

Abstract
Human African trypanosomiasis (HAT), caused by infection with sub-species of Trypanosoma brucei (T. b.), manifests as a hemolymphatic stage followed by an encephalitic stage. The distinction of the two stages needs improvement as drugs used for the late stage are highly toxic. Transcripts encoding 16 secreted proteins differentially expressed in the brains of mice at late stage T. b. brucei infection when the early stage drug suramin is no longer effective and different to immunoglobulins, chemokines, and cytokines, were selected by microarray analysis. Lipocalin 2 and secretory leukocyte peptidase inhibitor (SLPI) mRNA showed the highest differential expression in mice. These transcripts were also upregulated in brains from infected rats. Lipocalin 2 was increased in cerebrospinal fluid (CSF) from rats during late stage T. b. brucei infection. Protein levels of lipocalin 2, SLPI, and the chemokine CXCL10 were found increased in CSF from Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense late stage HAT compared to early stage.
Introduction
Human African trypanosomiasis (HAT) or sleeping sickness is caused by subspecies of Trypanosoma brucei (T. b.) transmitted by infected tsetse flies. The disease is divided into two stages: 1) an early stage, in which the parasites are confined to the hemolymphatic system; and 2) a late, meningo-encephalitic stage with severe signs of central nervous system (CNS) involvement when parasites are presumed to have penetrated into the brain parenchyma.1,2 The two variants of HAT, which differ in duration of incubation period and progression to CNS involvement, are the chronic Gambian form (caused by Trypanosoma brucei gambiense in West and Central Africa) and the acute Rhodesian form (caused by Trypanosoma brucei rhodesiense in East and Southern Africa).
Accurate staging of HAT is critical because early stage drugs do not efficiently cross the blood-brain barrier (BBB), and treating early-stage patients with the more toxic drugs should be avoided.3,4 According to current World Health Organization (WHO) criteria, HAT patients in late stage are those with trypanosomes present in the cerebrospinal fluid (CSF) and/or an elevated leukocyte count (> 5 cells/mm3) or an increase in protein content of the CSF (> 37 mg/100 mL).5 However, diagnosis of late stage HAT based on these criteria is unsatisfactory because the number of white blood cells (WBCs) or parasites in the CSF may not be good indicators of passage of trypanosomes across the BBB.5–8 Thus, there is a critical need for biomarkers to efficiently and reliably stage HAT for treatment guidance. Recently, immunoglobulins,9 cytokines such as interleukin (IL)-10,9–11 and chemokines such as CXCL10 and CXCL1312–14 have been suggested as useful biomarkers.
In the current study, aimed at the discovery of new staging markers for HAT, genes differentially expressed in the brain of mice at the early and late stage of T. b. brucei infection were identified by transcriptome analysis. Because some molecules such as cytokines and chemokines, which may be secreted by WBC in the CSF, have been studied, molecules mainly secreted by WBCs were excluded from the selection. Instead, we focused on other molecules, e.g., those secreted from brain parenchymal cells, as potential novel markers for invasion of trypanosomes.
We report that the level of lipocalin 2 and secretory leukocyte peptidase inhibitor (SLPI) transcripts is elevated in the brain of mice during the late phase of T. b. brucei infection, when such infected mice cannot be cured with the early stage drug suramin. Levels of these molecules, together with CXCL10, were also increased in the CSF from late stage trypanosome-infected individuals. Thus, lipocalin 2 and SLPI might be considered as markers of the late stage of the disease to complement the use of chemokines or antibodies for better staging.
Materials and Methods
Patients and specimen.
Early and late stage HAT patients with T. b. gambiense were recruited from the area around Dipumba Hospital, Mbuji Mayi, Democratic Republic of the Congo (DRC), where sleeping sickness caused by T. b. gambiense is endemic.15 Briefly, individuals who were seropositive in the card agglutination test for trypanosomiasis (CATT) or who presented suggestive clinical signs were examined for trypanosomes in the blood, lymph node aspirate, and CSF. The presence of trypanosomes in at least one of these body fluids was evidence of infection. The late stage disease was defined as either WBC count > 5 cells/µL or detection of trypanosomes in CSF. The study protocol was approved by the Ministry of Health, Kinshasa, DRC, and the Ethical Committee of the University of Antwerp, Belgium. Patients were informed about the objectives and modalities of the study and were asked to provide consent. Patients younger than 12 years of age, moribund or with a “blood-contaminated” CSF were excluded from this study. In total, 180 patients (early stage [n = 90] and late stage [n = 90]) with T. b. gambiense HAT were considered for analyses reported in this study. Patients with T. b. rhodesiense infections (6 in early stage and 20 at late stage) were recruited in the Rumphi region of Malawi and were screened at the local hospitals after providing consent. Disease stage was determined as indicated previously, and the study protocol was approved by the Ministry of Health and Population, Lilongwe, Malawi and the Ethical Committee of the University of Antwerp, Belgium. Blood, CSF, saliva, and urine were collected from all consenting patients. The CSF and urine samples were spun briefly before the supernatant was snap frozen in liquid nitrogen, and remained at −80°C until testing. Blood samples were also spun and sera carefully pipetted out and snap frozen. Aliquots of all samples were kept at −80°C until testing. Body fluids from non-infected age and sex matched patients subjected to lumbar puncture for spinal anaesthesia in Heidelberg, Germany (n = 18), were used as control. Patients signed an informed consent form. The protocol was approved by the Ethical Committee of the Medical Faculty, University of Heidelberg, Germany.
Mice, rats, parasites, and infection.
The C57BL/6 mice (8–12 weeks old) and Sprague-Dawley rats (180–200 g), kept under specific pathogen-free conditions with food and water ad libitum were used throughout. All experiments received institutional approval by the local animal ethical committees.
Mice were infected by intraperitoneal (i.p.) injection with 2–3 × 103 parasites of the pleomorphic stabilate AnTat 1.1E T. b. brucei. Rats were infected i.p. with 25 × 103 of the same parasite strain. Infected mice were deeply anesthetized with isoflurane, sacrificed by decapitation, and brains dissected out at 6, 15, and 28 days post infection (d.p.i.), immediately snap frozen on dry ice, and kept at −70°C. Twenty-seven trypanosome-infected rats and four control rats were anesthetized with pentobarbital (50 mg/kg i.p.) and after CSF collection, the brains were dissected out at 6 (n = 6), 11 (n = 5), 16 (n = 4), 21 (n = 5), 26 (n = 4), and 31 (n = 3) d.p.i, deeply frozen in liquid nitrogen and stored at −80°C. The CSF collection was performed on rats anesthetized and positioned in a stereotaxic apparatus. A midline incision was made over the surface of the skull and along the posterior region of the neck. After the underlying muscles had been retracted and foramen magnum localized, a glass micropipette (about 0.5 mm in diameter) was used to gently penetrate the dura and collect the CSF (30–80 µL) without any blood contamination and within 5 min from the anesthesia.
cDNA microarray analysis.
Labeling, complementary DNA (cDNA) synthesis, hybridization, scanning, and image processing were done as previously described.16 All protocols are available by the Royal Institute of Technology, Stockholm (KTH) microarray core facility homepage.17 Preliminary data analysis was performed as described.16 Briefly, data analysis was performed in the R environment mainly using KTH and Bioconductor packages.17–19 After importing the data into R, four filters were used to remove spots flagged in GenePix, spots saturated in both cy3 and cy5, spots in which 70% of the pixels had below background intensity +2 standard deviations, and spots that were enlarged because of spotting artefacts.
On average ~84% of the spots were kept across all slides. Differentially expressed genes were identified using an empirical Bayes' moderated t test available in the LIMMA package.19–21 The t test statistics was performed on all features that had an inter-quartile range > 0.5 in any contrast (7,075 features). The Gene Ontology and functions available in the Bioconductor project 19,22 were used to search for genes over-represented among the differentially expressed genes.
Quantitative real time reverse transcriptase-polymerase chain reaction (RT-PCR) for microarray data validation.
Transcripts of selected genes were quantified by real time PCR in brains from mice uninfected or at different time points after T. b. brucei infection. Total RNA was extracted from half of the fresh frozen brain, reverse-transcribed, and the transcripts levels quantified on an ABI Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA), as previously described.23 The sequences of the primers used are listed in Table 1, the primer sequences of cyclophilin have been published previously.23 The amount of transcripts of individual animal samples (n = 4 per group) was normalized to cyclophilin. The relative amount of target gene transcripts was calculated using the 2–ΔΔCt method as described.24 These values were then used to calculate the mean and standard error of the relative expression of the target gene messenger RNA (mRNA) in the brains of uninfected and infected mice. The same procedure was used to analyze transcripts for CXCL10, Lipocalin 2, and SLPI in brains of T. b. brucei-infected rats.
Table 1.
PCR primer sequences*
| Gene | Polarity and sequence 5¢ to 3¢ | |
|---|---|---|
| Sense | Anti-sense | |
| MOUSE | ||
| Biglycan | AACCCTGGAGGAAGCTGGAA | TCTTGATAGCAGAGTATGAACCCTTTC |
| BCKDHA | TGCTGTGTACAATGCCACTAAGG | CAATGAGGAAGGGCTGGTTCT |
| Carbonic anhydrase 9 | AGTGCCAGGACTGAGGGTATGT | TTTGACTGCAGAATAAAAGCAAGAA |
| Ceruloplasmin | CTGCTGATGGAGGCTGAACA | TTTGGCGTCTGTGGTCCTTT |
| Cystatin C | AGGCAGGTTCTGCACATCTGA | GCATGGCAGGTACTGCAAGA |
| CRP61 | CAGAGGAGAAGCGCAAGCAT | TGAGCAAGGCACCATTCATC |
| Eph receptor A4 | CACTTGGCCCATTCATGCT | ACCCCCCCTATGTCATAAAAATC |
| GFAP | GAAGGTCCGCTTCCTGGAA | GGCTCGAAGCTGGTTCAGTT |
| Granulin | CCAAAAGCCCCGTATCAAAC | GAGCACCTGGCCACAAGGT |
| IP-30 | CAAACTCGTCCCAGGAAAAACT | ATAGCGAGGCAGCAGCTCTT |
| LRRC4 | TCACTGCGCCGCCTAGA | GAAGGCCGCCTCAGATATGTAC |
| Lipocalin 2 | TCTGTCCCCACCGACCAAT | GCATCCCAGTCAGCCACACT |
| Midkine | TCCCACAGGCCCAAGATATAA | GGACAGGCGTGATTGACAGA |
| Neuronal pentraxin 1 | TGGCACCACCTACCAATCAG | CCCTCAGATGTAGACAGCAGACA |
| Prominin 1 | GGGACTGCTGTTCATTATCCTCAT | TGCAGCAACGGCACATACA |
| Reelin | TGTGTTCTCTTCGTGGGTTGTT | GGCTCCACCTCATCAGAAGGT |
| SLPI | CCCCTGCCTTCACCATGA | GCAAGGAGCACCGTGAAAG |
| Serglycin | TGGCTTCCTAGGTGACATGGA | GTCAAAAGGCTTATAGTTGAAATAGACAAT |
| Serpina3n | GCCCTGCTGTCCTCTGCTT | GGTCTTCTTGGACTGCAGCAT |
| Spon1 | TCTGATTGTTGGTGCCATAAGTG | GATCTGCCGAGAAAAGGTTAGTG |
| ST6GALNAC5 | AACGCCGGCATCCTTTG | AGTGCTTTTGAGGCACAGAAATC |
| Syndecan 4 | TGCACTAGCCACACAAAATGC | TGACCTGCTAAACCAGCTTAACC |
| Translocator protein | GGAAGCCACCAGGTAGGTTAGG | AGTGCAGAAAGGCAGGTATAACTGT |
| Vimentin | TCCCTTGTTGCAGTTTTTCCA | CCTGGTAGACATGGCTTCGAA |
| RAT | ||
| CXCL10 | GAGAAGCCACTCGCCACAGT | GGGTAAAGGGAGGTGGAGAGA |
| Lipocalin 2 | CAGGGCAGGTGGTTCGTT | AGCGGCTTTGTCTTTCTTTCTG |
| SLPI | AGGTGCCTCAAATTTCAAGGAA | GGCCGTCATTCTGGCACTT |
BCKDHA = branched chain ketoacid dehydrogenase E1, alpha polypeptide; CRP61 = cysteine-rich protein 61; GFAP = glial fibrillary acidic protein; IP-30 = interferon gamma inducible protein 30; LRRC4 = leucine-rich repeat containing 4; SLPI = secretory leukocyte peptidase inhibitor; Serpina3n = serine (or cysteine) peptidase inhibitor, clade A, member 3N; Spon1 = Spondin 1, (f-spondin) extracellular matrix protein; ST6GALNAC5 = ST6 (alpha-N-acetyl-neuraminyl-2,3-beta-galactosyl-1,3)-N-acetylgalactosaminide alpha-2,6-sialyltransferase 5.
Measurement of selected molecules in body fluids of Trypanosoma-infected individuals.
Screening for candidate biomarkers in body fluids (CSF, sera, and saliva) from Trypanosoma-infected individuals was done by enzyme-linked immunosorbent assay (ELISA) following a procedure described by the manufacturer (R&D systems, Minneapolis, MN).
The levels of CXCL10, lipocalin 2, and SLPI were measured in the CSF of 180 patients with T. b. gambiense form of HAT, whereas the CSF and urine from patients with T. b. rhodesiense form of HAT were screened for CXCL10, lipocalin 2, and SLPI. Lipocalin 2 was also screened in the CSF from infected rats. The detection limits of CXCL10, SLPI, and lipocalin 2 were 8, 25, and 12 pg/mL, respectively.
Statistical analysis.
Significant differences between the study groups for each measured parameter were determined. Empirical Bayes moderated t test was used to determine false discovery rate for differentially expressed genes in microarray, and to confirm overexpression for each selected gene by real time RT-PCR, analysis of variance (ANOVA) followed by Newman Keuls post test was used to compare the time points. Significant differences in the levels of proteins measured by ELISA in body fluids were determined by the Mann-Whitney U-test.
Results
Identification of genes differentially expressed in the brains of mice at the late stage of infection with T. b. brucei.
To identify molecules that are highly expressed during the encephalitic phase of experimental African trypanosomiasis, a transcriptome comparison of brain tissues at 6, 15, and 28 d.p.i. was performed. Suramin cures T. b. brucei-infected mice treated at 6 and 15 but not at 28 d.p.i.25
A total of 5,617 genes were differentially expressed between 6 and 28 d.p.i. (Figure 1) and of these 3,215 had gene ontology (GO) annotation. The levels of 2,495 genes with GO annotation were increased and that of 720 genes were decreased in the brain of mice at 28 as compared to 6 d.p.i.; 5,037 genes were differentially expressed between 15 and 28 d.p.i. (Figure 1) and of these 2,853 had GO annotation. The expression of 2,191 transcripts with GO annotation was enhanced and 662 genes were decreased in the brains of mice at 28 as compared to 15 d.p.i. Most of the genes, which were quantitatively augmented at 28 compared to 6 or 15 d.p.i., are involved in inflammatory and immune responses such as complement classical pathway, chemotaxis, T cell differentiation, and antigen processing and presentation by MHC class II.
Figure 1.
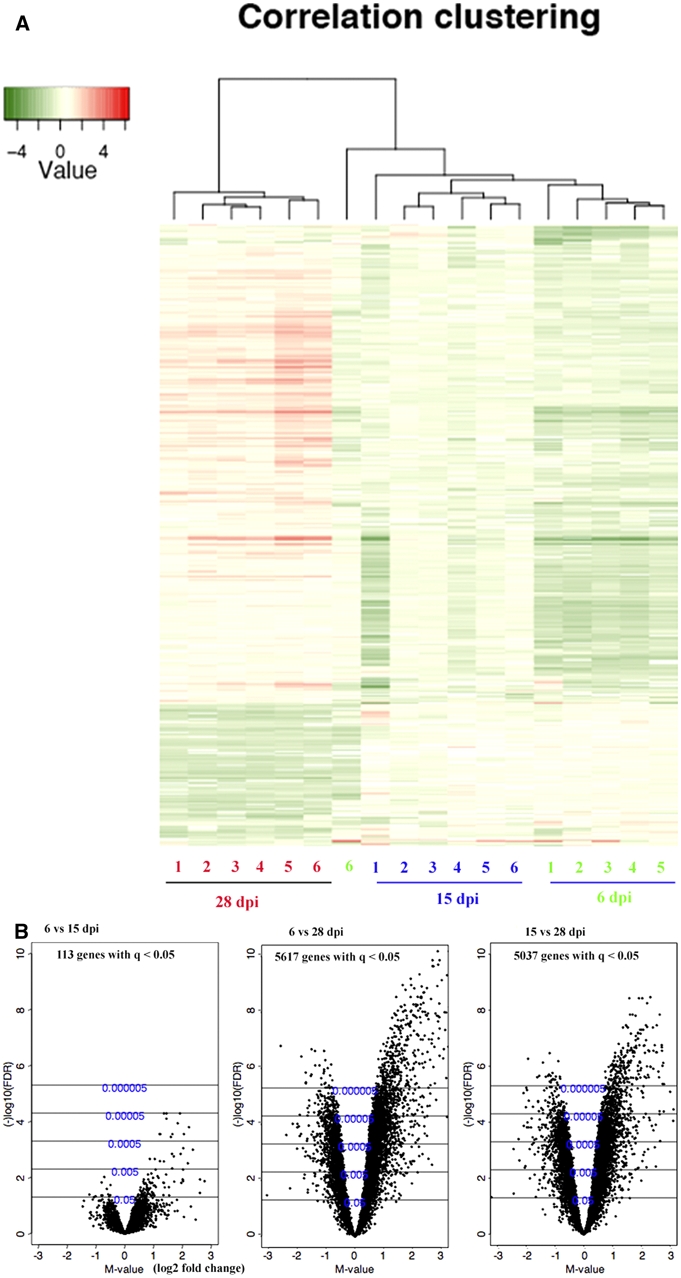
Heat map and volcano plots of complementary DNA (cDNA) microarray analysis of differentially expressed genes in the brains of T. b. brucei-infected mice between 6, 15, and 28 days post infection (d.p.i.). (A) Heat map of varying genes with dendrograms of hierarchical clustering for replicates of mice brain samples obtained at 6, 15, and 28 d.p.i. (n = 6 per time point). Red represents a higher expression level, green means a lower expression level, and white means no difference in expression level. (B) Volcano plots of differentially expressed genes in the brains of T. b. brucei-infected mice between 6 and 15 or 28 d.p.i. and 15 and 28 d.p.i. The X axis shows log2 of the fold-change differences in gene expression (M-value) and the Y axis the Q-value in -log10 (false discovery rate, FDR) scale calculated by an empirical Bayes moderated t test. This figure appears in color at www.ajtmh.org.
A subset of 24 differentially expressed genes was selected for further analysis based on the following criteria: 1) more than 1.5-fold change in gene expression by microarray; 2) the encoded protein is secreted and detectable in body fluids; 3) not produced or not known to be predominantly produced by WBC; 4) not a chemokine, cytokine, or an antibody; 5) expressed in the brain; and 6) transcribed in humans. The differential gene expression of these genes in brains was confirmed for 16 of the 24 molecules by real time PCR (Table 2). The levels of lipocalin 2 and SLPI mRNA, the two most highly differentially expressed transcripts, are shown Figure 2.
Table 2.
Microarray and real time PCR data of differential gene expression of genes in brains of T. b. brucei infected mice*
| Molecule | Fold changes in gene expression | |||
|---|---|---|---|---|
| Microarray | Real time PCR | |||
| 6 vs. 28 d.p.i. | 15 vs. 28 d.p.i. | 6 vs. 28 d.p.i. | ||
| Biglycan | 6.16 | 4.23 | 1.71 | |
| BCKDHA | 4.50 | 4.46 | NDE | |
| Carbonic anhydrase 9 | 2.13 | 2.11 | NDE | |
| Ceruloplasmin | 3.49 | 2.25 | 3.03 | |
| Cystatin C | 3.80 | 2.10 | 1.5 | |
| CRP61 | 2.65 | 2.40 | NDE | |
| Eph receptor A4 | 1.52 | 2.12 | NDE | |
| GFAP | 2.49 | 2.66 | 3.18 | |
| Granulin | 2.63 | 2.36 | 4.99 | |
| IP-30 | 4.15 | 2.92 | 7.43 | |
| LRRC4 | 1.57 | 1.84 | NDE | |
| Lipocalin 2 | 1.80 | 1.96 | 15.50 | |
| Midkine | 2.13 | 1.54 | 1.83 | |
| Neuronal pentraxin 1 | 1.56 | 2.00 | NDE | |
| Prominin 1 | 2.09 | 2.08 | 2.33 | |
| Reelin | 1.42 | 1.63 | 4.01 | |
| SLPI | 2.54 | 1.72 | 84.78 | |
| Serglycin | 1.98 | 2.80 | 5.94 | |
| Serpina3n | 3.30 | 3.37 | 3.96 | |
| Spon1 | 1.90 | 1.70 | 1.433 | |
| ST6GALNAC5 | 1.82 | 2.06 | NDE | |
| Syndecan 4 | 2.18 | 1.55 | 1.69 | |
| Translocator protein | 3.63 | 2.17 | 5.00 | |
| Vimentin | 4.26 | 3.57 | NDE | |
BCKDHA = branched chain ketoacid dehydrogenase E1, alpha polypeptide; CRP61 = cysteine-rich protein 61; GFAP = glial fibrillary acidic protein IP-30 - Interferon gamma inducible protein 30; LRRTM4 = leucine-rich repeat containing 4; SLPI = secretory leukocyte peptidase inhibitor; Serpina3n = serine (or cysteine) peptidase inhibitor, clade A, member 3N; Spon1 = spondin 1, (f-spondin) extracellular matrix protein; ST6GALNAC5 = ST6 (alpha-N-acetyl-neuraminyl-2,3-beta-galactosyl-1,3)-N-acetylgalactosaminide alpha-2,6-sialyltransferase 5; NDE = no differential expression.
Figure 2.
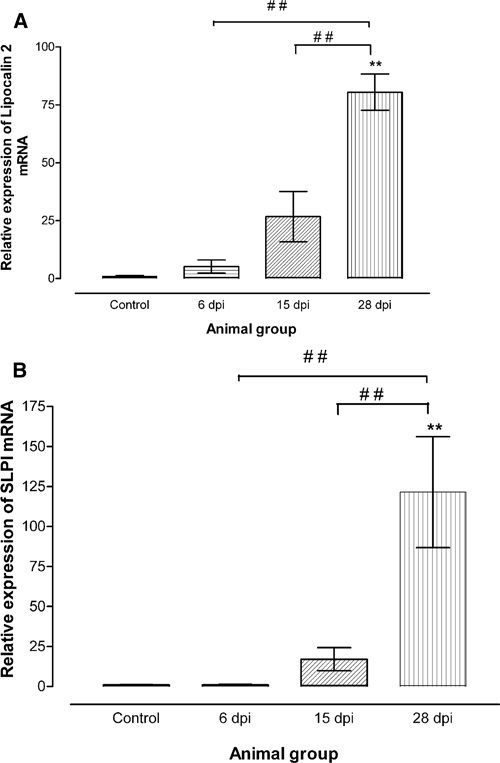
(A) Relative expression of lipocalin 2 and (B) secretory leukocyte peptidase inhibitor (SLPI) messenger RNA (mRNA) in brains of uninfected (control) and infected mice at 6, 15, and 28 days post infection (d.p.i.). Each bar represents the mean ± S.E.M of values obtained from four animals. Differences with uninfected mice are significant: **P < 0.01; and between infected mice sacrificed at 6 or 15 and 20 d.p.i.: # #P < 0.01 (one-way analysis of variance [ANOVA] followed by Newman Keuls post test).
Late stage T. b. brucei-infected rats show high levels of lipocalin 2 in CSF.
For technical reasons, we measured lipocalin 2 in CSF of T. b. brucei-infected rats instead of mice, harvested at different time points. Mice and rats show a similar outcome of T. b. brucei infection, and the penetration of parasites into the brain parenchyma occurs between 12 and 22 dpi.26,27 It was observed that all transcripts for CXCL10, lipocalin 2, and SLPI were differentially upregulated in brains of infected rats at late stage similar to what was observed in mice (Figure 3A–C). Because of limitations in commercially available antibodies against rat proteins, only lipocalin 2 was analyzed in rat CSF. As shown in Figure 3D, there was a significant increase in lipocalin 2 in the CSF from rats with late stage compared with early disease.
Figure 3.
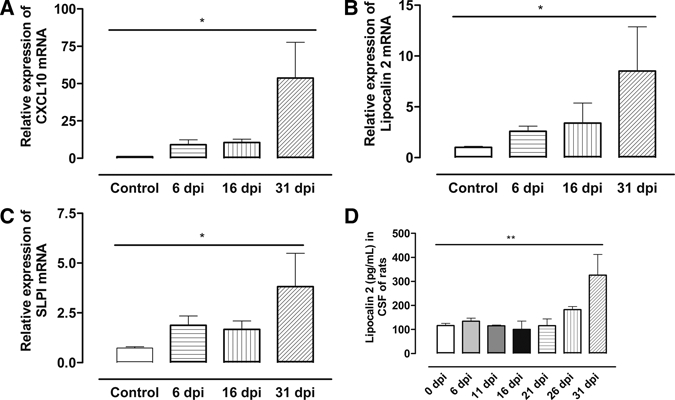
(A) Histogram showing relative expression of CXCL10, (B) lipocalin 2, and (C) secretory leukocyte peptidase inhibitor (SLPI) messenger RNA (mRNA) in brains of uninfected (control) and infected rats at 6, 16, and 31 days post infection (d.p.i.). Each bar represents the mean ± S.E.M of values obtained from four animals. Differences with uninfected rats are significant: *P < 0.05 (one-way analysis of variance [ANOVA] followed by Newman Keuls post test). Increased protein levels of lipocalin 2 in CSF from infected rats at late stage are shown in (D). Differences with uninfected rats and with infected rats sacrificed at 6 (n = 6), 11 (n = 5), 16 (n = 4), 21 (n = 5), and 26 (n = 4) d.p.i. are significant: **P < 0.01 (one-way analysis of variance [ANOVA]).
Levels of lipocalin 2 and SLPI are differentially increased in the CSF, but not sera, urine, or saliva, of HAT patients.
We next studied if protein levels of lipocalin 2 and SLPI are increased in the body fluids from late stage HAT. The concentration of lipocalin 2, SLPI, and CXCL10 in the CSF of 180 early (n = 90) and late stage (n = 90) T. b. gambiense HAT patients from DR Congo was measured by ELISA. We have recently shown in a smaller HAT patient sample originating from the same region that levels of CXCL10 were elevated.12 Higher levels of CXCL10, lipocalin 2, and SLPI in the CSF of late stage HAT patients were detected compared with early stage patients (Figure 4A–C). No correlation or association between SLPI and lipocalin 2 levels with other clinical (body mass index, somnolence, etc.) or laboratory parameters (number of trypanosomes in the CSF, intrathecal IgM titers, etc) was found. We performed the goodness-of-fit test, and obtained r2 < 1, with P value > 0.05 for all parameters. Similarly, higher levels of CXCL10, lipocalin 2, and SLPI were detected in CSF from late compared with early stage T. b. rhodesiense HAT patients from Malawi (Figure 4D–F). There was no difference in the levels of these molecules in sera and saliva of late stage compared with early stage T. b. gambiense HAT patients. Because we had urine samples from T. b. rhodesiense patients, we measured the levels of CXCL10, lipocalin 2, and SLPI in the urine samples but did not observe any significant differences in the levels of these molecules with disease stage (Figure 5). However, the results suggest that it may be necessary for future studies to be conducted on a large sample size of urine from HAT patients because relatively few sample sizes were analyzed in this study.
Figure 4.
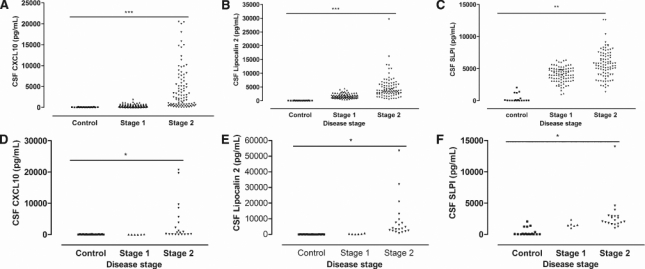
Levels of candidate biomarkers in the cerebrospinal fluid (CSF) of human African trypanosomiasis (HAT) patients. Scatter diagram of concentrations of (A) CXCL10, (B) lipocalin 2, and (C) secretory leukocyte peptidase inhibitor in the CSF of early (stage 1) and late stage (stage 2) T. b. gambiense HAT patients (n = 90 for each stage). Increased levels of (D) CXCL10, (E) lipocalin 2, and (F) SLPI were also measured in stage 2 T. b. rhodesiense HAT patients (6 in stage 1 and 20 in stage 2) compared with stage 1 and controls. Differences with the early stage patients are significant: *P < 0.05 and **P < 0.01 (Mann-Whitney U test).
Figure 5.
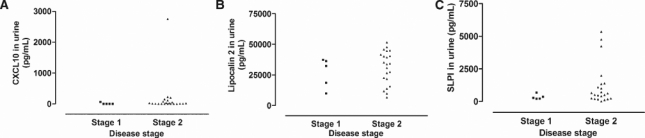
Scatter diagram of concentrations of (A) CXCL10, (B) lipocalin 2, and (C) SLPI in urine from T. b. rhodesiense HAT patients (5 in stage 1 and 22 in stage 2). There were no significant differences between disease stages.
Discussion
Here, we show that microarray gene expression analysis using animal models, in which individuals requiring late stage drugs are distinguished from those that are susceptible to early stage drugs can be used to predict molecules that are elevated in the CSF of HAT patients. Thus, lipocalin 2 and SLPI transcripts were elevated in brains of mice at a late compared with an early stage of T. b. brucei infection (as defined by susceptibility to suramin) and could be used to predict increased levels of these proteins in the CSF of late stage compared with early stage HAT patients.
In animal models, including non-human primates, early detection of trypanosomes and WBC in the CSF has been reported28 at a stage when they are most likely absent in the brain parenchyma. One suggestion from this is that cells may readily cross the blood-CSF barrier, but crossing of the BBB requires more complex mechanisms, which have been in part recently elucidated.26,29 Thus, there is most likely a delay in gaining access into brain parenchyma compared with CSF, implying that finding trypanosomes combined with mild pleocytosis (< 20 WBC/µL of CSF) in the CSF may not be a reliable parameter for disease staging in African trypanosomiasis. Perhaps, molecules that are released from brain parenchymal cells as a consequence of events taking place behind the BBB is better indicators of late stage infection.
We have focused on molecules mainly secreted by cells different from WBC, and different to chemokines and cytokines, which have been previously identified as candidate molecules for improved staging of HAT. Lipocalin 2, which plays a role in eukaryotic and bacterial cell death caused by iron sequestration,30 has been reported to be produced by epithelial cells in the choroid plexus and cerebral endothelial cells during inflammation.31 Moreover, astrocytes and microglia also express lipocalin 2 after inflammatory stimulation in vitro.32,33 The SLPI, an anti-inflammatory protease inhibitor,34 is expressed in neurons and astrocytes, but not in macrophages/microglia. The localization of SLPI in the peri-infarct zone after middle cerebral artery occlusion in rats, suggested its role in the suppression of the inflammatory response to the ischemia.35
We have previously reported that CXCL10 is increased in the brain parenchyma of T. brucei-infected mice when suramin is no longer curative and that this chemokine co-localizes with astrocytes.12 In this study, and confirming previous findings,12,14 higher CXCL10 levels were detected in the CSF of late stage compared with early stage T. b. gambiense and T. b. rhodesiense HAT patients. Thus, lipocalin 2, SLPI, and CXCL10 can be expressed by brain parenchymal cells, and, therefore, may be markers for inflammatory events occurring behind the BBB during T. brucei infections.
It should be noted that in several of the patients classified as stage 2 because of presence of WBC in the CSF, no increase in levels of lipocalin 2, SLPI, or CXCL10 was found. The question, therefore, arises whether these patients should have been subjected to toxic stage 2 drug treatment or not. Therefore, complementary to the present approach, the levels of molecules identified by a microarray could be measured in the CSF of experimental animals at time points when suramin or pentamidine are curative or not. In line with this, we observed a significant increase in levels of lipocalin 2 in the CSF from T. b. brucei-infected rats at a late stage. More data using animal models infected with human pathogenic subspecies of trypanosomes are needed to evaluate potential markers in HAT diagnosis and treatment follow-up.
In conclusion, this study shows that levels of lipocalin 2 and SLPI as well as CXCL10, identified using expression microarray analysis in the brain from infected mice, may contribute to differentiating between early and late stage HAT disease.
Acknowledgments
We are grateful to all the T. b. gambiense patients in Mbuji Mayi, DR Congo, and the T. b. rhodesiense patients in the Rumphi region, Malawi, who participated in the study. Special thanks to the medical staff at the various hospitals in these regions for their technical assistance.
Footnotes
Financial support: This work was supported by a grant from the European Union (FP6-2004-INCO-DEV-3 032324; NEUROTRYP). DNA received a post-doctoral research fellowship from the International Brain Research Organization. DMN received a PhD grant from the Belgian Directorate General for Development Cooperation.
Authors' addresses: Daniel Ndem Amin, Martin Rottenberg, and Krister Kristensson, Department of Neuroscience, Karolinska Institutet, Stockholm, Sweden, E-mails: ndemamin@yahoo.co.uk, martinrottenberg@ki.se, and krister.kristensson@ki.se. Dieudonné Mumba Ngoyi, Institut National de Recherche Biomédicale, Kinshasa, DR Congo, E-mail: mumbadieudonne@yahoo.fr. Gondwe-Mphepo Nhkwachi, Centre for Ticks and Tick-Borne Diseases, Lilongwe, Malawi, E-mail: ncgondwe@yahoo.com. Maria Palomba, Section of Anatomy and Histology, Faculty of Medicine, University of Verona, Verona, Italy, E-mail: mariella@anatomy.univr.it. Philippe Büscher, Institute of Tropical Medicine, Department of Parasitology, Antwerp, Belgium, E-mail: PBuscher@itg.be. Willias Masocha, Department of Applied Therapeutics, Faculty of Pharmacy, Kuwait University, Safat, Kuwait, E-mail: masocha@hsc.edu.kw.
References
- 1.Dumas M, Preux PM, Sagui E. Neurology in developing countries. Med Trop. 2009;69:5–6. [PubMed] [Google Scholar]
- 2.Kristensson K, Mhlanga JD, Bentivoglio M. Parasites and the brain: neuroinvasion, immunopathogenesis and neuronal dysfunctions. Curr Top Microbiol Immunol. 2002;265:227–257. doi: 10.1007/978-3-662-09525-6_12. [DOI] [PubMed] [Google Scholar]
- 3.Bouteille B, Oukem O, Bisser S, Dumas M. Treatment perspectives for human African trypanosomiasis. Fundam Clin Pharmacol. 2003;17:171–181. doi: 10.1046/j.1472-8206.2003.00167.x. [DOI] [PubMed] [Google Scholar]
- 4.Kennedy PG. Human African trypanosomiasis of the CNS: current issues and challenges. J Clin Invest. 2004;113:496–504. doi: 10.1172/JCI21052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization (WHO) Control and surveillance of African trypanosomiasis. Report of a WHO Expert Committee. World Health Organ Tech Rep Ser. 1998;881:1–114. [PubMed] [Google Scholar]
- 6.Lejon V, Büscher P. Stage determination and follow-up in sleeping sickness. Med Trop. 2001;61:355–360. [PubMed] [Google Scholar]
- 7.Bisser S, Lejon V, Preux PM, Bouteille B, Stanghellini A, Jauberteau MO, Büscher P, Dumas M. Blood-cerebrospinal fluid barrier and intrathecal immunoglobulins compared to field diagnosis of central nervous system involvement in sleeping sickness. J Neurol Sci. 2002;193:127–135. doi: 10.1016/s0022-510x(01)00655-4. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy PG. The continuing problem of human African trypanosomiasis (sleeping sickness) Ann Neurol. 2008;64:116–126. doi: 10.1002/ana.21429. [DOI] [PubMed] [Google Scholar]
- 9.Lejon V, Roger I, Mumba Ngoyi D, Menten J, Robays J, N'siesi FX, Bisser S, Boelaert M, Büscher P. Novel markers for treatment outcome in late-stage Trypanosoma brucei gambiense trypanosomiasis. Clin Infect Dis. 2008;47:15–22. doi: 10.1086/588668. [DOI] [PubMed] [Google Scholar]
- 10.Ngotho M, Kagira JM, Jensen HE, Karanja SM, Farah IO, Hau J. Immunospecific immunoglobulins and IL-10 as markers for Trypanosoma brucei rhodesiense late stage disease in experimentally infected vervet monkeys. Trop Med Int Health. 2009;14:736–747. doi: 10.1111/j.1365-3156.2009.02285.x. [DOI] [PubMed] [Google Scholar]
- 11.Lejon V, Robays J, N'Siesi FX, Mumba D, Hoogstoel A, Bisser S, Reiber H, Boelaert M, Büscher P. Treatment failure related to intrathecal immunoglobulin M (IgM) synthesis, cerebrospinal fluid IgM, and interleukin-10 in patients with hemolymphatic-stage sleeping sickness. Clin Vaccine Immunol. 2007;14:732–737. doi: 10.1128/CVI.00103-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amin DN, Rottenberg ME, Thomsen AR, Mumba D, Fenger C, Kristensson K, Büscher P, Finsen B, Masocha W. Expression and role of CXCL10 during the encephalitic stage of experimental and clinical African trypanosomiasis. J Infectious Dis. 2009;200:1556–1565. doi: 10.1086/644597. [DOI] [PubMed] [Google Scholar]
- 13.Courtioux B, Pervieux L, Vatunga G, Marin B, Josenando T, Jauberteau-Marchan MO, Bouteille B, Bisser S. Increased CXCL-13 levels in human African trypanosomiasis meningo-encephalitis. Trop Med Int Health. 2009;14:529–534. doi: 10.1111/j.1365-3156.2009.02263.x. [DOI] [PubMed] [Google Scholar]
- 14.Hainard A, Tiberti N, Robin X, Lejon V, Ngoyi DM, Matovu E, Enyaru JC, Fouda C, Ndung'u JM, Lisacek F, Müller M, Turck N, Sanchez JC. A combined CXCL10, CXCL8 and H-FABP panel for the staging of human African trypanosomiasis patients. PLoS Negl Trop Dis. 2009;16:e459. doi: 10.1371/journal.pntd.0000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mumba Ngoyi D, Lejon V, Pyana P, Boelaert M, Ilunga M, Mertens J, Mulunda J-P, Van Nieuwenhove S, Muyembe Tamfum J-J, Büscher P. How to shorten patient follow-up after treatment for Trypanosoma brucei gambiense sleeping sickness. J Infectious Dis. 2010;201:453–463. doi: 10.1086/649917. [DOI] [PubMed] [Google Scholar]
- 16.Lindberg J, af Klint E, Ulfgren AK, Stark A, Andersson T, Nilsson P, Klareskog L, Lundeberg J. Variability in synovial inflammation in rheumatoid arthritis investigated by microarray technology. Arthritis Res Ther. 2006;8:R47. doi: 10.1186/ar1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.KTH microarray core facility home page. 2007. http://www.ktharray.se/ Available at. Accessed October 2007.
- 18.R: A language and environment for statistical computing. 2007. http://www.cran.r-project.org/ Available at. Accessed October 2007.
- 19.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. doi:10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 21.Smyth GK, Speed T. Normalization of cDNA microarray data. Methods. 2003;31:265–273. doi: 10.1016/s1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- 22.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masocha W, Rottenberg ME, Kristensson K. Minocycline impedes African trypanosome invasion of the brain in a murine model. Antimicrob Agents Chemother. 2006;50:1798–1804. doi: 10.1128/AAC.50.5.1798-1804.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Amin DN, Masocha W, Ngan'dwe K, Rottenberg M, Kristensson K. Suramin and minocycline treatment of experimental African trypanososmiasis at an early stage of parasite brain invasion. Acta Trop. 2008;106:72–74. doi: 10.1016/j.actatropica.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 26.Masocha W, Robertson B, Rottenberg ME, Mhlanga J, Sorokin L, Kristensson K. Cerebral vessel laminins and IFN-gamma define Trypanosoma brucei brucei penetration of the blood-brain barrier. J Clin Invest. 2004;114:689–694. doi: 10.1172/JCI22104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulenga C, Mhlanga JD, Kristensson K, Robertson B. Trypanosoma brucei brucei crosses the blood-brain barrier while tight junction proteins are preserved in a rat chronic disease model. Neuropathol Appl Neurobiol. 2001;27:77–85. doi: 10.1046/j.0305-1846.2001.00306.x. [DOI] [PubMed] [Google Scholar]
- 28.Thuita JK, Kagira JM, Mwangangi D, Matovu E, Turner CM, Masiga D. Trypanosoma brucei rhodesiense transmitted by a single Tsetse fly bite in Vervet Monkeys as a model of human African trypanosomiasis. PLOS Negl Trop Dis. 2008;2:e238. doi: 10.1371/journal.pntd.0000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, Sorokin LM. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med. 2006;203:1007–1019. doi: 10.1084/jem.20051342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:811–813. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 31.Marques F, Rodrigues AJ, Sousa JC, Coppola G, Geschwind DH, Sousa N, Correia-Neves M, Palha JA. Lipocalin 2 is a choroid plexus acute-phase protein. J Cereb Blood Flow Metab. 2008;28:450–455. doi: 10.1038/sj.jcbfm.9600557. [DOI] [PubMed] [Google Scholar]
- 32.Lee S, Lee J, Kim S, Park JY, Lee WH, Mori K, Kim SH, Kim IK, Suk K. A dual role of lipocalin 2 in the apoptosis and deramification of activated microglia. J Immunol. 2007;179:3231–3241. doi: 10.4049/jimmunol.179.5.3231. [DOI] [PubMed] [Google Scholar]
- 33.Lee S, Park JY, Lee WH, Kim H, Park HC, Mori K, Suk K. Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J Neurosci. 2009;29:234–249. doi: 10.1523/JNEUROSCI.5273-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, DeWitt DL, McNeely TB, Wahl SM, Wahl LM. Secretory leukocyte protease inhibitor suppresses the production of monocyte prostaglandin H synthase-2, prostaglandin E2, and matrix metalloproteinases. J Clin Invest. 1997;99:894–900. doi: 10.1172/JCI119254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Li X, Xu L, Zhan Y, Yaish-Ohad S, Erhardt JA, Barone FC, Feuerstein GZ. Up-regulation of secretory leukocyte protease inhibitor (SLPI) in the brain after ischemic stroke: adenoviral expression of SLPI protects brain from ischemic injury. Mol Pharmacol. 2003;64:833–840. doi: 10.1124/mol.64.4.833. [DOI] [PubMed] [Google Scholar]