

Abstract
Over time, natural selection molds every gene into a unique mosaic of sites evolving rapidly or resisting change—an “evolutionary fingerprint” of the gene. Aspects of this evolutionary fingerprint, such as the site-specific ratio of nonsynonymous to synonymous substitution rates (dN/dS), are commonly used to identify genetic features of potential biological interest; however, no framework exists for comparing evolutionary fingerprints between genes. We hypothesize that protein-coding genes with similar protein structure and/or function tend to have similar evolutionary fingerprints and that comparing evolutionary fingerprints can be useful for discovering similarities between genes in a way that is analogous to, but independent of, discovery of similarity via sequence-based comparison tools such as Blast.
To test this hypothesis, we develop a novel model of coding sequence evolution that uses a general bivariate discrete parameterization of the evolutionary rates. We show that this approach provides a better fit to the data using a smaller number of parameters than existing models. Next, we use the model to represent evolutionary fingerprints as probability distributions and present a methodology for comparing these distributions in a way that is robust against variations in data set size and divergence. Finally, using sequences of three rapidly evolving RNA viruses (HIV-1, hepatitis C virus, and influenza A virus), we demonstrate that genes within the same functional group tend to have similar evolutionary fingerprints. Our framework provides a sound statistical foundation for efficient inference and comparison of evolutionary rate patterns in arbitrary collections of gene alignments, clustering homologous and nonhomologous genes, and investigation of biological and functional correlates of evolutionary rates.
Keywords: adaptive evolution, codon models, evolutionary distance, machine classification
Introduction
Bioinformatic algorithms based on genetic or structural sequence similarity, for example, “Blast” (Altschul et al. 1997), are routinely used to identify paralogous and orthologous sequences for comparative analyses or to classify sequences of unknown origin or function. However, sequence and evolutionary similarities are not equivalent as the former makes a statement about extant sequences and the latter about their evolutionary history. For instance, envelope glycoprotein sequences of HIV type 1 experience vastly different selective pressures in early and late stages of infection within a single patient (Shankarappa et al. 1999). Conversely, highly divergent sequences can evolve under very similar evolutionary contexts, as is the case for mammalian proteins involved in sperm–egg interactions (Swanson et al. 2003). This suggests that an information layer is left untapped by straightforward sequence comparisons that may be revealed by an “evolutionary Blast”—that is, an analysis that infers and compares signatures of mutational, selective, and other evolutionary processes acting on both homologous and nonhomologous genes.
A plethora of bioinformatics techniques aimed at inferring various components of the evolutionary process have been developed in the past two decades, but little attention has been devoted to the explicit comparison of the evolutionary processes acting on different sets of genes. We hypothesize that such comparisons can be useful in that similarities in how sequences evolve may reflect homology of protein structure and/or function, and we propose a qualitatively new statistical approach to perform such comparisons rapidly and efficiently.
We formulate the concept of an evolutionary fingerprint based on the probability distribution of site-to-site of synonymous (α) and nonsynonymous (β) substitution rates in an alignment. Methods that exploit the ω = β/α ratio as an indicator of positive or negative selection have become a mainstay of modern evolutionary analyses (Swanson 2003). For example, large-scale molecular studies of adaptive evolution in primate lineages (Nielsen et al. 2005, Arbiza2006uq) have used estimates of ω on a gene-by-gene basis to identify those functional groups of genes that are enriched for positively selected constituents. Although clearly logical, this approach discards a great deal of information. On the one hand, a positively selected gene may be rapidly accumulating amino acid replacements along its entire length, that is, have a consistently high ω for a large proportion of sites, as is the case with the APOBEC gene family (Sawyer et al. 2004). On the other hand, a positively selected gene could be mostly conserved (ω < 1) except for a relatively minor proportion of rapidly evolving sites (ω≫1), such as interaction domains that are involved in an evolutionary arms race, for example, the sperm lysin gene (Yang and Swanson 2002). An accurate estimate of the strength and extent of selective forces acting on a gene, represented by a distribution of α and β, is undoubtedly more informative than a simple binary test for any kind of positive selection. Now that large samples of functionally related genes are becoming available from multiple taxonomic families (for instance, genes encoding surface proteins of different viruses), similarities in evolutionary rates between groups of organisms may correlate with hypothesized or novel functional and evolutionary requirements. Unusual evolutionary patterns in a gene when compared with taxonomically or functionally related genes may point to a functional or environmental shift.
To mirror biological reality, an evolutionary model would need to endow every site with its own set of rates (α, β), which will vary over time with changes in the environmental and genetic contexts. However, such model would be statistically unjustifiable as the number of its parameters would match or exceed the sample size. Therefore, the underlying continuous distribution of rates must be approximated in a way which is both accurate and parsimonious in the number of model parameters. This is commonly done by assuming that rates are constant over time (within the phylogeny describing a homologous alignment) and by aggregating sites into bins of a discrete distribution. Published phylogenetic methods assume independent univariate discrete distributions for β (e.g., a discretized gamma distribution) and α, where the latter is often assumed to be constant, that is, a probability point mass at one. We show that as the information content of a sequence alignment grows to support an increasingly finer resolution of rate classes, the use of independent univariate distributions becomes increasingly inefficient. This shortcoming can be remedied with a general bivariate (GB) discrete distribution (GDD(α,β)) that restricts the number of rate classes to that which is supported by the data. We propose that this distribution makes an informative evolutionary fingerprint, capable not only of distinguishing between genes with similar mean ω values but also of revealing similarities between nonhomologous genes due to similarities of protein function and evolutionary environments.
The degree to which GDD(α,β) can be resolved (i.e., the number of distinct rate classes) is a function of the amount of information contained in the data set, which in turn is primarily affected by the number of sequences and genetic divergence. For this reason, using point estimates (e.g., maximum likelihood [ML]) of GDD(α,β) as the evolutionary fingerprint of selection could potentially lead to divergent fingerprints for different samples of the same gene. We address this issue by approximating the posterior distribution of GDD(α,β) in the neighborhood of our point estimates and show that these distributions exhibit a strong tendency to overlap for different-sized alignments of the same gene.
Having established the notion of evolutionary fingerprints, we proceed to define a distance metric—the evolutionary selection distance (ESD)—which can be used to quantify the similarity of two evolutionary fingerprints. Importantly, because the distance is measured between evolutionary fingerprints rather than alignments, the genes being compared need not be homologous. ESD can be applied to fingerprint point estimates to yield a point estimate of ESD; alternatively, by calculating the ESD for a set of fingerprints sampled from the posterior fingerprint distribution, we obtain a posterior distribution for the ESD. Relying on the posterior ESD distribution rather than the ESD point estimate is a way of taking the variance of the estimate into account and affords us robustness against variations in the information content of the data set. Using simulation, we validate this methodology by demonstrating that it succeeds in clustering alignments together that were generated using similar evolutionary parameters (but different data set size and divergence) while distinguishing between alignments that were generated using different evolutionary parameters.
Lastly, we investigate the extent to which similarity of evolutionary processes, as measured by ESD, reflect similarity in biological function in several viral species. We find that genes within the same functional group tend to exhibit similar evolutionary patterns in HIV-1 virus, hepatitis C virus (HCV), and influenza A virus (IAV).
Materials and Methods
Our comparative metric is based on distributions of site-specific synonymous and nonsynonymous substitution rates, where for tractability we make the usual assumption that codon sites are evolving independently of one another. We begin by defining a modeling framework for inferring these rates from coding sequence alignments.
Basic Model of Codon Substitution
We use a generalized form of the Muse–Gaut model of codon substitution (Muse and Gaut 1994; Kosakovsky Pond and Muse 2005), which represents a continuous time-reversible Markov substitution process stipulating the probability of a substitution from codon x to codon y occurring within a finite time interval t ≥ 0. This model is defined by an instantaneous rate matrix Q whose entries are given by
 |
where πy(n)n refers to the frequency of the nucleotide found in the nth position of codon y (n = 1,2,3), tabulated over all nth positions in the alignment. The nine independent frequency parameters are customarily estimated by observed counts in the alignment. α and β denote the synonymous and nonsynonymous substitution rate, respectively. θ(x,y) represents nucleotide substitution biases (e.g., transition–transversion bias), which are symmetrically constrained so that θ(x,y) = θ(y,x) for all x,y ∈ {A,C,G,T}. Note that because the mean nucleotide substitution rate is confounded with evolutionary time, one of the bias parameters cannot be estimated and is arbitrarily set to one.
Under this model, the stationary frequency of a nonstop codon composed of nucleotides ijk is given by the product of its constituent nucleotide frequencies, normalized by the sum over all nonstop codons:
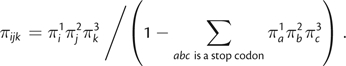 |
The probability of a substitution from codon x to codon y occurring within a time interval t ≥ 0 is given by the x,yth element of the transition matrix T(t), which is obtained by exponentiating the instantaneous rate matrix, that is, T(t) = exp(Qt).
A GB Model of Site-to-Site Variation in Substitution Rates
We make the common simplifying assumption that both rates remain constant at a given site throughout the phylogeny (Nielsen and Yang 1998; Yang et al. 2000). This assumption can be justified biologically if the sequences are sampled from a reasonably uniform selective environment, for example, from viruses that evolve in the same species. In the cases when selective pressures vary over time, the rates estimated under this assumption can be thought of as mean values over time.
To model variable substitution rates across S codon sites, we assume that the pair of rates (αs,βs) at codon s is a random variable drawn from the GB discrete distribution with D≪S categories, which we denote as GDD(α,β). This probability distribution function can be fully defined by D triplets (αk,βk,pk) such that Pr{α = αk,β = βk} = pk, where k = 1,…,D, αk ≥ 0,βk ≥ 0, pk > 0, ∑kpk = 1, and E[α] = ∑kαkpk = 1. The last constraint is included to ensure identifiability because either E[α] or E[β] is confounded with evolutionary time (Kosakovsky Pond and Muse 2005). pk should be strictly positive (otherwise the distribution has fewer than D identifiable rates). We estimate the 3D − 2 free parameters of this distribution from the alignment using numerical nonlinear maximization of model parameters in the standard phylogenetic random-effects likelihood framework (Nielsen and Yang 1998; Kosakovsky Pond and Muse 2005; Kosakovsky Pond and Frost 2005c). The optimization requires a value of D and starting values for the 3D − 2 parameters; the procedure for choosing the values is described in Inferring the Number of Rate Classes (Supplementary Material online).
To date, random-effects models have assumed either that α≡1 (Yang et al. 2000)—that is, constant synonymous substitution rate across sites and over time—or that α and β are drawn independently from univariate distributions (Kosakovsky Pond and Muse 2005). The first assumption now appears unrealistic because synonymous rates are variable across sites in a single gene due to a variety of biological processes (Conrad et al. 1983; DeBry and Marzluff 1994; Hurst and Pal 2001; Tuplin et al. 2002; Kosakovsky Pond and Muse 2005, Chamary et al. 2006). The second assumption, although more realistic, is potentially inefficient because it stipulates that nonsynonymous and synonymous rates are uncorrelated. To illustrate, consider a hypothetical gene comprising sites that experience one of two selective and mutational regimes: one of purifying selection and slow mutation (α = 0.1 and β = 0.01) and one neutrally evolving (α = 1.5 and β = 1.5; Supplementary fig. S1, Supplementary Material online). Under the second assumption, all four combinations of α and β are under consideration of which two are superfluous, namely (α = 0.1, β = 1.5) and (α = 1.5, β = 0.01). In contrast, the bivariate GD distribution can match the hypothetical distribution of rates exactly using a smaller number of rate classes and model parameters—an improvement of computational and statistical efficiency. An illustration of increased model efficiency in describing the distribution of substitution rates in biological data can be found in figure 1.
FIG. 1.

Modeling synonymous and nonsynonymous rate distributions in abalone sperm lysin. Rate plots are drawn on the log scale, with the diagonal line corresponding to values α = β: a proxy for neutral evolution. Colors are used to indicate the ratio β/α. Areas of shaded circles are proportional to the weights of corresponding rate classes. The patterns of variation in α and β can be described more efficiently with the GB distribution, as shown by the improved AICc at each value of D. The optimal value of D for the respective model is shown in boldface.
Inferring the Number of Rate Classes
For a gene with S sites, the number of rate classes in the underlying GDD distribution of rates can vary from 1 (constant rates across sites) to S (unique rates for every site). Given an alignment of practically available size, we can expect to identify a small number of rates 1 ≤ D≪S. To automatically determine this number, we make use of a simple stepwise heuristic. Initially, D is set to 1, corresponding to the basic model of constant rates along the entire gene. Now suppose that D rate classes have been fitted and we wish to determine if stepping up to D + 1 classes is warranted, without incurring the computational expense of ML estimation of D + 1 parameter triplets de novo. Intuitively, if an additional rate class is needed, then at least one of the sites in the alignment is not well explained by any of the existing D rate classes (Heckman and Singer 1984). In other words, including a rate class rD + 1 = (αD + 1,βD + 1) with an initial setting of pD + 1 = 1/S should immediately improve the log-likelihood of the model (the extent of such an improvement is immaterial at this stage).
We evaluate this for rD + 1 on a predefined grid of points in (α,β) rate parameter space; details and formal motivation for this procedure are given in the Supplementary Material online. If any of the proposed rate pairs rD + 1 yields an improvement in likelihood, we select the rate pair with the largest increase and seed the optimization for D + 1 rate categories from the optimized parameter values of the model with D rate categories (see Supplementary Material online), ensuring that the log-likelihood of the former model will be greater than that for the latter.
The step-up terminates if either 1) none of the proposed rates yield an increase in likelihood or 2) the small sample Akaike Information Criterion (AIC) (AICc) score of the model with D + 1 rates (and three additional parameters) does not improve upon the AICc score of the model with D rates.
The AICc score of model M with p adjustable parameters, fitted to a sample with s > p + 1 independent observations, is given by
where logL(M) is the log-likelihood of the model and s is estimated by the number of codons in alignment 𝒟, that is, s≈sites×sequences.
An example of this fitting procedure applied to an alignment of 25 sperm lysin sequences on 134 codons is shown in the left column of figure 1.
Accounting for Recombination
Recombination can severely mislead phylogenetic methods which assume that a single topology can adequately model shared ancestry for the entire gene, including methods for detecting positive selection (Anisimova et al. 2003; Shriner et al. 2003). To correct for this effect, we screen each gene alignment using GARD—a powerful and accurate likelihood-based method for estimating the number and location of recombination breakpoints in a sequence alignment (Kosakovsky Pond, Posada, et al. 2006). When identified, the breakpoints partition an alignment into contiguous nonrecombinant fragments; within each fragment, the original assumption of a singletree topology is exceedingly more likely to be a valid one. Fixing the fragment-specific tree topologies inferred by GARD, we jointly estimate all other parameters of the codon model over the entire alignment (described in preceding sections). We pool nucleotide frequencies across the entire alignment to estimate the stationary distribution of codon frequencies and adopt the branch length approximation procedure, whereby tree branch lengths for a given fragment are first fitted using an appropriate nucleotide model, and then their relative lengths are held constant for the rest of the optimization process, whereas the total length of the tree under the full codon model is estimated for each nonrecombinant fragment (Kosakovsky Pond and Frost 2005c). A similar procedure has been shown to perform well in the context of likelihood ratio tests for detecting positive selection using the ratio-parameterized Muse–Gaut codon model (Scheffler et al. 2006).
Assessing Uncertainty in Parameter Estimates
In order to assess the variances of parameter estimates and to incorporate them in the distance and similarity measures defined in the following sections, we approximate the posterior distribution of a set of model parameters ΘI (the distribution of substitution rates), given the data 𝒟 (sequence alignment) and the rest of the model parameters ΘR (such as branch lengths and nucleotide substitution biases or nuisance parameters), that is,
where L(ΘR,ΘI) is the standard phylogenetic likelihood function evaluated at given model parameters and h is a proposal distribution—the starting approximation to the posterior.
The computation of posterior distributions is an inherently Bayesian procedure—we start out with a prior distribution (h) that represents our knowledge of ΘI, which we update by the incorporation of data 𝒟 to yield the posterior Pr(ΘI|𝒟,ΘR). However, a complete Bayesian treatment of this computation is prohibitively expensive, so we rely on the approximate and fast technique of sampling importance resampling (SIR; Rubin 1987), described in the Supplementary Material online. Examples of ML point estimates and corresponding posterior densities for the distribution of substitution rates approximated by SIR in several alignments of influenza A (H3N2) virus hemagglutinin (HA) gene can be found in the left and middle columns of figure 2.
FIG. 2.

Evolutionary fingerprints of IAV HA alignments of varying sizes (10, 25, 50, 100, and 349 sequences top to bottom). The left panel shows point ML estimates. The middle panel depicts the approximate sampling distributions; color intensity reflects the density assigned to a particular square of the rate distribution; the ellipses are centered on approximate sampling means of corresponding rate estimates, and each axis is drawn as 1.96× the sampling standard deviation for α (horizontal) or β (vertical) rates. The rightmost panel shows approximate sampling distributions for ESD between the corresponding IAV subset (for the full alignment, this can be interpreted as the variance of the posterior ESD distribution), and the full IAV alignment (red), sperm lysin (blue), and primate COXI (green).
ESD: Computing the Distance between Two Discrete Probability Distributions
We aim to define a metric that quantifies the similarity between the evolutionary models fitted to different data sets to allow us to group and segregate genes with similar or different patterns of evolutionary rates. The main challenge is to find a metric that captures the different ways that the models can differ in a statistically robust and computationally efficient fashion, but one that has also a relatively clear interpretation.
ESD, formally introduced below, is based on an intuitively simple concept of “earth mover's distance.” We visualize an evolutionary rate distribution on D rates as D piles of sand on a plane, where the amount of sand in each pile is proportional to the weight of the corresponding rate class and the distance between piles reflects how similar the values of the rates are. Rate distribution A can be “transformed” into another rate distribution (B) by rearranging the sand from the piles from the initial configuration (defined by A) to the configuration defined by B. The cost of moving sand from one pile to the another is proportional to the amount being moved and the distance between the pile. “Earth mover's distance” between A and B seeks to find the transformation of A and B that incurs the minimum cost. As we show below, in the evolutionary context, this distance has two desirable properties: It can be computed very quickly and it obeys the three axioms of a proper distance metric.
Consider two GB discrete distributions, f1(α,β) and f2(α,β), each specified by a set of triplets (αid,βid,pid),d = 1,2. Let the first distribution have D1 ≥ 1 points and the second have D2 ≥ 1 points. We adapt the Monge–Kantorovich distance (MKD; Ambrosio 2003; Kantorovich 2006) to the problem of comparing two such distributions.
MKD on a pair of general discrete distributions is defined as follows:
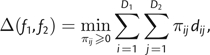 |
(1) |
where dij can be any metric that provides a distance between pairs of substitution rates (α,β) and πij denote weights subject to the following D1 + D2 linear constraints:
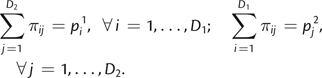 |
Here, pim denotes the estimated proportion of sites assigned to the ith rate class within the mth distribution. Equation (1) therefore presents a minimization problem in which one must compute weights πij that minimize the sum of distances dij while subject to these constraints. The minimum can be found using standard techniques of linear programming, for example, the simplex algorithm. Because the sum of the first D1 constraints is equal to the sum of the remaining D2 constraints (∑ipi1 = ∑jpj2 = 1), at least one of the constraints is a linear combination of other constraints and can be omitted. Formal definitions of dij and πij are provided as supplementary information, Supplementary Material online. Intuitively, each πij describes a component of the transport schedule: Take mass πij from source category i and move it to destination category j (at a cost dij). A valid transport schedule would use up all the weight from each source category and deliver the requisite weight to each of the destination categories.
We define the ESD as an instance of MKD that is obtained by choosing d to be the Euclidean distance in ℝ2. In practice, substitution rate estimates are not uniformly distributed in a subspace of ℝ2; rather, the rate values tend to cluster near the origin (0,0) with a long tail extending to outlying values. In these circumstances, the Euclidean distance will have a strongly L-shaped distribution that is not informative. Consequently, we apply a modified log-transformation that maps the rate values to a more amenable outcome space before computing d:
where ϵ is a small “shift” term that prevents a singularity when α = 0 and/or β = 0. We assign the value ϵ = e − 8, which is generally smaller than the attainable precision of numerical parameter estimates.
A work-through example in which we compute ESD for two rate distributions is provided in Supplementary table S5, Supplementary Material online and discussed in the Supplementary Material online.
Incorporating Parameter Estimate Uncertainty into ESD Measurement
In Assessing Uncertainty in Parameter Estimates (Supplementary Material online), we used the improved SIR scheme to sample a set of M points from an approximation of the joint posterior distribution over the parameter subspace ΘI. This sample provides an approximate posterior distribution for an evolutionary fingerprint. It remains to devise a scheme for incorporating this sample into our ESD metric. Consider a set of k evolutionary fingerprint distributions {f1,f2,…,fk} with a corresponding set of samples of parameter vectors, {ΘI,1M,ΘI,2M,…,ΘI,kM}. For every pairing of fingerprint distributions fi and fj, we draw 1,000 pairs of parameter vectors with replacement from ΘI,iM and ΘI,jM and compute the ESD metric on each pair. The resulting set of 1,000 ESD values, denoted by Δs(fi,fj), is subsequently handled as though it were a sample from the true distribution of ESD, Δ(fi,fj). Depending on the classification method being used (see below), the ESD value is represented by one of the following: 1) the sample mean, E[Δs(fi,fj)], 2) the sample median, μ1/2[Δs(fi,fj)], 3) the 95% interquantile range interval, or 4) some other function of the sample, g(Δs(fi,fj)).
Classification
We are finally in a position where we can compute the ESD metric for any pairwise comparison of gene sequence alignments. Having this metric enables us to exploit any of a vast library of statistical techniques developed in the field of machine learning. In this study, we draw upon kernel-based methods, which comprise a powerful family of techniques for generating nonlinear classifiers for supervised and unsupervised learning (see, e.g., Shawe-Taylor and Cristianini 2004). For instance, given a sample of N evolutionary fingerprints {fi,i = 1,…,N} and a symmetric N×N matrix of ESD values (Δ(fi,fj), which can be either ML estimates or statistics of the approximate posterior ESD distributions), kernel methods enable us to classify the fingerprints into M≪N groups, in a high-dimensional feature space defined by the mapping kernel. In this manuscript, we use the feature space induced by a Gaussian radial basis function (RBF) that can be written as
where kij corresponds to the i,jth entry of an N×N kernel matrix K and σ > 0 is a smoothing parameter, set to σ = 1 for the analyses in this study. Where necessary, we apply the shift transformation (see Supplementary Material online) to ensure that the kernel matrix is positive semidefinite.
In this section, we review a selection of nonlinear classification algorithms (K-nearest neighbors [KNNs], kernel K-means [KKM], and kernel principal component analysis [KPCA]) that we have employed in this study.
K-Nearest Neighbors
One of the simplest classification algorithms, the KNN algorithm classifies data point x by assigning the label according to a majority-rule consensus of the k nearest points in feature space that have been classified beforehand. In this case, the high-dimensional feature space (i.e., rate distribution parameters) has already been reduced using the ESD metric. This method is analogous to classifying a sequence according to the majority label in the top k Blast matches based on sequence similarity. The disadvantage of the method is that it requires supervised training data in the sense that labels for the KNN need to be available.
K-Means
The objective of a k-means algorithm is to identify the optimal clustering function C mapping N objects to K groups while minimizing the cumulative intracluster distance, E, which we quantify as
| (2) |
where fi denotes the ith evolutionary fingerprint that has been mapped to the feature space such that ‖fi − fj‖ = Δ(fi,fj) is the ESD between fingerprints i and j. C(n) = k indicates that fn is assigned to the kth category.
Let ck denote the centroid of the kth cluster, {fi:C(i) = k}, such that ck = ∑i:C(i) = kfi/|i:C(i) = k|, where |S| denotes the size of set S. Substituting this equality into equation (2) gives
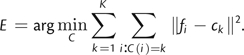 |
(3) |
The k-means algorithm specified by equation (3) does not perform well if the distribution of points in the feature space cannot be partitioned into clusters by simple convex polygons. In such circumstances, one can use a modification of the k-means algorithm that permits nonlinear clustering (see chapter 8 in Shawe-Taylor and Cristianini 2004; we also provide a brief description of such an algorithm in Kernel K-means, Supplementary Material online).
The k-means clustering algorithm performs a greedy climb toward the nearest extremum given a starting cluster allocation. Consequently, the end point depends critically on the starting point and many random restarts may be required to find the optimal (or near optimal) solution. We adopted a more robust approach of using a genetic algorithm (GA) to systematically explore the space of clustering functions C:{1,…,N}→{1,…,K} using a population-based hill-climber (Eshelman 1991), described in Kernel K-means (Supplementary Material online). This family of GAs has previously proven to be successful in complex optimization problems arising in molecular evolution applications (Kosakovsky Pond and Frost 2005a, Kosakovsky Pond, Posada, et al. 2006, Kosakovsky Pond et al. 2007).
Kernel Principal Component Analysis
Another venerable statistical technique that can be extended to nonlinear classification problems using the kernel techniques is principal component analysis (PCA). Traditional PCA seeks to find latent factors in a sample of N data points x1,…,xN centered in ℝd, by diagonalizing the estimated covariance matrix C = N − 1∑i = 1NxixiT and projecting the data onto the principal components—that is, the eigenvectors associated with the n largest eigenvalues of C. An analogous procedure can be performed in feature space (Scholkopf et al. 1999) by diagonalizing the centered kernel matrix that is defined as
where 1N is an N×N matrix populated with 1/N. If yk denotes the eigenvector of 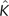 (corresponding to the eigenvalue λk > 0 and subject to ‖yk‖2 = 1/λk), then the image of data point xj in the feature space can be projected onto the kth principal component Vk using the relation
(corresponding to the eigenvalue λk > 0 and subject to ‖yk‖2 = 1/λk), then the image of data point xj in the feature space can be projected onto the kth principal component Vk using the relation
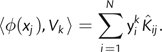 |
Because KPCA operates in a high-dimensional linear subspace that contains the feature map projections of all data points, the algorithm can identify up to N components, that is, all eigenvectors corresponding to nonzero eigenvalues of .
Simulated Data
To evaluate the performance of our inference methods, we generated a set of alignments from the parameterized models by simulation. We generated 500 codon alignments using five different evolutionary rate scenarios (100 replicates per scenario), with parameters drawn from our collection of biological alignments and summarized in Supplementary table S1, Supplementary Material online. Simulations 1–3 include a level of randomization not commonly included for simulation studies: The size of the data set (sequences and codons) and the level of sequence divergence were varied among replicates, to assess the reliability of ESD-based techniques to detect rate similarities in samples of different information content. Pairwise ESDs between all generating rate distributions can be found in Supplementary table S2, Supplementary Material online. We applied several clustering techniques to evaluate how well the ESD metric, represented by the median of the approximate posterior ESD distribution, was able to allocate each of the replicates to the appropriate cluster (a comma-separated text file with the 500×500 matrix of pairwise ESDs can be downloaded from http:// www.viralevolution.org/paper/simulated_distances.bz2).
Implementation
Evolutionary fingerprint inference, ESD estimation, and KPCA and k-means procedures were implemented in HyPhy v2.0 (Kosakovsky Pond et al. 2005), which can be downloaded from http://www.hyphy.org. An evolutionary fingerprint/ESD module for the Datamonkey (Kosakovsky Pond and Frost 2005b) Web server is currently under development and should be online when this manuscript is accepted.
Biological Data
We reanalyzed ten gene alignments representing different taxonomic levels that have been previously examined for evolutionary rate variation, most recently in Kosakovsky Pond and Muse (2005). In addition, we investigated gene alignments from three rapidly evolving RNA viruses:
We downloaded full-length gene alignments of the IAV from the NCBI Influenza resource (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html; Bao et al. 2007). Each gene was stratified by its HN serotype and human/avian/swine hosts. Alignments with fewer than ten sequences were discarded as too small for reliable rate inference, and those with more than 100 sequences were randomly subsampled, yielding 221 alignments. These sequence limits were also enforced for other alignments collected from public databases.
We utilized the Los Alamos Hepatitis C virus database (http://hcv.lanl.gov/), to retrieve full-length sequences for each HCV and stratifying them by subtype. Eighty-four HCV alignments were analyzed.
Three primary sources were used for the alignments of HIV-1 sequences. First, we extracted gene alignments stratified by viral subtype from the Los Alamos HIV sequence database (http://hiv.lanl.gov/). Second, we incorporated subtype B gene-by-gene alignments from a previous paper on selective pressures in HIV (Pillai et al. 2005). Third, we mined the Stanford Drug Resistance database (http://hivdb.stanford.edu) for protease, reverse transciptase (RT), and integrase sequences sampled from drug-naive patients stratified by source publication (often a region-specific sequence diversity survey) and subtype. Overall, 80 HIV-1 gene alignments were obtained.
Results
Simulation Study of GB Discrete Model and ESD
First, we used the simulated data sets to verify that we can infer the parameters of the GB discrete model. The results of our rate estimation procedures are summarized in Supplementary table S3, Supplementary Material online. In each scenario, the majority of replicates (74–92%) were assigned the correct number of rates, with an overall tendency of the method to be slightly conservative (i.e., erring on the side of too few rates), except in Simulation 5, where three replicates were underfitted and five overfitted.
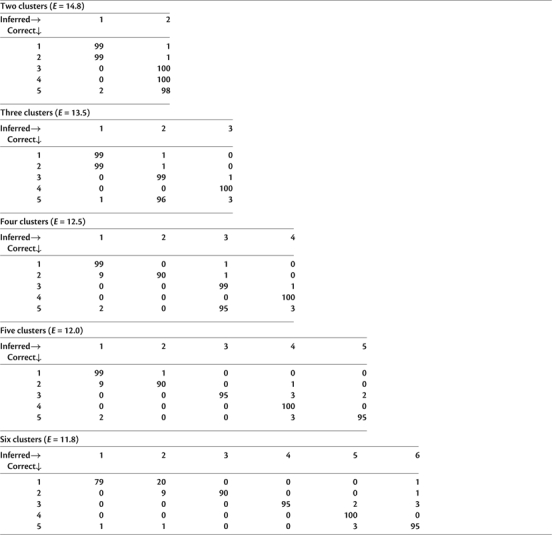
We applied several machine learning approaches to partition the 500 simulations into groups based on ESD. The simple nearest neighbor clustering, which relied on the a priori knowledge of the correct allocation to clusters, based either on the identity of the nearest neighbor or on the identity of a majority of five nearest neighbors showed a very high accuracy of predicted cluster membership, with 98% (1-NN) or 96.6% (5-NN) of simulated data sets allocated to the correct clusters. A more realistic benchmark is achieved using an unsupervised learning algorithm, namely KKM (Table 1). For the correct number of clusters (K = 5), the algorithm (based on the RBF kernel with σ = 1) showed an excellent overall 95.8% classification accuracy. When fewer than the correct number of five clusters are allowed, KKM separates the most dissimilar groups of simulations. Simulations 1, 2 are distinguished from 3, 4, 5 for K = 2, as can be expected because Simulation 1 has the largest mean ESD compared with other distributions, and Simulation 2 is its nearest ESD neighbor (Table 1). Simulation 4 receives its own cluster for K = 3 and Simulation 2 is further split off with K = 4. Because the algorithm imposes no additional penalties with increasing number of clusters, incrementing K up to the number of independent points would result in smaller cumulative distances from cluster centers (0 for K = 500). For instance, for K = 6 (Table 1), the simulations from Scenario 1 were split into two subclusters.
Table 1.
KKM Clustering Results, for K = 2, 3, 4, 5, and 6. Clustering Error E (eq. 3) Is Also Shown for Each K
 |
KPCA is able to visually separate all five groups of simulations based on just the two principal components corresponding to the largest eigenvalues of the centered kernel matrix as shown in figure 3.
FIG. 3.

KPCA of simulated data. Projections of the 500 simulation data points on pairs drawn from five simulation scenarios projected onto the eigenvectors corresponding to two largest eigenvalues. Each simulation scenario is color coded: Set 1—black, Set 2—red, Set 3—blue, Set 4—yellow, and Set 5—magenta.
The analysis of simulated data clearly demonstrates the power of ESD to cluster and discriminate alignments on the basis of the underlying distribution of substitution rates in the presence of confounding factors, such as variable sequence length, phylogenetic tree, alignment size, and divergence level.
GB Discrete Distributions Provide an Efficient Description of Rate Heterogeneity in Biological Data
We re-examined ten sequence alignments (Table 2) analyzed in Kosakovsky Pond and Muse (2005) to investigate whether the use of the GB discrete distribution to model the variation of synonymous (αs) and nonsynonymous (βs) rates across sites improves upon the fit of models that assume independent univariate general discrete distributions for αs and βs.
Table 2.
Data Sets from Kosakovsky Pond and Muse (2005) Reanalyzed Using the GB Discrete Rate Distribution
| MG94 × REV IB |
MG94 × REV GB |
|||||||||||
| Data | N | L | Log L | AICc | E[β – α] | Pr{β > α} | D | Log L | AICc | E[β – α] | Pr{β > α} | Δ AICc |
| Sperm lysin | 25 | 134 | − 4,340.6 | 8,832.6 | − 0.02 | 0.34 | 5 | − 4,340.3 | 8,832.6 | 0.07 | 0.37 | 3.1 |
| Primate COXI | 21 | 510 | − 11,985.6 | 24,095.9 | − 0.18 | 0.013 | 4 | − 11,975.5 | 24,077.7 | − 0.98 | 0.00 | 18.2 |
| Drosophila adh | 23 | 254 | − 4,583.41 | 9,300.36 | − 0.91 | 0 | 3 | − 4,0583.57 | 9,296.58 | − 0.90 | 0 | 3.8 |
| HIV-1 vif | 29 | 192 | − 3,334.37 | 6,826.98 | − 0.24 | 0.28 | 5 | − 3,331.17 | 6,828.82 | − 0.24 | 0.22 | − 1.9 |
| β-globin | 17 | 144 | − 3,649.66 | 7,409.81 | − 0.78 | 0.06 | 4 | − 3,650.3 | 7,413.2 | − 0.77 | 0.08 | − 3.4 |
| IAV HA | 349 | 329 | − 10,891.1 | 23,227.2 | − 0.86 | 0.12 | 7 | − 10,837.9 | 23,141.1 | − 0.52 | 0.27 | 86.0 |
| Camelid VHH | 212 | 96 | − 16,382.2 | 33,672.3 | − 0.27 | 0.37 | 10 | − 16,283.6 | 33,514.9 | − 0.25 | 0.28 | 157.4 |
| Encephalitis env | 23 | 500 | − 6,752.7 | 13,638.2 | − 0.95 | 0.02 | 4 | − 6,750.1 | 13,635 | − 0.95 | 0 | 3.3 |
| Flavivirus NS5 | 18 | 342 | − 9,144.33 | 18,402.9 | − 0.65 | 0.01 | 4 | − 9,109.04 | 18,333.2 | − 0.96 | 0 | 69.7 |
| Hepatitis D virus Ag | 33 | 196 | − 5,078.3 | 10,330.9 | − 0.56 | 0.44 | 5 | − 5,074.15 | 10,330.9 | − 0.56 | 0.30 | 0.03 |
NOTE.—The optimal number of rate classes D for the GB model was inferred using the step-up procedure described in this manuscript, whereas the total number of rate classes for the IB (dual model in Kosakovsky Pond and Muse 2005) analog were fixed at 3 synonymous × 3 nonsynonymous = 9. N refers to the number of sequences and L to the number of codons in each alignment. E[β – α] is the mean of the estimated β – α distribution, whereas Pr{β > α} is the cumulative probability of all rate classes with β > α in the respective distribution. ΔAICc is computed between the GB model with D rate classes and the 3 × 3 IB model.
We will refer to these models as general bivariate (GB) and independent bivariate (IB) models, respectively. IB models allocate rate classes to a rectangular grid of points and are also subject to constraints on rate class weights, which are the products of corresponding weights for the α and β distributions, whereas GB models are free to place rate classes anywhere on the plane and estimate weights of each class directly (see, e.g., fig. 1). Recall that, given D independent values of αs and βs, the IB model introduces D2 rate classes (αs,βs), whereas the GB model only introduces D rate classes. The number of rate classes has a significant impact on the cost of likelihood evaluations, which is linear in the number of rate classes. Also, GB models are more parsimonious than IB models. Indeed, because the number of free parameters for GB models is pGB(D) = 3D − 2, whereas its counterpart for IB models is pIB(D) = 4D − 3, we have pIB(D) − pGB(D) = D − 1 > 0 for D > 1.
Consider the example of sperm abalone lysin protein (fig. 1)—a well-characterized gene subject to strong selective forces to restrict heterospecific hybridization during sperm–egg interaction (Lee et al. 1995, Yang and Swanson 2002). A GB model with five rate classes provides the best fit to the data and also outperforms the 3×3 = 9 IB model but is 9/5 = 1.8 times less computationally expensive. As can be seen in figure 1, substitution rates inferred by the GB model are not arranged on a grid, providing an intuitive explanation of why this model provides a better fit to the data than the more computationally expensive IB model. Looking down the left column of this figure, it is apparent that the GB model begins by describing the major features of the evolutionary process, such as large proportions of nearly neutral sites and weakly negatively selected sites for D = 2, and successively adds new features, for example, a class of strongly positively selected sites when moving from D = 3 to D = 4, until no new features are supported by the data.
A similar pattern emerges for the other nine data sets (cf. Table 2). GB models return better (except in the cases of HIV-1 vif and β-globin where GB models are slightly worse) AICc scores with fewer than the nine rate classes stipulated by a 3×3 IB model, except for the large IAV HA data set, where the best-fitting GB model has 10 rate classes but improves AICc by 86 points. More importantly, this pattern confirms the reasonable a priori expectation that the model which allows synonymous and nonsynonymous rates to covary should perform as well as or better than the model that imposes the artificial constraint of independence between rates.
ESD Estimates Between the Empirical Data Sets
To introduce the reader to the empirical applications of ESD and the types of inference that it enables, we computed the matrix of pairwise ESD estimates between the point estimates of their evolutionary fingerprints for the data sets in Table 2 and constructed the neighbor-joining tree on these 10 nonhomologous genes (Saitou and Nei 1987). The use of simple point estimates of ESD is the least computationally demanding way to implement the method and is applicable to data sets which are of comparable sizes and large enough to obtain stable point estimates of evolutionary fingerprints. For general analyses, it is advisable to make use of posterior estimates of the ESD distribution, as discussed below. The tree (fig. 4) clusters the genes into those that are subject, as previously reported, to strong diversifying selection (camelid VHH, HIV-1 vif, IAV HA, sperm lysin, and hepatitis D Ag), those that are subject to strong purifying selection (Drosophila adh, primate COX I, Flavivirus NS5, Encephalitis env) and beta-globin that is subject to weaker positive selection. This broad separation mirrors (Table 2) the obvious groupings based on the mean β − α estimate (an analog to ω = β/α, but more appropriate for models where α can be small or zero) and the proportion of sites where β > α (analogous to those with ω > 1). Simple machine classification techniques, such as KKM, can identify patterns in the data automatically; for example, Drosophila adh, primate COX I, Flavivirus NS5, and Encephalitis env form one cluster detected by KKM, whereas the other genes constitute the second cluster.
FIG. 4.

Point estimates of evolutionary fingerprints for ten empirical data sets from Table 2 labeling a neighbor-joining tree built from pairwise ESDs between them. Branches are scaled on ESDs, rate plots are drawn on the log scale that is consistent between all ten data sets, with α on the x axis and β on the y axis, and the diagonal line corresponding to values α = β: a proxy for neutral evolution. Colors are used to indicate the ratio β/α. Areas of shaded circles are proportional to the weights of corresponding rate classes.
Because ESD is based on comparing entire rate distributions and not their summary statistics, it can exploit considerably more of the structure in the data than traditional methods can. For example, HIV vif, IAV HA, and hepatitis D Ag all encode viral proteins that come under considerable selective pressure from the host immune system, and traditional tests for positive selection (e.g., PARRIS; Scheffler et al. 2006) show strong evidence for selection in each gene. However, their evolutionary fingerprints are quite different. Indeed, ESD (vif, HA) = 0.47, whereas ESD (vif, Ag) = 1.35 and ESD (HA, Ag) = 1.30, and an inspection of fingerprint plots for the three genes in figure 4 reveals that one of the features of hepatitis D Ag evolutionary rate distribution, namely the rate with nearly zero α and nonzero β (red circle near the origin of the hepatitis D Ag plot), is missing from the fingerprints of the other two genes. Of course, there are other differences between distributions, for example, the proportions of nearly neutral sites in all three genes, but the magnitude of these differences is considerably less between HIV vif and IAV HA than it is between either gene and hepatitis D Ag. ESD is equally adept at differentiating the genes that are conserved, for example, primate cytochrome oxidase I (COX I) versus flavivirus NS5 (ESD = 0.62) and flavivirus NS5 versus encephalitis envelope (ESD = 0.92).
An immediate application of ESD is to rank all genes by how close they are to a gene of interest, as this may reveal similarities and dissimilarities in the evolutionary process, analogous to how the ranking of Blast database hits can reveal close and distant homologies. For instance, the ordering of the genes with respect to IAV HA is as follows: HIV vif (ESD = 0.48), camelid VHH (0.67), beta globin (0.90), sperm lysin (1.05), encephalitis env (1.09), hepatitis D antigen (1.30), Flavivirus NS5 (1.77), primate COX I (1.84), and Drosophila adh (2.01).
ESD Robustness against Estimation Errors: An Empirical Example
When comparing data sets with different information content, as measured for example by the number of sites, sequences, and divergence levels, it is essential to incorporate a measure of estimation uncertainty into the calculations. A straightforward way to do this is to approximate the sampling (posterior) distribution of ESD between any two alignments (using importance sampling, see Materials and Methods) and then compare those distributions instead of the corresponding ML point estimates (fig. 2). The goals of this procedure are analogous to those of the more specialized Bayes Empirical Bayes approach (Yang et al. 2005) introduced in the context of detecting individual codon sites under selection to reduce the effect of noisy parameter estimates, but our approach is more general. It is also important to realize that some parameter estimates (e.g., α and β for rate classes with small weights) will remain noisy even for large data sets because the effective sample size for these parameters is a fraction of the size of the data set, as evidenced by sampling variances shown in figure 2. The figure shows the ML evolutionary fingerprints for four alignments comprising N = 10,25,50,and 100 randomly selected (without replacement) sequences from an IAV HA alignment with 347 sequences and the full alignment. Increasing the number of sequences improves the resolution of the evolutionary fingerprint from two to seven rates. Just as was the case for sperm lysin (fig. 1), simpler rate distributions inferred from sparse data sets capture the main features of the more complex distributions. The rightmost panel of figure 2 shows approximate sampling distributions for ESD between each of the four IAV subsets and the full IAV alignment (red), sperm lysin (blue), and primate COXI (green). We highlight several features of ESD that are illustrated in this figure.
ESD very clearly separates a positively selected alignment (IAV HA) from a positively selected alignment with a distinct fingerprint (sperm lysin, which has more sites under selection, weaker constraint) and even more clearly from an alignment under strong purifying selection (mammalian COX I).
Mean ESD estimates from subsets of IAV to lysin and COXI are quite consistent despite the dramatic differences in fingerprint complexity between small and large IAV HA alignments. These estimates (and corresponding standard deviations) between IAV and lysin are 0.94 (0.11), 0.95 (0.12), 0.89 (0.11), 0.82 (0.11), and 0.79 (0.11), for N = 10,25,50,100,and 349, respectively. For COXI, the corresponding estimates are 1.99 (0.13), 1.86 (0.10), 1.88 (0.12), 1.85 (0.10), and 1.83 (0.09).
The mean ESDs from subsets of IAV to the complete data set are reduced as the number of sequences is increased: 0.62 (0.06), 0.56 (0.05), 0.54 (0.09), and 0.43 (0.006). The sampling mean of ESD between the full IAV alignment and itself (0.20) provides a reasonable indication of the precision that can be achieved using ESD on a given data set.
Analysis of Viral Genes
ESD enables the previously unavailable comparison of nonhomologous genes sampled from the same virus or from different viruses. When multiple alignments of a single viral gene were available, as was the case with most HIV, HCV, and IAV genes, we represented all alignments of a given gene (e.g., HIV-1 RT alignments from different subtypes, geographic regions, and treatment histories) by an “average gene,” defined as the centroid of all the individual samples (here, a sample denotes an alignment of homologous sequences) in the kernel feature space to minimize sampling effects (see Gene trees based on multiple samples; Supplementary Material online). The analogy for this in traditional sequence analysis is taking the consensus sequence of an alignment to represent the “average” sequence. KPCA and hierarchical clustering applied to the ESD matrix suggested that genes within the same functional group tended to cluster together (fig. 5). For example, polymerase, transcription, and assembly complex genes (protease, RT, integrase, and gag in HIV; PA [acidic polymerase], PB2 [basic polymerase], and NP [nucleoprotein] in IAV; and nonstructural proteins in HCV [except NS5B]) form clusters within their respective genomes. NS5B in HCV is highly conserved and evolves more similarly to the Core protein than to other nonstructural genes, consistent with extensive RNA secondary structure in these regions (Tuplin et al. 2004). Envelope and fusion proteins were similarly grouped: E1 and E2 envelope proteins in HCV, HA, and neuraminidase in IAV. Genes with multiple regulatory functions, such as nef and vif in HIV, nonstructural (NS1) protein and the matrix polyprotein in IAV, did not appear to have close evolutionary neighbors.
FIG. 5.

Clustering of viral genes in HIV, HCV, and IAV based on the similarities in inferred substitution rates, as measured by ESD. Some genes were represented by averaging over several alignments (samples) as indicated in square brackets following the standard abbreviation for a gene. Hierarchical clustering (left column) was derived using neighbor joining on pairwise distance matrices constructed in the feature space of the RBF kernel. It is important to remember that these “trees” represent evolutionary process–based clustering and do not imply a particular “gene phylogeny” and that branch lengths are measured in arbitrary units. Projections on the two principal components in the feature space with the largest corresponding eigenvalues are shown in the right column.
We also inquired to what extent ESD mirrored genetic distances for homologous genes sampled from the same virus. For example, our collection included alignments representing the envelope glycoprotein E1 gene for six subtypes of HCV and RT genes (all from drug-naive subjects) for seven subtypes of HIV. We analyzed ESD matrices and genetic distances calculated using the TN93 (Tamura and Nei 1993) model by generating neighbor-joining trees (Saitou and Nei 1987) and performing KPCA (fig. 6), with each subtype represented by the centroid of all alignments sampled from that subtype. In the case of HCV, there was a perfect correspondence between trees constructed using ESD and those constructed using genetic distances; this is not likely a chance coincidence as there are 105 possible unrooted trees on six sequences and because ESD and genetic distance matrices for the six subtypes were significantly correlated (exact Mantel test P = 0.045). For HIV RT, this correspondence did not hold; overall, the ESD and molecular trees were discordant, and there was little correlation between the two distance matrices (P = 0.28). This does not appear to be due simply to inadequate statistical power to detect clustering as subtype A and the circulating recombinant form AE, which is subtype A in the RT gene, clustered together. Interestingly, subtypes B and D, which are closely related at the sequence level, were highly divergent at the level of selection pressures; this may reflect differences in the host population, as has previously been shown for divergent evolution of subtype C sequences (Kosakovsky Pond, Frost, et al. 2006). These analyses are consistent with current knowledge of HCV and HIV subtypes; biological and clinical differences between HCV subtypes have been well characterized (Smith and Simmonds 1998), whereas for HIV, there appear to be fewer such differences, making the selective patterns among HIV subtypes relatively uniform (Choisy et al. 2004).
FIG 6.

Comparisons of subtype clustering using genetic and ESDs. When multiple alignments of the same alignment were available, they were represented by the “centroid” in the feature space (see text) as indicated in square brackets following the standard abbreviation for a gene. Hierarchical ESD clustering (left column) was generated by neighbor joining on pairwise distance matrices constructed in the feature space of the RBF kernel, whereas molecular phylogenies (middle column) were constructed using neighbor joining on inter-subtype Tamura–Nei genetic distances, computed as means of all pairwise sequence distances with appropriate subtypes. Procrustes mapping of KPCA projections for ESD (black dots) and genetic (arrows) distances on the two principal components in the feature space are shown in the right column; it is useful for assessing the degree of similarity between two different PCA projections.
Conclusion
Our understanding of the processes that have shaped genetic variation is limited by the predominance of methods that rely on sequence similarity, which cannot easily compare distantly related genes. This study describes the novel concept of evolutionary fingerprints, which flexibly captures the imprint of natural selection on an individual gene. These fingerprints can be rapidly compared against both closely related and highly divergent genes, enabling what can be thought of as evolutionary Blast.
To estimate an evolutionary gene fingerprint that is encoded by the distribution of synonymous and nonsynonymous substitution rates across sites, we developed a likelihood-based random-effects model that can efficiently, and reliably (based on simulated data), capture a wide range of patterns with a relatively small number of parameters. Our model extends exiting models which postulate somewhat restrictive parametric distribution for rates (Yang et al. 2000), disallow variable synonymous rates (Yang et al. 2000; Huelsenbeck et al. 2006), posit that nonsynonymous and synonymous rate distributions are independent (Kosakovsky Pond and Frost 2005c; Kosakovsky Pond and Muse 2005), or require many parameters to capture covariation of nonsynonymous and synonymous rates (Kosakovsky Pond 2003). Using ten previously analyzed data sets, we demonstrate that the new model attains a better or comparable fit relative to the previous state of the art models and provides a more efficient description of the distribution of evolutionary rates. Based on the MKD (or earth mover's), used in a wide range of pattern-matching applications (e.g., Rubner et al. 1998), we define an ESD and show that it forms a proper metric on the space of evolutionary rate distributions and can be computed in a negligible amount of time using standard linear programming algorithms.
Because sampling errors in rate estimates from finite alignments can be significant, we replace the point estimate of an evolutionary rate distribution with the posterior density on such distributions approximated using an SIR procedure—a statistically sound and computationally attractive alternative to a full Bayesian treatment. The use of full posterior distributions of ESD to compare evolutionary fingerprint alignments helps mitigate the effect of information content in an alignment on inference and permits a robust comparison of disparately sized alignments. This assertion is supported by a simulation study where ESD-based machine learning and clustering techniques reliably (>95% accuracy) discriminated data sets simulated using five different rate distributions in the presence of significant statistical noise.
We show the considerable inferential potential of ESD by applying it to an analysis of approximately 400 alignments representing genes from three rapidly evolving RNA viruses (HIV-1, HCV, and IAV). Evolutionary fingerprinting establishes that genes within the same functional group (e.g., polymerases, envelopes) tend to exhibit similar evolutionary patterns within a viral genome. Our methodology provides a first tractable treatment that is able to quantify these differences between nonhomologous genes and smooth the sampling effects by combining multiple gene alignments into a “centroid” gene. When many samples of the same gene are available, ESD can also be useful for comparing different samples for the effect of sampling epoch, geographic region, viral subtype, and other external factors on evolutionary rates. As an example, we contrast hierarchical clusterings based on evolutionary fingerprints and genetic sequences among HCV and HIV-1 subtypes and obtain patterns consistent with known population dynamics and evolutionary history of viral subtypes—concordance for HCV and discordance for HIV-1.
In conclusion, we have developed a “meta-evolutionary” approach for quantifying and comparing selection pressures in coding regions and demonstrated its utility through simulation and by comparative analysis of viral and other genes. Our approach is immediately applicable to any collection of coding sequence alignments and can be used to conduct large-scale analyses of, for example, coevolution of host and pathogen genes, the comparative evolution of different gene families or genome-wide screens of mammalian species. The inference and comparison of evolutionary fingerprints open a new pathway toward an evolutionary Blast and can be extended to incorporate many other evolutionary features, such as tree shape and length, amino acid frequencies and substitutional preferences, codon bias, and numbers and locations of coevolving sites. By comparing genes at the level of the evolutionary processes rather than the pattern of sequence variation, we are no longer restricted to analyzing closely related genes or motifs but can compare both closely and distantly related genes in a single analysis, potentially revealing the guiding principles underlying the evolution of genetic diversity.
Supplementary Material
Supplementary figure S1 and tables S1–S3, S5 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Supplementary Material
Acknowledgments
We thank Alex Smola for pointing out the connection between ESD and the MKD and Jeffrey Thorne for comments on a previous version of this manuscript. This research was supported by the Joint DMS/NIGMS Mathematical Biology Initiative through Grant NSF-0714991, the National Institutes of Health (AI43638, AI47745, and AI57167), the University of California Universitywide AIDS Research Program (grant number IS02-SD-701), a University of California, San Diego Center for AIDS Research/NIAID Developmental Award to S.D.W.F. and S.L.K.P (AI36214). S.D.W.F. is supported in part by a Royal Society Wolfson Research Merit Award. This work was facilitated by IBM Deep Computing.
References
- Altschul S, Madden T, Schäffer A, Zhang J, Zhang Z, Miller W, Lipman D. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosio L. Berlin (Germany): Springer Verlag. 2003. Lecture notes on optimal transport problems. In: Mathematical aspects of evolving interfaces. p. 1–52. [Google Scholar]
- Anisimova M, Nielsen R, Yang Z. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics. 2003;164(3):1229–1236. doi: 10.1093/genetics/164.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbiza L, Dopazo J, Dopazo H. Positive selection, relaxation, and acceleration in the evolution of the human and chimp genome. PLoS Comput Biol. 2006 doi: 10.1371/journal.pcbi.0020038. 2(4):e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. The influenza virus resource at the national center for biotechnology information. J Virol. 2007;82(2):596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary JV, Parmley JL, Hurst LD. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet. 2006;7(2):98–108. doi: 10.1038/nrg1770. [DOI] [PubMed] [Google Scholar]
- Choisy M, Woelk CH, Guegan JF, Robertson DL. Comparative study of adaptive molecular evolution in different human immunodeficiency virus groups and subtypes. J Virol. 2004;78:1962–1970. doi: 10.1128/JVI.78.4.1962-1970.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, Friedlander C, Goodman M. Evidence that natural selection acts on silent mutation. Biosystems. 1983;16(2):101–111. doi: 10.1016/0303-2647(83)90031-x. [DOI] [PubMed] [Google Scholar]
- DeBry RW, Marzluff WF. Selection on silent sites in the rodent H3 histone gene family. Genetics. 1994;138(1):191–202. doi: 10.1093/genetics/138.1.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshelman LJ. The CHC adaptive search algorithm. In: Rawlins GJE, editor. Foundations of genetic algorithms. Vol. 1991. pp. 265–283. I. San Francisco (CA): Morgan Kaufman. [Google Scholar]
- Heckman J, Singer B. A method for minimising the impact of distributional assumptions in econometric models for duration data. Econometrica. 1984;52(2):271–320. [Google Scholar]
- Huelsenbeck JP, Jain S, Frost SWD, Pond SLK. A Dirichlet process model for detecting positive selection in protein-coding DNA sequences. Proc Natl Acad Sci USA. 2006;103(16):6263–6268. doi: 10.1073/pnas.0508279103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst L, Pal C. Evidence for purifying selection acting on silent sites in BRCA1. Trends Genet. 2001;17:62–65. doi: 10.1016/s0168-9525(00)02173-9. [DOI] [PubMed] [Google Scholar]
- Kantorovich L. On the translocation of masses. J Math Sci. 2006;133(4):1381–1382. [Google Scholar]
- Kosakovsky Pond SL . Modeling evolution of protein coding DNA sequences. University of Arizona; 2003. [ Ph.D. thesis] [Tucson (AZ)] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. A genetic algorithm approach to detecting lineage-specific variation in selection pressure. Mol Biol Evol. 2005a;22(3):478–485. doi: 10.1093/molbev/msi031. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005b;21(10):2531–2533. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005c;22(5):1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW, Grossman Z, Gravenor MB, Richman DD, Leigh Brown AJ. Adaptation to different human populations by HIV-1 revealed by codon-based analyses. PLoS Comput Biol. 2006;2 doi: 10.1371/journal.pcbi.0020062. e62(6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW, Muse SV. hypothesis testing using phylogenies. Bioinformatics. 2005;21(5):676–679. doi: 10.1093/bioinformatics/bti079. HyPhy. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Mannino FV, Gravenor MB, Muse SV, Frost SDW. Evolutionary model selection with a genetic algorithm: a case study using stem RNA. Mol Biol Evol. 2007;24(1):159–170. doi: 10.1093/molbev/msl144. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Muse SV. Site-to-site variation of synonymous substitution rates. Mol Biol Evol. 2005;22(12):2375–2385. doi: 10.1093/molbev/msi232. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol. 2006;23(10):1891–1901. doi: 10.1093/molbev/msl051. [DOI] [PubMed] [Google Scholar]
- Lee YH, Ota T, Vacquier VD. Positive selection is a general phenomenon in the evolution of abalone sperm lysin. Mol Biol Evol. 1995;12(2):231–238. doi: 10.1093/oxfordjournals.molbev.a040200. [DOI] [PubMed] [Google Scholar]
- Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates with application to the chloroplast genome. Mol Biol Evol. 1994;11:715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Bustamante C, Clark AG, et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005 doi: 10.1371/journal.pbio.0030170. 3(6):e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Yang ZH. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai SK, Kosakovsky Pond SL, Woelk CH, Richman DD, Smith DM. Codon volatility does not reflect selective pressure on the HIV-1 genome. Virology. 2005;336(2):137–143. doi: 10.1016/j.virol.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Rubin D. A noniterative sampling/importance resampling alternative to the data augmentation algorithm for creating a few imputations when fractions of missing information are modest: the SIR algorithm. J Am Stat Assoc. 1987;82(398):543–546. [Google Scholar]
- Rubner Y, Tomasi C, Guibas LJ. A metric for distributions with applications to image databases. Sixth International Conference on Computer Vision. New Delhi: India; 1998. 1998 Jan 4 Bombay (India): Narosa Publishing House p. 59–66. [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sawyer SL, Emerman M, Malik HS. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004;2(9):E275. doi: 10.1371/journal.pbio.0020275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffler K, Martin DP, Seoighe C. Robust inference of positive selection from recombining coding sequences. Bioinformatics. 2006;22(20):2493–2499. doi: 10.1093/bioinformatics/btl427. [DOI] [PubMed] [Google Scholar]
- Scholkopf B, Smola A, Muller KR. Cambridge. MIT Press; 1999. Kernel principal component analysis. (MA) [Google Scholar]
- Shankarappa R, Margolick J, Gange S, et al. 12 co-authors. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol. 1999;73(12):10489–10502. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawe-Taylor J, Cristianini N. Cambridge. (UK): Cambridge University Press; 2004. Kernel methods for pattern analysis. p. 484. [Google Scholar]
- Shriner D, Nickle DC, Jensen MA, Mullins JI. Potential impact of recombination on sitewise approaches for detecting positive natural selection. Genet Res. 2003;81(2):115–121. doi: 10.1017/s0016672303006128. [DOI] [PubMed] [Google Scholar]
- Smith D, Simmonds P. Hepatitis C virus: types, subtypes, and beyond. In: Lau JYN, editor. Methods in molecular medicine. Totowa (NJ) Vol. 19. Humana Press Inc; 1998. pp. 133–146. Hepatitis C protocols. [DOI] [PubMed] [Google Scholar]
- Swanson WJ. Adaptive evolution of genes and gene families. Curr Opin Genet Dev. 2003;13(6):617–622. doi: 10.1016/j.gde.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Swanson WJ, Nielsen R, Yang Q. Pervasive adaptive evolution in mammalian fertilization proteins. Mol Biol Evol. 2003;20(1):18–20. doi: 10.1093/oxfordjournals.molbev.a004233. [DOI] [PubMed] [Google Scholar]
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Tuplin A, Evans DJ, Simmonds P. Detailed mapping of RNA secondary structures in core and NS5B-encoding region sequences of hepatitis C virus by RNase cleavage and novel bioinformatic prediction methods. J Gen Virol. 2004;85(Pt 10):3037–3047. doi: 10.1099/vir.0.80141-0. [DOI] [PubMed] [Google Scholar]
- Tuplin A, Wood J, Evans D, Patel A, Simmonds P. Thermodynamic and phylogenetic prediction of RNA secondary structures in the coding region of hepatitis C virus. RNA. 2002;82(6):824–841. doi: 10.1017/s1355838202554066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen AMK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155(1):431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Swanson WJ. Codon-substitution models to detect adaptive evolution that account for heterogeneous selective pressures among site classes. Mol Biol Evol. 2002;19:49–57. doi: 10.1093/oxfordjournals.molbev.a003981. [DOI] [PubMed] [Google Scholar]
- Yang Z, Wong WSW, Nielsen R. Bayes Empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22(4):1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.