

Abstract
Rapidly evolving viruses such as HIV-1 display extensive sequence variation in response to host-specific selection, while simultaneously maintaining functions that are critical to replication and infectivity. This apparent conflict between diversifying and purifying selection may be resolved by an abundance of epistatic interactions such that the same functional requirements can be met by highly divergent sequences. We investigate this hypothesis by conducting an extensive characterization of sequence variation in the HIV-1 nef gene that encodes a highly variable multifunctional protein. Population-based sequences were obtained from 686 patients enrolled in the HOMER cohort in British Columbia, Canada, from which the distribution of nonsynonymous substitutions in the phylogeny was reconstructed by maximum likelihood. We used a phylogenetic comparative method on these data to identify putative epistatic interactions between residues. Two interactions (Y120/Q125 and N157/S169) were chosen to further investigate within-host evolution using HIV-1 RNA extractions from plasma samples from eight patients. Clonal sequencing confirmed strong linkage between polymorphisms at these sites in every case. We used massively parallel pyrosequencing (MPP) to reconstruct within-host evolution in these patients. Experimental error associated with MPP was quantified by performing replicates at two different stages of the protocol, which were pooled prior to analysis to reduce this source of variation. Phylogenetic reconstruction from these data revealed correlated substitutions at Y120/Q125 or N157/S169 repeated across multiple lineages in every host, indicating convergent within-host evolution shaped by epistatic interactions.
Keywords: coevolution, epistasis, HIV-1, next-generation sequencing, ancestral reconstruction, sequencing error
INTRODUCTION
Many RNA viruses exhibit tremendously high rates of molecular evolution (Jenkins et al. 2002) that underlie their facility to outpace the immune responses of their hosts. How are these viruses able to maintain the functions that are essential to replication and infectivity in the face of such extensive divergence in their genome sequences? There is accumulating experimental (Nijhuis et al. 2001; Poon and Chao 2005; Poon et al. 2005) and comparative evidence (Shapiro et al. 2006) that epistatic interactions between different sites of a genome sequence may be sufficiently abundant to provide a resolution of this evolutionary conflict. Epistasis is the phenomenon in which the contribution of a given site to one or more phenotypes is dependent on its genetic context. This context dependence is taken to an extreme under a specific form of epistasis denoted as “compensatory” or “sign” epistasis, in which the combination of two or more loss-of-function substitutions at different sites into the same genome can restore function to wild-type levels (Weinreich et al. 2005). The influence of compensatory epistasis potentially creates a complex many-to-one mapping of sequence variation to phenotype space; in other words, such interactions can enable the same functions to be accomplished by highly divergent sequences.
Compensatory epistasis appears to play an important role in shaping the evolution of HIV-1. For instance, HIV-1 populations rapidly acquire mutations conferring resistance in the presence of antiretroviral drugs, which are often followed by additional mutations that compensate for the fitness costs incurred by acquiring resistance (Nijhuis et al. 2001). Similarly, escape mutations in HIV-1 that impede recognition and/or processing of human leukocyte antigen (HLA) class I–restricted epitopes by the cytotoxic T lymphocyte (CTL)–mediated immune response in exchange for reduced viral replicative capacity can induce further compensatory mutations that restore viral fitness (Crawford et al. 2007; Troyer et al. 2009). Many of these examples stem from direct observation of sequence evolution over the course of in vitro passaging or longitudinal samples from clinical trials. In the absence of a modeling framework, however, we are limited to subjective criteria that identify only the most unambiguous epistatic interactions and are unable to capitalize on increases in sample size. Hence, the development of statistical comparative methods to detect epistatic interactions has flourished alongside the accumulation of extensive sequence data from HIV-1 (Korber et al. 1993; Hoffman et al. 2003; Gilbert et al. 2005; Carlson et al. 2007; Poon et al. 2007; Rhee et al. 2007) and other RNA viruses as well (Shapiro et al. 2006).
All comparative methods in this context generally rely on the assumption that correlated evolution among sites is the product of epistatic interactions. It is also common practice to disregard the evolutionary relatedness between sequences for the convenience of applying association test statistics directly to the sequence alignment (e.g., Hoffman et al. 2003; Rhee et al. 2007). However, a substantial number of the significantly covariant sets of sites that derive from this practice may reflect identity by descent, not independently evolved adaptations under the influence of epistatic interactions (Felsenstein 1973). Phylogenetic comparative methods (PCMs) address this confounding effect by explicitly modeling the evolutionary history of the extant sequences (Shapiro et al. 2006; Carlson et al. 2007; Poon et al. 2007). Although PCMs tend to be more computationally demanding, they can also greatly reduce an exceedingly high false discovery rate when there is substantial phylogenetic structure in the data (Poon et al. 2007).
A limitation common to both types of comparative methods is that our ability to detect epistatic interactions is strongly dependent on the sample size. To date, all studies of HIV-1 utilizing comparative methods to identify epistatic interactions have been directed at sequence variation at the level of the patient population. As a result of the HIV-1 pandemic and routine subtyping and drug resistance genotypic testing, large data sets comprising on the order of 104 sequences have become available through public HIV sequence repositories (Kuiken et al. 2003). The molecular evolution of HIV-1 among hosts, however, can be remarkably distinct from its within-host evolution. For instance, intra-host phylogenies reconstructed from serially sampled HIV-1 env sequences tend to assume a “ladder-like” shape that implies an ongoing turnover of sequence variants under immune selection, whereas inter-host phylogenies show little evidence of a similar mechanism operating in the patient population (Lemey et al. 2006). Likewise, there is no guarantee that the epistatic interactions affecting sequence divergence among hosts coincide with the interactions that shape within-host evolution. Whereas selection by antiretroviral drugs is largely indifferent to host environment, selection by neutralizing antibodies and CTLs is host specific by definition, and epistatic interactions induced by the latter may not necessarily emerge at the level of the patient population. Therefore, we are interested in applying comparative methods for detecting epistatic interactions directly to sequence variation at the within-host level. This has only recently become feasible with the emergence of so-called “next-generation” sequencing (NGS) technologies.
NGS is generally distinguished from conventional capillary-based (i.e., Sanger) sequencing by its high-throughput capacity for processing potentially millions of sequence reads in a single run. Current platforms for NGS are hampered by short sequence read lengths ranging from 35 to 400 bp (Voelkerding et al. 2009). For rapidly evolving RNA viruses, this limited read length can nonetheless suffice to render an informative sample of sequence diversity within a narrowly defined portion of the genome, that is, “deep sequencing.” As a result, the massively parallel pyrosequencing (MPP) platform distributed by Roche/454 Life Sciences, which offers the longest mean read length (100– 450 bp) at the expense of a reduced throughput capacity (≈105 reads), has been adopted by many investigators in HIV research (Hoffmann et al. 2007; Wang, Ciuffi, et al. 2007; Wang, Mitsuya, et al. 2007; Rozera et al. 2009), where NGS is most often used to detect minority variants in the virus population. Although such data might carry a sufficiently strong phylogenetic signal to infer within-host evolutionary processes such as sequence coevolution, this application has only begun to be pursued (Campbell et al. 2008; Tsibris et al. 2009). Additionally, the reproducibility and error distribution of NGS has only recently been assessed on control populations comprising a single HIV-1 clone or a known mixture of clones (Tsibris et al. 2006; Huse et al. 2007; Wang, Mitsuya, et al. 2007). Even so, there remain several sources of error (e.g., polymerase chain reaction [PCR] amplification, alignment uncertainty) to be quantified in an experimental setting, and there is an unsettling lack of routine replication in NGS experiments.
In this study, we employ a PCM in concert with MPP to investigate epistatic interactions in the HIV-1 accessory protein Nef. HIV-1 Nef enhances virus replication and infectivity via several mechanisms, including downregulating the expression of cell surface receptors (e.g., CD4, MHC-I) to facilitate immune evasion and virion release (Roeth and Collins 2006), interfering with signal transduction pathways to stimulate T-cell activation and enhance viral replication (Baur et al. 1994), and reshaping the actin cytoskeleton (Campbell et al. 2004). Several of these functions are conserved in homologs in other primate lentiviruses despite as much as 70% amino acid sequence divergence (Munch et al. 2007). Nevertheless, HIV-1 Nef displays extensive genetic variability both within and among human hosts (Zanotto et al. 1999; Yusim et al. 2002) that is likely driven by the cellular immune response, given its high density of HLA class I–restricted epitopes (Yusim et al. 2009), high level of expression at an early stage of the infection cycle (Hewlett et al. 1991), and high frequency of targeting by CTL in early disease stages (Lichterfeld et al. 2004). For instance, Noviello et al. (2007) demonstrated that HIV-1 Nef is able to maintain two genetically independent functions (downregulation of CD4 and MHC-I) despite sequence diversification during sexual transmission. Hence, HIV-1 Nef provides a useful case study to assess whether the conflict between function and variability is resolved by compensatory interactions.
To identify targets for deep sequencing analysis, we analyzed 686 bulk HIV-1 nef sequences sampled from a patient population in British Columbia, Canada. Two putative epistatic interactions (Y120/Q125 and N157/S169) identified by PCM were selected for a detailed investigation of within-host evolution in eight patients. Reproducibility of deep sequencing was quantified by replicating the experiment at two stages of the protocol: 1) extraction of viral RNA from plasma samples and 2) second-round PCR amplification of the reverse transcription (RT)–PCR product. We observed substantial variability in allele frequencies stemming from the sample preparation stages of the parallel pyrosequencing protocol—a source of variance that cannot be ascertained from sequencing control mixtures of clonal populations. Nevertheless, phylogenetic inferences based on these data were robust to experimental error. By reconstructing within-host phylogenies from these data, we found that correlated substitutions at putative interacting sites arose in multiple independent lineages within each host. These results indicate that a comparative analysis of sequence variation in the patient population can provide a convenient and reliable measure of epistatic interactions shaping within-host evolution. Our analysis also identifies new functional clusters of residues in HIV-1 Nef and serves as an informative case study on the practical application of massively parallel sequencing in the study of RNA virus evolution.
MATERIALS AND METHODS
Data Collection
Baseline plasma samples were obtained from 686 antiretroviral-naive subjects, who were enrolled in the HOMER (HAART Observational Medical Evaluation and Research) cohort (Hogg et al. 2001) comprising participants in British Columbia, Canada, predominantly infected by HIV-1 subtype B and commencing triple combination therapy. Extraction of HIV-1 RNA from plasma was performed either using an automated QIAGEN viral RNA kit and BioRobot 9600/9604 workstation or manually using guanidinium-based buffer followed by isopropanol/ethanol washes or using automated guanidinium-based methods on a NucliSens easyMAG (bioMérieux). Full-length sequences of HIV-1 nef were amplified using nested RT-PCR and sequenced following the procedure described previously by Alexander et al. (2001). Further details and GenBank accession numbers for the bulk sequences can be found in Brumme et al. (2007).
A total of 85 clonal sequences were obtained from eight of the plasma samples, which were selected on the basis of mixtures (ambiguous base calls as designated by an ABI 3730xl DNA Analyzer) present in the corresponding bulk sequences, thereby indicating within-host polymorphisms. Cloning was performed using a TOPO TA cloning kit (Invitrogen, Burlington, Ontario, Canada) containing the PCR 2.1-TOPO vector with chemically competent TOP10F′ one shot cells, according to the manufacturer's instructions. HIV RNA levels for the eight plasma samples were determined using a Roche Amplicor version 1.5 assay (Roche Diagnostics, Mississauga, Ontario, Canada).
Detecting Sequence Covariation
Automated alignment of HIV-1 nef bulk sequences was carried out using ClustalW and manually refined using Se-Al (Andrew Rambaut, http://tree.bio.ed.ac.uk/software). Unless noted otherwise, all analyses described in this section were carried out in the phylogenetic software package HyPhy (Kosakovsky Pond et al. 2005). We reconstructed a phylogeny from this alignment by a neighbor-joining (NJ) method with Tamura–Nei (Tamura and Nei 1993) distances and rate variation across sites under a one-parameter gamma distribution (α = 0.5). Branch length estimates were refined by fitting a reversible model of nucleotide substitution rates (encoded by PAUP* model specification string 012232), which was determined by an automated model selection procedure as described in Kosakovsky Pond and Frost (2005a). Ancestral sequences at internal nodes of this phylogeny were reconstructed by fitting a codon substitution model (MG94; Muse and Gaut 1994) crossed with the nucleotide substitution model (012232) using joint maximum likelihood (ML) methods (Pupko et al. 2000). Nonsynonymous substitutions were mapped to specific branches of the phylogeny wherever ancestral or extant sequences occupying adjacent nodes of the phylogeny encoded different residues at a given codon site (Kosakovsky Pond and Frost 2005b).
If nonsynonymous substitutions affecting two or more sites in the gene sequence occurred in the same branches of the phylogeny significantly more often than expected by chance, then we would infer an interaction between those sites. We converted the entire map of substitutions to the phylogeny into a binary-valued matrix in which each row corresponded to a branch in the phylogeny, each column corresponded to a codon site in the alignment, and a “1” entry denoted the assignment of a nonsynonymous substitution to the corresponding site and branch (Poon et al. 2007). We discarded highly conserved sites at which nonsynonymous substitutions occurred at fewer than 1% of the branches in the tree (retaining n = 101 codon sites) to minimize the computational cost of the Bayesian graphical model analysis (described below). Additionally, we performed a similar analysis on the distribution of synonymous substitutions, so that we could address the potentially confounding influence of functional motifs in the nef nucleotide sequence, for example, 3′ long terminal repeat (LTR) region (see Ngandu et al. 2008, and references therein). To identify correlations in the occurrence of nonsynonymous mixtures among sites, we used a custom Python script to convert the entire alignment of nef bulk sequences into another binary-valued matrix in which “1”s encoded the presence of a nonsynonymous mixture and each row corresponded to a sequence in the alignment.
Both the substitutions- and mixtures-based matrices were analyzed separately using Bayesian graphical models (BGMs), following the procedure described in Poon et al. (2007). A BGM is a compact graphical representation of a joint probability distribution such that each random variable is represented by a node (Pearl 1988). Using BGMs to analyze coevolving sites confers the advantages of 1) generating a natural graphical representation of interactions; 2) minimization of the number of model parameters, which makes tractable the analysis of large systems given a limited number of observations, and 3) the potential to distinguish indirect correlations from direct causative relationships between sites. Results from BGM analysis were corroborated against results from a similar analysis of this data based on a phylogenetic correction for pairwise correlations between sites (Carlson et al. 2008).
Deep Sequencing
To further investigate covariant polymorphisms within patients detected as mixtures, we selected eight plasma samples for MPP using a Genome Sequencer FLX System (Roche/454 Life Sciences). We performed three replicate second-round PCR amplifications on the products of the initial RT-PCR product for each isolate, using primers spanning the HXB2 reference sequence coordinates 9121–9521. Each second-round PCR product was bar coded using a unique ten-nucleotide sequence tag for multiplexed pyrosequencing, that is, parallel tagged sequencing (PTS) (Meyer et al. 2008). Performing deep sequencing in triplicate enabled us to quantify the variability in the sequence composition of sample populations that was introduced by the second-round PCR and MPP steps. This procedure would not completely eliminate experimental bias, however, as it fails to address any variability introduced by the preliminary sample extraction and RT-PCR steps. Consequently, we took the additional measure of replicating the first-round RT-PCR and second-round PCR steps in triplicate, using a second extraction of viral RNA from the frozen plasma samples, followed by pyrosequencing of the pooled amplicons. Due to the limited availability of plasma samples, we were only able to carry out this level of replication for three out of eight patients.
The MPP data generated by the Genome Sequencer FLX base calling algorithm were partitioned by patient sequence tags and by forward/reverse primers and aligned using a set of custom HyPhy batch language and Python scripts (available upon request). For each data set, a subsample of 100 sequences were aligned pairwise to the HXB2 reference sequence using an implementation of the Gotoh algorithm (Myers and Miller 1988) in HyPhy to generate a patient-specific consensus sequence (J. Archer, M. Lewis, and D.L. Robertson, presented at the 15th International Workshop on HIV Dynamics & Evolution, Santa Fe, NM, 27–30 April 2008). Subsequently, all sequences were aligned pairwise to this consensus sequence. The resulting alignments were post-processed with a Python script and manually refined in Seaview (Galtier et al. 1996) or Se-Al (A. Rambaut, http://tree.bio.ed.ac.uk/software/seal). Columns in refined alignments whose nonprefix/suffix gap frequencies exceeded a threshold of 99% (e.g., singleton nucleotide insertions) were stripped out. The final MPP alignments were validated by manually aligning the MPP consensus sequence against bulk and clonal sequences from the same plasma sample.
To characterize sequence evolution within the eight patients, we analyzed each alignment of MPP reads using phylogenetic methods. Because phylogenetic methods generally rely on the pattern of sequence divergence to infer rates of molecular evolution, they do not make use of allele frequency information, that is, the number of instances of a given sequence variant in a population sample. Moreover, the proportion of pyrosequencing reads that are redundant tends to be considerable, reflecting both the real composition of the viral population as well as the limited mean read length of the NGS platform. Each alignment was therefore reduced to a minimal set of longest nonredundant sequences using a custom Python script by matching sequences that were identical over any length of the longest nonredundant sequence and recording the corresponding number of matches. Sequences containing mixtures or partial sequences were partitioned equally over matching nonredundant sequence variants as fractional increments to each total.
A phylogeny was reconstructed from each alignment by the NJ method with Tamura–Nei distances (Tamura and Nei 1993). Branch length estimates in the NJ phylogeny were refined by fitting a full parameterization of the time-reversible nucleotide substitution model (REV; Tavaré 1986) by ML. The resulting phylogeny was used to fit a Muse–Gaut codon substitution model (Muse and Gaut 1994) crossed with the REV nucleotide substitution model by ML. These analyses were carried out on a high-performance computing Linux cluster (comprising 260 processors) using a message passing interface–enabled version of HyPhy. ML parameter estimates were used to reconstruct ancestral sequences and map nonsynonymous substitution events to individual branches of each within-host phylogeny. Correlated substitutions at potentially interacting sites were visualized by color-annotating a radial tree in PostScript using a custom HyPhy batch language script. All scripts used to perform this analysis are available on the Web at http://www.hyphy.org/pubs/Nef.
RESULTS
Genetic Variation in Nef
We observed extensive genetic variation in HIV-1 nef bulk sequences isolated from 686 patients enrolled in the HOMER cohort. The mean Shannon entropy per site was approximately 0.75, commensurate with the overall level of amino acid variability across HIV-1 M group nef sequences (Yusim et al. 2002). In-frame deletions affected 226 (33%) of the sequences, clustering predominantly in three regions spanning codon sites 8–12, 48–50, and 151–158. Using a conservative Bonferroni correction for multiple comparisons (α = 2.43×10 − 4), we found a significant excess of nonsynonymous substitutions above the expected values (i.e., scaled dN − dS values ranging from 0.82 to 4.2) at 18 different codon sites, namely, V10, V11, P14, A15, A23, V33, G83, K92, K94, E98, I101, I133, P150, V153, K178, V182, R188, and L198 (alignment consensus residues and coordinates; see supplementary table S1, Supplementary Material online).
Sequencing mixtures (i.e., ambiguous bases due to multiple peaks in the sequence chromatogram) were abundant, averaging 7.0 mixtures per bulk sequence, suggesting that genetic variation within patients was commensurate with the extensive variation in the patient population. Any given mixture was equally likely to be nonsynonymous (i.e., the ambiguous codon could encode more than one amino acid) as synonymous (odds 2450:2385 = 1.03). Considering that the neutral expectation for these odds is closer to 3 (392:134 = 2.92 for two-way mixtures given the universal genetic code), we interpreted the observed odds as a significant depletion of nonsynonymous mixtures, consistent with purifying selection as a dominant force shaping within-host polymorphism. The difference in the site-specific frequencies of nonsynonymous and synonymous mixtures, normalized, respectively, by their expected values based on the genetic code and empirical base frequencies, was significantly correlated with the quantity dN − dS representing diversifying selection at the level of the host population (Spearman's ρ = 0.803,P < 10 − 15). This correlation implied that the distribution of nonsynonymous mixtures in our sequence alignment was predominantly the outcome of site-specific selection, rather than being an artifact of sequencing error.
Among-Host Sequence Coevolution
We used BGM methods to infer a network of interactions between 63 different residues in Nef from the distribution of nonsynonymous substitutions reconstructed from the sequence alignment (fig. 1). A network is a set of nodes that each represent a random variable, in our case being the presence or absence of a nonsynonymous substitution at a given codon site and branch of the phylogeny (Poon et al. 2007). Connections between nodes, termed “edges,” are used to represent interactions, that is, a conditional dependence between variables. Figure 1 depicts a model-averaged network (obtained by sampling from the posterior probability distribution using a Bayesian Monte Carlo Markov chain method) comprising 40 edges whose marginal posterior probabilities exceeded a threshold of 0.95. The majority (88%) of edges in the network connected residues located in the same domain of the Nef protein (namely, anchor versus core domains) with only five edges connecting residues located in different domains (P14/Y203, V16/E151, R19/M173, A50/H116, and T51/Q170; fig. 1). These interdomain edges are consistent with previous observations that anchor residues that interact with the core tend to occur either in close proximity to the protein N-terminus or within the interval bounded by residues 49 and 60 (Groesch and Freire 2007).
FIG. 1.

Schematic approximating the anchor and core structures of the HIV-1 Nef protein (thick black lines), annotated with residue–residue interactions from a BGM analysis of a phylogenetic reconstruction of nonsynonymous substitutions. Amino acid sites implicated in epistatic interactions (rectangular nodes) are labeled with the alignment consensus residue and position. Thirty-eight sites that were not implicated in interactions (G3, V10, G12, A15, M20, A23, R35, E38, K39, I43, S46, A49, A53, C55, A56, E62, E63, E65, R71, I101, Y102, Q107, P129, V148, P150, V153, A156, N161, C163, M168, P176, K178, V182, K184, R196, L198, E201, and D205) are omitted for clarity. Putative interactions are depicted as connections (solid or dashed lines) between nodes, labeled with marginal posterior probabilities × 100, using a cutoff value of 0.95. Dashed lines connect sites separated by more than 15 residues in the protein sequence, though the respective amino acids are frequently clustered in the folded protein structure (e.g., 8.3Å from E98 to F143; 13.1Å from A50 to H116). Thirty-nine conditionally independent sites were omitted for clarity. Node names in italics are used to indicate long-distance interactions (> 100 residue separation). Interactions supported by significant correlations in bulk sequencing mixtures are indicated by double lines.
RNA virus genomes experience selection on the nucleotide level owing to functional stem-loop structures and binding motifs. To determine whether coevolution at the level of the nucleotide sequence (e.g., compensatory substitutions in RNA secondary structures) may have generated potentially confounding edges between codon sites in this network, we performed a similar analysis on the distribution of synonymous substitutions. We obtained the following significant edges, that is, with marginal posterior probabilities exceeding a cutoff of 0.9: T51/K204, L76/R77, D86/L110, D108/S187, V148/G119/I133, Q125/P136, D174/F191/A190, H192/M194, R196/L198, and H199/P200. None of these edges was present in the network estimated from nonsynonymous substitutions. However, four of these edges connected codon sites within or adjacent to the core negative response element (overlapping codons A190 to V194) of the 3′ LTR region, which suggests that this analysis of synonymous variation was able to detect functional interactions at the nucleotide level. In addition, we determined whether the residue–residue interactions presented here coincided with any of the putative base-pairing interactions recently reported in a complete structural model of the HIV-1 RNA genome (Watts et al. 2009). We found no overlap between these sets of interactions, lending further support to the conjecture that our results were exempt from confounding by selection at the nucleotide level.
To select a subset of putative interactions in HIV-1 Nef that potentially carried over to the within-host level for further investigation by deep sequencing, we applied the same BGM procedure to the distribution of nonsynonymous mixtures in the alignment of bulk sequences. A significant positive correlation in mixtures suggests that the sites coevolve at the within-host level. We identified ten positively correlated pairs of sites (supplementary table S2, Supplementary Material online), of which four coincided with edges in figure 1 (K92/E93, Y120/Q125, N157/S169, and R188/H192). The residue pairs Y120/Q125 and N157/S169 were chosen for further analysis—Y120 and Q125 reside in several CTL epitopes (Yusim et al. 2009) and the substitutions Y120F and Q125H have been associated with escape from CTL recognition in vitro (Culmann et al. 1991), whereas N157 and S169 have been implicated in the rate of progression to AIDS (Kirchhoff et al. 1999). Moreover, these pairs fell within the expected range of sequence coverage for Roche/454 MPP. We selected eight baseline plasma samples in which nonsynonymous mixtures occurred at both sites for one of the two pairs (denoted P3, P5, and P6 for Y120/Q125; and P1, P2, P4, P7, and P8 for N157/S169) for deep sequencing analysis.
Deep Sequencing Analysis
We carried out three replicate second-round PCR amplifications on the RT-PCR products obtained from the eight plasma samples and ran PTS on a GS-FLX Sequencer (Roche/454 Life Sciences) for all 24 amplification products. We also replicated the full experimental protocol for three patient isolates (P2, P4, and P8), namely, a second extraction of RNA from the plasma sample, followed by RT-PCR amplification in triplicate, second-round PCR amplification in triplicate, and MPP of the pooled amplification product. The mean viral load per sample was approximately 2.5×105 copies/ml. Individual viral load measurements and mean forward read depths for the eight baseline plasma samples are presented in table 1. As the input reverse transcribed RNA copy number was unknown, there was a significant possibility that some nucleic acids were resampled by the sequencing process. However, because we restricted our analysis of reproducibility (see below) to the upper portion of the frequency distribution (0.1–70%), resampling was likely a relatively minor confounding factor.
Table 1.
Characterization of Eight HIV-1 Populations Subjected to MPP Analysis
| Mean Read | COT | Root | Correlated | |||
| Patient | Viral Load | Depth | Codon Sites | Genotype | Genotype | Substitutions |
| P1 | 300,000 | 2,797 | 157,169 | S,N | S,N | 2(2,0) |
| P2 | 130,000 | 4,306 | 157,169 | N,S | N,S | 2(1,1) |
| P3 | 750,000 | 2,717 | 120,125 | F,H | Y,Q | 2(0,1) |
| P4 | 130,000 | 3,181 | 157,169 | T,S | N,S | 3(2,1) |
| P5 | 170,000 | 3,232 | 120,125 | F,H | F,H | 9(3,6) |
| P6 | 200,000 | 3,927 | 120,125 | F,H | Y,D | 6(1,2) |
| P7 | 270,000 | 4,377 | 157,169 | N,S | N,S | 7(5,1) |
| P8 | 76,000 | 2,849 | 157,169 | T,N | T,N | 4(2,2) |
Viral load is measured in copies per milliliter of baseline plasma. Mean forward read depth was averaged across three second-round PCR replicates. The number of correlated substitutions in each phylogeny is followed by the number of instances that the first substitution was mapped at the first or second site (enclosed in parentheses). If both substitutions were mapped to the same branch, then substitution order could not inferred.
The average read depth per product was n = 8,167 sequences (range 5,543–12,180) with an average read length of 247 nt (range 201–341 nt). Forward reads covered Nef codon sites 109–192, and the reverse-primed reads covered sites 164–206 and approximately 120 nucleotides further into the 3′ LTR as well. Because the reverse reads failed to extend into the region of interest, we directed our analysis specifically on the forward reads. Forward reads comprised about 43.7% of sample depth, averaging approximately 3400 forward reads per plasma sample. As anticipated for this sequencing technology, single nucleotide insertions occurred abundantly in forward read alignments; on average, 22% of alignment columns corresponded to insertions present in less than 5% of sequences and often a single sequence only. Even after removing spurious gaps attributable to errors in alignment and the expected baseline indel rate of the MPP platform (≈1%; Huse et al. 2007; Wang, Mitsuya, et al. 2007), an average of 5.5 columns with low-frequency ( < 5% of sequences) insertions remained (patient-specific means ranged from 1.0 to 9.1 insertions). The insertions did not discernibly coincide with homopolymeric regions in the alignment. When we mapped the affected alignment columns to an NJ tree reconstructed from the remainder of the alignment, we found that the putative insertions failed to cluster in the tree. This outcome suggested that the insertions represented experimental artifacts rather than defective nef sequences that can nevertheless be propagated in viable HIV-1 lineages (Salvi et al. 1998). We omitted these columns from alignments in subsequent analyses such that the resulting sequences remained in-frame over their entire length.
To quantitatively assess the reproducibility of frequency estimates by MPP, we calculated the variance-to-mean ratio for each sequence variant shared across replicates. This dispersion statistic was more robust than the coefficient of variation to differences in expected values (supplementary text S1, Supplementary Material online), making it the more appropriate choice to apply across the entire frequency distribution of sequence variants to yield a meaningful summary statistic. The average variance-to-mean ratio from second-round amplification and onward was 3.7×10 − 4. For a given sequence variant with expected frequency x, the predicted standard deviation (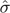 ) in frequency estimates can be obtained from this ratio by multiplying by x and taking the square root; for example, a minority variant expected to be observed in 1% of MPP reads has = 0.19%. The average variance-to-mean ratio for fully replicated experiments was 7.6×10 − 3; for example, frequency estimates of a 1% variant would have = 0.87%. An extended discussion of this reproducibility analysis for MPP data is provided as supplementary text S1, Supplementary Material online.
) in frequency estimates can be obtained from this ratio by multiplying by x and taking the square root; for example, a minority variant expected to be observed in 1% of MPP reads has = 0.19%. The average variance-to-mean ratio for fully replicated experiments was 7.6×10 − 3; for example, frequency estimates of a 1% variant would have = 0.87%. An extended discussion of this reproducibility analysis for MPP data is provided as supplementary text S1, Supplementary Material online.
Because phylogenetic analyses do not incorporate frequency information (i.e., identical sequences are not phylogenetically informative) and are therefore robust to uncertainty in frequency estimates (except for zero versus nonzero frequencies), we pooled these replicates for each of the eight baseline plasma samples to maximize our sample of sequence variation. To summarize this variability, we present the ten most abundant amino acid sequences for each plasma sample in figures 2 and 3. On average, the most frequent amino acid sequence from a patient was present at a frequency of 19.7% (range 10.0–30.5%). We confirmed that the consensus sequence from each pooled alignment of MPP reads matched the corresponding bulk sequence. Additionally, we obtained 85 clonal full-length nef sequences in total from these baseline plasma samples (median 11 clones per sample). Alignments of these clonal sequences against the MPP consensus nucleotide sequence and of the five most frequent amino acid sequences from MPP against the patient bulk sequence are provided in supplementary text S2 and S3 (Supplementary Material online), respectively. Both the bulk sequence and the composition of clonal sequences corresponded well with the MPP data. We also observed that minority variants represented by mixtures in bulk sequences were present in the corresponding MPP data at frequencies ranging from 20% to 50%. For example, the bulk sequence from patient P1 contained four mixtures within the interval covered by MPP, namely, R(A/G)375, R470, R506, and M(A/C)531. At each position, the frequency of the subdominant (i.e., second-most abundant) allele was 42.2% G375; 43.1% G470; 42.8% G506; and 23.8% C531. MPP also revealed six other polymorphic nucleotide sites with subdominant allele frequencies between 10% and 25% within this patient population, consistent with 25% as a likely limit for detection of minority variants by mixtures in bulk sequences in our experiment.
FIG. 2.

Alignments of the ten most frequent amino acid sequences from five patients with polymorphisms at HIV-1 Nef residues N157 and S169 (highlighted columns). Alignments are truncated to display variable residues only. Frequencies within each patient data set (combining three replicate RT-PCRs and Roche/454 pyrosequencing runs) are listed to the right of each sequence. A dot “.” indicates that the residue is identical to that of the most frequent sequence. An “X” represents a premature stop codon. During translation, nucleotide sequences containing an ambiguous character (“N”) or a frame-shift deletion were discarded. The frequency of any incomplete sequence (with gap prefix or suffix) was partitioned equally among all sequences containing the sequence fragment, which was then discarded. Frequencies shown here were rounded to the nearest integer.
FIG. 3.

Alignments of the ten most frequent amino acid sequences from three patients with polymorphisms at HIV-1 Nef residues Y120 and Q125 (highlighted columns). Alignments are truncated to display variable residues only. Processing and interpretation are identical to figure 2.
Within-Host Sequence Coevolution
In the pooled MPP data, linkage disequilibria between amino acid site pairs N157/S169 (fig. 2) and Y120/Q125 (fig. 3) was largely consistent with coevolution due to residue–residue interactions, that is, substitutions from consensus residues occurred predominantly in cis-linkage. On the other hand, intermediate variants containing a mixture of consensus and nonconsensus residues were found at substantial frequencies (≈10–20%) in patient plasma samples P3 (F120/Q125) and P4 (T157/S169). Because the complements to these intermediate variants (Y120/H125 and N157/N169, respectively) occurred at very low frequencies ( < 0.1%) in the corresponding samples, overall linkage disequilibrium remained high nonetheless. However, linkage disequilibrium does not provide sufficient evidence of coevolution at these sites within patients as such patterns may be confounded by the phylogeny, that is, when infrequent substitutions at independently evolving sites are by chance transmitted jointly or in mutual exclusion to large numbers of descendents (Felsenstein 1973). A stronger case can be made for sequence coevolution within patients if correlated substitutions at the sites occur multiply in independent lineages of the phylogeny.
To assess the extent of sequence coevolution within patients, we reconstructed the distribution of nonsynonymous substitutions throughout each within-patient phylogeny from the respective alignments of MPP nucleotide sequences using an ML procedure (Kosakovsky Pond and Frost 2005b). The phylogenies were color-annotated with respect to residue combinations at the target sites with reference to the ancestral reconstruction at the central node of the radial tree (i.e., center of the tree [COT]; Nickle et al. 2003) to provide a visual representation of within-host evolution. Annotated phylogenies for patients P1, P5, and P8 are shown in figure 4, whereas the remaining phylogenies are provided as Supplementary figure S4, Supplementary Material online. We found that substitution maps to the phylogeny were correlated for the target sites such that a substitution at one site was quickly followed by another substitution at the second site. More importantly, we observed multiple instances of correlated substitutions in phylogenetically independent clades in all patient phylogenies, that is, convergent evolution. Overall, the number of independent lineages carrying correlated substitutions ranged among patients from 2 to 9 with a mean of 4.6 (table 1). Note that these estimates were conservative because all sites in each alignment of pyrosequencing reads were used to reconstruct the phylogeny. Otherwise, convergent evolution would tend to collapse independent lineages into a single clade, leading to an underestimate of the number of instances of correlated substitution.
FIG. 4.

Radial phylogenies reconstructed by ML from nonredundant sequences obtained from MPP of HIV-1 Nef from three patient baseline plasma samples. Branches are colored with respect to substitutions away from the ancestral genotype reconstructed at the inferred root (which coincides with the genotype at the COT): grey to indicate no substitutions; red to indicate a substitution at one site only; and blue to indicate substitutions at both sites. Terminal branches corresponding to more than one identical sequence are labeled with open triangles scaled in proportion to the number of identical sequences, N (height 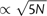 , base
, base 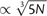 ). A legend with triangles corresponding to N = {2, 10, 100, 500} is provided in the figure to facilitate interpretation.
). A legend with triangles corresponding to N = {2, 10, 100, 500} is provided in the figure to facilitate interpretation.
While the ancestral sequence inferred at the COT provided a convenient reference point, we were cautious about interpreting this sequence as reconstructing the root ancestor as these phylogenies were unrooted. To illustrate, we rooted each within-host phylogeny using a bulk sequence from another patient that was closely related in the among-host phylogeny. Ancestral state reconstructions at these roots are reported alongside reconstructions at the COT from the corresponding unrooted trees for each patient in table 1. Reconstructions at the root matched the COT for five out of eight patients, whereas both sites were unmatched in two patients (P3 and P6). For patient P4, the COT reconstruction comprised one consensus and one nonconsensus residue (T157/S169), a combination that was present at an unusually high frequency in P4 when compared with other patient samples (fig. 2). In contrast, the consensus genotype (N157/S169) was reconstructed at the inferred root in P4, suggesting that the COT genotype corresponded to a prolonged intermediate stage in the evolution of HIV-1 in this patient. We surmised that the placement of roots in these phylogenies was accompanied with a high level of uncertainty due to the nature of these data. Therefore, we have avoided drawing any conclusions from our results that would be sensitive to uncertainty in rooting of within-host phylogenies.
Because we considered two target sites in each patient, there was always more than one mutational pathway to evolve from one residue combination to another. Previous investigators have observed that HIV-1 evolution can be constrained to certain mutational pathways; for example, substitutions at reverse transcriptase T215 tend to precede those at M41 in the evolution of resistance to zidovudine (Kellam et al. 1994). We examined the substitution maps to determine whether HIV-1 evolution was significantly biased toward one mutational pathway over another. We found no evidence of a significant bias: site 120 acquired a substitution before site 125 in 4 out of 13 cases, and site 157 before site 169 in 14 of 20 cases (table 1; in nearly all cases, the inferred order of substitutions was unambiguous en route to a terminal sequence). In most cases, evolution proceeded through one of two intermediates to an end point with paired consensus or nonconsensus residues. For example, in patient P1, the mutational pathways were comprised as follows: SN → (NN or SS) → NS (omitting site coordinates for compact notation). Although several other intermediate genotypes emerged in P1 (e.g., GN, AN, SD; fig. 5), those lineages never acquired a substitution at the second site. However, we found alternative end points in other patients. For example, the following pathways were both reconstructed in patients P4 and P7: 1) NS → (NN or SS) →SN and2) NS → (TS or NN) → TN.
FIG. 5.

Circle plots summarizing proportions of tree length by genotype. Each circle corresponds to a unique combination of residues at the target sites (see labels), with its area proportional to the total evolutionary time occupied by that genotype in the within-host phylogeny. Circles were arranged in each plot with respect to the minimum height of the genotype in the tree (y axis) and order that the genotype was first encountered during preorder traversal of the tree (x axis). The genotype reconstructed at the inferred root of each tree is indicated in bold text. Evolutionary time was calculated in units of expected substitutions by summing the respective branch lengths in the phylogeny. Confidence intervals were estimated by resampling ancestral reconstructions from each posterior probability distribution but were too small to appear in this figure. The majority of evolutionary time is occupied by the ancestral genotype at the root (bolded circle) or a single predominant genotype in which both sites have substitutions from the ancestor. In contrast, nearly all genotypes with only one substitution away from the ancestor occupy relatively little evolutionary time, including those intermediate of the majority genotypes.
To further characterize sequence evolution within patients at the target sites, we calculated the total branch lengths in each patient phylogeny with respect to residue combinations at the target sites. These results are summarized in figure 5, where each circle corresponds to a given residue combination with its area proportional to the branch length total. Branch lengths in the phylogenies are scaled in units of the expected number of substitutions per site, which is confounded with chronological time. Put another way, these quantities provide a crude estimate of the proportion of time that any given HIV-1 lineage has assumed a particular configuration of residues, throughout the entire history of sequence evolution in that patient. Confidence intervals on the estimates of branch length totals, generated by resampling ancestral sequences from the posterior probability distribution, were too small to be distinguished in these plots. We observed that, in most cases, the total branch lengths of genotypes comprising one consensus and one nonconsensus residue (i.e., intermediate genotypes) were substantially less than either the consensus or the predominating nonconsensus genotypes (fig. 5). This trend suggested that correlated substitutions occurred rapidly within patients, either through selection against intermediate genotypes or with synergistic epistasis accompanying a shift in host environments that conferred a disproportionate selective advantage to the double-mutant genotype. In patient P3, however, representation of the intermediate genotype FQ was slightly greater than the consensus genotype inferred at the root (YQ). Similarly, the total branch length associated with intermediate genotype TS in P4 was slightly greater than the predominating nonconsensus genotype SN. These discrepancies indicate that coevolution at these sites by rapid transitions through intermediate genotypes do not necessarily apply to every host, possibly due to either infrequent host-specific factors or higher-order interactions involving additional sites outside of nef.
DISCUSSION
NGS technologies, such as MPP, are a promising development that offer a fine-scale resolution of the genetic composition of a virus population, that is, deep sequencing. The clinical utility of such methods has been demonstrated with respect to the detection of drug-resistant variants in HIV-1 populations at frequencies well below the detection threshold of conventional sequencing (Tsibris et al. 2006; Hoffmann et al. 2007; Wang, Mitsuya, et al. 2007). Because many RNA viruses such as HIV-1 evolve so rapidly, deep sequencing might also provide valuable insight into the parameters of the molecular evolution of virus populations. We have demonstrated that MPP can be used to reconstruct within-host evolution of HIV-1, specifically to characterize the correlated substitution process affecting sites in the accessory protein Nef. The distribution of multiple instances of correlated substitutions at the sites Y120/Q125 and N157/S169 along several independent lineages in the phylogenies provided strong evidence that putative residue–residue interactions estimated from population-based (bulk) sequence variation also shaped variation within hosts. This result is encouraging because deep sequencing via NGS is not yet capable of spanning the entire length of most genes; instead, we show that information contained in conventional full-length bulk sequences (namely, mixtures, phylogenetic analysis of correlated substitutions) can be used to pinpoint the most informative gene intervals for deep sequencing.
However, NGS is also fraught with commensurately novel sources of experimental error whose parameters have only recently been assessed in the context of deep sequencing of virus populations. Deep sequencing attempts to measure the genetic composition of a population, which comprises a set of nucleotide sequences variants and the number of instances of each variant. Previous studies have focused on one specific source of error, namely, the surfeit of single nucleotide indels introduced by MPP (approximately 1 for every 100 nucleotides) that can interfere with the estimation of variant frequencies (Huse et al. 2007; Wang, Mitsuya, et al. 2007). This must be considered alongside several other sources of error, some of which are not specific to NGS but compounded by NGS-specific error:
(i) Sampling error. Sequence variant frequencies in the plasma sample may not be representative of the true frequencies in the virus population, due to random variation or compartmentalization. Extraction of HIV-1 RNA is another potential source of sampling error.
(ii) RT and amplification. These processes contribute two different types of error. First, misincorporation of nucleotides during primer extension may either introduce a new spurious variant or misclassify the sequence as another variant. Second, stochastic variability among templates in the waiting time until a doubling event can skew the frequency distribution, particularly during the initial amplification cycles (Peccoud and Jacob 1996). Variability in the observation of mixtures in HIV-1 pol bulk sequences has been attributed largely to this source (Galli et al. 2003). Recombination at the RT-PCR step may also constitute a significant source of error.
(iii) Sequencing error. The majority (≈60% − 90%) of sequencing errors introduced by MPP are insertions and deletions, which tend to be associated with homopolymeric regions (Huse et al. 2007; Wang, Mitsuya, et al. 2007). Some of these errors are attributed to optical signal bleed between the picoliter wells that separate emulsion PCR products derived from single nucleic acids (Margulies et al. 2005). (Other NGS platforms such as Solexa or SOLiD are reported to yield more accurate homopolymer run lengths.) Because of this high error rate, investigators have proposed null models that define allele frequency significance cutoffs, ranging in complexity from a Poisson distribution (Wang, Mitsuya, et al. 2007) to population genetic models (Johnson and Slatkin 2008). In addition, the frequency of indel and mismatch errors can be exacerbated by poor read quality, though this can be prescreened to some extent by automated algorithms (Brockman et al. 2008).
-
(iv) Alignment error. An alignment is a hypothesis of evolutionary homology relating sites in nucleotide or protein sequences. In the presence of a high indel rate due to pyrosequencing error, accurate alignment of reads has become a highly active area of bioinformatics research. Genetic analyses based on frequency information (e.g., characterization of “mutation spectra”; Wang, Mitsuya, et al. 2007) are especially prone to misclassification of sequence variants due to alignment error, such as nucleotides that toggle on either side of a gap. This source of error is arguably the least understood, but it can have severe consequences on large-scale sequence analyses (Wong et al. 2008). We found that results based on phylogenetic analyses (e.g., number of correlated substitutions, branch length distribution of genotypes) were quite robust to uncertainty in sequence alignment, whereas results based on variant frequencies were more sensitive.
A potentially powerful approach to deal directly with alignment error would be to integrate alignments over the posterior probability distribution. Bayesian methods such as MCMC sampling make joint estimation of the alignment and phylogeny attainable (Redelings and Suchard 2005), but such approaches remain computationally costly and impractical for deep sequencing data.
Due to its current prohibitive cost, MPP experiments are rarely replicated in practice. Instead, sequencing error rates have been estimated by pyrosequencing control populations (Huse et al. 2007; Wang, Mitsuya, et al. 2007); however, this procedure is unable to quantify other sources of error such as PCR amplification variance. To address this we have performed replicate pyrosequencing experiments at two different steps of the protocol. We found that the RT and amplification steps contributed substantial error variation to frequency estimates, which is consistent with previous findings in the context of mixtures (Galli et al. 2003). Additionally, replication of the sample extraction step revealed disproportionately greater reductions in reproducibility of frequency estimates, relative to contributions from RT-PCR and second-round PCR amplifications (supplementary text S1 and fig. S3, Supplementary Material online). Such discrepancies have critical implications for the clinical use of MPP and other NGS platforms to screen for minority HIV-1 variants. We therefore propose that a minimum of two replicates from the RNA extraction step become standard procedure in the application of NGS technology to genotypic resistance testing.
The MPP experiments presented here provide an important source of validation for phylogenetic comparative methods that predict epistatic interactions between protein residues. Specifically, we observed patterns of correlated substitution within patients that was consistent with an interaction between the residue pairs Y120/Q125 and N157/S169. Our analysis of HIV-1 nef bulk sequences from 686 patients predicted 38 other interactions (fig. 1), many of which are consistent with prior observations in the experimental HIV-1 literature. First, Nef-induced activation of p21-activated kinase 2 (PAK-2) is one of the most intensively investigated functions of Nef. PAK-2 participates in the regulation of cellular processes that reshape the cytoskeleton and modulate gene transcription, apoptosis, and more. O'Neill et al. (2006) used site-directed mutagenesis to demonstrate that the subtype B consensus residues L85, H89, and F191 were collectively involved in PAK-2 activation. Our finding that these sites coevolve is consistent with this observation (fig. 1). Second, posttranslational ubiquitination of multiple lysine residues in Nef has been implicated in the Nef-induced downregulation of CD4 (Jin et al. 2008). We observed interactions among many of these lysine-enriched sites (K82, K92, K94, and K105; fig. 1), and K144, on which this putative function is conditioned, is highly conserved in our study population. Alternatively, K92 and K94 coincide with the polypurine tract (where DNA synthesis of the plus strand is selectively initiated) so that this putative lysine cluster may be confounded by selection on the nucleotide sequence. However, synonymous sites within these codons were completely conserved in the study population. Finally, several interactions described here are consistent with CTL epitope polymorphisms identified by Brumme et al. (2007) as significantly associated with HLA class I variation. The residues and corresponding HLA allele (in brackets) are as follows: D28/V33 (A11), A50/T51/H116 (B58), G83/L85 (C07), and R188/M194 (A31).
Though the results from our comparative analysis of bulk HIV-1 nef sequences are consistent with experimental data, we caution that the phylogenetic method employed here does not account for recombination. Indeed, not one of the currently available comparative methods for inferring epistatic interactions from sequence data explicitly addresses recombination, and the effect of recombination on such estimates has not yet been quantified. This may be problematic because the HIV-1 genome undergoes frequent recombination as reverse transcriptase switches between genomic templates during replication, resulting in one of the highest rates of recombination found in nature (Zhuang et al. 2002). Recombination has the effect of partitioning a sequence alignment into two or more segments with discordant phylogenies (Posada and Crandall 2002). Because our analysis takes substitutions mapped to the joint tree (i.e., a tree estimated from the entire alignment) as data, recombination will induce some degree of error almost surely. However, the net effect of recombination is to break up statistical associations between loci (i.e., restore linkage equilibrium); on average, recombination should make sequence coevolution more difficult to detect. It is therefore less likely that recombination has generated spurious associations between sites in our data. Nevertheless, incorporating recombination into phylogenetic comparative methods remains a critical objective for future research.
In summary, deep sequencing can provide valuable information about the molecular evolution of RNA virus proteins at the within-host level. Our phylogenetic analysis of deep sequencing data targeting HIV-1 nef from a selection of baseline plasma samples substantiated the existence of epistatic interactions shaping within-host evolution, which were predicted from an analysis of among-host sequence variation. More importantly, our application of NGS revealed intriguing patterns of convergent evolution in sequence within hosts. In this study, we have developed analytical methods to exploit the information-rich data generated by NGS of an RNA virus beyond the detection of minority variants. Future work will be directed on developing models to infer parameters of molecular evolution (e.g., directional selection) and population dynamics from NGS data.
SUPPLEMENTARY MATERIAL
Supplementary tables S1 and S2, figures S1–S4, and text S1–S3 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Supplementary Material
Acknowledgments
This work was supported by grants AI69432 (ACTG), AI043638 (AIEDRP), MH62512 (HNRC), MH083552 (Clade), AI077304 (Dual Infection), AI36214 (the Viral Pathogenesis Core of the UCSD Center for AIDS Research), AI047745 (Dynamics), AI57167, and AI74621 (Transmission) and AI27757 (Computational Biology Core of the University of Washington Center for AIDS Research) from the National Institutes of Health and the California HIV/AIDS Research Program RN07-SD-702. S.D.W.F. and S.L.K.P. received support from University of California San Diego Centers for AIDS Research/National Institute of Allergy and Infectious Disease developmental awards AI36214. A.F.Y.P. is supported by Canadian Institutes of Health Research Fellowships Award in HIV/AIDS Research (200802HFE). S.D.W.F. is supported in part by a Royal Society Wolfson Research Merit Award. P.R.H. is supported by a Canadian Institutes of Health Research/GlaxoSmithKline research chair in clinical virology. Z.L.B. is supported by a CIHR New Investigator Award. Our computing cluster is funded in part by National Science Foundation award 0714991.
References
- Alexander CS, Dong W, Chan K, Jahnke N, O'Shaughnessy MV, Mo T, Piaseczny MA, Montaner JS, Harrigan PR. HIV protease and reverse transcriptase variation and therapy outcome in antiretroviral-naive individuals from a large North American cohort. AIDS. 2001;15:601–607. doi: 10.1097/00002030-200103300-00009. [DOI] [PubMed] [Google Scholar]
- Baur AS, Sawai ET, Dazin P, Fantl WJ, Cheng-Mayer C, Peterlin BM. HIV-1 Nef leads to inhibition or activation of T cells depending on its intracellular localization. Immunity. 1994;1:373–384. doi: 10.1016/1074-7613(94)90068-x. [DOI] [PubMed] [Google Scholar]
- Brockman W, Alvarez P, Young S, Garber M, Giannoukos G, Lee WL, Russ C, Lander ES, Nusbaum C, Jaffe DB. Quality scores and SNP detection in sequencing-by-synthesis systems. Genome Res. 2008;18:763–770. doi: 10.1101/gr.070227.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumme ZL, Brumme CJ, Heckerman D, et al. (14 co-authors. Evidence of differential HLA class I-mediated viral evolution in functional and accessory/regulatory genes of HIV-1. PLoS Pathog. 2007;3 doi: 10.1371/journal.ppat.0030094. e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EM, Nunez R, Hope TJ. Disruption of the actin cytoskeleton can complement the ability of Nef to enhance human immunodeficiency virus type 1 infectivity. J Virol. 2004;78:5745–5755. doi: 10.1128/JVI.78.11.5745-5755.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PJ, Pleasance ED, Stephens PJ, Dicks E, Rance R, Goodhead I, Follows GA, Green AR, Futreal PA, Stratton MR. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci USA. 2008;105:13081–13086. doi: 10.1073/pnas.0801523105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson J, Kadie C, Mallal S, Heckerman D. Leveraging hierarchical population structure in discrete association studies. PLoS ONE. 2007;2 doi: 10.1371/journal.pone.0000591. e591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson JM, Brumme ZL, Rousseau CM, et al. (8 co-authors) Phylogenetic dependency networks: inferring patterns of CTL escape and codon covariation in HIV-1 Gag. PLoS Comput Biol. 2008;4 doi: 10.1371/journal.pcbi.1000225. e1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford H, Prado JG, Leslie A, et al. (16 co-authors) Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J Virol. 2007;81:8346–8351. doi: 10.1128/JVI.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmann B, Gomard E, Kiény MP, Guy B, Dreyfus F, Saimot AG, Sereni D, Sicard D, Lévy JP. Six epitopes reacting with human cytotoxic CD8+ T cells in the central region of the HIV-1 NEF protein. J Immunol. 1991;146:1560–1565. [PubMed] [Google Scholar]
- Felsenstein J. Maximum likelihood and minimum-steps methods for estimating evolutionary trees from data on discrete characters. Syst Zool. 1973;22:240–249. [Google Scholar]
- Galli RA, Sattha B, Wynhoven B, O'Shaughnessy MV, Harrigan PR. Sources and magnitude of intralaboratory variability in a sequence-based genotypic assay for human immunodeficiency virus type 1 drug resistance. J Clin Microbiol. 2003;41:2900–2907. doi: 10.1128/JCM.41.7.2900-2907.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Gouy M, Gautier C. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- Gilbert P, Novitsky V, Essex M. Covariability of selected amino acid positions for HIV type 1 subtypes C and B. AIDS Res Hum Retroviruses. 2005;21:1016–1030. doi: 10.1089/aid.2005.21.1016. [DOI] [PubMed] [Google Scholar]
- Groesch TD, Freire E. Characterization of intramolecular interactions of HIV-1 accessory protein Nef by differential scanning calorimetry. Biophys Chem. 2007;126:36–42. doi: 10.1016/j.bpc.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Hewlett IK, Geyer SJ, Hawthorne CA, Ruta M, Epstein JS. Kinetics of early HIV-1 gene expression in infected H9 cells assessed by PCR. Oncogene. 1991;6:491–493. [PubMed] [Google Scholar]
- Hoffman NG, Schiffer CA, Swanstrom R. Covariation of amino acid positions in HIV-1 protease. Virology. 2003;314:536–548. doi: 10.1016/s0042-6822(03)00484-7. [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Minkah N, Leipzig J, Wang G, Arens MQ, Tebas P, Bushman FD. DNA bar coding and pyrosequencing to identify rare HIV drug resistance mutations. Nucleic Acids Res. 2007;35 doi: 10.1093/nar/gkm435. e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg RS, Yip B, Chan KJ, Wood E, Craib KJ, O'Shaughnessy MV, Montaner JS. Rates of disease progression by baseline CD4 cell count and viral load after initiating triple-drug therapy. JAMA. 2001;286:2568–2577. doi: 10.1001/jama.286.20.2568. [DOI] [PubMed] [Google Scholar]
- Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins GM, Rambaut A, Pybus OG, Holmes EC. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J Mol Evol. 2002;54:156–165. doi: 10.1007/s00239-001-0064-3. [DOI] [PubMed] [Google Scholar]
- Jin Y-J, Cai CY, Zhang X, Burakoff SJ. Lysine 144, a ubiquitin attachment site in HIV-1 Nef, is required for Nef-mediated CD4 down-regulation. J Immunol. 2008;180:7878–7886. doi: 10.4049/jimmunol.180.12.7878. [DOI] [PubMed] [Google Scholar]
- Johnson PLF, Slatkin M. Accounting for bias from sequencing error in population genetic estimates. Mol Biol Evol. 2008;25:199–206. doi: 10.1093/molbev/msm239. [DOI] [PubMed] [Google Scholar]
- Kellam P, Boucher CA, Tijnagel JM, Larder BA. Zidovudine treatment results in the selection of human immunodeficiency virus type 1 variants whose genotypes confer increasing levels of drug resistance. J Gen Virol. 1994;75(Pt 2):341–351. doi: 10.1099/0022-1317-75-2-341. [DOI] [PubMed] [Google Scholar]
- Kirchhoff F, Easterbrook PJ, Douglas N, Troop M, Greenough TC, Weber J, Carl S, Sullivan JL, Daniels RS. Sequence variations in human immunodeficiency virus type 1 Nef are associated with different stages of disease. J Virol. 1999;73:5497–5508. doi: 10.1128/jvi.73.7.5497-5508.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber BT, Farber RM, Wolpert DH, Lapedes AS. Covariation of mutations in the V3 loop of human immunodeficiency virus type 1 envelope protein: an information theoretic analysis. Proc Natl Acad Sci USA. 1993;90:7176–7180. doi: 10.1073/pnas.90.15.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. A simple hierarchical approach to modeling distributions of substitution rates. Mol Biol Evol. 2005a;22:223–234. doi: 10.1093/molbev/msi009. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005b;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Kuiken C, Korber B, Shafer RW. HIV sequence databases. AIDS Rev. 2003;5:52–61. [PMC free article] [PubMed] [Google Scholar]
- Lemey P, Rambaut A, Pybus OG. HIV evolutionary dynamics within and among hosts. AIDS Rev. 2006;8:125–140. [PubMed] [Google Scholar]
- Lichterfeld M, Yu XG, Cohen D, et al. (14 co-authors) HIV-1 Nef is preferentially recognized by CD8 T cells in primary HIV-1 infection despite a relatively high degree of genetic diversity. AIDS. 2004;18:1383–1392. doi: 10.1097/01.aids.0000131329.51633.a3. [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, et al. (53 co-authors) Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Stenzel U, Hofreiter M. Parallel tagged sequencing on the 454 platform. Nat Protoc. 2008;3:267–278. doi: 10.1038/nprot.2007.520. [DOI] [PubMed] [Google Scholar]
- Munch J, Rajan D, Schindler M, et al. (11 co-authors) Nef-mediated enhancement of virion infectivity and stimulation of viral replication are fundamental properties of primate lentiviruses. J Virol. 2007;81:13852–13864. doi: 10.1128/JVI.00904-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol. 1994;11:715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- Myers EW, Miller W. Optimal alignments in linear space. Comput Appl Biosci. 1988;4:11–17. doi: 10.1093/bioinformatics/4.1.11. [DOI] [PubMed] [Google Scholar]
- Ngandu NK, Scheffler K, Moore P, Woodman Z, Martin D, Seoighe C. Extensive purifying selection acting on synonymous sites in HIV-1 Group M sequences. Virol J. 2008;5:160. doi: 10.1186/1743-422X-5-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickle DC, Jensen MA, Gottlieb GS, Shriner D, Learn GH, Rodrigo AG, Mullins JI. Consensus and ancestral state HIV vaccines. Science. 2003;299:1515–1518. doi: 10.1126/science.299.5612.1515c. author reply 1515–1518. [DOI] [PubMed] [Google Scholar]
- Nijhuis M, Deeks S, Boucher C. Implications of antiretroviral resistance on viral fitness. Curr Opin Infect Dis. 2001;14:23–28. doi: 10.1097/00001432-200102000-00005. [DOI] [PubMed] [Google Scholar]
- Noviello CM, Pond SLK, Lewis MJ, Richman DD, Pillai SK, Yang OO, Little SJ, Smith DM, Guatelli JC. Maintenance of Nef-mediated modulation of major histocompatibility complex class I and CD4 after sexual transmission of human immunodeficiency virus type 1. J Virol. 2007;81:4776–4786. doi: 10.1128/JVI.01793-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill E, Kuo LS, Krisko JF, Tomchick DR, Garcia JV, Foster JL. Dynamic evolution of the human immunodeficiency virus type 1 pathogenic factor, Nef. J Virol. 2006;80:1311–1320. doi: 10.1128/JVI.80.3.1311-1320.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl J. San Mateo (CA): Morgan Kaufmann Publishers; 1988. Probabilistic reasoning in intelligent systems: networks of plausible inference. 552 p. [Google Scholar]
- Peccoud J, Jacob C. Theoretical uncertainty of measurements using quantitative polymerase chain reaction. Biophys J. 1996;71:101–108. doi: 10.1016/S0006-3495(96)79205-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon A, Chao L. The rate of compensatory mutation in the DNA bacteriophage φX174. Genetics. 2005;170:989–999. doi: 10.1534/genetics.104.039438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon A, Davis BH, Chao L. The coupon collector and the suppressor mutation: estimating the number of compensatory mutations by maximum likelihood. Genetics. 2005;170:1323–1332. doi: 10.1534/genetics.104.037259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon AFY, Lewis FI, Kosakovsky Pond SL, Frost SDW. An evolutionary-network model reveals stratified interactions in the V3 loop of the HIV-1 envelope. PLoS Comput Biol. 2007;3 doi: 10.1371/journal.pcbi.0030231. e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D, Crandall KA. The effect of recombination on the accuracy of phylogeny estimation. J Mol Evol. 2002;54:396–402. doi: 10.1007/s00239-001-0034-9. [DOI] [PubMed] [Google Scholar]
- Pupko T, Pe'er I, Shamir R, Graur D. A fast algorithm for joint reconstruction of ancestral amino acid sequences. Mol Biol Evol. 2000;17:890–896. doi: 10.1093/oxfordjournals.molbev.a026369. [DOI] [PubMed] [Google Scholar]
- Redelings BD, Suchard MA. Joint Bayesian estimation of alignment and phylogeny. Syst Biol. 2005;54:401–418. doi: 10.1080/10635150590947041. [DOI] [PubMed] [Google Scholar]
- Rhee S-Y, Liu TF, Holmes SP, Shafer RW. HIV-1 subtype B protease and reverse transcriptase amino acid covariation. PLoS Comput Biol. 2007;3 doi: 10.1371/journal.pcbi.0030087. e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeth JF, Collins KL. Human immunodeficiency virus type 1 Nef: adapting to intracellular trafficking pathways. Microbiol Mol Biol Rev. 2006;70:548–563. doi: 10.1128/MMBR.00042-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozera G, Abbate I, Bruselles A, Vlassi C, D'Offizi G, Narciso P, Chillemi G, Prosperi M, Ippolito G, Capobianchi MR. Massively parallel pyrosequencing highlights minority variants in the HIV-1 env quasispecies deriving from lymphomonocyte sub-populations. Retrovirology. 2009;6:15. doi: 10.1186/1742-4690-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi R, Garbuglia AR, Di Caro A, Pulciani S, Montella F, Benedetto A. Grossly defective nef gene sequences in a human immunodeficiency virus type 1-seropositive long-term nonprogressor. J Virol. 1998;72:3646–3657. doi: 10.1128/jvi.72.5.3646-3657.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro B, Rambaut A, Pybus OG, Holmes EC. A phylogenetic method for detecting positive epistasis in gene sequences and its application to RNA virus evolution. Mol Biol Evol. 2006;23:1724–1730. doi: 10.1093/molbev/msl037. [DOI] [PubMed] [Google Scholar]
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Tavaré S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect Math Life Sci. 1986;17:57–86. [Google Scholar]
- Troyer RM, McNevin J, Liu Y, et al. (11 co-authors) Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog. 2009;5 doi: 10.1371/journal.ppat.1000365. e1000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsibris AMN, Korber B, Arnaout R, et al. (14 co-authors) Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One. 2009;4 doi: 10.1371/journal.pone.0005683. e5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsibris AMN, Russ C, Lee W, Paredes R, Arnaout R, Honan T, Cahill P, Nusbaum C, Kuritzkes DR. Detection and quantification of minority HIV-1 env V3 loop sequences by ultra-deep sequencing: preliminary results. Antivir Ther. 2006;11:S74. [Google Scholar]
- Voelkerding KV, Dames SA, Durtschi JD. Next-generation sequencing: from basic research to diagnostics. Clin Chem. 2009;55:641–658. doi: 10.1373/clinchem.2008.112789. [DOI] [PubMed] [Google Scholar]
- Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res. 2007;17:1195–1201. doi: 10.1101/gr.6468307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007;17:1186–1194. doi: 10.1101/gr.6286907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JWJ, Swanstrom R, Burch CL, Weeks KM. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–716. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich DM, Watson RA, Chao L. Perspective: Sign epistasis and genetic constraint on evolutionary trajectories. Evolution. 2005;59:1165–1174. [PubMed] [Google Scholar]
- Wong KM, Suchard MA, Huelsenbeck JP. Alignment uncertainty and genomic analysis. Science. 2008;319:473–476. doi: 10.1126/science.1151532. [DOI] [PubMed] [Google Scholar]
- Yusim K, Kesmir C, Gaschen B, Addo MM, Altfeld M, Brunak S, Chigaev A, Detours V, Korber BT. Clustering patterns of cytotoxic T-lymphocyte epitopes in human immunodeficiency virus type 1 (HIV-1) proteins reveal imprints of immune evasion on HIV-1 global variation. J Virol. 2002;76:8757–8768. doi: 10.1128/JVI.76.17.8757-8768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusim K, Korber BTM, Brander C, Haynes BF, Koup R, Moore JP, Walker BD, Watkins DI. Los Alamos (NM): Los Alamos National Laboratory, Theoretical Biology and Biophysics; 2009. HIV Molecular Immunology 2009. [Google Scholar]
- Zanotto PM, Kallas EG, de Souza RF, Holmes EC. Genealogical evidence for positive selection in the nef gene of HIV-1. Genetics. 1999;153:1077–1089. doi: 10.1093/genetics/153.3.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, Jetzt AE, Sun G, Yu H, Klarmann G, Ron Y, Preston BD, Dougherty JP. Human immunodeficiency virus type 1 recombination: rate, fidelity, and putative hot spots. J Virol. 2002;76:11273–11282. doi: 10.1128/JVI.76.22.11273-11282.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.