

Abstract
A method for studying the divergence of multiple closely related populations is described and assessed. The approach of Hey and Nielsen (2007, Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc Natl Acad Sci USA. 104:2785–2790) for fitting an isolation-with-migration model was extended to the case of multiple populations with a known phylogeny. Analysis of simulated data sets reveals the kinds of history that are accessible with a multipopulation analysis. Necessarily, processes associated with older time periods in a phylogeny are more difficult to estimate; and histories with high levels of gene flow are particularly difficult with more than two populations. However, for histories with modest levels of gene flow, or for very large data sets, it is possible to study large complex divergence problems that involve multiple closely related populations or species.
Keywords: divergence population genetics, coalescent, gene flow, speciation
Introduction
By including both population splitting and gene flow, isolation-with-migration (IM) models hold the potential to help investigators disentangle two of the main factors that determine how fast populations diverge (Latter 1973; Wakeley and Hey 1998; Nielsen and Wakeley 2001). In the absence of gene exchange, the expected level of divergence between populations or species will be proportional to the amount of time since they shared a common ancestor. But if gene exchange has been occurring since the separation of populations, it can greatly shape the dynamics of the divergence process (Dobzhansky 1951; Endler 1977; Arnold 1997; Barton 2001). In the absence of gene exchange, speciation is a byproduct of multiple independent selective fixations in the separate populations (Dobzhansky 1936; Muller 1940; Orr 1995). But when genes have been moving as divergence accumulates, speciation is a more complex processes in which natural selection drives the divergence process to a degree that overcomes the unifying effects of gene exchange (Millicent and Thoday 1961; Maynard Smith 1966; Felsenstein 1981; Rice and Hostert 1993).
Nielsen and Wakeley (2001) developed the first general procedure for estimating population size, migration and splitting time parameters for an IM model with two populations. They adapted the Bayesian Markov chain Monte Carlo (MCMC) approach devised by Wilson and Balding (1998) to estimate the posterior probability of the parameters and the genealogy, , where X is the data, G is the genealogy, and Θ is a vector of population size, migration rate and population splitting parameters. However, this approach suffers because the state space of the Markov chain simulation is very large (i.e., it includes all parameters and genealogies) and because the primary results are a list of recorded values of parameters, rather than an estimate of a posterior density function. These difficulties can be partly overcome by a method that uses direct calculation of the prior probability of the genealogy to run a Markov chain simulation over a space that includes only G and population splitting times, t. This MCMC simulation generates samples from the posterior density , and these are then used to build an estimate of the joint posterior probability of the model parameters, (Hey and Nielsen 2007).
A major limitation of current IM-based applications is the restriction to samples from two populations. Analyses of pairs of populations assume that each is the other's closest relative and that no other populations have contributed to the divergence process of the sampled populations (e.g., by exchanging genes with them). However, many, if not most, cases of divergence that are of interest to investigators involve more than two related populations or species. To get around this limitation, studies typically report analyses of multiple pairwise combinations of sampled populations and then try to make some, usually qualitative, sense of the overall pattern present in the multiple separate analyses (Hoelzel et al. 2007; Lucas et al. 2009; Pinho et al. 2008).
In this paper, the method of Hey and Nielsen (2007) is extended to the case of multiple populations that have a known phylogenetic history.
Methods
A basic two-population IM model includes three population size parameters (for the two sampled populations and the ancestral population), two migration rates (for migration in each direction between sampled populations), and a time since the ancestral population split into two. An IM model of k > 2 sampled populations will include these same types of parameters, albeit in greater abundance, as well as some kind of representation of the population phylogeny. Figure 1 shows an IM model for three sampled populations in which sampled populations 1 and 2 are more recently diverged from each other than either are with respect to population 3. In figure 1 and throughout this paper, the populations in a model with k sampled populations are numbered first from 1 through k, for the sampled populations, and then from k + 1 (for the most recent ancestral population) to 2k − 1 (for the oldest ancestral population at the root of the phylogeny).
FIG. 1.

An isolation-with-migration model for three sampled populations.
The general k-population model includes the following assumptions:
The history of the sampled populations can be represented by a bifurcating phylogenetic tree.
The population phylogeny is rooted, and the topology of the tree and the sequence of splitting events in time is known.
Each sampled population, as well as each ancestral population, is constant in size and follows Fisher–Wright population assumptions (Ewens 1979).
Gene flow may have occurred, in either or both directions, between each pair of populations that coexist over one or more time intervals.
No gene flow occurred between unsampled populations and sampled populations or their ancestors.
The individual loci sampled from these populations are assumed 1) to not have had recombination since the most recent common ancestor, 2) to be effectively unlinked from each other, and 3) to have a history that has not been shaped by natural selection (Hey and Nielsen 2004).
As in the case of a two-population IM model, multipopulation models have three main types of unknown, each of which is scaled by the rate of neutral mutations per generation, u (Hey and Nielsen 2004). Every population has a population mutation rate, θ or 4Niu (Ni is the effective population size for population i), as well as a rate of mutation-scaled migration to each population with which it coexists in time m = M/u (M is the migration rate per generation per gene copy). The root population at the base of the population tree has a population size parameter but no migration parameters because there is only one population in the model at that point in time. In addition to population size and migration parameters, we have for every internal node of the population tree a population splitting time, t = Tu, where T is the time since common ancestry in generations.
A k-population IM model will have k − 1 splitting events and a total of 2k-1 populations (i.e., k sampled populations plus k − 1 ancestral populations). The splitting events are numbered beginning with the most recent, as are the time intervals, or periods, such that period i extends between ti-1 and ti. In the case of time period 1, the interval extends from the time of sampling to t1 and in the case of time period k, the interval extends from tk-1 to infinity.
The number of distinct migration parameters can be found by considering that the model will have two migration rates for every distinct pair of populations that coexist during at least one time period. During period 1, when there are k populations, there will be migration rates. Moving down the tree to period 2, two populations merge to form an ancestral population and so, of the migration rates in period 1, only also apply over period 2. However, for period 2, we also need to consider the migration rates between the ancestral population that first appears in period 2 and the other populations that are present at that time. Therefore, for period i, where i > 1, we need to introduce migration parameters to the model. Summing the count for period 1 to the sum over periods 2 through k − 1, we find the total number of migration parameters to be
| (1) |
Unlike the count of population size parameters, which increases by only two for every additional sampled population added to the model, the number of migration parameters goes up rather quickly with increasing k. For k = 3, expression (1) yields 8; for k = 4, it is 18; and for k = 10, the IM model described here will have 162 migration parameters. There are various ways to simplify the model, with regard to migration, that will reduce this total. For example, it can be assumed that migration rates are equal in both directions between populations, which will reduce the number of parameters by a factor of two. Alternatively, one can assume that migration occurs only between sister populations so that in any time period, there are only two migration terms. Figure 2 shows the number of parameters under a full model as a function of the number of sampled populations.
FIG. 2.

Numbers of parameters of different types and total numbers, as a function of the number of sampled populations in the model.
The method of Hey and Nielsen (2007) generates Bayesian estimates of the model parameters by approximating the integration over genealogies.
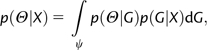 |
(2) |
where G is the genealogy (i.e., gene tree history) of the data set X, ψ is the set of possible genealogies, and Θ is the set of model parameters (excluding t). The method proceeds in two steps. First, a sample of genealogies and splitting time values is generated, using a MCMC simulation, from the posterior density, . Second, the posterior density of the set of remaining model parameters Θ is estimated using the genealogies saved in the first step,
| (3) |
Although originally described for a two-population IM model, the approach can be directly extended to k sampled populations with a given population tree. The key to the method is the integration, over the elements of Θ, to obtain the prior probability of G as given by (1):
| (4) |
Adding more populations to the model causes the number of parameters to increase considerably, but the form and tractability of this integration are unaffected by the increase (see Appendix).
Prior Distributions for Multiple Splitting Times
Heretofore, two-population IM models have generally assumed a constant prior distribution on the parameters for population sizes, migration rates and splitting times (Nielsen and Wakeley 2001; Hey and Nielsen 2004; Becquet and Przeworski 2007; Hey and Nielsen 2007). However, with k > 2 populations and a population tree, it is no longer feasible to have uniform prior distributions for individual splitting time parameters. To see this, consider two possible ways to parameterize splitting times.
First, consider a model in which the splitting times are randomly distributed over the range from zero to a maximum value specified by the investigator, . And assume that we know the order of the splitting events, such that . The joint prior distribution of intervals between splitting times will then follow a Dirichlet distribution on the interval {0,tm}, and the joint prior density of splitting times (i.e., the joint set of time values, not the intervals between them) will have a uniform density over a portion of a hypercube. Consider that for a k = 3 population model, there will be two splitting times, and the constraint of results in a uniform distribution over one half of the area of a square with sides tm in length. The general form for the marginal prior density of in a k-population model is a beta distribution on the interval {0,tm}:
| (5) |
For example, the marginal priors for a model with k = 5 and are shown in figure 3.
FIG. 3.

Marginal prior distributions for splitting times in a model with five splitting events for a shared common maximum time of 10.
An alternative approach to parameterizing splitting times is to let the individual time parameters in the model each follow a uniform distribution over a common interval and then to have the splitting times be sums of these terms. For example, if we let be the duration of period i, then the splitting time associated with period i, would be
Clearly, the prior density of is uniform under this approach, but for higher values of i, the marginal prior for ti is a convolution of two or more uniform variables and is not uniform. The first approach, in which the set of splitting times follows a uniform density, subject to the constraint that , has been used here.
Exponential Priors for Migration
Since Nielsen and Wakeley (2001), IM analyses have been based on uniform prior distributions for migration parameters, just as for population size and splitting time parameters. However, there are two reasons why investigators might wish to consider a prior distribution for migration that has the highest probability at zero. The first stems from the fact that IM analyses are generally directed at populations that exhibit at least some divergence. Given that divergence is not expected if gene flow is high, the very observation of divergence can motivate an expectation that gene flow has been low.
A second reason is that many data sets do not provide a lot of information on both splitting time and migration. When analyzed under a model with a uniform and wide prior distribution on migration, such data may reveal a posterior density that has its highest values for histories in which both the migration rates and the splitting time are at the upper bounds of their respective prior distributions. Unless the upper bound on splitting time was low, such a history is effectively that of an island model, in which the divergence is at an equilibrium due to a balance between genetic drift within populations and gene exchange between them. Although such a history might indeed be correct, IM analyses are generally conducted because of a prior expectation that an IM model is suitable and thus that the data did not arise from an equilibrium island model. One possible way to deal with this would be to specify a joint prior distribution in which histories with both high gene flow and high splitting times are unlikely. However, having the priors of the different parameters not independent of one another raises a number of complications. It is in this context, where limited data suggests an island model when wide priors are used, that it may be useful to consider a prior for migration that has lower probabilities for higher values of migration.
Both of these considerations suggest using an exponential prior distribution on migration. Unlike a uniform prior with an upper bound, , specified by the user, an exponential prior will extend from zero to infinity, with an expected value,  , that is specified by the user:
, that is specified by the user:  . Calculation of the prior probability of the genealogy is even simpler for an exponential prior than for a constant prior (see Appendix). However, with exponential priors, the posterior density will no longer be uniformly proportional to the likelihood, a key assumption for likelihood-ratio tests (Hey and Nielsen 2007).
. Calculation of the prior probability of the genealogy is even simpler for an exponential prior than for a constant prior (see Appendix). However, with exponential priors, the posterior density will no longer be uniformly proportional to the likelihood, a key assumption for likelihood-ratio tests (Hey and Nielsen 2007).
Population Migration Rates
For migration, the relevant parameterization in most population genetic contexts is the population migration rate, the product of the effective number of gene copies and the migration rate per gene per generation. Given an estimate of the posterior probability of the population size and migration rate parameters, , we can generate an estimate of the posterior density for a function of multiple elements of Θ. For the population migration rate into population i from population j, we first find the marginal density for the two parameters:
| (6) |
where is Θ with θi and mi→j removed. Letting R = 2NM = θ × m/2,
| (7) |
where mmax and θmax are the upper bounds on the uniform prior density of these parameters.
IM Analyses with Multiple Populations
A computer program was written (available from http://genfaculty.rutgers.edu/hey/home) to implement the method of Hey and Nielsen (2007) for an arbitrary number of sampled populations and a known phylogeny. The major changes, with respect to an earlier program for the analyses of two populations, involved designing and implementing algorithms for the MCMC simulation of genealogies over multiple time periods. In addition, to improve mixing of the MCMC simulation, two algorithms for updating the population splitting time terms were included. The first method is that of Rannala and Yang (2003), in which branch lengths above and below the splitting time being altered are stretched and contracted reciprocally (much like moving the center point of a rubber band with fixed end points). The second method is the original approach of Nielsen and Wakeley (2001) extended to multiple populations. This method involves the removal and resimulation of migration events when a splitting time is moved.
Simulations
To explore sampling and estimation issues when more than two populations are studied, data sets were simulated and analyzed under several models. Table 1 shows the different simulation models that were considered, each of which includes 20 independent simulated data sets generated under the infinite-sites mutation model (Kimura 1969). Simulation models 1 and 4 are baselines for many comparisons, such that other simulation models typically vary from 1 or 4 in just one or two ways. All simulation models except 2, 3, and 6 are based on a three-population phylogeny in which two of the three sampled populations (populations 1 and 2) join at the most recent splitting event, and then the ancestor of these populations, population 4, joins with sampled population 3 at the older splitting time. To simplify interpretations, all population size parameters (for sampled and ancestral populations) in each simulation were set to a single value (though this value varied among some simulation models). In all three-population simulations, the ratio of splitting time to population size (i.e., tu/4Nu) for the most recent split was set to 0.2 and to 0.5 for the older splitting event. For each simulated data set, the program was run using a uniform prior distribution for population size parameters, with an upper bound for population size that is three times the true value and an upper bound on the oldest splitting time that is two times the true value. The prior distribution for migration parameters is shown in table 1. Preliminary analyses were used to estimate the necessary duration of a run and the number of Metropolis-coupled chains required to achieve a well-mixed Markov chain, based on visual observation of trend plots for splitting time values and on autocorrelations of splitting time values over the course of the run (Hey and Nielsen 2004). Marginal posterior probability density estimates for population size and migration parameters were based on 20,000 sampled genealogies.
Table 1.
Simulation Models.
| Model No. | 1 | 2a | 3a | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11b | 12b | 13b |
| No. of sampled populationsc | 3 | 2 | 2 | 3 | 3 | 4 | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| θd | 10 | 10 | 10 | 10 | 10 | 10 | 1 | 1 | 10 | 10 | 10 | 10 | 10 |
| t1e | 2 | 2 | 5 | 2 | 2 | 2 | 0.2 | 0.2 | 2 | 2 | 2 | 2 | 2 |
| t2e | 5 | — | — | 5 | 5 | 4 | 0.5 | 0.5 | 5 | 5 | 5 | 5 | 5 |
| t3e | — | — | — | — | — | 5 | — | — | — | — | — | — | — |
| mf | — | — | — | — | — | — | — | — | m1>2 = 0.02 m3>(1,2) = 0.02 | m1>2 = 0.02 m3>(1,2) = 0.02 | m1>2 = 0.2 m3>(1,2) = 0.2 | m1>2 = 0.2 m3>(1,2) = 0.2 | m1>2 = 0.2 m3>(1,2) = 0.2 |
| No. of loci | 10 | 10 | 10 | 50 | 10 | 50 | 50 | 50 | 10 | 50 | 50 | 50 | 50 |
| No. of gene copies per locus | 5 | 5 | 5 | 5 | 20 | 5 | 5 | 15 | 5 | 5 | 5 | 5 | 5 |
| m prior termg | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0.5 | 5.0 | 0.05h |
| False positivesi | 0.050 | 0.0 | 0.0 | 0.0 | 0.0 | 0.019 | 0.012 | 0.056 | 0.025 | 0.042 | 0.075 | 0.200 | 0.0130 |
| Mean run duration (h)j | 3 | 1 | 1 | 12 | 48 | 96 | 6 | 20 | 3 | 12 | 48 | 96 | 96 |
| Figure no. | 4 | 4 | 4 | 5 | 5 | 6 | 7 | 7 | 8 | 8 | 9 | 9 | 9 |
Data for models 2 and 3 were taken from data sets simulated for model 1.
Models 11, 12, and 13 all used the same data sets.
Population trees for two-population models, (1,2):3; for three-population models, ((1,2):4,3):5; and for four-population models, ((1,2):5,(3,4):6):7.
The true value for all population size parameters in the model.
Population splitting time values.
Migration rate parameters with nonzero values, rates for all parameters not shown were set to zero.
The upper bound on the uniform prior distribution for migration parameters.
The mean of the exponential prior distribution for migration parameters.
The proportion of those migration parameters that have true values of zero and that are estimated to be significantly different from zero.
The average time required for an analysis on a single PC processor. Run duration depends on the size of the data set, the burn-in duration, the rate of sampling genealogies, and the number of Metropolis-coupled chains specified by the investigator (Hey and Nielsen 2004).
For each simulation model, the results for a given parameter are summarized by plotting the sum of the 20 marginal posterior densities for that parameter. Thus, each curve shown in the figs. 4–9 is actually the sum of 20 curves, each of which was estimated from one of the simulated data sets for that simulation model. These summed posteriors are useful for looking at overall patterns of estimator variance and bias; however, they necessarily combine two sources of variance (within data sets and among data sets) in the posterior density for a given parameter. For simulation models 1, 4, and 10, curves for all data sets are provided in Supplementary Material online.
FIG. 4.

Marginal posterior probability density estimates for simulated data for models 1, 2, and 3 (left column, center column, and right column, respectively). As described under Methods, each curve is the sum of 20 curves from the analysis of 20 independently simulated data sets. Each row of panels shows the results for a different set of parameters (top row: population size; middle row: splitting time; and bottom row: migration rates). For migration rates, only four or two of the possible migration rates are shown.
FIG. 5.

Marginal posterior probability density estimates for models 4 and 5. For migration rates, only four of the eight migration rates in a three-population IM model are shown.
FIG. 6.

Marginal posterior probability density estimates for simulations under a four-population model (model 6). For migration rates, only 4 of the 18 migration rates in a four-population IM model are shown.
FIG. 7.

Marginal posterior probability density estimates for models 7 and 8.
FIG. 8.

Marginal posterior probability density estimates for models 9 and 10.
FIG. 9.

Marginal posterior probability density estimates for models 11, 12, and 13.
Tests of Migration
One of the main questions that arise in IM analyses is whether the estimated migration rates are significantly different from zero (Nielsen and Wakeley 2001; Hey 2006; Hey and Nielsen 2007; Becquet and Przeworski 2009). Nielsen and Wakeley (2001) proposed a simple likelihood-ratio test for which the test statistic is twice the difference in the logarithms of the maximal posterior probability for a migration parameter and the posterior probability when that parameter is at zero. This likelihood-ratio statistic is tested against a mixture of distributions,  (where is equal to zero) (Nielsen and Wakeley 2001). Tests of the null hypothesis, that the true migration rate is zero, were conducted for all migration parameters in all the models. For most of the simulation models, most or all the true values for migration rates were zero, and for these, the estimated false-positive rate (i.e., the proportion of migration parameter estimates with log-likelihood-ratio statistics greater than 2.74, i.e., P < 0.05 under a distribution that is 50% zero and 50% ) was calculated. For migration rates with nonzero true values, the proportion of statistically significant values (i.e., statistical power) was also determined.
(where is equal to zero) (Nielsen and Wakeley 2001). Tests of the null hypothesis, that the true migration rate is zero, were conducted for all migration parameters in all the models. For most of the simulation models, most or all the true values for migration rates were zero, and for these, the estimated false-positive rate (i.e., the proportion of migration parameter estimates with log-likelihood-ratio statistics greater than 2.74, i.e., P < 0.05 under a distribution that is 50% zero and 50% ) was calculated. For migration rates with nonzero true values, the proportion of statistically significant values (i.e., statistical power) was also determined.
Results
One of the general questions that become possible with multipopulation models is whether subsets of the data, for just pairs of populations, provide a different picture than that obtained with the full multipopulation analysis. Figure 4 compares results for a three-population model (simulation model 1) with those for two two-population models (models 2 and 3), each of which is based upon excluding all the samples from one of the three populations in model 1. In this case, the two-population simulated data sets, even though they are sampled from larger three-population data sets, do not in fact violate the assumptions of the IM model. This is because their histories do not include gene flow and do not have any population size change. Given the uniformity of these models (i.e., all population sizes equal) and the absence of gene flow, it is not surprising that the modes of the curves for the two-population models are close to the true values. However, the same data fitted to a three-population model shows more bias in the size of population 4, more bias in the splitting time estimates, and flatter curves for t2 and the migration parameters (only four of the eight migration curves are shown for simulation model 1 in fig. 4). A full three-population model has 15 parameters, compared with just 6 for a two-population model, and for these parameter values, it appears that these sample sizes are insufficient for the full three-population model. The individual curves for each of the 20 data sets for model 1 are provided in Supplementary Material online.
Results for models with larger three-population data sets are shown in figure 5. Comparison of models 4 (50 loci, 5 gene copies per locus) and 5 (10 loci, 20 gene copies per locus) with model 1 (fig. 4; 10 loci, 5 gene copies per locus) shows that both 4 and 5 have summed curves that are much closer to the true value. For these parameter values, having more loci (model 4) rather than more gene copies per locus (model 5) seems to provide narrower posterior density estimates and less bias.
To see results with the same sampling scheme as model 4, but for a four-population model, data were simulated under a phylogenetic tree in which populations 1and 2 join at t1 = 2, populations 3 and 4 join at t2 = 4, and then populations 5 (the ancestor of 1 and 2) and 6 (the ancestor of 3 and 4) join to form population 7 at t3 = 5. Results are shown in figure 6. A four-population model has 28 parameters, and not surprisingly, we again see some of the patterns observed with smaller data sets under a three-population model (e.g., simulation model 1; fig. 4) in which some parameter estimates are biased (most notably the size of ancestral population 6) and some curves that extend all the way to the upper boundary of the parameter.
For models 1–6, all the population size parameters had true values of 10.0. To see the effect of having a much smaller value, and thus much less variation in the data set, simulations were done using θ = 1.0, with splitting times scaled accordingly. Two models were considered, one with a data set the same size as for model 4 (model 7 with 50 loci, 5 gene copies per locus) and one with a larger data set (model 8 with 50 loci and 15 gene copies per locus). Both models 7 and 8 returned summed curves for migration parameters that are very flat, suggesting that for low values of θ and these parameters values, much more data will be required to estimate migration parameters with confidence (fig. 7).
Models with Gene Flow
Migration between populations causes those populations to resemble each other and to appear more like a single population. If it is high enough, migration can obliterate any population genetic appearance of a history of population splitting. In terms of Bayesian estimation of a divergence model, data from histories with migration will yield flatter posterior densities for population size parameters, splitting times, and migration rate parameters. In each of the simulation models with migration, migration events were added to the simulations both from population 1 to 2, in period 1, and from population 3 to population 4 (i.e., the ancestor of 1 and 2), in period 2. Note that because migration rates are defined in terms of the coalescent process, these rates refer to time as it proceeds back into the past. Defined this way, movement of a gene from population 1 to 2 is equivalent to the movement of a gene in the reverse direction (population 2 to 1) as time moves chronologically, from the past to the present. Similarly, population migration in the coalescent from population 1 to 2 (i.e., 2N1M1→2), when interpreted as time moves from the past to the present, is the rate at which population 1 receives migrants from population 2.
Simulation models with migration varied for sample size, amount of migration, and the prior distribution on the migration parameters (table 1). Figure 8 shows the results for simulation models that are similar to models 1 and 4 with the addition of a small amount of migration (m = 0.02, 2NM = 0.1). In the simulations in figure 8, there were on average 0.25 migration events per locus; so, the 10 locus data sets (model 9) had on average 2.5 actual migrations, whereas the 50 locus data sets had on average 7.5 migration events. Comparisons of models 9 and 10 to 1 and 4 (figs. 4 and 5), respectively, shows more variance (i.e., flatter curves) in the models with migration. The summed posterior densities for migration show either no peak or a very slight peak near the true migration rate for those nonzero migration parameters. However, these curves do not reveal the variation among the different replicates, some of which showed clear nonzero peaks for migration parameters and many of which that did not (see Supplementary Material online). Table 2 shows the estimated statistical power of detecting nonzero migration using the test of Nielsen and Wakeley (2001); for m1>2, migration was detected 10% of the time in model 9 (i.e., 2 of the 20 simulated data sets) and 25% of the time in model 10. For m4>3, estimated statistical power was zero in both models 9 and 10. Clearly, for these parameter values and data set sizes, low levels of migration are difficult to detect, particularly for migration events prior to the most recent population splitting.
Table 2.
False-Positive Rates and Statistical Power for Detecting Migration.
| Simulation Model No. | 9 | 10 | 11 | 12 | 13 |
| No. of loci | 10 | 50 | 50 | 50 | 50 |
| m prior term | 1 | 1 | 0.5 | 5.0 | 0.05a |
| False positivesb | 0.025 | 0.042 | 0.075 | 0.200 | 0.0130 |
| m1 > 2 powerc | 0.1 | 0.25 | 0.95 | 0.667 | 0.600 |
| m2 > (1,2) powerc | 0.0 | 0.0 | 0.70 | 1.0 | 0.050 |
The mean of the exponential prior distribution for migration parameters.
The proportion of migration rates having true values of zero and for which the likelihood-ratio statistic did indicate that the estimated migration rate was significantly different from zero.
The proportion of migration rates having true values greater than zero and for which the likelihood-ratio statistic was statistically significant so that a rate of zero is rejected.
Results for moderately high levels of gene flow (m = 0.2, 2NM = 1.0) are shown in figure 9 for data sets with 50 loci. This figure compares results for a common set of simulations for three different types of prior distributions for migration parameters, including a uniform distribution with a low upper bound (model 11 with an upper bound on migration of ), a uniform distribution with a high upper bound (model 12, ), and an exponential distribution with a low mean value (model 13, = 0.05). For model 11, the statistical power for detecting recent gene flow was quite high (0.95; table 2) and fairly high for gene flow that predated the most recent population split (0.7).
However, the results when using a high prior on migration are very different from those found with a low prior. For model 12, we find that the curves for t2 and for migration rates in period 2 fall virtually at the upper bound for the respective parameter (i.e., barely visible at the right side of the middle and lower chart in the middle column of fig. 9). We also find a very narrow peak for t1 that is far to the left of its true value and broader distributions for ancestral population sizes. These analyses also returned a strikingly high false-positive rate of 20% (table 1) for those migration parameters that had a true value of zero. Much of this false-positive rate was due to statistically significant estimates for m4>3 (the true value of which was zero), for which the curve was always abutting the upper bound (fig. 9). What appears to be happening for these data sets is that histories with large splitting times and high migration rates are found to have high posterior probability. In effect, the method has returned estimates that encroach on an island model in which population splitting was long ago and divergence is the result of an equilibrium between genetic drift and migration. This tendency for the analyses to indicate (wrongly) a history with ancient splitting time and high gene flow seems to be a result of three things: having limited data for a model with many migration parameters, such that the data does not dominate the prior distribution; having a high amount of gene flow in the true history; and having high values for the upper bounds on migration and splitting time prior distributions.
The contrast between simulation models 11 and 12 shows how estimated posterior densities can be very sensitive to the prior distribution for migration parameters. If data sets are very large, or migration rates are low, this is less likely to be a problem, but going into an analysis, an investigator may not know whether their data is sufficient for the history they are trying to discern. It is partly for this reason that the use of exponential priors for migration rates were explored (simulation model 13). An exponential prior has the advantage that it ranges from zero to infinity and so an investigator can use it without explicitly ruling out high migration rates. In this case, we find that the summed curves for population size and splitting time are similar to that for a low prior (model 11); however, the migration curves are noticeably shifted to the left, as expected if the prior distribution is having a large effect on the posterior distribution. If we apply the test of Nielsen and Wakeley (2001) to the case with exponential priors, the statistical power for recent gene flow is 0.6, but for gene flow older than the most recent population split, it is only 0.05 (table 2). It must be noted that an exponential prior necessarily shifts all migration curves closer to zero and will make this test even more conservative than it is under a uniform prior.
Likelihood-Ratio Tests of Migration Parameters
With an estimate of the marginal posterior density for a migration rate m or a population migration rate 2NM, obtained using a uniform prior on m, it is straightforward to calculate a likelihood-ratio statistic and to assess the statistical significance with a mixed χ2 distribution (Nielsen and Wakeley 2001). This test was examined in several ways using the analyses on simulated data, for both m and 2NM.
To assess the rate at which the test is significant, when the true rate is zero (i.e., false positives), tests were conducted for all the migration parameter estimates in all the data sets listed in table 1, with the exceptions of models 12 and 13 and those migration parameters in models 9–11 with nonzero true values. Variation in the false-positive rate among models (table 1) suggests that false positives are more likely when data sets are small (either in terms of number of gene copies or amount of variation in the data). Thus, we see that models 1 and 6, which have small samples for the number of sampled populations, and models 7 and 8, which have low polymorphism, return nonzero false-positive rates in contrast to models 2, 3, 4, and 5. Simulation models with true nonzero migration also have higher rates of false positives (i.e., models 9, 10, and 11). Part of this may be due to insufficiency of data, as more data are generally required for histories with gene flow. Not shown in table 1 but apparent in a summary of all the migration rate tests (Supplementary Material online) is that migration rates between closely related populations are much more likely to be falsely identified as significantly different from zero than are migration rates between distantly related populations.
To assess the power of the likelihood-ratio test, it was also applied to all those migration parameters with nonzero true values in models 9–11, and these results are shown in table 2. For low migration rates (models 7 and 8), statistical power is estimated to be low (gene flow in period 1) or zero (gene flow in period 2). For high gene flow, when a low prior is used on migration rates, statistical power is fairly high in both time periods (model 11) , whereas for model 13 with an exponential prior, statistical power was reduced, especially in period 2.
To see if the distribution of the likelihood-ratio test statistic actually follows that suggested by Nielsen and Wakeley, the cumulative distribution of observed values was plotted alongside that for the distribution in figure 10. As suggested by its authors, the test is fairly conservative; however, the observed and expected cumulative distributions are similar for higher values of the test statistic.
FIG. 10.

Cumulative distributions of the log-likelihood-ratio (LLR) statistic calculated for estimated posterior densities for the migration rate (m) and the population migration rate (i.e., 2Nm) for all migration parameters with nonzero true values in models 1 through 11. Also plotted is the distribution.
Discussion
Joining Population Genetics and Phylogenetics
The discernment of recent evolutionary history for closely related populations or species presents challenges that fall in the area of population genetics (Gillespie and Langley 1979; Pamilo and Nei 1988; Hudson 1992; Nielsen 1998; Rannala and Yang 2003; Degnan and Salter 2005), and evolutionary biologists have long recognized that investigators cannot address the phylogenetic history of closely related species without simultaneously considering their population genetic history (Gillespie and Langley 1979; Tajima 1983; Felsenstein 1988; Hey 1994; Maddison 1997; Avise 1998; Arbogast et al. 2002; Knowles and Maddison 2002; Maddison and Knowles 2006). The extension of IM analyses to cases with multiple populations, for a known phylogenetic tree, is one step toward a general statistical framework that merges phylogenetics and populations genetics.
Covariation of Migration and Splitting Time when Data is Limiting
Flat posteriors for migration parameters, which are commonly found in IM analyses, also present the difficulty that point estimates based on these posteriors will vary with the prior distribution that is used. A marked example of this is shown in figure 9. Traditionally uniform priors (so-called “uninformative priors”) have been used in IM analyses (Nielsen and Wakeley 2001), both for simplicity and so that the posterior probability will be proportional to the likelihood. The latter point means that likelihood-ratio tests of nested IM models can be done using the joint posterior density estimate (Hey and Nielsen 2007). However, a uniform prior is not literally uninformative if in fact the posterior density estimates change with the upper bound that is used. This is the case for many migration parameters in the analyses described here. In effect, what has been done here, by relying mostly on analyses that use an upper bound on m of 1.0, is to impose an informative prior in which we assume that migration has not had a high value. The use of exponential priors for migration can help in some circumstances, as shown in figure 9. However, an investigator using an exponential prior must still specify a mean for that distribution, and setting this value too high can lead to results similar to what is found for a high uniform prior for migration (e.g., like those for model 12 in fig. 9).
Contrasting Two-Population Models with Multipopulation Models
In some ways, multipopulation IM models are not much more complex than two-population models. The calculation of the prior probability of the genealogy turns out to be accessible under a wide class of models (wider than those considered here—see Appendix), and the Markov chain simulation over genealogies, while requiring more complex updating algorithms, does not change much conceptually when there are more than two populations. Nevertheless, the analysis of a data set under a highly parameterized IM model with multiple populations and a phylogeny is a complex endeavor. In the first place, there are the many practical issues that arise before analyses can even begin, including questions about the quantity of data needed, the shape of prior distributions, how to ensure sufficient Markov chain mixing, and dealing with long run times (average run times for the simulation models are given in table 1). However, we may expect that in the future, some of these questions will tend to go away as data acquisition and computing time continue to become less expensive.
A more interesting set of complexities arises when an investigator must make sense of a posterior density estimate in 15, 28, or 190 dimensions (3, 4, and 10 population models, respectively). Even if data and computing power are not limiting, large models can seem overwhelming, and investigators will be inclined to work with simple smaller models that are more easily interpreted. One way to proceed is to consider both a full k-population model and multiple small models each with data only from a subset with j of the sampled populations (where 2 ≤ j < k). The analyses on the reduced data sets will proceed much more quickly, and the comparison of results for small and large data sets can help make clear those aspects of the overall history that require the full analysis in order to be revealed. An example of this is provided in a four-population study of chimpanzee divergence (Hey, 2010). In cases where gene flow levels have been low, population splitting times are not very close to one another in time, and population sizes have not greatly changed over time periods, the assumptions of an IM analysis on a reduced set of populations will not be greatly violated, and the results for a full multipopulation analysis should be predictable on the basis of results on reduced samples. Thus, for example, in comparison of simulation models 1, 2, and 3, the use of a two-population model for models 2 and 3 (each based on subsets of the data in model 1) was not in violation of the general IM model assumptions.
The method presented here is intended for the analysis of recent divergence. The implicit assumption of recent divergence applies not only for the most closely related populations but for all the populations included in the analysis. Necessarily, genealogies will extend deeper (i.e., will include coalescent events and migration events from older times) the more populations that are included in an IM model. Every additional population entails an additional splitting event and additional sampled and ancestral population size parameters, as well as many additional migration parameters over all time intervals in the model. With more populations and parameters comes the need not only for more data to inform on recent processes involving the newly included populations but also an additional need for data that can inform on processes that occurred further back in time. Increasing the number of gene copies sampled per locus will not be of much help in accessing older time periods because the timing of coalescent events in populations are heavily weighted toward the recent (Felsenstein 2006). However, increasing the number of loci can provide access to older time periods, though the numbers of loci required may be very large (e.g., hundreds or thousands) for older histories and larger models.
Supplementary Material
Supplementary table S1 and supplementary data are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Supplementary Material
Acknowledgments
This work was supported by the United States National Oceanic and Atmospheric Administration Northwest Fisheries Science Center and by the National Institutes of Health (GM078204). Thanks very much to Rasmus Nielsen, Sang Chul Choi, and Yong Wang for discussion and helpful suggestions.
Appendix
Hey and Nielsen (2007) showed how the prior probability of a genealogy, f(G), could be calculated for a two-population IM model in which the population size and migration rate parameters in each time period are constant. In this appendix, the approach used in the 2007 paper is extended to a class of models that include multiple populations and time periods. As described below, this class of models is actually much broader than the bifurcating phylogenetic models with gene flow that are examined in this paper.
With more than two populations, and with multiple time periods, a general model can be described in terms of both its phylogenetic and population genetic components. The phylogenetic part of the model, P, which is assumed to be fixed, specifies the history of the populations from which genetic data are sampled, including 1) the number of time periods, 2) the durations and sequence in time of time periods, 3) the number of populations in each time period, and 4) and the ancestor/descendant identities of all populations in each time period (i.e., which population in period z is a descendant of which population in period z + 1). Importantly, P need not be bifurcating, nor indeed are there any constraints on the number of populations in any time period, so long as these conditions apply. The population genetic part of the model, Ω, specifies the kinds of event (including coalescent events and possibly events of other types) that can happen in a genealogy under the model, and it also specifies the prior probability distributions of all the rates at which each of these types of events can occur. These rates are the parameters of the population genetic model.
The purpose of this appendix is to develop an expression for the probability of a genealogy, given P, by solving . We call this the prior probability of G. But unlike the prior probabilities for the parameters in Ω, the prior probability of G is not directly specified by the investigator, but rather it is induced on G by the priors specified for Ω. In what follows, it is assumed that the prior probability distributions for each of the parameters in Ω are independent of each other so that the joint prior,, is just the product of the prior distributions for each element of Ω.
The key to the calculation is the Markov property of the genealogical process, by which the time interval between any two successive events in a genealogy follows a simple waiting time distribution that is independent of the time duration between previous events. Because the Markov property holds for models in which parameters and numbers of populations change over time periods, but are constant within time periods, it is possible to describe a general representation for the probability of a genealogy for a fairly broad class of models.
Let there be Y possible types of events in the genealogy. These must include coalescent events and for IM models will also include migration events. Other types of events that would fit this framework are those for which their occurrence does not disrupt the Markov property. All event types must be defined in such a way that for every event the number of edges in G and their population locations immediately after an event be completely determined by 1) that event and 2) the numbers and locations of edges immediately before the event. Recombination, in which a gene copy is split into two ancestral gene copies (Hudson 1983), is a type of event that, although not included in this paper, could be included in this general framework.
Let πy,z,j be the rate parameter for event type y during period z, in population j. If the event involves multiple populations, as is the case with migration, then a pair of populations can be indicated in the population subscript, e.g., πy,z,(jk). If events of type y are coalescent events in population j during period z, then , where Nz,j is the effective size of population j in period z. If by y we mean migration events, then for population j to k, .
At any point in time in the genealogy, there will be some number of gene copies in each population. These counts of edge numbers determine xy,z,j,i, the total number of events that are possible in population j of type y during time interval i in period z. For example, if there are n gene copies in population j during interval i in period 1 and events of type 1 are coalescent events, then x1,1,j,i = n(n − 1)/2. Similarly, if type 2 events are migrations from population j to k, then x2,1,j,i = n.
The total rate of all events during interval i in period z is just a sum of products of x values and rates. For example, continuing the example of a model with coalescent parameters identified as type 1 and migration parameters identified as type 2, the total rate would be
| (A1) |
However, this kind of notation, that separates parameters by the number of populations to which they apply, is unnecessarily cumbersome for the present purpose. Hereafter, a single subscript j is used to indicate the population, or populations, to which a parameter applies. Then the total rate to the next event during interval i of time period z is
| (A2) |
The probability that this interval has duration τz,i follows an exponential density, .
The probability that time interval i in period z has a particular duration and that it ends in a specific event includes three components: 1) the probability that this interval has duration τz,i; 2) that it ends in an event of type y; and 3) that a particular one of the possible xy,z,j,i events of type y in population j occurred, assuming that all are equally likely. Because of cancellation of terms, this probability takes a fairly simple form:
| (A3.a) |
If interval i is the time interval immediately prior to the end of time period z, then we need to find the probability that none of the possible events occur in an interval of that duration. Letting refer to the case when none of the possible events occur and τz,i refer in this case to the duration of this last interval in period z,
| (A3.b) |
Together, (A3.a) and (A3.b) can be used to cover all the time intervals and events in period z. Under the Markov property, the total probability for the portion of the genealogy in a particular time period is the product of the probability for each time interval in that period (i.e., a product of (A3) terms). If we let cy,z,j be the total number of time intervals that end in an event of type y in population j during period z, then the total probability of the genealogy over all the intervals in this time period, including the interval preceding the end of the time period, is
| (A4) |
For a model with multiple time periods, such as a phylogeny P with multiple population splitting events, the total probability is the product of terms like (A4) over time periods. With Ω being the vector that includes all parameters (i.e., for all y, z, and j),
| (A5) |
where j and i are understood to index the populations and time intervals, respectively, in each time period (i.e., for each value of z). Substituting the right side of (A2) for rz,i and rearranging,
| (A6) |
where .
In (A6), the range of j is understood to depend on the value of z. But for some models, we may not wish to identify populations by time periods but rather to number all populations distinctly and keep track separately of those time periods in which a population occurs. If we do this, letting j range over populations (and pairs of populations as needed for migration parameters), then (A6) can be rewritten as
| (A7) |
where and .
For multiple unlinked loci, all sampled from the same model {P,Ω}, let g index the set G that contains the individual genealogies for each of the loci. Then,
 |
(A8) |
where and .
From (A6) and (A8), we see that the probability of a genealogy, or a set of genealogies drawn from the model, is just a product of terms, one for each parameter in the model. Expression (A8) also reveals that all the information in the genealogies that is relevant for likelihood or Bayesian calculations is contained in the C and H terms.
If the elements of Ω have independent uniform prior distributions, such that the prior probability for parameter πy,j is given by , where is the upper bound on the prior distribution, then the prior probability of the genealogy is
 |
(A9) |
For a parameter that has an exponential prior with an expected value of 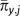 , the integration on the right side of the product in (A9) is even simpler:
, the integration on the right side of the product in (A9) is even simpler:
 |
(A10) |
References
- Arbogast BS, Edwards SV, Wakeley J, Beerli P, Slowinski JB. Estimating divergence times from molecular data on phylogenetic and population genetic timescales. Annu Rev Ecol Syst. 2002;33:707–740. [Google Scholar]
- Arnold ML. Natural hybridization and evolution. New York: Oxford University Press; 1997. [Google Scholar]
- Avise JC. The history and purview of phylogeography: a personal reflection. Mol Ecol. 1998;7:371–379. [Google Scholar]
- Barton NH. The role of hybridization in evolution. Mol Ecol. 2001;10:551–568. doi: 10.1046/j.1365-294x.2001.01216.x. [DOI] [PubMed] [Google Scholar]
- Becquet C, Przeworski M. A new approach to estimate parameters of speciation models with application to apes. Genome Res. 2007;17:1505–1519. doi: 10.1101/gr.6409707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becquet C, Przeworski M. Learning about modes of speciation by computational approaches. Evolution. 2009;63:2547–2562. doi: 10.1111/j.1558-5646.2009.00662.x. [DOI] [PubMed] [Google Scholar]
- Degnan JH, Salter LA. Gene tree distributions under the coalescent process. Evolution. 2005;59:24–37. [PubMed] [Google Scholar]
- Dobzhansky T. Studies of hybrid sterility. II. Localization of sterility factors in Drosophila pseudoobscura hybrids. Genetics. 1936;21:113–135. doi: 10.1093/genetics/21.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky T. Genetics and the origins of species. New York: Columbia University Press; 1951. [Google Scholar]
- Endler JA. Geographic variation, speciation, and clines. Princeton (NJ): Princeton University Press; 1977. [PubMed] [Google Scholar]
- Ewens WJ. Mathematical population genetics. New York: Springer Verlag; 1979. [Google Scholar]
- Felsenstein J. Skepticism towards Santa Rosalia, or why are there so few kinds of animals. Evolution. 1981;35:124–138. doi: 10.1111/j.1558-5646.1981.tb04864.x. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Phylogenies from molecular sequences: inference and reliability. Annu Rev Genet. 1988;22:521–565. doi: 10.1146/annurev.ge.22.120188.002513. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Accuracy of coalescent likelihood estimates: do we need more sites, more sequences, or more loci? Mol Biol Evol. 2006;23:691–700. doi: 10.1093/molbev/msj079. [DOI] [PubMed] [Google Scholar]
- Gillespie JH, Langley CH. Are evolutionary rates really variable? J Mol Evol. 1979;13:27–34. doi: 10.1007/BF01732751. [DOI] [PubMed] [Google Scholar]
- Hey J. Bridging phylogenetics and population genetics with gene tree models. In: Schierwater B, Streit B, Wagner G, DeSalle R, editors. Molecular approaches to ecology and evolution. Basel (Switzerland): Birkhäuser-Verlag; 1994. pp. 435–449. [Google Scholar]
- Hey J. Recent advances in assessing gene flow between diverging populations and species. Curr Opin Genet Dev. 2006;16:592–596. doi: 10.1016/j.gde.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Hey J. Isolation with migration models for more than two populations. Mol Biol Evol. 2010 doi: 10.1093/molbev/msp296. Advance Access published December 2, 2009, doi:10.1093/molbev/msp296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167:747–760. doi: 10.1534/genetics.103.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc Natl Acad Sci USA. 2007;104:2785–2790. doi: 10.1073/pnas.0611164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelzel AR, Hey J, Dahlheim ME, Nicholson C, Burkanov V, Black N. Evolution of population structure in a highly social top predator, the killer whale. Mol Biol Evol. 2007;24:1407–1415. doi: 10.1093/molbev/msm063. [DOI] [PubMed] [Google Scholar]
- Hudson RR. Properties of a neutral allele model with intragenic recombination. Theor Popul Biol. 1983;23:183–201. doi: 10.1016/0040-5809(83)90013-8. [DOI] [PubMed] [Google Scholar]
- Hudson RR. Gene trees, species trees and the segregation of ancestral alleles. Genetics. 1992;131:509–513. doi: 10.1093/genetics/131.2.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics. 1969;61:893–903. doi: 10.1093/genetics/61.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles LL, Maddison WP. Statistical phylogeography. Mol Ecol. 2002;11:2623–2635. doi: 10.1046/j.1365-294x.2002.01637.x. [DOI] [PubMed] [Google Scholar]
- Latter BDH. The island model of population differentiation: a general solution. Genetics. 1973;73:147–157. doi: 10.1093/genetics/73.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas L, Gompert Z, Ott J, Nice C. Geographic and genetic isolation in spring-associated Eurycea salamanders endemic to the Edwards Plateau region of Texas. Conserv Genet. 2009;10:1309–1319. [Google Scholar]
- Maddison W, Knowles L. Inferring phylogeny despite incomplete lineage sorting. Syst Biol. 2006;55:21–30. doi: 10.1080/10635150500354928. [DOI] [PubMed] [Google Scholar]
- Maddison WP. Gene trees in species trees. Syst Biol. 1997;46:523–536. [Google Scholar]
- Maynard Smith J. Sympatric speciation. Am Nat. 1966;100:637–650. [Google Scholar]
- Millicent E, Thoday JM. Effects of disruptive selection. Heredity. 1961;16:199–217. [Google Scholar]
- Muller HJ. Bearings of the Drosophila work on systematics. In: Huxley J, editor. The new systematics. Oxford: Clarendon Press; 1940. pp. 185–268. [Google Scholar]
- Nielsen R. Maximum likelihood estimation of population divergence times and population phylogenies under the infinite sites model. Theor Popul Biol. 1998;53:143–151. doi: 10.1006/tpbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Wakeley J. Distinguishing migration from isolation. A Markov chain Monte Carlo approach. Genetics. 2001;158:885–896. doi: 10.1093/genetics/158.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. The population genetics of speciation: the evolution of hybrid incompatibilities. Genetics. 1995;139:1805–1813. doi: 10.1093/genetics/139.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamilo P, Nei M. Relationships between gene trees and species trees. Mol Biol Evol. 1988;5:568–583. doi: 10.1093/oxfordjournals.molbev.a040517. [DOI] [PubMed] [Google Scholar]
- Pinho C, Harris DJ, Ferrand N. Non-equilibrium estimates of gene flow inferred from nuclear genealogies suggest that Iberian and North African wall lizards (Podarcis spp.) are an assemblage of incipient species. BMC Evol Biol. 2008;8:63. doi: 10.1186/1471-2148-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannala B, Yang Z. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics. 2003;164:1645–1656. doi: 10.1093/genetics/164.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WR, Hostert EF. Laboratory experiments on speciation: what have we learned in 40 years? Evolution. 1993;47:1637–1653. doi: 10.1111/j.1558-5646.1993.tb01257.x. [DOI] [PubMed] [Google Scholar]
- Tajima F. Evolutionary relationships of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley J, Hey J. Testing speciation models with DNA sequence data. In: DeSalle R, Schierwater B, editors. Molecular approaches to ecology and evolution. Basel (Switzerland): Birkhäuser Verlag; 1998. pp. 157–175. [Google Scholar]
- Wilson IJ, Balding DJ. Genealogical inference from microsatellite data. Genetics. 1998;150:499–510. doi: 10.1093/genetics/150.1.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.