., Downing K.H. The beginning of kinesin's force-generating cycle visualized at 9-A resolution. J. Cell Biol. 2007;177:377–385. doi: 10.1083/jcb.200612090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naber N., Minehardt T.J., Pate E. Closing of the nucleotide pocket of kinesin-family motors upon binding to microtubules. Science. 2003;300:798–801. doi: 10.1126/science.1082374. [DOI] [PubMed] [Google Scholar]
- 25.Kaan H.Y., Ulaganathan V., Kozielski F. An allosteric transition trapped in an intermediate state of a new kinesin-inhibitor complex. Biochem. J. 2009;425:55–60. doi: 10.1042/BJ20091207. [DOI] [PubMed] [Google Scholar]
- 26.Krzysiak T.C., Wendt T., Hoenger A. A structural model for monastrol inhibition of dimeric kinesin Eg5. EMBO J. 2006;25:2263–2273. doi: 10.1038/sj.emboj.7601108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sindelar C.V., Budny M.J., Cooke R. Two conformations in the human kinesin power stroke defined by X-ray crystallography and EPR spectroscopy. Nat. Struct. Biol. 2002;9:844–848. doi: 10.1038/nsb852. [DOI] [PubMed] [Google Scholar]
- 28.Maliga Z., Mitchison T.J. Small-molecule and mutational analysis of allosteric Eg5 inhibition by monastrol. BMC Chem. Biol. 2006;6:2. doi: 10.1186/1472-6769-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krzysiak T.C., Grabe M., Gilbert S.P. Getting in sync with dimeric Eg5. Initiation and regulation of the processive run. J. Biol. Chem. 2008;283:2078–2087. doi: 10.1074/jbc.M708354200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rice S., Cui Y., Cooke R. Thermodynamic properties of the kinesin neck-region docking to the catalytic core. Biophys. J. 2003;84:1844–1854. doi: 10.1016/S0006-3495(03)74992-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan Y., Sardana V., Kuo L.C. Inhibition of a mitotic motor protein: where, how, and conformational consequences. J. Mol. Biol. 2004;335:547–554. doi: 10.1016/j.jmb.2003.10.074. [DOI] [PubMed] [Google Scholar]
- 32.Parsegian V.A., Rand R.P., Rau D.C. Osmotic stress for the direct measurement of intermolecular forces. Methods Enzymol. 1986;127:400–416. doi: 10.1016/0076-6879(86)27032-9. [DOI] [PubMed] [Google Scholar]
- 33.Cochran J.C., Gatial J.E., Gilbert S.P. Monastrol inhibition of the mitotic kinesin Eg5. J. Biol. Chem. 2005;280:12658–12667. doi: 10.1074/jbc.M413140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim K.S., Lu S., Roussell D.L. Synthesis and SAR of pyrrolotriazine-4-one based Eg5 inhibitors. Bioorg. Med. Chem. Lett. 2006;16:3937–3942. doi: 10.1016/j.bmcl.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 35.Rice S., Lin A.W., Vale R.D. A structural change in the kinesin motor protein that drives motility. Nature. 1999;402:778–784. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.