

Abstract
Prolonged seizures (status epilepticus) can activate apoptosis-associated signaling pathways. The extent to which such pathways contribute to cell death might depend on the insult intensity, whereby the programmed or apoptotic cell death component is reduced when seizures are more severe or protracted. We recently showed that mice lacking the pro-apoptotic Bcl-2 homology domain 3-only protein Puma (Bbc3) were potently protected against damage caused by status epilepticus. In the present study we examined whether Puma deficiency was protective when the seizure episode was more severe. Intra-amygdala microinjection of 1 μg kainic acid (KA) into C57BL/6 mice triggered status epilepticus that lasted about twice as long as with 0.3 μg KA prior to lorazepam termination. Hippocampal damage was also significantly greater in the higher-dose group. Over 80 % of degenerating neurons after seizures were positive for DNA fragmentation assessed by terminal deoxynucleotidyl dUTP nick end labeling (TUNEL). Microscopic analysis of neuronal nuclear morphology in TUNEL-positive cells revealed the proportion displaying large rounded clumps of condensed chromatin was ∼50 % lower in the high-dose versus low-dose KA group. Nevertheless, compared to heterozygous and wild-type mice subject to status epilepticus by high-dose KA, neuronal death was reduced by ∼50 % in the hippocampus of Puma-deficient mice. These data suggest aspects of the apoptotic component of seizure-induced neuronal death are insult duration- or severity-dependent. Moreover, they provide further genetic evidence that seizure-induced neuronal death is preventable by targeting so-called apoptosis-associated signaling pathways and Puma loss likely disrupts caspase-independent or non-apoptotic seizure-induced neuronal death.
Keywords: Apoptosis, Bcl-2, Caspase, Epilepsy, Hippocampal sclerosis, Necrosis
A prolonged seizure (status epilepticus) or repeated brief seizures over time can cause neuronal death in both animal models and humans (Meldrum, 1993, Wasterlain et al., 1993, Fujikawa et al., 2000a). Seizure-induced neuronal death is mainly triggered by prolonged over-activation of glutamate receptors which results in loss of cellular ion homeostasis, generation of free radicals, edema and rupture of various cell membranes, and necrosis (Choi, 1988, Meldrum, 1991, Fujikawa, 2005). However, coordinated (programmed) cell death pathways underlie a major component of the seizure-induced neuronal death process, in as much as there is an ordered cascade of signaling events which progress in a predictable manner that are amenable to pharmacologic or genetic disruption (Liou et al., 2003, Henshall and Murphy, 2008). A molecular signature of apoptosis-associated signaling pathways is found within the hippocampus and other brain regions in several experimental models of seizure-induced neuronal death, and also in resected human temporal lobe material from patients with refractory epilepsy. For example, induction of Bcl-2 family proteins, release of apoptogenic molecules from mitochondria, activation of caspases and DNA fragmentation (for review see (Engel and Henshall, 2009)). Caspase-independent apoptosis signaling pathways (Cheung et al., 2005, Heo et al., 2006, Zhao et al., 2010) and non-apoptotic proteases such as calpains (Araujo et al., 2008, Wang et al., 2008), also contribute to excitotoxic neuronal death. Apoptosis-associated genes such as bcl-2 have potent effects not only against caspase-dependent apoptosis but also caspase-independent apoptosis and necrotic cell death (Zhong et al., 1993, Kane et al., 1995, Susin et al., 1999, Single et al., 2001).
The extent of involvement of programmed or apoptotic signaling may be dictated by the severity of the insult, whereby milder insults produce more apoptotic cell death (Ankarcrona et al., 1995, Bonfoco et al., 1995, Ward et al., 2007). Studies of status epilepticus have also suggested that when seizure development is delayed, the apoptotic molecular signature is more pronounced (Kondratyev and Gale, 2004). Our group has shown that focal-onset status epilepticus in mice induced by intra-amygdala kainic acid (KA) results in nuclear morphology consistent with an apoptotic or programmed form of cell death in ∼30 % of neurons positive for DNA fragmentation (Shinoda et al., 2004a). Various biochemical features are also present in this model, including p53 involvement, release of cytochrome c and caspase activation (Murphy et al., 2007, Engel et al., 2010). Increasing the dose of KA in this model produces greater cumulative seizure time and a larger CA3-hilar lesion (Hatazaki et al., 2007, Tanaka et al., 2010). It is unknown whether this affects the relative contribution of apoptosis-associated processes.
We recently reported that mice deficient in puma (bbc3), a potent pro-apoptotic Bcl-2 family protein, were protected against seizure-induced neuronal death in the lower-dose KA model (Engel et al., 2010). In the present study we investigated whether the higher-dose KA model produces a different apoptotic contribution, and the extent to which Puma deficiency may or may not be protective under those conditions.
Experimental Procedures
Mouse model of focal-onset status epilepticus
All animal procedures were performed in accordance with the principals of the European Communities Council Directive (86/609/EEC) and National Institute of Health's Guide for the Care and Use of Laboratory Animals. Procedures were reviewed and approved by the Research Ethics Committee of the Royal College of Surgeons in Ireland, under license from the Department of Health, Dublin, Ireland. Studies were performed according to previously described techniques (Engel et al., 2010). Adult male C57BL/6 mice (20-25 g) (Harlan, UK) and puma+/+, +/-, -/- mice on a C57BL/6 background (Villunger et al., 2003) were used. Briefly, mice were anesthetized using isoflurane (3-5%) and maintained normothermic by means of a feedback-controlled heat blanket (Harvard Apparatus Ltd, Kent, England). A catheter was inserted into the femoral vein for administration of anticonvulsant. Mice were next placed in a stereotaxic frame and following a midline scalp incision three partial craniotomies were performed. Mice were affixed with three recording electrodes (Bilaney Consultants Ltd, Sevenoaks, UK) to record surface electroencephalogram (EEG). Electrodes were placed above dorsal hippocampus and a third over frontal cortex. A guide cannula was affixed over the dura (coordinates from Bregma: AP = -0.94; L = -2.85 mm) (Paxinos and Franklin, 2001) and the entire skull assembly fixed in place with dental cement. Anesthesia was discontinued, EEG recordings were commenced using a Grass Comet XL lab-based EEG (Medivent Ltd, Lucan, Ireland), and then a 31-gauge internal cannula (Plastic Ones Inc.) was inserted into the lumen of the guide to inject KA (Ocean Produce International, Nova Scotia, Canada) [0.3 μg or 1.0 μg in 0.2 μl of vehicle; phosphate-buffered saline (PBS), pH adjusted to 7.4] into the amygdala. The EEG was recorded until intravenous lorazepam (6 mg/kg) administration at 40 min and then recorded 1 h thereafter. Seizure time was calculated from the EEG as the duration of high amplitude and high frequency discharges (HAHFDs) during the 40 min experimental period, as described (Shinoda et al., 2004a).
Histopathology and immunohistochemistry
Mice were euthanized 72 h after anticonvulsant and brains were flash-frozen whole in 2-methylbutane at -30°C and sectioned in the coronal plane at 12 μm. Sections were defrosted, air-dried and post-fixed in 10 % formalin for 30 min, followed by washes in PBS. For assessment of overall (total) neuronal death, sections were stained using Fluoro-Jade B (FJB) (Millipore, Cork, Ireland), as described (Tanaka et al., 2010). For immunohistochemistry, sections were permeabilized with 0.3 % Triton X-100, blocked in 5 % goat serum for 1 h and then incubated overnight at 4°C with antibodies against NeuN (1:400, Chemicon, Hampshire, UK). Sections were washed again in PBS and incubated in goat anti-mouse AlexaFluor 568 (Bio Sciences Ltd, Dun Laoghaire, Ireland). DNA fragmentation was detected using a Fluorescein-based terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) kit according to manufacturer's instructions (Promega, Madison, WI) (Engel et al., 2010). Sections were mounted with medium containing 4, 6 diamidino-2-phenylindole (Vector Laboratories Ltd, Peterborough, UK) to visualize nuclei. Imaging was performed using a Nikon 2000s epifluorescence microscope with a Hamamatsu Orca 285 camera (Micro-optica, Dublin, Ireland). Cell counts were performed under 40× or 60× magnification and were the average of two adjacent sections assessed by an observer blinded to experimental treatment. NeuN or TUNEL-stained cells were counted along the entire CA3 subfield from border of CA3a with CA2 through to the hilar region, according to standard demarcations. For assessment of TUNEL nuclear morphology, 90-210 cells were examined per slide on sections from mice from each dose group (n = 3-4 each). Assignment of apoptosis-like or non-apoptotic features was according to the presence or absence of rounded or spherical clumps (< 5 per cell) within TUNEL-positive nuclei of cells in the CA3 subfield.
Statistical analysis
Data are presented as means ± S.E.M. Data were analyzed using analysis of variance with post hoc Fisher's PLSD test or for two group comparison Student's t-test (StatView software; SAS Institute, Inc., Cary, NC). Significance was accepted at p < 0.05.
Results
KA dose-dependency of seizures
We recently reported that seizure duration was related to KA dose during status epilepticus induced by intra-amygdala KA in SJL mice (Tanaka et al., 2010). Here, we began by examining the relationship between seizure damage and seizure duration for the 0.3 and 1 μg KA doses in C57BL/6 mice. Status epilepticus induced by 0.3 μg KA mainly caused damage to the ipsilateral CA3a subfield of the hippocampus, as assessed by TUNEL staining of cells containing fragmented DNA (Fig. 1A, B). TUNEL-positive cells were also occasionally found in sectors CA3b and CA3c, but rarely in the hilus (Fig. 1A, B). In contrast, TUNEL staining in mice injected with 1 μg KA was typically more extensive in CA3a and extended into CA3b and CA3c subfields (Fig. 1A). TUNEL-positive hilar neurons were also present in mice given 1 μg KA (Fig. 1A, B). Counts of TUNEL-positive cells revealed a significant difference between the doses in both CA3 and hilar subfields (Fig. 1B).
Fig. 1. Influence of KA dose on seizure-damage.

(A) Representative photomicrographs showing TUNEL-stained mouse hippocampus 72 h following status epilepticus induced by intra-amygdala KA at different doses. Note enhanced cell death in the 1 μg group in each region. Scale bar for CA3a, 60 μm; hilus, 120 μm. (B) Graphs showing TUNEL counts at 72 h for CA3 and the hilus for each group (n = 3-5 per group). **p < 0.05 compared to 0.3 μg group.
To investigate the cause of the additional damage in the higher dose group we examined EEG records. Total seizure time was significantly longer in mice subject to status epilepticus induced by 1 μg KA compared to 0.3 μg-injected mice (Fig. 2A). Further analysis of the EEG indicated that HAHFDs (i.e. polyspike seizure discharges) lasted longer in the higher dose group (Fig. 2A). There was also a strong trend to earlier seizure onset in 1 μg KA-injected mice, although this did not reach statistical significance (data not shown). Representative EEG traces from both groups are shown in Fig. 2B.
Fig. 2. Influence of KA dose on seizure time.
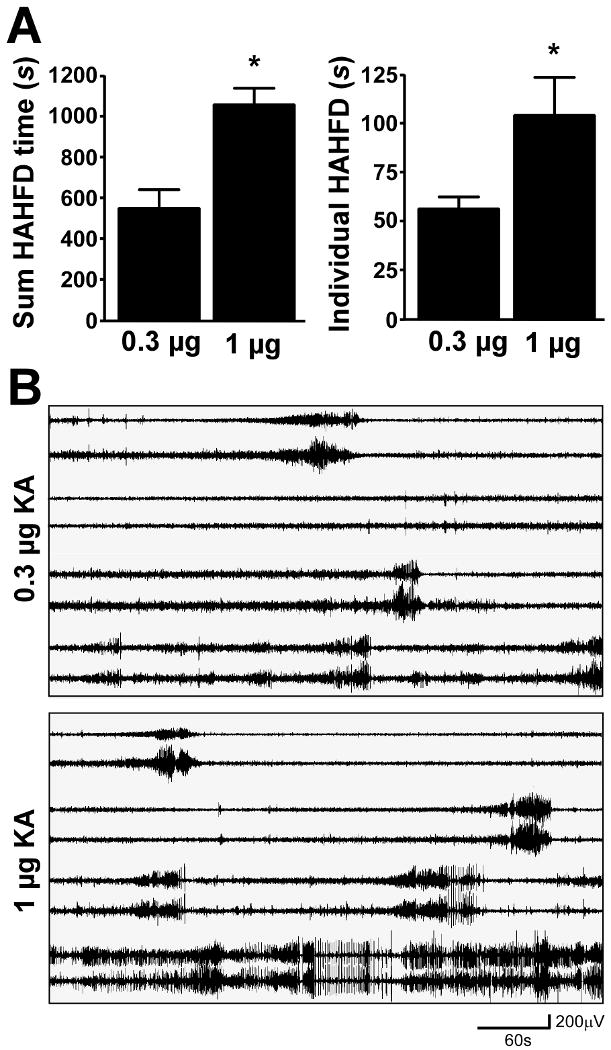
(A) Semi-quantitative EEG analysis of HAHFDs (high amplitude high frequency discharges) in mice given either 0.3 μg or 1 μg KA demonstrating increased overall seizure time and increased duration of individual HAHFDs at the higher dose (n = 3-4 per group). *p < 0.05 compared to 0.3 μg group. (B) Representative EEG traces following injection of KA. Note tendency to earlier seizure onset and earlier development of protracted HAHFDs.
Majority of degenerating neurons following status epilepticus are positive for DNA fragmentation
To assess the proportion of dying neurons which were positive for DNA fragmentation we stained tissue sections from 0.3 μg KA-injected mice for FJB to label all irreversibly damaged neurons, and adjacent sections for TUNEL (Note, double-staining of FJB and TUNEL on the same sections was not technically feasible). Hippocampal FJB counts in the CA3 subfield were higher than TUNEL counts in all mice studied. The proportion of FJB-stained degenerating neurons which were TUNEL positive was 83.5 % (range 62.5 – 92 %, n = 7 mice).
Reduced apoptosis-like nuclear morphology following more prolonged status epilepticus
To explore whether the balance between apoptosis-like and non-apoptotic or necrotic cell death differed between the seizure models, we examined the morphology of TUNEL-positive nuclei. TUNEL-positive nuclei were considered not to have apoptosis-like morphology if fragmented nuclear material was predominantly evenly dispersed or the TUNEL-positive nucleus had a “bean-shaped” appearance (Fig. 3A). In contrast, apoptosis-like morphology included nuclei displaying one or more large spherical clumps within the TUNEL-positive nucleus (Fig. 3B). Mice subjected to status epilepticus induced by 1 μg KA had a significantly lower proportion of TUNEL-positive nuclei displaying apoptosis-like features compared to 0.3 μg KA mice (Fig. 3C). However, raw counts of TUNEL-positive CA3 cells with apoptosis-like features was very similar between models (average total of 34 and 31 cells per CA3 subfield for 0.3 μg vs. 1 μg, respectively).
Fig. 3. Influence of KA dose on proportion of TUNEL-positive cells displaying apoptosis-like nuclear morphology.
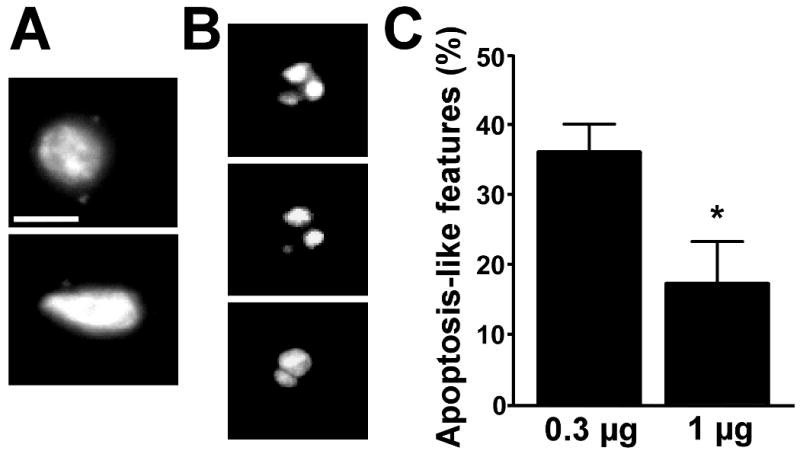
(A) Representative photomicrographs (60× lens) of individual TUNEL-positive nuclei displaying non-apoptotic or necrotic features. Bar in A, 7 μm; B, 6 μm. (B) Representative photomicrographs of individual TUNEL-positive nuclei displaying apoptosis-like morphology. (C) Graph showing percentage of TUNEL-positive cells displaying apoptosis-like nuclear features for each KA dose. *p < 0.05 compared to 0.3 μg group.
Neuroprotection in Puma-deficient mice subjected to 1 μg KA-induced status epilepticus
Genetic ablation of bim or puma, members of the pro-apoptotic BH3-only proteins, confers neuroprotection against status epilepticus (Engel et al., 2010, Murphy et al., 2010). These studies were performed with the injection of 0.3 μg KA. Since Puma deficiency provided a stronger neuroprotection than Bim deficiency we reasoned that Puma deficiency might also protect against a more necrotic insult. To explore this, we compared 1 μg KA-induced seizure damage in wild-type mice to puma+/- and puma-/- mice.
Seizure durations were not significantly different between the three genotypes (data not shown). Counts of TUNEL-positive cells 72 h after status epilepticus within the CA3 subfield of the hippocampus revealed ∼50 % less in puma-/- mice compared to wild-type and puma+/- mice (Fig. 4A, B). To support these data we compared neuronal survival at 72 h between the different genotypes by assessing numbers of NeuN-stained CA3 cells displaying normal neuronal morphology. Counts of surviving CA3 neurons were significantly higher in puma-/- mice when compared to wild-type and puma+/- mice (Fig. 4C, D). Representative immunofluorescence microscopy images from TUNEL- and NeuN-stained tissue sections are shown in Figures 4B and 4D. Typically, wild-type mice displayed extensive loss of CA3 neurons throughout the subfield. In contrast, Puma-deficient mice had fewer dying cells in the CA3a and CA3b/c regions.
Fig. 4. Neuroprotection in puma-/- mice after status epilepticus induced by intra-amygdala microinjection of 1 μg KA.

(A) Graph showing TUNEL counts in ipsilateral CA3 for each genotype 72 h after status epilepticus induced by 1 μg intra-amygdala KA. Note reduced TUNEL in puma-/- mice compared to puma+/- and wild-type (+/+) mice. (B) Representative photomicrographs showing TUNEL (green) in ipsilateral CA3 subfields from wild-type and puma-/- mice at 72 h. Scale bar = 60 μm. (C) Graph showing NeuN counts in the ipsilateral CA3 for each genotype 72 h after status epilepticus induced by 1 μg intra-amygdala KA. Note higher counts in puma-/- mice compared to puma+/- and wild-type mice. (D) Representative photomicrographs showing NeuN (red) immunostaining in ipsilateral CA3 from wild-type and puma-/- mice at 72 h. *p < 0.05 compared to wild-type and puma+/- mice (n = 4–7 per group).
Discussion
In the present study we found that increasing status epilepticus duration led to increased seizure damage and a lower percentage of cells displaying apoptosis-like nuclear TUNEL-positive morphology. Puma deficiency nevertheless conferred strong protection against seizure-induced neuronal death in vivo. These data provide genetic evidence that Puma influences seizure-induced neuronal death and suggest this likely extends beyond caspase-dependent apoptosis to caspase-independent apoptotic or non-apoptotic pathways.
The present study explored the relationship between seizure duration, hippocampal damage and mode of cell death in a mouse model of status epilepticus. This remains an important issue since it has the potential to influence how to protect brain against seizure damage (Meldrum, 2002, Henshall and Murphy, 2008). Here, we attributed the mode of cell death as apoptosis on the basis of nuclear morphology in TUNEL-positive cells. Previously we used the term programmed cell death positive/negative (Shinoda et al., 2004a) but the criteria here were the same. Over 80 % of dying neurons were TUNEL-positive in our study and nuclear morphology consistent with apoptosis-like cell death was evident in a subpopulation of these cells, in agreement with our previous reports (Araki et al., 2002, Shinoda et al., 2004a, Murphy et al., 2007). Occasional cells also display TUNEL-positive material dispersed throughout the soma in this model (Murphy et al., 2007), probably the result of nuclear membrane breakdown during apoptosis (Taylor et al., 2008). Chromatin masses have been reported to be TUNEL-positive during physiological apoptosis in the developing rat brain (Ishimaru et al., 1999) and we found here and previously that rounded nuclear clumps were invariably TUNEL-positive (Araki et al., 2002, Shinoda et al., 2004a, Murphy et al., 2007). This contrasts reports that heavily condensed chromatin in apoptotic cells remains TUNEL-negative (Barth et al., 2002, Fujikawa et al., 2010). Whether this represents a failure of the TUNEL stain to penetrate, different stages of an essentially similar process, or a substantive and important difference in the form of cell death, is not certain. While we did not use electron microscopy in the present study to determine morphology, multiple independent markers of apoptosis are present in the model including caspase cleavage, cytochrome c release and modulation by pharmacologic and genetic disruption of specific pathways (Araki et al., 2002, Murphy et al., 2007, Engel et al., 2010, Murphy et al., 2010). The TUNEL-positive cells not showing apoptosis-like nuclear morphology in our study presumably represent DNA fragmentation arising from non-caspase-dependent signaling (e.g. apoptosis-inducing factor, AIF), or non-apoptotic cell death pathways (Dong et al., 1997, Cheung et al., 2005, Kroemer and Martin, 2005, Taylor et al., 2008, Zhao et al., 2010). The remaining ∼20 % of TUNEL-negative dead neurons presumably represent necrotic cells in which programmed pathways were minimally activated or absent (Fujikawa, 2005, Fujikawa et al., 2010).
Little doubt remains that activation of ordered cell death signaling cascades make a major contribution to seizure-induced neuronal death (Liou et al., 2003, Engel and Henshall, 2009). The relative contribution of caspase-dependent versus -independent pathways or non-apoptotic pathways is less understood and seems strongly model-, species- and development-stage dependent (Fujikawa, 2005, Engel and Henshall, 2009). The present study explored whether a more severe seizure insult generates a less “apoptotic” profile of cell death. This concept has support from several studies (Ankarcrona et al., 1995, Bonfoco et al., 1995, Du et al., 1996, Portera-Cailliau et al., 1997, Ward et al., 2007) and has previously been examined in the setting of seizure-induced neuronal death where it was affirmed (Kondratyev and Gale, 2004) but also rejected (Fujikawa et al., 2010). Our data show that when status epilepticus was more severe there was a reduction in the relative proportion of dying cells displaying apoptosis-like nuclear TUNEL-positive morphology. Critically, however, absolute numbers of such cells remained about the same. If we assume caspase-dependent signaling is important for apoptosis-like morphology (Kuida et al., 1998, Kroemer and Martin, 2005, Taylor et al., 2008), which is not certain (Joza et al., 2001), then extending seizure severity or duration does not appear to alter this component. Instead, only the non-caspase-dependent apoptotic or non-programmed necrotic component is enhanced. Kondratyev et al. reported that caspase-dependent apoptosis signaling increased when status epilepticus was slower to develop, but since that study only investigated molecular pathways we cannot be certain of whether the morphology component changed or simply the biochemical signature (Kondratyev and Gale, 2004). Likewise, we cannot directly compare our data to Fujikawa and coworkers since they found neither mild nor severe seizures produced any apoptotic morphology or biochemical signature (Fujikawa et al., 2010). Indeed, little or no TUNEL staining was reported in their study, nor previously using either pilocarpine or systemic kainate models (Fujikawa et al., 2000b, Fujikawa et al., 2007). This differs from findings by many other groups including ours, where TUNEL staining is widespread (Faherty et al., 1999, Shinoda et al., 2004a, Heo et al., 2006, Mikati et al., 2008). However, the conclusions drawn by Fujikawa et al. are apt given their data and it remains difficult to reconcile that substantive differences would exist in the mechanism of seizure-induced neuronal death between models. Debate over which animal model best represents the pathology of human temporal lobe epilepsy continues (Sloviter, 2008). Notably, altered Bcl-2 family proteins, caspase processing and DNA fragmentation are all evident in resected material from patients with temporal lobe epilepsy (Shinoda et al., 2004b, Schindler et al., 2006, Yamamoto et al., 2006, Narkilahti et al., 2007, Yang et al., 2008). A human case of extensive apoptosis as a result of status epilepticus has also been documented (Cherian et al., 2009).
Puma is a potent activator of the mitochondrial cell death pathway via neutralization of anti-apoptotic Bcl-2 family proteins and possibly direct Bax-activating effects, leading to mitochondrial outer membrane permeabilization, caspase activation and apoptosis (Villunger et al., 2003, Cartron et al., 2004, Willis et al., 2007). However, caspase-independent apoptosis such as mediated by AIF is also regulated by Bcl-2 and certain BH3-only proteins (Susin et al., 1999, Culmsee et al., 2005, Liou et al., 2005, Landshamer et al., 2008). It is possible that Puma can influence AIF-induced cell death (Wang et al., 2007). Moreover, Bcl-2 has also been shown to protect against necrotic cell death as mitochondrial permeabilization also leads to mitochondrial dysfunction (Zhong et al., 1993, Kane et al., 1995, Single et al., 2001). We previously reported that seizure-induced neuronal death was reduced by over 70 % in Puma-deficient mice (Engel et al., 2010). The present study demonstrated that seizure-induced neuronal death was again strongly inhibited in Puma-deficient mice despite a reduced overall apoptosis component in this model. Protection was however less than when compared to our previous study (Engel et al., 2010). Although the influence of Puma was reduced, our data provide evidence that Puma influences both caspase-dependent and caspase-independent apoptosis and perhaps has effects on non-apoptotic pathways or necrosis, e.g. by protecting mitochondrial integrity (Kroemer and Martin, 2005). Last, it is noteworthy that in human epileptic brain tissue, cells displaying active caspase-3 are negative for nuclear AIF and vice versa (Schindler et al., 2006), implying autonomous apoptosis-related signaling pathways can be activated in neighboring cells subject to the same insult.
While the mechanism by which Puma mediates its effects in the present study is unknown, Puma induction after seizures is chiefly p53-dependent (Engel et al., 2010). Beyond the therapeutic potential for neuroprotection in status epilepticus, the wider value of targeting Puma in neurologic disorders is uncertain. Despite induction following cerebral ischemia, Puma appears to be dispensable for neuronal death in in vivo mouse models of focal cerebral ischemia (Kuroki et al., 2009). Puma is also not required for neuronal death following intra-hippocampal N-methyl-d-aspartate injection (Concannon et al., 2007).
One potential caveat in the present study is that sampling at a single time point may miss key features in the high dose group which develop earlier and disappear, rather than are absent. This is unlikely however, and we have not seen obvious temporal shifts in cell death signaling pathway activation between doses in the model. Of additional note, our studies here establish that seizure duration and seizure damage in C57BL/6 mice are broadly related to KA dose. This supports earlier work in SJL mice that highlighted this relationship in the same model (Tanaka et al., 2010). The higher KA dose appears to induce an earlier onset of electrographic seizures and somewhat longer electrographic discharges (present data and (Tanaka et al., 2010). While varying seizure time by anticonvulsant administration is common among models and effective in the present model (Henshall et al., 2000, Shinoda et al., 2004a), this KA dose-dependency is a useful property and provides versatility for studying gene or drug manipulations which may increase as well as decrease cell death.
In conclusion, we find that absolute apoptosis-like morphology varies little in relation to status epilepticus duration but rather its relative contribution is reduced. We find that absence of pro-apoptotic Puma provides robust protection against seizure-induced neuronal death even where the contribution of apoptosis is effectively lower, implying effects on caspase-independent and non-apoptotic cell death.
Acknowledgments
The authors thank Andreas Strasser and Andreas Villunger for providing the Puma-deficient mice and Nikolaus Plesnila for useful discussion of the manuscript. We thank Ina Woods for animal husbandry and Brona Murphy and Heiko Duessmann for technical support. This work was supported by grants from Health Research Board Ireland (RP/2005/24, RP/2007/37, RP/2008/69), Science Foundation Ireland (04/IN3/B466, 08/IN1/B1875), Marie Curie ToK FP-14499, National Institutes of Health (NS39016 and NS46722) and fellowships (to T.E.) from the Irish Research Council for Science Engineering and Technology and the Health Research Board (Ireland).
Abbreviations
- Bcl-2
B cell lymphoma
- BH3
Bcl-2 homology domain
- CA
cornu ammonis
- EEG
electroencephalogram
- FJB
Fluoro-Jade B
- KA
kainic acid
- NeuN
neuron-specific nuclear protein
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Araki T, Simon RP, Taki W, Lan JQ, Henshall DC. Characterization of neuronal death induced by focally evoked limbic seizures in the C57BL/6 mouse. J Neurosci Res. 2002;69:614–621. doi: 10.1002/jnr.10356. [DOI] [PubMed] [Google Scholar]
- Araujo IM, Gil JM, Carreira BP, Mohapel P, Petersen A, Pinheiro PS, Soulet D, Bahr BA, Brundin P, Carvalho CM. Calpain activation is involved in early caspase-independent neurodegeneration in the hippocampus following status epilepticus. J Neurochem. 2008;105:666–676. doi: 10.1111/j.1471-4159.2007.05181.x. [DOI] [PubMed] [Google Scholar]
- Barth M, Oulmi Y, Ehrenreich H, Schilling L. Pre-embedding immunogold labeling of TUNEL stain enables evaluation of DNA strand breaks and ultrastructural alterations in individual cells of neuronal tissue. Acta Neuropathol. 2002;104:621–636. doi: 10.1007/s00401-002-0595-8. [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartron PF, Gallenne T, Bougras G, Gautier F, Manero F, Vusio P, Meflah K, Vallette FM, Juin P. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol Cell. 2004;16:807–818. doi: 10.1016/j.molcel.2004.10.028. [DOI] [PubMed] [Google Scholar]
- Cherian KA, Weidenheim K, Legatt AD, Shifteh K, Abbott IR, Moshe SL. Extensive apoptosis in a case of intractable infantile status epilepticus. Epilepsy Res. 2009;85:305–310. doi: 10.1016/j.eplepsyres.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Melanson-Drapeau L, Cregan SP, Vanderluit JL, Ferguson KL, McIntosh WC, Park DS, Bennett SA, Slack RS. Apoptosis-inducing factor is a key factor in neuronal cell death propagated by BAX-dependent and BAX-independent mechanisms. J Neurosci. 2005;25:1324–1334. doi: 10.1523/JNEUROSCI.4261-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Concannon CG, Ward MW, Bonner HP, Kuroki K, Tuffy LP, Bonner CT, Woods I, Engel T, Henshall DC, Prehn JH. NMDA receptor-mediated excitotoxic neuronal apoptosis in vitro and in vivo occurs in an ER Stress and PUMA independent manner. J Neurochem. 2007;105:891–903. doi: 10.1111/j.1471-4159.2007.05187.x. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu C, Landshamer S, Becattini B, Wagner E, Pellecchia M, Blomgren K, Plesnila N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci. 2005;25:10262–10272. doi: 10.1523/JNEUROSCI.2818-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Saikumar P, Weinberg JM, Venkatachalam MA. Internucleosomal DNA cleavage triggered by plasma membrane damage during necrotic cell death. Involvement of serine but not cysteine proteases. Am J Pathol. 1997;151:1205–1213. [PMC free article] [PubMed] [Google Scholar]
- Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis? J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- Engel T, Henshall DC. Apoptosis, Bcl-2 family proteins and caspases: the ABCs of seizure-damage and epileptogenesis? Int J Physiol Pathophysiol Pharmacol. 2009;1:97–115. [PMC free article] [PubMed] [Google Scholar]
- Engel T, Murphy BM, Hatazaki S, Jimenez-Mateos EM, Concannon CG, Woods I, Prehn JH, Henshall DC. Reduced hippocampal damage and epileptic seizures after status epilepticus in mice lacking proapoptotic Puma. FASEB J. 2010;24:853–861. doi: 10.1096/fj.09-145870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faherty CJ, Xanthoudakis S, Smeyne RJ. Caspase-3-dependent neuronal death in the hippocampus following kainic acid treatment. Brain Res Mol Brain Res. 1999;70:159–163. doi: 10.1016/s0169-328x(99)00143-6. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG. Prolonged seizures and cellular injury: understanding the connection. Epilepsy Behav. 2005;7 3:S3–11. doi: 10.1016/j.yebeh.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Itabashi HH, Wu A, Shinmei SS. Status epilepticus-induced neuronal loss in humans without systemic complications or epilepsy. Epilepsia. 2000a;41:981–991. doi: 10.1111/j.1528-1157.2000.tb00283.x. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Shinmei SS, Cai B. Kainic acid-induced seizures produce necrotic, not apoptotic, neurons with internucleosomal DNA cleavage: implications for programmed cell death mechanisms. Neuroscience. 2000b;98:41–53. doi: 10.1016/s0306-4522(00)00085-3. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Shinmei SS, Zhao S, Aviles ER., Jr Caspase-dependent programmed cell death pathways are not activated in generalized seizure-induced neuronal death. Brain Res. 2007;1135:206–218. doi: 10.1016/j.brainres.2006.12.029. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Zhao S, Ke X, Shinmei SS, Allen SG. Mild as well as severe insults produce necrotic, not apoptotic, cells: Evidence from 60-min seizures. Neurosci Lett. 2010;469:333–337. doi: 10.1016/j.neulet.2009.12.022. [DOI] [PubMed] [Google Scholar]
- Hatazaki S, Bellver-Estelles C, Jimenez-Mateos EM, Meller R, Bonner C, Murphy N, Matsushima S, Taki W, Prehn JH, Simon RP, Henshall DC. Microarray profile of seizure damage-refractory hippocampal CA3 in a mouse model of epileptic preconditioning. Neuroscience. 2007;150:467–477. doi: 10.1016/j.neuroscience.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshall DC, Murphy BM. Modulators of neuronal cell death in epilepsy. Curr Opin Pharmacol. 2008;8:75–81. doi: 10.1016/j.coph.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Sinclair J, Simon RP. Spatio-temporal profile of DNA fragmentation and its relationship to patterns of epileptiform activity following focally evoked limbic seizures. Brain Res. 2000;858:290–302. doi: 10.1016/s0006-8993(99)02452-x. [DOI] [PubMed] [Google Scholar]
- Heo K, Cho YJ, Cho KJ, Kim HW, Kim HJ, Shin HY, Lee BI, Kim GW. Minocycline inhibits caspase-dependent and -independent cell death pathways and is neuroprotective against hippocampal damage after treatment with kainic acid in mice. Neurosci Lett. 2006;398:195–200. doi: 10.1016/j.neulet.2006.01.027. [DOI] [PubMed] [Google Scholar]
- Ishimaru MJ, Ikonomidou C, Tenkova TI, Der TC, Dikranian K, Sesma MA, Olney JW. Distinguishing excitotoxic from apoptotic neurodegeneration in the developing rat brain. J Comp Neurol. 1999;408:461–476. [PubMed] [Google Scholar]
- Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW, Zuniga-Pflucker JC, Kroemer G, Penninger JM. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- Kane DJ, Ord T, Anton R, Bredesen DE. Expression of bcl-2 inhibits necrotic neural cell death. J Neurosci Res. 1995;40:269–275. doi: 10.1002/jnr.490400216. [DOI] [PubMed] [Google Scholar]
- Kondratyev A, Gale K. Latency to onset of status epilepticus determines molecular mechanisms of seizure-induced cell death. Brain Res Mol Brain Res. 2004;121:86–94. doi: 10.1016/j.molbrainres.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Martin SJ. Caspase-independent cell death. Nat Med. 2005;11:725–730. doi: 10.1038/nm1263. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Kuroki K, Virard I, Concannon CG, Engel T, Woods I, Taki W, Plesnila N, Henshall DC, Prehn JH. Effects of transient focal cerebral ischemia in mice deficient in puma. Neurosci Lett. 2009;451:237–240. doi: 10.1016/j.neulet.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Landshamer S, Hoehn M, Barth N, Duvezin-Caubet S, Schwake G, Tobaben S, Kazhdan I, Becattini B, Zahler S, Vollmar A, Pellecchia M, Reichert A, Plesnila N, Wagner E, Culmsee C. Bid-induced release of AIF from mitochondria causes immediate neuronal cell death. Cell Death Differ. 2008;15:1553–1563. doi: 10.1038/cdd.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou AK, Zhou Z, Pei W, Lim TM, Yin XM, Chen J. BimEL up-regulation potentiates AIF translocation and cell death in response to MPTP. FASEB J. 2005;19:1350–1352. doi: 10.1096/fj.04-3258fje. [DOI] [PubMed] [Google Scholar]
- Liou AKF, Clark RS, Henshall DC, Yin XM, Chen J. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol. 2003;69:103–142. doi: 10.1016/s0301-0082(03)00005-4. [DOI] [PubMed] [Google Scholar]
- Meldrum B. Excitotoxicity and epileptic brain damage. Epilepsy Res. 1991;10:55–61. doi: 10.1016/0920-1211(91)90095-w. [DOI] [PubMed] [Google Scholar]
- Meldrum BS. Excitotoxicity and selective neuronal loss in epilepsy. Brain Pathol. 1993;3:405–412. doi: 10.1111/j.1750-3639.1993.tb00768.x. [DOI] [PubMed] [Google Scholar]
- Meldrum BS. Implications for neuroprotective treatments. Prog Brain Res. 2002;135:487–495. doi: 10.1016/S0079-6123(02)35046-5. [DOI] [PubMed] [Google Scholar]
- Mikati MA, Zeinieh M, Habib RA, El Hokayem J, Rahmeh A, El Sabban M, Usta J, Dbaibo G. Changes in sphingomyelinases, ceramide, Bax, Bcl(2), and caspase-3 during and after experimental status epilepticus. Epilepsy Res. 2008;81:161–166. doi: 10.1016/j.eplepsyres.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Murphy B, Dunleavy M, Shinoda S, Schindler C, Meller R, Bellver-Estelles C, Hatazaki S, Dicker P, Yamamoto A, Koegel I, Chu X, Wang W, Xiong Z, Prehn J, Simon R, Henshall D. Bcl-w protects hippocampus during experimental status epilepticus. Am J Pathol. 2007;171:1258–1268. doi: 10.2353/ajpath.2007.070269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BM, Engel T, Paucard A, Hatazaki S, Mouri G, Tanaka K, Tuffy LP, Jimenez-Mateos EM, Woods I, Dunleavy M, Bonner HP, Meller R, Simon RP, Strasser A, Prehn JH, Henshall DC. Contrasting patterns of Bim induction and neuroprotection in Bim-deficient mice between hippocampus and neocortex after status epilepticus. Cell Death Differ. 2010;17:459–468. doi: 10.1038/cdd.2009.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkilahti S, Jutila L, Alafuzoff I, Karkola K, Paljarvi L, Immonen A, Vapalahti M, Mervaala E, Kalviainen R, Pitkanen A. Increased expression of caspase 2 in experimental and human temporal lobe epilepsy. Neuromolecular Med. 2007;9:129–144. doi: 10.1007/BF02685887. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. second. Elsevier; San Diego, CA: 2001. [Google Scholar]
- Portera-Cailliau C, Price DL, Martin LJ. Excitotoxic neuronal death in the immature brain is an apoptosis- necrosis morphological continuum. J Comp Neurol. 1997;378:70–87. [PubMed] [Google Scholar]
- Schindler CK, Pearson EG, Bonner HP, So NK, Simon RP, Prehn JH, Henshall DC. Caspase-3 cleavage and nuclear localization of caspase-activated DNase in human temporal lobe epilepsy. J Cereb Blood Flow Metab. 2006;26:583–589. doi: 10.1038/sj.jcbfm.9600219. [DOI] [PubMed] [Google Scholar]
- Shinoda S, Araki T, Lan JQ, Schindler CK, Simon RP, Taki W, Henshall DC. Development of a model of seizure-induced hippocampal injury with features of programmed cell death in the BALB/c mouse. J Neurosci Res. 2004a;76:121–128. doi: 10.1002/jnr.20064. [DOI] [PubMed] [Google Scholar]
- Shinoda S, Schindler CK, Meller R, So NK, Araki T, Yamamoto A, Lan JQ, Taki W, Simon RP, Henshall DC. Bim regulation may determine hippocampal vulnerability after injurious seizures and in temporal lobe epilepsy. J Clin Invest. 2004b;113:1059–1068. doi: 10.1172/JCI19971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Single B, Leist M, Nicotera P. Differential effects of bcl-2 on cell death triggered under ATP-depleting conditions. Exp Cell Res. 2001;262:8–16. doi: 10.1006/excr.2000.5059. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Hippocampal epileptogenesis in animal models of mesial temporal lobe epilepsy with hippocampal sclerosis: the importance of the “latent period” and other concepts. Epilepsia. 2008;49 9:85–92. doi: 10.1111/j.1528-1167.2008.01931.x. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Jimenez-Mateos EM, Matsushima S, Taki W, Henshall DC. Hippocampal damage after intra-amygdala kainic acid-induced status epilepticus and seizure preconditioning-mediated neuroprotection in SJL mice. Epilepsy Res. 2010;88:151–161. doi: 10.1016/j.eplepsyres.2009.10.012. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Wang S, Wang S, Shan P, Song Z, Dai T, Wang R, Chi Z. Mu-calpain mediates hippocampal neuron death in rats after lithium-pilocarpine-induced status epilepticus. Brain Res Bull. 2008;76:90–96. doi: 10.1016/j.brainresbull.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Wang YF, Jiang CC, Kiejda KA, Gillespie S, Zhang XD, Hersey P. Apoptosis induction in human melanoma cells by inhibition of MEK is caspase-independent and mediated by the Bcl-2 family members PUMA, Bim, and Mcl-1. Clin Cancer Res. 2007;13:4934–4942. doi: 10.1158/1078-0432.CCR-07-0665. [DOI] [PubMed] [Google Scholar]
- Ward MW, Huber HJ, Weisova P, Dussmann H, Nicholls DG, Prehn JH. Mitochondrial and plasma membrane potential of cultured cerebellar neurons during glutamate-induced necrosis, apoptosis, and tolerance. J Neurosci. 2007;27:8238–8249. doi: 10.1523/JNEUROSCI.1984-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasterlain CG, Fujikawa DG, Penix L, Sankar R. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia. 1993;34:S37–53. doi: 10.1111/j.1528-1157.1993.tb05905.x. [DOI] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, Strasser A, Kluck RM, Adams JM, Huang DC. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Murphy N, Schindler CK, So NK, Stohr S, Taki W, Prehn JH, Henshall DC. Endoplasmic reticulum stress and apoptosis signaling in human temporal lobe epilepsy. J Neuropathol Exp Neurol. 2006;65:217–225. doi: 10.1097/01.jnen.0000202886.22082.2a. [DOI] [PubMed] [Google Scholar]
- Yang T, Hsu C, Liao W, Chuang JS. Heat shock protein 70 expression in epilepsy suggests stress rather than protection. Acta Neuropathol. 2008;115:219–230. doi: 10.1007/s00401-007-0297-3. [DOI] [PubMed] [Google Scholar]
- Zhao S, Aviles ER, Jr, Fujikawa DG. Nuclear translocation of mitochondrial cytochrome c, lysosomal cathepsins B and D, and three other death-promoting proteins within the first 60 minutes of generalized seizures. J Neurosci Res. 2010 doi: 10.1002/jnr.22338. [DOI] [PubMed] [Google Scholar]
- Zhong LT, Sarafian T, Kane DJ, Charles AC, Mah SP, Edwards RH, Bredesen DE. bcl-2 inhibits death of central neural cells induced by multiple agents. Proc Natl Acad Sci USA. 1993;90:4533–4537. doi: 10.1073/pnas.90.10.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]