

Abstract
The spontaneously hypertensive rat (SHR) is the most widely studied animal model of hypertension. Scores of SHR quantitative loci (QTLs) have been mapped for hypertension and other phenotypes. We have sequenced the SHR/OlaIpcv genome at 10.7-fold coverage by paired-end sequencing on the Illumina platform. We identified 3.6 million high-quality single nucleotide polymorphisms (SNPs) between the SHR/OlaIpcv and Brown Norway (BN) reference genome, with a high rate of validation (sensitivity 96.3%–98.0% and specificity 99%–100%). We also identified 343,243 short indels between the SHR/OlaIpcv and reference genomes. These SNPs and indels resulted in 161 gain or loss of stop codons and 629 frameshifts compared with the BN reference sequence. We also identified 13,438 larger deletions that result in complete or partial absence of 107 genes in the SHR/OlaIpcv genome compared with the BN reference and 588 copy number variants (CNVs) that overlap with the gene regions of 688 genes. Genomic regions containing genes whose expression had been previously mapped as cis-regulated expression quantitative trait loci (eQTLs) were significantly enriched with SNPs, short indels, and larger deletions, suggesting that some of these variants have functional effects on gene expression. Genes that were affected by major alterations in their coding sequence were highly enriched for genes related to ion transport, transport, and plasma membrane localization, providing insights into the likely molecular and cellular basis of hypertension and other phenotypes specific to the SHR strain. This near complete catalog of genomic differences between two extensively studied rat strains provides the starting point for complete elucidation, at the molecular level, of the physiological and pathophysiological phenotypic differences between individuals from these strains.
The laboratory rat was the first mammalian species domesticated for scientific research and has been used as an animal model for physiology, toxicology, nutrition, behavior, immunology, and neoplasia for >150 yr (Jacob 1999; Aitman et al. 2008). Since development of the first inbred rat strain by King in 1909 (Lindsey 1979), over 500 inbred rat strains have been developed for a wide range of physiological phenotypes and different disease models. The spontaneously hypertensive rat (SHR) has been inbred over 130 generations and is the most widely studied animal model of human hypertension. In addition to hypertension, the SHR displays many other physiological and pathophysiological phenotypes, scores of which have been mapped to the genome as quantitative trait loci (QTLs) (Rat Genome Database; http://rgd.mcw.edu/). For some of the QTLs, the underlying genes and nucleotide variants have been identified (Aitman et al. 2008).
In the early 1980s, the SHR/Ola strain was crossed with the normotensive Brown Norway (BN.Lx) strain to create the BXH/HXB panel of recombinant inbred (RI) strains (Pravenec et al. 1989, 2004). RI panels are powerful and renewable resources for genetic mapping that offer the opportunity to accumulate genetic and physiological data over time. By integrating gene expression profiling and linkage analysis, thousands of expression quantitative trait loci (eQTLs) have been mapped in several tissues in the BXH/HXB RI panel (Hubner et al. 2005; Petretto et al. 2006). Subsequently, the integration of eQTL and physiological data led to identification of Cd36, Ogn, and Ephx2 as genes for insulin resistance, cardiac hypertrophy, cardiac failure, and hypertension in SHR or SHR-derived strains (Aitman et al. 1999; Monti et al. 2008; Petretto et al. 2008; Pravenec et al. 2008a).
The draft genome sequence of the reference rat strain, BN/SsNHsd/Mcwi, which is phylogenetically closely related to BN.Lx (The STAR Consortium 2008), was sequenced using a combined whole-genome shotgun and BAC sequencing strategy (Gibbs et al. 2004). Although sequencing of the BN genome has facilitated genetic studies in the rat (Aitman et al. 2008), progress in identifying SHR QTL genes, and therefore understanding the molecular basis of SHR phenotypes, has been limited by the continuing absence of the SHR genome sequence.
A number of human genomes have recently been resequenced using “next-generation” sequencing, showing the high quality and accuracy that can be achieved with these sequencing platforms (Bentley et al. 2008; Campbell et al. 2008; Ley et al. 2008; Wang et al. 2008; Wheeler et al. 2008; Ahn et al. 2009). As humans are outbred and have diploid genomes, relatively high coverage levels are required, in particular for accurate and complete calling of heterozygous nucleotide variants. However, laboratory rat strains such as SHR have been inbred for more than 130 generations and are therefore homozygous at almost all loci across the genome. Lower depth of sequence coverage may therefore be required for such inbred genomes, because a single allele is present at almost all genomic locations, providing a high likelihood at relatively low coverage that almost all allelic variation can be detected compared with other inbred strains.
Here, we report the genome sequence of the spontaneously hypertensive rat using short-read next-generation sequence technology. We illustrate that with paired-end sequencing on the Illumina platform, a near complete catalog of variants between two rat strains can be generated. The data will greatly accelerate progress in identifying the genes, genetic variants, and molecular mechanisms underlying the complex phenotypes manifested in SHR and SHR-derived strains, and provide a first step toward a complete description of functional variations in this extensively studied model organism.
Results
Sequencing
We sequenced the SHR/OlaIpcv genome using paired-end sequencing on the Illumina Genome Analyzer (GAII). To minimize systematic bias in library preparation, three different paired-end libraries were prepared, two with a short insert size (∼200 bp) and one with a longer insert size (∼2000 bp). Post quality filtering, we sequenced a total of 33.78 Gb from 816.5 million reads with an average read length of 41 bases. Approximately 715.5 million (87.63%) reads were mapped to the BN reference genome (RGSC-3.4) using MAQ-0.6.6 (Li et al. 2008), of which 684.7 million reads were mapped to the 20 autosomes or X chromosome, while 30.83 million reads were mapped to the unassigned contigs. Approximately 33.71 million reads were exact duplicates of other sequences from the respective library, which may have arisen due to PCR amplification during library production. We excluded these reads from further analysis as they may affect the accuracy of single nucleotide polymorphism (SNP) calling and structural variant (SV) calling. After filtering duplicate reads, 681.8 million reads (28.17 Gb) could be mapped to the reference genome, equating to 10.7-fold coverage of the SHR/OlaIpcv genome (Table 1; Supplemental Fig. 1). In total, 99.22% of non-gap, non-N bases of the reference genome were covered by at least one read, and 97.70% of the non-gap, non-N bases of the reference genome were covered by at least three reads. There was no significant difference in mapping quality between the different libraries.
Table 1.
Sequencing and mapping statistics of the three libraries used for sequence generation

Calling and accuracy of single nucleotide and short indel variants
We identified single nucleotide variants between the SHR/OlaIpcv and BN reference genomes using the MAQ program and evaluated the reliability of variant calls by comparing the data generated on the Illumina platform, at varying read depths and varying consensus quality scores, with two complementary SNP data sets. We first evaluated the Illumina data against a set of 108 SNPs previously identified in this strain combination from 66,052 bp in 94 different genomic regions by conventional capillary sequencing (C Morrissey, M Johnson, and T Aitman, unpubl.), and, second, against a set of 20,283 SNPs genotyped in multiple rat strains by the STAR consortium using Affymetrix or Illumina SNP genotyping microarrays (The STAR Consortium 2008). Of 108 SNPs detected by capillary sequencing between SHR/OlaIpcv and BN.Lx, 104 (96.3%) were confirmed by Illumina GAII sequencing at read density ≥3 and consensus quality ≥30 (Fig. 1A; Supplemental Table 1). The four SNPs that could not be confirmed by Illumina sequencing either had read depth <3 (two SNPs) or the consensus quality was <30 (two SNPs). No false-positive SNPs were called in the GAII sequencing within the 66,052 bp of capillary sequence at this read depth and consensus quality score.
Figure 1.

Receiver operating characteristic (ROC) curves to determine optimal thresholds for SNP calling. Evaluation of sensitivity and specificity of SNP prediction using Illumina paired-end sequencing at various read depths and quality scores compared with 108 SNPs predicted in a 66-kb region of the genome sequenced using capillary sequencing (A) and the STAR SNP data set (B). Each curve represents a different read depth, and each point represents different consensus quality scores.
Of the 20,283 STAR consortium SNPs, 13,627 had been previously determined to be polymorphic between SHR/OlaIpcv and the BN reference, while 6461 were nonpolymorphic and the remainder had not been genotyped in SHR/OlaIpcv. At consensus score ≥30 and read depth ≥3, the GAII data detected 13,349 of the 13,627 SNPs previously assigned as polymorphic (97.96% sensitivity). Of the 6461 positions previously assigned as nonpolymorphic, the GAII data correctly assigned 6399 as nonpolymorphic (99% specificity; Fig. 1B; Supplemental Table 2), whereas 62 were apparent false positives. However, for the 62 apparent false-positive SNPs, the GAII consensus quality score was high (mean score 59.3) with high read depth (mean depth 12.3 reads). In addition, for 51 of these SNPs, the STAR BN/SsNHsd/Mcwi allele was discordant with the BN reference genome allele (Supplemental Table 3), suggesting that these apparent false-positive SNPs arose due to sequencing errors in the BN reference genome (RGSC-3.4). We therefore assessed the extent to which errors in the BN reference genome sequence could have accounted for false-positive SNPs between SHR/OlaIpcv and the BN/SsNHsd/Mcwi strain. STAR genotypes for the 16,920 SNPs showed a concordance rate of 99.2% between the BN/SsNHsd/Mcwi genotype and the BN reference sequence, indicating that sequence errors in the BN reference sequence are likely to have a small effect (<1%) on the specificity and accuracy of SNPs detected in this study between SHR/OlaIpcv and BN.
The high sensitivity and specificity obtained at read density ≥3 and consensus quality ≥30 suggested that these thresholds were optimal for variant calling. Using these thresholds, 3,642,090 genomic locations (0.15%) were determined to be variant between the SHR/OlaIpcv and the reference BN sequence, 2,416,312,168 (97.35%) genomic positions were identical between the two genomes, and 61,981,672 (2.5%) positions remain undetermined. Of 3,642,090 genomic locations that were variant between the SHR/OlaIpcv and reference BN genomes, 3,590,437 SHR/OlaIpcv alleles were homozygous and 51,653 were heterozygous. Therefore, the proportion of all nucleotides in the SHR genome that were called as heterozygous is 2.1 × 10−5. The average read depth was 10× for homozygous SNP locations and 24× for heterozygous SNP locations.
To predict short indels, reads that remained unmapped to the reference BN genome by MAQ alignment were mapped to the reference sequence using BLAT (Kent 2002), which allows gapped alignment. Using the reads aligned with gaps to the reference BN genome, either by BLAT or MAQ, we identified 343,243 short indels of ≤15 bp.
Structural variant calling and validation
Structural variants (SVs), defined as insertions or deletions >50 bp, were predicted using mapping information of abnormally mapped read pairs either with improper span size or orientation. We predicted a total of 13,438 deletions in SHR/OlaIpcv compared with the BN reference. An illustration of the schema and data underlying prediction of deletions are shown in Figure 2A. The majority of deletions (n = 10,416) are <1 kb in size, while 3022 deletions were >1 kb (Fig. 2B), with a maximum deletion size of 1.2 Mb. The majority of deleted sequences were repeat elements, as previously found in human genome resequencing projects (Campbell et al. 2008; Wang et al. 2008). We also found that the distribution of the type of repeat in deleted sequences is nonrandom and depends on the size of the deletion, with simple sequence repeats and short interspersed nuclear elements (SINEs) mostly represented in deletions <270 bp, and long interspersed nuclear elements (LINEs) and long terminal repeats (LTRs) in deletions >270 bp (Fig. 2C).
Figure 2.

Method of calling, distribution, and repeat content of SHR/OlaIpcv deletions. (A) Deletion calling in SHR/OlaIpcv compared with the reference BN genome. SHR/OlaIpcv read pairs (dark blue) align with expected span size to the BN reference (top black line). (Red curve) SHR/OlaIpcv read pairs that align with span size greater than expected, (red box) region of deletion, (brown vertical lines) illustrative SNPs between the SHR/OlaIpcv and BN reference genomes, (green vertical lines) short indels, (light blue arrow) a gene in the BN reference. (B) Distribution of length of deletions identified in the SHR/OlaIpcv genome. (C) Rank analysis of repeat content in SHR deletions; each line represents a different type of repeat element. Deletions were ranked according to size and separated into bins of 500 deletions. Median size of deletion within each bin was plotted against content for each type of repeat element.
To validate SVs we randomly selected 79 deletions from the SHR/OlaIpcv compared with the BN reference for validation by PCR and, where required, conventional capillary sequencing. PCR primers were designed outside the region of the deletion in both the SHR and BN genomes (Supplemental Table 4). The genomic sequence was determined in both strains, and 75 of the 79 deletions were confirmed. Functioning PCR assays could not be designed for the remaining four deletions.
We also predicted 835 insertions in SHR/OlaIpcv compared with the BN reference from the long insert library (Supplemental Table 5) and a further 4933 probable insertions from the short insert libraries using reads where only one read of the read pair is mapped to the reference genome (hanging reads). We predicted a smaller number of insertions as compared with deletions, first, because of the smaller number of paired-end reads from the long insert library, and, second, because we can only predict insertions with a length smaller than the insert size. In addition, prediction of insertions from hanging reads is uncertain because hanging reads may arise due to other mechanisms such as high sequence variability or reduced sequence quality in one of the reads. Finally, we predicted 366 inversions using read pairs that mapped with abnormal orientation.
Copy number variation
Using a simulation method based on read coverage (Campbell et al. 2008), we predicted 588 copy number variants (CNVs) between the SHR/OlaIpcv and BN genomes, ranging in length from 7 kb to 1.4 Mb (Supplemental Table 6). To validate our CNV prediction, we compared the CNVs predicted using read coverage with a set of 134 CNVs (Supplemental Table 7) identified by array comparative genome hybridization (aCGH).
The 588 CNVs detected by read coverage included 101 of the 134 CNVs observed in the aCGH data, including the CNV previously reported at the Cd36 locus (Supplemental Fig. 2; Aitman et al. 1999; Glazier et al. 2002). Because our CNV predictions could have been affected by collapses in the BN reference genome assembly, we determined how many of the predicted CNVs fell within regions of reported collapse (Guryev et al. 2008). We found that only 84 of the 588 CNVs fell within these regions.
Distribution and density of variants
The origin of the laboratory rat has been partially documented (Lindsey 1979; Krinke 2000), with increasing genetic evidence showing that BN is the most divergent amongst laboratory inbred strains (The STAR Consortium 2008). Our sequence data allowed us to revisit this question. We will use the term “observed strain difference” (OSD) to describe the mean density of single nucleotide variants between two strains adjusted for the ability to discover each variant. This term is therefore analogous to heterozygosity measured as the proportion of segregating polymorphic sites in a freely mating population.
We correlated the SNP-based OSD index across the genome with the density of short indels and SVs per megabase. Short indels and SVs both showed strongly positive correlation with OSD (R2 = 0.79 and R2 = 0.35, respectively; Fig. 3A,B). Short indels were also correlated with SV density (R2 = 0.43; Fig. 3C).
Figure 3.

Relationship between distribution of different types of sequence variants. Density of sequence variants was calculated in 1-Mb windows of the BN reference genome, normalized to the number of bases in the window that were covered at threefold coverage or greater in the SHR/OlaIpcv sequence. (A) Correlation between SNP density and indel density. (B) Correlation between number of deletions per megabase and SNP density. (C) Correlation between number of deletions per megabase and indel density.
The distribution of OSD, normalized over 100-kb windows, shows striking dichotomy into regions with low mean SNP density (0–0.0004) and regions with high OSD (0.0004–0.0032), with a long tail with much higher OSD (up to 0.01; Fig. 4A). The regions of low SNP density are mainly accounted for by three large chromosomal regions of near identity between SHR/OlaIpcv and BN. These three regions, on chromosome 2 (124.6 Mb to 133.8 Mb), chromosome 13 (92.2 Mb to the telomere [111.1 Mb]) and chromosome 15 (32.8 Mb to 53.4 Mb), each has a mean OSD < 0.00008 and show very low density for all types of genomic variation (Fig. 4B). Since there were few if any SNPs between the SHR/OlaIpcv and the BN reference sequence in these regions, we looked at the distribution of SNPs genotyped across Wistar-derived strains (including SHR and SHRSP) by The STAR Consortium (2008) to provide information about the ancestry of these segments in SHR. On chromosome 2, SHR alleles were mostly identical to alleles in SHRSP and WKY strains. On chromosome 13, SHR alleles were identical to SHRSP alleles, whereas WKY alleles mostly differed from SHR or SHRSP alleles. On chromosome 15, SHR alleles differed consistently from SHRSP alleles, whilst WKY alleles were separated into two distinct haplotypes, some of which were identical to SHR, the remainder being identical to SHRSP (Supplemental Table 8).
Figure 4.

Distribution and density of sequence variants. (A) Distribution of SNP density across the SHR/OlaIpcv genome, in 100-kb windows, calculated as in Figure 3. (B) Distribution of density of SNPs (green), indels (red), and larger deletions (blue inner circle) on SHR/OlaIpcv chromosomes 2, 13, and 15. (Blue outer circle) Sequence coverage of the SHR/OlaIpcv genome, (outermost circle) chromosomal banding, (innermost red bars) copy number variations.
The proportion of single base pair differences provides an estimate of the divergence between two strains. The divergence between SHR/OlaIpcv and reference genome BN was 0.0015. The divergence between the reference human genome (NCBI build 36.1) and each of the five personal sequenced genomes (Levy et al. 2007; Bentley et al. 2008; Wang et al. 2008; Wheeler et al. 2008; Ahn et al. 2009) was in the range of 0.00108–0.00145 (Supplemental Table 9). However, the divergence between the 11 laboratory mouse strains (Frazer et al. 2007) was in the range of 0.00061–0.00080 (Supplemental Table 10). This shows that at least between the SHR and BN rat strains there is higher diversity than the divergence between a range of different laboratory strains of mouse, and that the divergence of these two rat strains is comparable to the divergence between human populations.
Functional significance of coding sequence variants
The possible functional effects of both small and large variants can be categorized according to their location in the BN reference sequence in relation to annotated genes. We used Ensembl build 54 to determine likely consequences of genetic variation on the gene set. The genomic variants in the coding sequence were prioritized as they may explain some of the previously observed phenotypic differences between the BN and SHR.
A total of 60 genes was completely deleted from the SHR/OlaIpcv genome sequence compared with BN (Supplemental Table 11). While the majority of these genes are either genes with unknown function (33 genes), genes encoding ribosomal proteins (11 genes), or olfactory receptors (one gene), 15 of the genes have been assigned distinct rat gene symbols for which there is at least a partial functional characterization. Of these 15 genes, although some may be incorrectly annotated as protein-coding genes in Ensembl, the majority would be expected to code for functional proteins.
Mouse orthologs (one-to-many or many-to-many) are reported in Ensembl for 33 of the 60 genes that are completely deleted in SHR, and for six of these (Ybx1, Dstn, Il13ra1, Gapdh, Ppp2ca, and Nlk), the respective mouse orthologs have been deleted in knockout mouse models. Deletion of all six of the SHR genes was confirmed by PCR and direct sequencing (data not shown). Three of the six genes show strong homology (>90% amino acid identity) across mammals (Supplemental Table 12); one of the genes, Ppp2ca, shows >85% amino acid identity down to Caenorhabditis elegans and Drosophila. The functional implications of these SHR gene deletions and their potential effects on cardiovascular and metabolic phenotypes in the respective knockout mice are presently unclear but merit further investigation, as do the partial gene deletions in a further 47 genes that show deletion of one or more coding exons from the SHR/OlaIpcv genome (Supplemental Table 13).
A total of 1,103,149 SNPs were located within the transcribed portion of the genome, of which 27,340 SNPs were located in gene coding regions, 8782 were in 5′ or 3′ untranslated regions (UTRs), and the remainder were lying within introns. Of the coding SNPs, a total of 11,542 were nonsynonymous SNPs and 15,798 were synonymous. Within the set of nonsynonymous SNPs, a total of 161 SNPs create or abolish stop codons, 153 result in gain of a stop codon, and eight result in loss of a stop codon in the SHR/OlaIpcv compared with the BN reference (Supplemental Table 14). 3358 SNPs were located within splice sites (1–3 bp into exons or 3–8 bp into introns), with 213 of those affecting essential splice sites (first 2 bp or last 2 bp of an intron) (Table 2).
Table 2.
Classification of SNPs in different categories based on location in the genome

We detected a total of 343,243 short indels of <15 bp, of which 629 affected the coding sequence of 771 transcripts from 550 genes (Supplemental Table 15). The length of 67 indels was an exact multiple of 3 nucleotides (nt), resulting in in-frame deletion/insertion in 87 transcripts from 67 genes, whereas 562 indels were not in multiples of 3 nt, leading to predicted frameshifts in the open reading frame of 483 genes. Considering all the frameshift and stop codon changes together, 165 of these resulted in a stop gain/loss or frameshift in the first 20% of the open reading frame, suggesting that for these genes at least, the encoded proteins would have a major loss of function.
Gene Ontology (GO) analysis of the 788 distinct genes affected by complete or partial deletion of gene, stop gain/loss, frameshifts, or inframe indels showed enrichment of genes related to ion transport (P = 6.51 × 10−6, false discovery rate [FDR] = 0.0099) and transport (P = 1.91 × 10−5, FDR = 0.029; Supplemental Table 16). The cellular component ontology showed enrichment for gene products localized to the plasma membrane (P = 6.44 × 10−5, FDR = 0.087; Supplemental Table 17).
688 genes partially or completely overlapped with copy number variable regions (CNVR) (Supplemental Table 6). GO analysis of these genes showed significant enrichment of genes related to (1) antigen processing and presentation of peptide antigen (P = 2.40 × 10−17, FDR = 3.68 × 10−14), (2) regulation of neurological process (P = 9.82 × 10−12, FDR = 1.51 × 10−8), (3) myeloid leukocyte activation (P = 7.07 × 10−12, FDR = 1.09 × 10−8), (4) response to mechanical stimulus (P = 1.16 × 10−10, FDR = 1.79 × 10−7), and (5) regulation of transport (P = 1.32 × 10−9, FDR = 2.03 × 10−6).
Identification of transcription start sites
To date, transcription start sites (TSSs) have been poorly defined in the rat genome. Cap analysis of gene expression (CAGE) tags are 25-nt sequence tags derived from mRNA sequenced in proximity of the cap site, and their mapping to unique genomic locations provides an accurate localization of TSSs (Carninci et al. 2006). Approximately 16.4 million CAGE tags were sequenced from two rat tissues (adult brain and rat embryonic tissue), of which 10.7 million tags were mapped to the BN reference genome. Clustering of overlapping tags resulted in 1,819,003 tag clusters, and 353,987 unique tag clusters had more than three tags, each cluster representing a unique TSS. Of 31,099 transcripts represented on the Affymetrix rat 230 2.0 array, 26,754 transcripts could be mapped reliably to a unique location on autosomal chromosomes in the BN reference genome (RGSC-3.4). We were able to adjust the transcription start site for 13,664 genes using rat CAGE tags (Supplemental Table 18). For the remainder, either the TSS in brain and embryos are located >1 kb upstream or 500 bp downstream of the known annotated genes, or the genes are not sufficiently expressed in these tissues to generate CAGE tags.
Genome diversity and distribution of cis-eQTLs
Expression QTLs (eQTLs) are genetic loci that have been shown by gene expression and linkage analysis to affect variation in gene expression levels. Cis-eQTLs are caused by genomic sequence variants that reside within or close to the gene itself (Hubner et al. 2005; Petretto et al. 2008; Cookson et al. 2009). We used the large eQTL data sets (Petretto et al. 2006) mapped across seven tissues in the BXH/HXB panel of rat RI strains that are of SHR and BN ancestry, to explore further how sequence variation correlates with variation in gene expression. Of the 26,754 transcripts on the Affymetrix 230 2.0 array that mapped reliably to a unique location on autosomal chromosomes in the BN reference genome, 3045 transcripts represent cis-eQTL genes (physical location of gene within 10 Mb of the peak of linkage), detected with genome-wide significance PGW = 0.05 and FDR of 0.05, as previously described (Hubner et al. 2005; Petretto et al. 2006).
We defined the gene region as the region between the TSS and the annotated transcript end site with 2 kb of flanking region upstream and downstream and estimated density of SNPs, short indels, and larger deletions (>50 bp) in the gene regions of all 26,754 genes. The TSS was defined by CAGE tag analysis as above, or, if not so defined, as in Ensembl build 54. The SNP density (Fig. 5A) and indel density (Fig. 5B) in cis-eQTL gene regions were significantly higher than in non-cis-eQTL gene regions (Mann–Whitney–Wilcoxon [MWW] test P-value for both comparisons < 2.2 × 10−16). In addition, 552 of 3045 cis-eQTL genes (18.1%) overlap with one or more larger deletions, compared with 2861 of 23,709 non-cis-eQTL genes (12.1%; Fisher's exact test, P = 2.02 × 10−19). The enrichment of SNPs, indels, and deletions in cis-eQTL gene regions remained highly significant (all P-values < 10−10) after removing so-called “spurious” eQTLs (Doss et al. 2005) from the data set. Cis-eQTL gene regions are therefore significantly enriched for SNPs, short indels, and larger deletions, suggesting that some of these variants have functional effects on gene expression.
Figure 5.

SNP and indel enrichment in cis-eQTL gene regions and promoters. (A) Boxplot showing SNP density in gene regions containing cis-eQTL genes compared with regions containing only non-cis-eQTL genes. (B) Boxplot showing indel density in cis-eQTL-containing gene regions compared with non-cis-eQTL-containing gene regions. (C) Boxplot showing SNP density in a 10-kb region of the promoter, centered on the transcription start site, of cis-eQTL genes and non-cis-eQTL genes. (D) Distribution of SNP density between cis-eQTL genes and non-cis-eQTL genes in a 40-kb region surrounding the transcription start site. (Boxes and whiskers) Median, interquartile range, and 95th percentile.
Within the promoter regions (defined here as 5 kb upstream and downstream of the TSS) of cis-eQTL genes, there was significant SNP enrichment as compared with promoter regions of non cis-eQTL genes (MWW test, P < 2.2 × 10−16; Fig. 5C). Interestingly, SNP density was highest in the 1 kb upstream of the TSS and fell inconsistently further upstream of the TSS. However, at a distance ≥10 kb downstream of the promoter, SNP density fell more consistently, though not to levels seen in the regions of non-cis-eQTL genes (Fig. 5D).
Discussion
Sequencing and variant calling
We have sequenced the SHR/OlaIpcv genome at ∼10.7-fold coverage and identified 3.6 million SNPs between the SHR/OlaIpcv and BN reference genomes. Independent, de novo sequencing in this study of 66 kb of BN and SHR/OlaIpcv genomic DNA and previous independent genotyping (The STAR Consortium 2008) confirmed that >99% of these SNPs are true-positive sequence variants between these two inbred strains. The SHR/OlaIpcv strain is highly inbred, and therefore almost all of the nucleotides in the SHR/OlaIpcv genome are anticipated to be homozygous at each individual genomic location. It has previously been reported in sequencing studies of human genomes that most homozygous SNPs can be predicted with a very low error rate using paired-end reads at 10-fold coverage (Wang et al. 2008). Taken together, these data suggest that this level of coverage is sufficient to determine a very high proportion of all sequence variants between SHR/OlaIpcv and the BN reference strain at high accuracy and high sensitivity.
We observed a low but finite frequency of heterozygosity in the SHR/OlaIpcv genome sequence. This may be true heterozygosity, caused either by incomplete fixation during inbreeding or new mutations in the SHR/OlaIpcv strain. Alternatively, apparent heterozygosity may have arisen by collapsing reads from regions of segmental duplication in the SHR/OlaIpcv sequence, as appears likely from the increased read depth observed here and previously at heterozygous SNP locations (Sudbery et al. 2009). It is also possible that recent segmental duplications have been collapsed in the BN assembly, resulting in incorrect mapping of near-identical reads.
Paired-end sequencing provides an advantage over single-fragment sequencing in being able to predict short indels (1–15 bp) and SVs (>50 bp). The combination of different length insert libraries is complementary for SV prediction and allows accurate identification of shorter as well as longer size deletions. In addition, large insert-size libraries permit prediction of insertions that are smaller than the size of the insert. Probable regions of insertions can also be predicted using hanging reads. However, such reads can also arise either due to regions of very high diversity between the two strains or due to the regions of low-quality sequence in the reference genome. The majority of deletions that we detected were deletions of repeat elements. The identification of deletions or insertions of repeat elements is of particular interest in light of the recently identified role of retrotransposons in the regulation of gene expression (Faulkner et al. 2009). Specific difference in expression of protein-coding genes between the SHR/OlaIpcv and reference BN may therefore be influenced by the action of expressed repeat elements.
In our data set, we identified 588 CNVs, the smallest of which were 7 kb in size. This included 101 of 134 CNVs detected previously by aCGH, the smallest of which was 22 kb. A large proportion (244 of 588) of the CNVs that were not detected by aCGH were <22 kb in length, indicating that the present platform provides higher resolution for CNV prediction than the current generation of aCGH platforms for the rat (Supplemental Fig. 3). An additional feature of this analysis is that the breakpoints can be detected at nucleotide resolution, which provides a simple route to validation. This level of resolution may also help to understand further the mechanisms that are proposed to lead to complex structural rearrangements (Lee et al. 2007; Hastings et al. 2009; Zhang et al. 2009). By combining SV and CNV prediction algorithms, we believe that most deletions, short insertions, and CNVs in the SHR compared with the genome BN have been detected, though detection of medium-sized deletions (15–50 bp) and large insertions in the SHR genome would require additional sequencing or comparative analyses.
The BN reference sequence contains only limited amounts of finished sequence, with relatively low overall coverage (∼7×) (Gibbs et al. 2004) and areas of reported sequence collapse (Guryev et al. 2008). Incompleteness of the BN sequence or errors in the BN genome assembly may therefore affect the accuracy of indels and CNVs detected between SHR/OlaIpcv and BN. We believe we have filtered out most of the likely sources of errors by removing deletions that were detected around BN assembly gaps, filtering out insertions that were predicted from hanging reads around BN assembly gaps, and highlighting CNVs that overlap with reported collapse or overprediction in the BN assembly. This assertion is justified by our validation data on indels and CNVs.
Distribution and density of sequence variants
The density of SNPs and indels, and the number of SVs per megabase between SHR/OlaIpcv and BN, were strongly positively correlated with one another across the genome. This correlation is most likely biologically driven rather than due to technical confounding, because in the case that paired-reads that lead to prediction of a SV would not align to the reference genome in highly divergent regions, this would tend to have the opposite effect, i.e., a negative correlation between SNP density and SV density. The simplest explanation is that the SVs were present in the founding common population of SHR/OlaIpcv and BN, and that the process of selecting for an inbred strain has captured more variants at both the small and larger scales when by chance the haplotypes selected had a deeper coalescence in this population. However, we cannot rule out other processes, for example that the mechanism for generating structural variation is mechanistically correlated with changes in single nucleotide mutation rate.
The presence of large runs of near identity on chromosomes 2, 13, and 15, referred to as “SNP deserts” (Miller et al. 2001), most likely represents recent common ancestry of these genomic segments in the two strains, probably due to introgression of BN chromosomal segments at an early stage in the derivation of current Wistar-derived stocks. The retention of different BN haplotype blocks on different chromosomal segments in the current Wistar-derived strains is consistent with this hypothesis. Anecdotally, SNP deserts have been previously recognized between Wistar-derived strains (of which SHR is one) and the reference BN genome (The STAR Consortium 2008). The present SHR/OlaIpcv genome sequence confirms this observation and suggests that recent introgression of BN genomic DNA during the derivation of Wistar-derived strains provides the likely explanation.
Overall, we estimate that the sequence diversity between SHR/OlaIpcv and the BN reference is comparable to that shown in human populations. In contrast to mouse strains, which show a complex pattern of diversity between any two strains due to the breeding history from isolated subspecies in the mouse, the SHR/OlaIpcv and BN genomes show a smooth distribution of diversity outside the three large regions of close sequence identity.
Genome diversity and regulation of gene expression
Our extensive gene expression and eQTL data sets in the SHR/OlaIpcv × BN strain combination (Hubner et al. 2005; Petretto et al. 2006) permit a preliminary analysis of the functional significance of sequence variations found here between the SHR/OlaIpcv and BN genomes. We found that the density of SNPs, indels, and SVs was significantly higher in genomic regions containing cis-eQTL genes than in regions that contained non-cis-eQTL genes. This result was unchanged after exclusion of genes that had SNPs in the gene expression microarray probe-binding regions. An increase in SNP density in the region of cis-eQTL genes has previously been reported in yeast, although this was restricted to the region immediately upstream of the TSS. Our data showed high sequence diversity across the entire regions of cis-eQTL genes, with only modest enrichment in the immediate vicinity of the TSS (Fig. 5D). The strong association that we observed between SNP density and regions containing cis-eQTLs is consistent with previous observations of high sequence diversity in cis-eQTL regions in the mouse (Doss et al. 2005), and with the view that sequence variants in the vicinity of cis-eQTL genes are likely to have functional effects on interstrain variability in gene expression.
Functional significance of detected genomic variants
Our data provide a unique resource for investigation of the ways in which interindividual genomic differences can influence whole organism phenotypes. We found 788 SHR genes affected by major coding sequence mutations, including 60 genes that were completely deleted from the SHR/OlaIpcv genome. Of the 788 genes showing a coding region frameshift or alteration of stop codon usage, there was striking enrichment for genes related to ion transport, transport, and plasma membrane localization. Additionally, of the 688 genes that overlap with regions showing CNV, there was a strong overrepresentation of genes for immunological, neurological, or mechanical functions. Enrichment of these gene classes could be a general feature of variation between rat strains that would be found in a sequence comparison of any two strains. However, the high statistical significance of these functional enrichments, coupled with the known metabolic, cardiovascular, and neurobehavioral phenotypes described in SHR (Okamoto 1972), suggests that enrichment of these gene classes may relate causally to the phenotypes manifested by SHR/OlaIpcv and other SHR-derived strains.
Previously, integration of microarray expression data with QTL linkage mapping and physiological studies in experimental crosses and congenic strains led to identification of CNV in the rat Cd36 gene as a cause of CD36 deficiency, insulin resistance, dyslipidaemia, and hypertension in the SHR strain (Aitman et al. 1999; Pravenec et al. 2001, 2008a). The present sequence data also demonstrated CNV in the SHR Cd36 gene, confirming the previous report of CNV at this locus (Glazier et al. 2002) and supporting the hypothesis that the observed enrichment in membrane transport proteins, such as Cd36, plays a significant part in the development of SHR cardiovascular and metabolic phenotypes. It also highlights the potential functional significance of other CNVs detected in the SHR/OlaIpcv genome (Supplemental Table 6).
Osteoglycin (Ogn) was recently identified by combined use of linkage and eQTL analyses as a regulator of left ventricular mass in SHR/OlaIpcv (Petretto et al. 2008). The Ogn gene shows many sequence variants in SHR in the 5′ UTR, exon 3, and 3′ UTR, along with two insertions in introns and 2-bp and 47-bp deletions in the 3′ UTR. Our SHR/OlaIpcv sequence was able to detect all Ogn mutations and indels except the 47-bp deletion, which was not detected because it is out of the size range of both short indels and SVs that could be detected in this study. Similarly, we were able to detect several of the likely causal polymorphisms in the promoter or coding region of Gstm1 (McBride et al. 2005) and Srebf1 (Pravenec et al. 2008b), which were positionally cloned as QTL genes in SHR or the related SHRSP strain (Aitman et al. 2008).
In our sequence data, Cacna1d, a gene encoding the Cav1.3 (α1D) L-type Ca2+ channel, showed a 4-bp deletion at the start of exon 11 that includes 3 bp of the exon and 1 bp of the splice site. The Cacna1d gene also shows mutation in an essential splice site between exons 15 and 16. These mutations are likely to reduce greatly or even abolish the function of the encoded gene product. Cav1.3 (α1D) is a key regulator of calcium homeostasis, electrical activity, and maintenance of normal rhythm in the heart (Chahine et al. 2008; Mancarella et al. 2008), and in Cacna1d knockout mice, gene deletion is associated with hypoinsulinaemia, glucose intolerance, and decreased number and size of pancreatic islets (Namkung et al. 2001). It is therefore of interest that genome-wide association analysis in humans has shown an association between polymorphism in the Cacna1d gene and type 2 diabetes (Sookoian et al. 2009) and suggests that investigation of the functional consequences of Cacna1d mutations in SHR/OlaIpcv would be worthwhile.
We also found a single base pair deletion that results in a frameshift in the coding region of the Cacna1a gene, which encodes the voltage-dependent Cav2.1 (α1A), P/Q-type Ca2+ channel. This calcium channel is expressed in vascular smooth muscle cells, is important for the contraction of renal resistance vessels (Andreasen et al. 2006), and could therefore be important in blood pressure regulation in SHR/OlaIpcv, particularly given that calcium homeostasis is known to be dysregulated in SHR smooth muscle (Wilde et al. 1994; Manso et al. 1999).
Other transporters also show major coding sequence mutations in SHR. The SHR gene Kcnj1, which encodes the inwardly rectifying potassium channel, contains three different single-base deletions in its coding region. The impact of these mutations on hypertension in SHR/OlaIpcv is of particular interest as the human ortholog has been associated with systolic or diastolic blood pressure in human genome-wide association studies (Tobin et al. 2008).
Recent data from human genetic studies have shown that insertion of retrotransposons in regulatory regions or introns can markedly reduce expression of the primary transcript (Han et al. 2004; Ustyugova et al. 2006; Faulkner et al. 2009). Our data detected 13,438 chromosomal deletions, 73.7% of which are due to loss or gain of retrotransposons such as LINE, SINE, or endogenous retrovirus sequences. However, the pathophysiological significance of this class of structural variants in humans is not clear. We found an ∼6-kb intronic deletion of a LTR, class II endogenous retroviruses (ERV-Class II), in the SHR Echdc2 gene, whose product catalyzes the second step in the physiologically important beta-oxidation pathway of fatty acid metabolism (Agnihotri and Liu 2003). This gene, which has no mutations in the coding region, is overexpressed by up to 31-fold in various SHR tissues compared with BN.Lx (Hubner et al. 2005). Similarly, we found deletion of a LINE element between the fourth and fifth exon of the gene Cyp4a8 gene, a hypertension candidate gene that shows 2.5-fold higher expression in SHR kidney than in BN.Lx (Yamaguchi et al. 2002; Hubner et al. 2005; Dunn et al. 2008). It would seem worthwhile to investigate whether these alterations in gene expression are caused by the associated ERV or LINE deletions in the SHR genome.
The 3,642,090 polymorphisms, along with 343,243 indels, 19,572 SVs, and 588 CNVs observed between the SHR/OlaIpcv and Brown Norway genomes provide an almost complete catalog of genomic differences between these two strains with strikingly different physiology, particularly in cardiovascular phenotype. As these are experimentally amenable animals, present in renewable resources, and with a large number of pre-existing and ongoing functional studies, one can now consider the complete problem of assigning every functional change between these two strains to one or more polymorphic changes in their genomes, and, as a consequence, start to build a catalog of functional impacts for each described variant. Multiple approaches will be needed to achieve this assignment, but this sequence will provide the starting point for complete functional elucidation between two individuals at the molecular level, and thus provide an unprecedented tool for elucidation of the extensively studied pathophysiological phenotypes manifested by SHR.
Methods
Rat strains used for sequencing
The SHR/Ola strain was imported to the Institute of Physiology, Czech Academy of Sciences in 1980 from OLAC Blackthorn Bicester, UK at F48 to create the inbred SHR/OlaIpcv strain. Two SHR/OlaIpcv females at generation F130 were used for DNA isolation from liver tissue and subsequent sequencing.
10K genotyping
To confirm the identity of the SHR/OlaIpcv female rats used for sequencing, we performed genotyping using the 10K Rat Array (Affymetrix). Genomic DNA was extracted from SHR/Olapcv liver tissue using a standard phenol–chloroform protocol. Genotyping was carried out as previously described (The STAR Consortium 2008). Briefly, genotyping was carried out using the GeneChip Scanner 3000 Targeted Genotyping System protocol from Affymetrix, originally described as MIP technology (Hardenbol et al. 2005), and genotypes were compared with previously determined data for SHR/OlaIpcv (The STAR Consortium 2008).
Illumina genome sequencing library preparations
Ten micrograms of genomic DNA from liver tissue of two female SHR/OlaIpcv rats was used to construct paired-end whole-genome shotgun libraries (WGSS-PE) with a 200-bp insert size. For large-insert mate-pair whole-genome shotgun library (WGSS-MP) construction, 5–10 μg of genomic DNA was used and libraries were prepared according to the manufacturer's instructions (Illumina). The resulting libraries were sequenced on an Illumina Genome Analyzer II following the manufacturer's instructions. Further details of library construction and sequencing are given in the Supplemental Methods.
Mapping to reference genome
We used MAQ-0.6.6 (Li et al. 2008) to align chastity quality-filtered paired-end reads to the BN reference genome (RGSC-3.4), which includes unassigned contigs along with the 20 autosomes and X chromosome. We used default parameters for the short-insert libraries RN001 and RN009; paired-end reads from the long-insert library RN0010 were mapped with insert size parameter set to 2500 bp.
SNP detection
Using MAQ, we produced a consensus sequence from the reads aligned to the BN reference genome. SNPs were called using the cns2SNP module of MAQ-0.6.6 with the following filtering criteria: minimum read depth of 3, minimum consensus quality of 30, minimum mapping quality of 60 for at least one read covering SNP, and no SNP call within a 3-bp flanking region around the potential indel.
Short indel prediction
Single reads that showed a gap in sequence alignment to the reference BN genome when mapped with either MAQ or BLAT (Kent 2002) were selected for prediction of indels in the SHR/OlaIpcv genome. Reads that remained unmapped to the reference BN genome after MAQ alignment were mapped to the BN reference genome using BLAT. Reads were selected from BLAT alignment that mapped to a unique location in the genome with a maximum of two mismatches or a single indel up to 15 bp. To be selected for indel prediction, reads that showed a gap either by MAQ or BLAT also had to satisfy paired-end mapping of the second read of the read-pair to the reference genome, in keeping with the expected library insert size. Indels were called only when the predicted indel size was a maximum of 15 bp, was called by at least four nonredundant reads, and the number of reads called as a gap (by either MAQ or BLAT) exceeded the number of reads called as a mismatch.
Structural variant calling
Larger deletions (>50 bp) were predicted using the paired-end reads mapped with outer distance >250 bp (2 SD above average insert size) for the short-insert libraries and 2500 bp (2 SD above average insert size) for the long-insert size library. A deletion was called only when at least four overlapping read-pairs mapped with mapping quality >30 with the outer distance greater than expected. To discard potential false positives we filtered deletions using the following criteria: (1) the deletion completely overlapped with another deletion; (2) unambiguous reads were mapped within the deletion boundaries; (3) the BN reference genome has 126,672 gaps covering 237 Mb of the genome. Due to erroneous gap size estimation in the reference genome, many read pairs mapped with distance greater than expected. Such read pairs may result in false prediction of deletions, and we therefore discarded all deletions that spanned within 100 bp on both sides of the gap.
Insertions were predicted from the long-insert library where read pairs were mapped with distance ≤750 bp. An insertion was called only when at least four reads mapped with mapping quality greater than 30 with distance ≤750 bp. Probable insertions were predicted from “hanging reads” in the short-insert library, where only one read of the read-pair mapped to the reference BN sequence. We filtered out all single reads that mapped around BN genome gaps and also chimeric reads. We called probable insertions only when the cluster of at least four single reads (mapping quality greater than 30) each on forward strand and reverse strand was observed with a distance between the forward and reverse cluster of <100 bp.
Copy number variation calling
When comparing a test to a reference genome sequence, the reference genome is not uniformly mappable across the genome and varies according to the degree of uniqueness and repetitiveness of individual chromosomal segments. To correct for varying levels of sequence uniqueness across the genome we simulated read pairs from the BN reference with varying length and insert size of 200 bp. These simulated reads were mapped back to the reference genome using MAQ-0.6.6. The reference genome was then divided into nonoverlapping, unequal-length windows in which a constant number of in silico-simulated bases were mapped (Campbell et al. 2008). We selected a constant number of 50,000 mappable bases per window, as it is equivalent to ∼5 kb of mappable sequence per window. Once the window boundaries were fixed, the number of bases from the SHR sequence mapping to each window was calculated. Based on the observed and expected number of bases mapping to each window the presence of CNVs was inferred using the DNAcopy program from R, which implements a circular segmentation algorithm (Venkatraman and Olshen 2007).
Array CGH data were generated on the NimbleGen platform using SHR/OlaIpcv and BN/SsNHSd/Mcwi genomic DNA. Data were normalized using the loess normalization method from the limma package in R. CNVs were called using the DNAcopy program in R.
Transcription start site identification
CAGE tag libraries were prepared from rat adult brain and embryonic tissue from Sprague–Dawley rats according to a novel nanoCAGE method (C Plessy, R Simone, N Bertin, G Pascarella, A Akalin, C Carrieri, A Vassalli, S Olivarius, J Severin, D Lazarevic, et al., in prep.). 16.4 million CAGE tags were mapped to the BN reference genome using MAQ-0.6.6. Tags mapping to multiple locations on the genome were discarded, as were tags that mapped to the reference genome with more than one mismatch. Tags that mapped less than 10 bp apart on the same strand were then built into clusters. To reduce false-positives, clusters having fewer than three tags were discarded. CAGE tag clusters were then selected for transcripts that were represented on the Affymetrix rat RAE 230 2.0 array that mapped within 1000 bases upstream or 500 bases downstream of the transcription start site as annotated (in order of priority) by Ensembl, NCBI, or Affymetrix. If a tag cluster was found to be present in this region, the maximum tag count position in the cluster was assigned as the new TSS.
Validation of SNP and accuracy
To validate SNPs, 94 targeted genomic regions of SHR/OlaIpcv and reference BN genome, covering 66,052 bp, were sequenced by traditional capillary sequencing, and SNPs were identified using Sequencher 4.8 software. We also used 20,283 STAR SNPs for validation. Sensitivity, specificity, and accuracy of SNP prediction were calculated as:
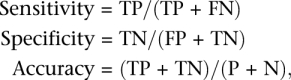 |
where TP = true-positive, TN = true-negative, FP = false-positive, FN = false-negative, P = total positives, and N = total negatives.
Validation of structural variants
A set of 79 deletions, ranging in size from 56 to 10,801 bp, was selected for validation by PCR. To generate PCR fragments, primer pairs outside the start and end position of all individual deletions were designed using the BN genomic reference sequence as template. Sites of deletions within the SHR/OlaIpcv genome versus the BN reference were sequenced using Sanger sequencing on an ABI 3730 sequencer using standard BigDye chemistry.
Detection of SNPs in Affymetrix probe-binding regions
To find probe sequences affected by variants within the probe binding regions of the Affymetrix rat RAE 230 2.0 array, we mapped all probes of the probe sets represented on the array to the BN reference genome using RMAP (Smith et al. 2008). Probe regions were then examined for the presence of SNPs.
Statistical and Gene Ontology analysis
All statistical analysis is carried out in R. Gene Ontology analysis was carried out using DAVID (Dennis et al. 2003; Huang et al. 2009).
Acknowledgments
T.J.A. received funding from the MRC, Wellcome Trust, British Heart Foundation Centre of Research Excellence, EU-funded EURATools Integrated Project, and the Leducq Foundation. M.P. is a Howard Hughes international research scholar and received grant (1M6837805002) support from the Ministry of Education of the Czech Republic. N.H. acknowledges National Genome Research Network (NGFN) Plus of the German Ministry for Science and Education for supporting this study in part. V.G. is supported by NWO Horizon grant no. 93519030.
Footnotes
[Supplemental material is available online at http://www.genome.org. The sequence data from this paper have been submitted to the EBI Sequence Read Archive (http://www.ebi.ac.uk/) under accession no. ERA000170. SNPs have been submitted to dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/) under batch id 2010-1_SHR. The complete set of SNPs is also available on Ensembl (ftp://ftp.ebi.ac.uk/pub/databases/ensembl/snp/rat/shr/). All variants are available in a custom database, SHRbase (http://shr.csc.mrc.ac.uk/) and will be available in the next release of Ensembl. The CGH array data have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE20102. The CAGE tag data have been submitted to the DDBJ Read Archive (http://trace.ddbj.nig.ac.jp/registered/) under accession no. DRA000155.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.103499.109.
References
- Agnihotri G, Liu HW 2003. Enoyl-CoA hydratase. Reaction, mechanism, and inhibition. Bioorg Med Chem 11: 9–20 [DOI] [PubMed] [Google Scholar]
- Ahn SM, Kim TH, Lee S, Kim D, Ghang H, Kim DS, Kim BC, Kim SY, Kim WY, Kim C, et al. 2009. The first Korean genome sequence and analysis: Full genome sequencing for a socio-ethnic group. Genome Res 19: 1622–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitman TJ, Glazier AM, Wallace CA, Cooper LD, Norsworthy PJ, Wahid FN, Al-Majali KM, Trembling PM, Mann CJ, Shoulders CC, et al. 1999. Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat Genet 21: 76–83 [DOI] [PubMed] [Google Scholar]
- Aitman TJ, Critser JK, Cuppen E, Dominiczak A, Fernandez-Suarez XM, Flint J, Gauguier D, Geurts AM, Gould M, Harris PC, et al. 2008. Progress and prospects in rat genetics: A community view. Nat Genet 40: 516–522 [DOI] [PubMed] [Google Scholar]
- Andreasen D, Friis UG, Uhrenholt TR, Jensen BL, Skott O, Hansen PB 2006. Coexpression of voltage-dependent calcium channels Cav1.2, 2.1a, and 2.1b in vascular myocytes. Hypertension 47: 735–741 [DOI] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, et al. 2008. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet 40: 722–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, Frith MC, et al. 2006. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet 38: 626–635 [DOI] [PubMed] [Google Scholar]
- Chahine M, Qu Y, Mancarella S, Boutjdir M 2008. Protein kinase C activation inhibits α1D L-type Ca channel: A single-channel analysis. Pflugers Arch 455: 913–919 [DOI] [PubMed] [Google Scholar]
- Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M 2009. Mapping complex disease traits with global gene expression. Nat Rev Genet 10: 184–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA 2003. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: 3 doi: 10.1186/gb-2003-4-5-p3 [PubMed] [Google Scholar]
- Doss S, Schadt EE, Drake TA, Lusis AJ 2005. Cis-acting expression quantitative trait loci in mice. Genome Res 15: 681–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KM, Renic M, Flasch AK, Harder DR, Falck J, Roman RJ 2008. Elevated production of 20-HETE in the cerebral vasculature contributes to severity of ischemic stroke and oxidative stress in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 295: H2455–H2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T, et al. 2009. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet 41: 563–571 [DOI] [PubMed] [Google Scholar]
- Frazer KA, Eskin E, Kang HM, Bogue MA, Hinds DA, Beilharz EJ, Gupta RV, Montgomery J, Morenzoni MM, Nilsen GB, et al. 2007. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature 448: 1050–1053 [DOI] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, et al. 2004. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493–521 [DOI] [PubMed] [Google Scholar]
- Glazier AM, Scott J, Aitman TJ 2002. Molecular basis of the Cd36 chromosomal deletion underlying SHR defects in insulin action and fatty acid metabolism. Mamm Genome 13: 108–113 [DOI] [PubMed] [Google Scholar]
- Guryev V, Saar K, Adamovic T, Verheul M, van Heesch SA, Cook S, Pravenec M, Aitman T, Jacob H, Shull JD, et al. 2008. Distribution and functional impact of DNA copy number variation in the rat. Nat Genet 40: 538–545 [DOI] [PubMed] [Google Scholar]
- Han JS, Szak ST, Boeke JD 2004. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature 429: 268–274 [DOI] [PubMed] [Google Scholar]
- Hardenbol P, Yu F, Belmont J, Mackenzie J, Bruckner C, Brundage T, Boudreau A, Chow S, Eberle J, Erbilgin A, et al. 2005. Highly multiplexed molecular inversion probe genotyping: Over 10,000 targeted SNPs genotyped in a single tube assay. Genome Res 15: 269–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR 2009. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327 doi: 10.1371/journal.pgen.1000327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Hubner N, Wallace CA, Zimdahl H, Petretto E, Schulz H, Maciver F, Mueller M, Hummel O, Monti J, Zidek V, et al. 2005. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat Genet 37: 243–253 [DOI] [PubMed] [Google Scholar]
- Jacob HJ 1999. Functional genomics and rat models. Genome Res 9: 1013–1016 [DOI] [PubMed] [Google Scholar]
- Kent WJ 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krinke GJ 2000. History, strains and models. In The laboratory rat (Handbook of experimental animals) (ed. Bullock G, Bunton TE), pp. 3–16 Academic Press, London [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247 [DOI] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254 doi: 10.1371/journal.pbio.0050254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. 2008. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 456: 66–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ruan J, Durbin R 2008. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18: 1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey JR 1979. Historical foundations in the laboratory rat. In The laboratory rat (ed. Baker HJ et al. ), pp. 1–36 Academic Press, New York [Google Scholar]
- Mancarella S, Yue Y, Karnabi E, Qu Y, El-Sherif N, Boutjdir M 2008. Impaired Ca2+ homeostasis is associated with atrial fibrillation in the α1D L-type Ca2+ channel KO mouse. Am J Physiol Heart Circ Physiol 295: H2017–H2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manso AM, Encabo A, Ferrer M, Balfagon G, Salaices M, Marin J 1999. Changes of cardiac calcium homeostasis in spontaneously hypertensive rats. J Auton Pharmacol 19: 123–130 [DOI] [PubMed] [Google Scholar]
- McBride MW, Brosnan MJ, Mathers J, McLellan LI, Miller WH, Graham D, Hanlon N, Hamilton CA, Polke JM, Lee WK, et al. 2005. Reduction of Gstm1 expression in the stroke-prone spontaneously hypertension rat contributes to increased oxidative stress. Hypertension 45: 786–792 [DOI] [PubMed] [Google Scholar]
- Miller RD, Taillon-Miller P, Kwok PY 2001. Regions of low single-nucleotide polymorphism incidence in human and orangutan Xq: Deserts and recent coalescences. Genomics 71: 78–88 [DOI] [PubMed] [Google Scholar]
- Monti J, Fischer J, Paskas S, Heinig M, Schulz H, Gosele C, Heuser A, Fischer R, Schmidt C, Schirdewan A, et al. 2008. Soluble epoxide hydrolase is a susceptibility factor for heart failure in a rat model of human disease. Nat Genet 40: 529–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung Y, Skrypnyk N, Jeong MJ, Lee T, Lee MS, Kim HL, Chin H, Suh PG, Kim SS, Shin HS 2001. Requirement for the L-type Ca2+ channel α1D subunit in postnatal pancreatic β cell generation. J Clin Invest 108: 1015–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K 1972. Spontaneous hypertension: Its pathogenesis and complications Igaku Shoin, Tokyo [Google Scholar]
- Petretto E, Mangion J, Dickens NJ, Cook SA, Kumaran MK, Lu H, Fischer J, Maatz H, Kren V, Pravenec M, et al. 2006. Heritability and tissue specificity of expression quantitative trait loci. PLoS Genet 2: e172 doi: 10.1371/journal.pgen.0020172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petretto E, Sarwar R, Grieve I, Lu H, Kumaran MK, Muckett PJ, Mangion J, Schroen B, Benson M, Punjabi PP, et al. 2008. Integrated genomic approaches implicate osteoglycin (Ogn) in the regulation of left ventricular mass. Nat Genet 40: 546–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravenec M, Klir P, Kren V, Zicha J, Kunes J 1989. An analysis of spontaneous hypertension in spontaneously hypertensive rats by means of new recombinant inbred strains. J Hypertens 7: 217–221 [PubMed] [Google Scholar]
- Pravenec M, Landa V, Zidek V, Musilova A, Kren V, Kazdova L, Aitman TJ, Glazier AM, Ibrahimi A, Abumrad NA, et al. 2001. Transgenic rescue of defective Cd36 ameliorates insulin resistance in spontaneously hypertensive rats. Nat Genet 27: 156–158 [DOI] [PubMed] [Google Scholar]
- Pravenec M, Zidek V, Landa V, Simakova M, Mlejnek P, Kazdova L, Bila V, Krenova D, Kren V 2004. Genetic analysis of “metabolic syndrome” in the spontaneously hypertensive rat. Physiol Res 53: S15–S22 [PubMed] [Google Scholar]
- Pravenec M, Churchill PC, Churchill MC, Viklicky O, Kazdova L, Aitman TJ, Petretto E, Hubner N, Wallace CA, Zimdahl H, et al. 2008a. Identification of renal Cd36 as a determinant of blood pressure and risk for hypertension. Nat Genet 40: 952–954 [DOI] [PubMed] [Google Scholar]
- Pravenec M, Kazdova L, Landa V, Zidek V, Mlejnek P, Simakova M, Jansa P, Forejt J, Kren V, Krenova D, et al. 2008b. Identification of mutated Srebf1 as a QTL influencing risk for hepatic steatosis in the spontaneously hypertensive rat. Hypertension 51: 148–153 [DOI] [PubMed] [Google Scholar]
- Smith AD, Xuan Z, Zhang MQ 2008. Using quality scores and longer reads improves accuracy of Solexa read mapping. BMC Bioinformatics 9: 128 doi: 10.1186/1471-2105-9-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sookoian S, Gianotti TF, Schuman M, Pirola CJ 2009. Gene prioritization based on biological plausibility over genome wide association studies renders new loci associated with type 2 diabetes. Genet Med 11: 338–343 [DOI] [PubMed] [Google Scholar]
- The STAR Consortium 2008. SNP and haplotype mapping for genetic analysis in the rat. Nat Genet 40: 560–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudbery I, Stalker J, Simpson JT, Keane T, Rust AG, Hurles ME, Walter K, Lynch D, Teboul L, Brown SD, et al. 2009. Deep short-read sequencing of chromosome 17 from the mouse strains A/J and CAST/Ei identifies significant germline variation and candidate genes that regulate liver triglyceride levels. Genome Biol 10: R112 doi: 10.1186/gb-2009-10-10-r112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin MD, Tomaszewski M, Braund PS, Hajat C, Raleigh SM, Palmer TM, Caulfield M, Burton PR, Samani NJ 2008. Common variants in genes underlying monogenic hypertension and hypotension and blood pressure in the general population. Hypertension 51: 1658–1664 [DOI] [PubMed] [Google Scholar]
- Ustyugova SV, Lebedev YB, Sverdlov ED 2006. Long L1 insertions in human gene introns specifically reduce the content of corresponding primary transcripts. Genetica 128: 261–272 [DOI] [PubMed] [Google Scholar]
- Venkatraman ES, Olshen AB 2007. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 23: 657–663 [DOI] [PubMed] [Google Scholar]
- Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Guo Y, et al. 2008. The diploid genome sequence of an Asian individual. Nature 456: 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. 2008. The complete genome of an individual by massively parallel DNA sequencing. Nature 452: 872–876 [DOI] [PubMed] [Google Scholar]
- Wilde DW, Furspan PB, Szocik JF 1994. Calcium current in smooth muscle cells from normotensive and genetically hypertensive rats. Hypertension 24: 739–746 [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Kirita S, Hasegawa H, Aoyama J, Imaoka S, Minamiyama S, Funae Y, Baba T, Matsubara T 2002. Contribution of CYP4A8 to the formation of 20-hydroxyeicosatetraenoic acid from arachidonic acid in rat kidney. Drug Metab Pharmacokinet 17: 109–116 [DOI] [PubMed] [Google Scholar]
- Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR 2009. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet 41: 849–853 [DOI] [PMC free article] [PubMed] [Google Scholar]