

Abstract
For template-based modeling in the CASP8 Critical Assessment of Techniques for Protein Structure Prediction, this work develops and applies six new full-model metrics. They are designed to complement and add value to the traditional template-based assessment by GDT (Global Distance Test) and related scores (based on multiple superpositions of Cα atoms between target structure and predictions labeled “model 1”). The new metrics evaluate each predictor group on each target, using all atoms of their best model with above-average GDT. Two metrics evaluate how “protein-like” the predicted model is: the MolProbity score used for validating experimental structures, and a mainchain reality score using all-atom steric clashes, bond length and angle outliers, and backbone dihedrals. Four other new metrics evaluate match of model to target for mainchain and sidechain hydrogen bonds, sidechain end positioning, and sidechain rotamers. Group-average Z-score across the six full-model measures is averaged with group-average GDT Z-score to produce the overall ranking for full-model, high-accuracy performance.
Separate assessments are reported for specific aspects of predictor-group performance, such as robustness of approximately correct template or fold identification, and self-scoring ability at identifying the best of their models. Fold identification is distinct from but correlated with group-average GDT Z-score if target difficulty is taken into account, while self-scoring is done best by servers and is uncorrelated with GDT performance. Outstanding individual models on specific targets are identified and discussed. Predictor groups excelled at different aspects, highlighting the diversity of current methodologies. However, good full-model scores correlate robustly with high Cα accuracy.
Keywords: homology modeling, protein structure prediction, all-atom contacts, full-model assessment
Introduction
The problem of protein structure prediction is certainly not yet “solved”. However, enormous progress has been made in recent years, with much credit due to the objective, double-blind assessments of the biennial CASP experiments1. In CASP8, even for difficult targets some individual predictions were very accurate, and for relatively easy targets many groups submitted good models, as seen for T0512 and its 354 predicted models in Figure 1. As assessors, we had the task of evaluating the 55,000 models submitted for CASP8 template-based modeling (TBM). Descriptions, statistics, and results for CASP8 are available at http://www.predictioncenter.org/casp8/. The existence and relatively automated application2 of an appropriate, highly tuned, well accepted tool for assessing the overall success of TBM predictions – the GDT-TS Z-score for Cα superposition3,4 – has allowed us to explore new ways of adding information and value to the CASP TBM process. Specifically, since that primary GDT assessment uses only the Cα atoms, we have developed a set of full-model measures which take into consideration the other 90% of the protein that provides essentially all of the biologically relevant interactions.
Figure 1.
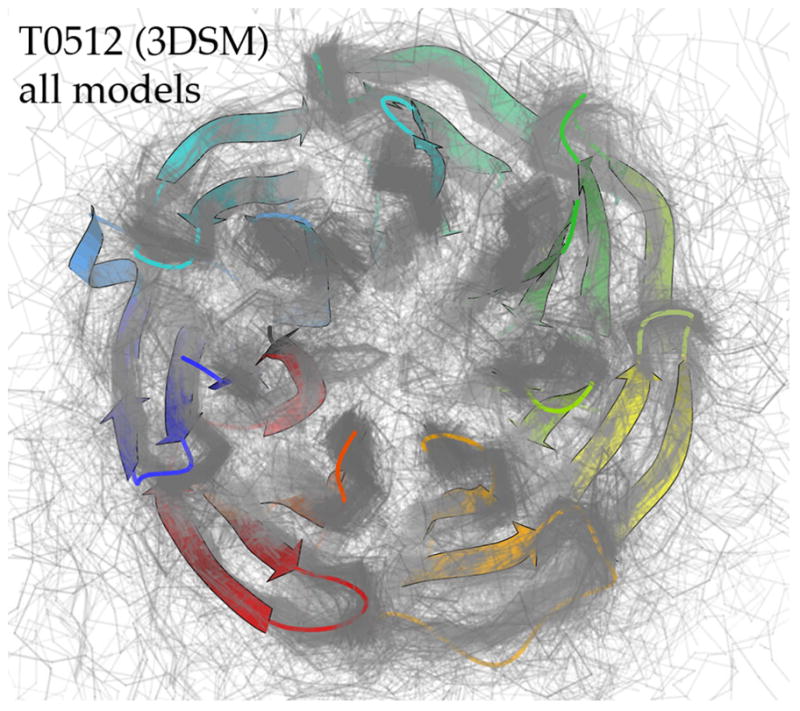
All 354 predicted models for T0512-D1. Target backbone is in ribbon representation colored blue to red in N- to C-terminal order; model Cα traces are in translucent gray. PDB code: 3DSM (NESG, unpublished).
In the long run, correct predictions will satisfy the same steric and conformational constraints that are satisfied by accurate experimental structures. One general question we addressed was whether the time has yet come when evaluating full-model details can contribute productively to achieving more correct predictions, by spurring methods development and by guiding local choices during individual model construction. This is not a foregone conclusion, since too much detail is irrelevant or even detrimental to judging model correctness if modeling remains very approximate. Our second general aim was to increase the diversity and specificity of assessment measures within the TBM category. The TBM prediction process encompasses many somewhat independent aspects, and both targets and methods are highly diverse. It seems likely, therefore, that future methods development could be catalyzed more effectively if more extensive separate evaluations of distinct aspects (such as template/fold recognition or sidechain rotamer correctness) were provided where feasible, in addition to the single, winner-take-all assessment of predictor groups. It is not a new idea to penalize backbone clashes5,6 or to include sidechain or H-bond assessment7,5,8, but the quantity and quality of models in CASP8 allow those things to be done more extensively than before and we have adopted a different perspective. For instance, we consider steric clashes for all atoms, using a well-validated physical model rather than an ad-hoc cutoff. These new full-model metrics provide a model-oriented rather than target-oriented version of a “high-accuracy” (HA) assessment for CASP8 predictions, as suggested for future development by the CASP7 HA assessor8. The scope here is over models accurate enough to score in the top section of the bimodal GDT-HA distribution, rather than over targets assigned as TBM-HA based on having a close template9, as was done for the HA assessment in CASP78.
The work described here, therefore, even further broadens the scope of assessment techniques and delves into finer atomic detail, by separately evaluating multiple aspects of TBM prediction, by identifying outstanding individual models, and especially by examining backbone sterics and geometry, sidechain placement, and hydrogen bond prediction in the CASP8 template-based models. Ultimately, our goal is to encourage fully detailed and “protein-like” models that can be used productively by experimental biologists. A relatively large number of prediction groups are found to score well on various of these measures, including the demanding new measures of full-model detail.
Methods
General Approach and Nomenclature
Previous assessments of CASP template-based models have focused primarily on GDT (global distance test) from the program LGA (local-global alignment)3. GDT is an excellent indicator of one structure’s similarity to another, applicable across the entire range of difficulty for TBM (template-based modeling) targets and to a large extent for FM (free modeling) as well. Its power derives primarily from its use of multiple superpositions to assess both high- and low-accuracy similarity, as opposed to more quotidian metrics like RMSD (root-mean-square deviation) which use a single superposition. Specifically, a version of GDT using relatively loose inter-atomic distance cutoffs of 1, 2, 4, and 8Å called GDT-TS (“total score”) has traditionally been the principal metric for correctness of predictions. However, a variant using stricter cutoffs of 0.5, 1, 2, and 4Å called GDT-HA (“high accuracy”) was used for much of the CASP7 TBM assessment because of its enhanced sensitivity to finer structural details6,8. We feel that GDT-HA probes a level of structural detail similar to our new measures (see below) and, therefore, continue to use it widely here.
Despite the power of LGA’s traditional scores, they consider only the Cα atoms – in other words, they ignore more than 90% of the protein. Many current prediction methods make use of all the atoms, and many of this year’s CASP models are accurate enough to make a broader assessment appropriate. Therefore, our primary contribution to CASP8 TBM assessment is additional full-model structure accuracy and quality metrics that are to some degree orthogonal to Cα coordinate superposition metrics like GDT. Our group has extensive experience in structure validation for models built using experimental data, mainly from X-ray crystallography and nuclear magnetic resonance (NMR), and has over the years developed strong descriptors of what makes a model “protein-like”10,11,12,13. Here we seek to apply some of those same rules to homology models in CASP8.
Two of the new full-model metrics evaluate steric, geometric, and conformational outliers in the model, and are normalized on a per-residue basis. The other four measure match of model to target on hydrogen bond or sidechain features, and are expressed as percentages. Raw scores, for these or other metrics, are the appropriate way to judge quality of an individual model. Averaging the raw scores of all models for an individual target provides a rough estimate of that target’s difficulty (which varies widely). Finally, in order to combine the six new metrics into a single full-model measure, or to evaluate relative performance between prediction groups, the metrics were converted into Z-scores measured in standard deviations above or below the mean, as has been standard practice in CASP for some time4. Group-average Z-scores are not reported here for groups that submitted usable models for fewer than 20 targets.
In the descriptions that follow, 3-digit target codes are written starting with “T0” (ranging from T0387 to T0514 for CASP8), while prediction groups are referred to by their brief names, except when making up part of a model number (e.g., 387_1 is model 1 from group 387). Name, identifying number, and participants for prediction groups can be looked up at http://www.predictioncenter.org/casp8/, as well as definitions, statistics, and results for CASP8. Groups are designated either as human or server. Server groups employ automated methods and are required to return a prediction within 3 days; that server may or may not be publicly accessible. Human groups need not use purely automated methods and are allowed 3 weeks to respond. Targets are also designated as either server or human (the latter are more difficult on average); typically, servers submit models for all targets and human groups submit for human targets only. When a target is illustrated or discussed individually, its 4-character PDB code will also be given (e.g., 3DSM for T0512), and those coordinates can be obtained from the Protein Data Bank14 at http://www.rcsb.org/pdb/.
Model File Pre-Processing
CASP8 TBM assessment involved evaluating over 55,000 whole-target predictions and over 77,000 target domain predictions (250–550 models per target, as shown for T0512 in Figure 1), which highlighted the importance of file management, clean formatting, and interpretable content. It was discovered early in our work that a surprisingly high percentage of the prediction files did not adhere to the PDB format14, even though CASP model files require only a very simple and limited subset of the format, with some checks done at submission. The commonest problems involved spacing, column alignment, or atom names, but there were a few global issues such as concatenated models, empty files, and even a set of files with the text “NAN” in place of all coordinates. General-purpose software, including our structural evaluation tools, must deal correctly with the full complexity of the PDB format and thus cannot be designed for tolerance of these errors in the simpler all-protein mode of CASP. Therefore, as noted also for CASP615, most format irregularities produce incorrect or skipped calculations, and the most inventive ones occasionally cause crashes.
To address the reparable issues, we created a Python script to “preprocess” and correct most of the formatting and typographical errors. Among the errors it can address are non-standard header tags, new (version 3.x) vs. old (version 2.3) PDB format, nonstandard hydrogen names, incorrect significant digits in numerical columns, and incorrectly justified columns, specifically the atom name, residue number, coordinate, occupancy, and B-factor fields. Unfortunately, due to the number and variety of model files, some formatting errors slipped past the preprocessing. One example discovered only later was a set of models with interacting errors both in column spacing and in chain-ID entries placed into the field normally containing the insertion code; these produced incorrect results even from LGA, which is admirably tolerant and only needs to interpret Cα records.
Beyond format are issues of incorrect or misleading content, which are nearly impossible to stipulate in advance and were usually discovered either by accident or by aberrant results from the assessment software. A few of the many cases in CASP8 TBM models were Cβs on glycines, multiple atoms with identical coordinates, and sidechain centroids left in as “CEN” atoms misinterpreted as badly clashing carbon atoms. Usually, format or content problems result in falsely poor scores, which should concern the predictor but did not worry the assessor except for distortions in the overall statistics. However, sometimes the errors produce falsely good scores (such as low clashscores from missing or incomplete sidechains), making their diagnosis and removal a very serious concern to everyone involved in CASP.
Hydrogen Atoms
Explicit hydrogens must be present for all-atom contact analysis to yield meaningful results. The program Reduce was used to add both polar and non-polar H atoms at geometrically ideal positions16. When H atoms were already present in the model or target file, we used them but standardized their bond lengths for consistency in evaluation. For all files, we optimized local H-bonding networks for the orientations of rotatable polar groups such as OH and NH3 and for the protonation pattern of His rings, but did not apply MolProbity’s usual automatic correction for 180° flips of Asn/Gln/His sidechains16.
Measure 1: MolProbity Score (MPscore)
The first two of the six new full-atom metrics, MolProbity score and mainchain reality score, are based only on properties of the predicted model. Previous work on all-atom contact analysis demonstrated that protein structures are exquisitely well packed, with interdigitating favorable van der Waals contacts and minimal overlaps between atoms not involved in hydrogen bonds10. Unfavorable steric clashes are strongly correlated with poor data quality, with clashes reduced nearly to zero in the well-ordered parts of very high-resolution crystal structures17. From this analysis – originally intended to improve protein core redesign, but since applied also to improving experimental structures – came the clashscore, reported by the program Probe10; lower numbers indicate better models.
In addition, the details of protein conformation are remarkably relaxed, such as staggered χ angles11 and even staggered methyls10. Forces applied to a given local motif in the crowded environment of a folded protein interior can result in a locally strained conformation, but evolution seems to keep significant strain near the minimum needed for function, presumably because protein stability is too marginal to tolerate more. In updates of traditional validation measures, we have compiled statistics from rigorously quality-filtered crystal structures (by resolution, homology, and overall validation scores at the file level, and by B-factor and sometimes by all-atom steric clashes at the residue level). After appropriate smoothing, the resulting multi-dimensional distributions are used to score how “protein-like” each local conformation is relative to known structures, either for sidechain rotamers11 or for backbone Ramachandran values12. Rotamer outliers asymptote to < 1% at high resolution, general-case Ramachandran outliers to < 0.05%, and Ramachandran favored to 98% (Fig. 2).
Figure 2.
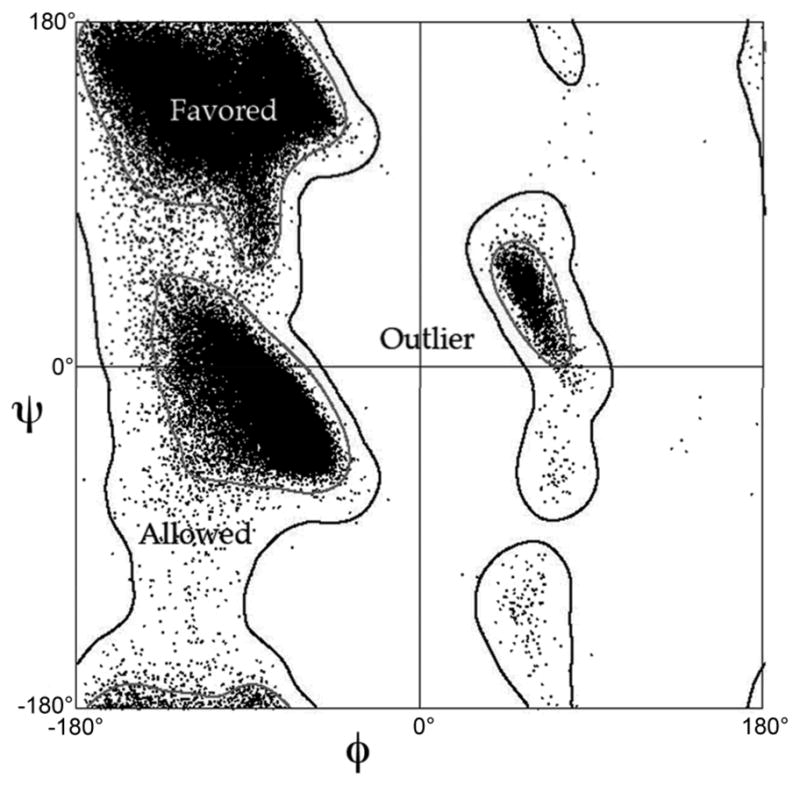
Empirical Ramachandran distribution12, one component in both MolProbity and mainchain reality scores. The data points are ϕ,ψ backbone dihedral angles for all general-case residues with maximum mainchain B-factor ≤ 30, from the Top500 quality-filtered set of crystal structures; Gly, Pro, and pre-Pro residues are analyzed separately. Contours are calculated with a density-dependent smoothing algorithm. 98% of the data fall within the favored region (inside gray contour), 99.95% within the allowed or favored regions (inside black contour), and 0.05% in the outlier region (outside black contour).
All-atom contact, rotamer, and Ramachandran criteria are central to the MolProbity structure-validation web site13, which has become an accepted standard in macromolecular crystallography: MolProbity hosted over 78,000 serious work sessions in the past year. To satisfy a general demand for a single composite metric for model quality, the MolProbity score (MPscore) was defined as:
where clashscore is defined as the number of unfavorable all-atom steric overlaps ≥ 0.4Å per 1000 atoms10; rota_out is the percentage of sidechain conformations classed as rotamer outliers, from those sidechains that can be evaluated; and rama_iffy is the percentage of backbone Ramachandran conformations outside the favored region, from those residues that can be evaluated. The coefficients were derived from a log-linear fit to crystallographic resolution on a filtered set of PDB structures, so that a model’s MPscore is the resolution at which its individual scores would be the expected values. Thus, lower MPscores are better.
CASP8 marks the first use of the MolProbity score for evaluation of non-experimentally-based structural models. It is a very sensitive and demanding metric, a fact also evident for low-resolution crystal structures or for NMR ensembles. It must be paired with a constraint on compactness, provided by the electron density in crystallographic use and approximately by the GDT score in CASP evaluation. Crystal contacts occasionally alter local conformation, but are too weak to sustain unfavorable strain. Those changes are much smaller than at multimer or ligand interfaces. For CASP8 targets, potential problems between chains or at crystal contacts were addressed as part of defining the assessment units9.
Measure 2: Mainchain Reality Score (MCRS)
To complement the MolProbity score, it seems desirable to have a model evaluation that (1) only uses backbone atoms in its analysis and (2) takes account of excessive deviations of bond lengths and bond angles from their chemically expected ideal values. For those purposes, the mainchain reality score (MCRS) was developed, defined as follows:
where spike is the per-residue average of the sum of “spike” lengths from Probe (indicating the severity of steric clashes) between pairs of mainchain atoms, rama_out is the percentage of backbone Ramachandran conformations classed as outliers (as opposed to favored or allowed; see Fig. 2), and length_out and angle_out are the percentages of mainchain bond lengths and bond angles respectively that are outliers > 4σ from ideal18. The perfect MCRS is 100 (achieved fairly often by predicted models), and any non-idealities are subtracted to yield less desirable scores. The coefficients were set manually to achieve a range of approximately 0–100 for each of the four terms, so that egregious errors in just one of these categories can “make or break” the score. To counter this and achieve a reasonable overall distribution, we truncated the overall MCRS at 0 (necessary for about 14% of all models); note that 0 is already such a bad MCRS that truncation isn’t unduly forgiving of the model. However, we did not discover any models as charmingly dreadful as in the first figure of CASP6 TBM 5.
Measures 3,4: Hydrogen Bond Correctness (HBmc and HBsc)
The last 4 of these 6 new full-model metrics are based on comparisons between the predicted model and the target structure. Knowing the importance of H-bonds in determining the specificity of protein folds19, the CASP7 TBM assessors examined H-bond correctness relative to the target6. We have followed their lead but separated categories for mainchain (HBmc: mainchain-mainchain only) and sidechain (HBsc: sidechain-mainchain and sidechain-sidechain), using Probe10 to identify the H-bonds.
Briefly, the approach was to calculate the atom pairs involved in H-bonds for the target, do the same for the model, and then score the percentage of H-bond pairs in the target correctly recapitulated in the model. Probe defines hydrogen bonding rather strictly, as donor-acceptor pairs closer than van der Waals contact. That definition was used for all target H-bonds and for mainchain H-bonds in the models, which often reached close to 100% match (see Results). However, it is more difficult to predict sidechain H-bonds, since they require accurately modeling both backbone and sidechains. Therefore, for HBsc model (but not target) H-bonds, we also counted donor-acceptor pairs ≤ 0.5Å beyond van der Waals contact; this raised the scores for otherwise good models from the 20–40% range to the 30–80% range. This extended H-bond tolerance was readily accomplished using Probe atom selections of “donor, sc” and “acceptor, sc” with the normal 0.5Å diameter probe radius, thus identifying these slightly more distant pairs as well as the usual H-bond atom pairs. Note that both HBmc and HBsc measure the match of model to target, since we (like the CASP7 assessors) explicitly required that a model H-bond be between the same pair of named atoms as in the target H-bond.
CASP7 excluded surface H-bonds, but we did not. We believe the best strategy would be in between those two extremes, where sidechain H-bonds would be excluded if they were in regions of uncertain conformation in the target. However, surface H-bonds are generally under- rather than over-represented in crystal structures (perhaps because of high ionic strength in many crystallization media), so prediction of those recognizable in the target should be feasible.
Measure 5: Rotamer Correctness (corRot)
For sidechain rotamers, MolProbity works from smoothed, contoured, multidimensional distributions of the high-quality χ-angle data11,13; the score value at each point is the percentage of good data that lies outside that contour level. For each individual sidechain conformation, MolProbity looks up the percentile score for its χ-angle values; if that score is ≥ 1%, it assigns the name of the local rotamer peak and if < 1%, it declares an outlier. Rotamer names use a letter for each χ angle (t = trans, m = near −60°, p = near +60°), or an approximate number for final χ angles that significantly differ from one of those 3 values. Using this mechanism, we can define rotamer correctness (corRot) as the match of valid rotamer names between model and target. Note that any model sidechain not in a defined rotamer (i.e., an outlier) is considered non-matching, unless the corresponding target rotamer is also undefined, in which case that residue is simply ignored for corRot. The sidechain rotamers used in SCWRL20 are quite similar to the MolProbity rotamers, since both are based on recent high-resolution data, quality-filtered at the residue level.
For X-ray targets, the target rotamer set consists of all residues for which a valid rotamer name could be assigned (i.e. not < 1% rotamer score and not undefined because of missing atoms). For NMR targets, we defined the target rotamer set to include only those residues for which one named rotamer comprised a specified percentage (85, 70, 55, and 40% for sidechains with one, two, three, and four χ angles respectively) of the ensemble. We also considered requiring a sufficient number of nuclear Overhauser effect (NOE) restraints for a residue for it to be included, but concluded that in practice this would be largely redundant with the simpler consensus criterion (data not shown).
Since incorrect 180° flips of Asn/Gln/His sidechains are caused by a systematic error in interpreting electron density maps, there is no reason for them to be wrong by 180° in predicted models, which could thus sometimes improve locally on the deposited target structure. However, we found that applying automatic correction of Asn/Gln/His flips in targets by MolProbity’s standard function yielded only 1% or less improvement in any group-average corRot score. We therefore chose not to apply target flips for the final scoring.
Using rotamer names based on multidimensional distributions rather than simple agreement of individual χ1, or χ1 and χ2, values7,5,8 has the advantage of favoring predictions in real local-minimum conformations and with good placement of the functional sidechain ends. However, a disadvantage is that matching is all-or-none; for example, model rotamers tttm and mmmm would be equally “wrong” matches to a target rotamer tttt in our formulation, meaning the corRot score is more stringent for long sidechains. An improved weighting system might be devised for future use.
Measure 6: Sidechain Positioning (GDC-sc)
In order to apply superposition-based scoring to the functional ends of protein sidechains, we developed a GDT-like score called GDC-sc (global distance calculation for sidechains), using a modification of the LGA program3. Instead of comparing residue positions on the basis of Cαs, GDC-sc uses a characteristic atom near the end of each sidechain type for the evaluation of residue-residue distance deviations. The list of 18 atoms is given by the -gdc_at flag in the LGA command shown below, where each one-letter amino-acid code is followed by the PDB-format atom name to be used:
-3 -ie -o1 -sda -d:4 -swap -gdc:10
-gdc_at:V.CG1,L.CD1,I.CD1,P.CG,M.CE,F.CZ,W.CH2,S.OG,T.OG1,C.SG,Y.OH, N.OD1,Q.OE1,D.OD2,E.OE2,K.NZ,R.NH2,H.NE2
or, alternatively with a new flag, just: -3 -ie -o1 -sda -d:4 -gdc_sc
Gly and Ala are not included, since their positions are directly determined by the backbone. The -swap flag takes care of the possible ambiguity in Asp or Glu terminal oxygen naming.
The traditional GDT-TS score is a weighted sum of the fraction of residues superimposed within limits of 1, 2, 4, and 8Å. For GDC-sc, the LGA backbone superposition is used to calculate fractions of corresponding model-target sidechain atom pairs that fit under 10 distance-limit values from 0.5Å to 5Å, since 8Å would be a displacement too large to be meaningful for a local sidechain difference. The procedure assigns each reference atom to the relevant bin for its model vs. target distance: < 0.5Å, < 1.0Å, … < 4.5Å, < 5.0Å; for each bin_i, the fraction (Pa_i) of assigned atoms is calculated. Finally the fractions are added and scaled to give a GDC-sc value between 0 and 100, by the formula:
The goal was a measure sensitive to correct placement of sidechain functional or terminal groups relative to the entire domain, both in the core and forming the surface that makes interactions. The three sidechain measures (HBsc, corRot, and GDC-sc) are meaningful evaluations only for models with an approximately correct overall backbone fold, and so we make use of them only for models with above-average GDT scores (see Model Selection, below).
Databases, Statistics, and Visualizations
We have made extensive manual use of the comprehensive summaries, charts, tables, and alignments provided on the Prediction Center web site21 for CASP8, now available at http://www.predictioncenter.org/casp8/. A MySQL22 database was constructed for storing and querying all the basic data needed for our TBM assessments. It was loaded with the full contents of the Prediction Center’s Results tables (including rerun values for Dali23 scores where format-error crashes had been incorrectly registered as zeroes), plus all of our own analyses and scores on all targets, models, and groups. Statistical properties were calculated in the R program24, and plots were made in pro Fit (Quantum Soft).
For model superpositions onto both whole targets and domain targets, we used the results from the standard LGA sequence-dependent analysis runs3 provided by the Prediction Center. The full set of superimposed models for each target was converted by a script into a kinemage file for viewing in KiNG13 or Mage25,26, organized by LGA score and arranged for animation through the models (e.g., Fig. 1). Structural figures were made in KiNG and plot figures in pro Fit, with some post-processing in PhotoShop (Adobe). Once targets were deposited, their electron density maps were obtained from the Electron Density Server27 (http://eds.bmc.uu.se/eds/). For many individual targets and models, multi-criterion kinemages that display clashes, rotamer, Ramachandran, and geometry outliers on the structure in 3D were produced in MolProbity13.
Model Selection and Filtering
Although predictors are allowed to submit up to five models per target, most statistics require the choice of one model per group per target for assessment. The central GDT-TS assessment in CASP has always used the first model, designated “model 1”; this is what predictors expect, and the precedent was followed again in CASP8 for the official group rankings2. This has the advantage of rewarding the groups that are best at self-scoring to decide which of their predictions is best, a skill of real value to end users. However, using model 1 comes at the expense of eliminating many of the very best models. So, for the full-model TBM assessments in this paper, we have instead chosen to assess success at self-scoring separately (see Results), allowing the main evaluations to use the best model (as judged by GDT-TS) for each group on each target.
Superposition-based scores (GDT-HA, GDT-TS, GDC-sc) were computed on domain targets because, as in past CASP TBM assessments, we wished not to penalize predictors that correctly modeled domain architectures but incorrectly modeled relative inter-domain orientations. Model quality and local match-to-target scores (MPscore, MCRS, corRot, HBmc, HBsc) were computed on whole targets, because such scores are approximately additive even across inaccurate domain orientations.
Some targets contain domains assigned to different assessment classes9; for example, 443-D1 is FM/TBM, 443-D2 is FM, and 443-D3 is TBM. For our scores computed on target domains, any FM domains were omitted. For scores computed on whole targets, any targets for which all domains were FM were omitted, but targets with at least one TBM or FM/TBM domain were retained.
We eliminated from assessment all models for canceled or reassigned targets (T0387, T0403, T0410, T0439, T0467, T0484, T0510) and from groups (067, 265, 303) that withdrew. The full-model measures are inappropriate for “AL” submissions (done by only two groups), which consist of a sequence alignment to a specified template, with coordinates then generated at the Prediction Center by taking the aligned parts from the template structure; therefore, only the usual “TS” models are assessed here, for which at least all backbone and usually also sidechain coordinates are directly predicted.
Predictors were allowed to submit a prediction model in multiple “segments”, which they believed to be likely domain divisions in the true target but which did not necessarily coincide with the official CASP8 domain boundaries9. Full-model scores additive across domains are also additive across segments. GDT or GDC scores are fundamentally non-additive, however, so we evaluated GDC-sc by domain, using whichever segment had the highest GDT-TS score for that domain.
After the segment selection/combination, we required that each model contain at least 40 residues in order to avoid artifacts from essentially partial predictions. For all sidechain-relevant metrics (including MolProbity score), a further filter was applied on a per-model basis requiring that at least 80% of the model Cα atoms be attached to sidechains that included coordinates for the residue-type-specific terminal atom defined for the GDC-sc metric (see above). This avoids misleadingly high or low sidechain scores on incomplete models.
As previously noted28, the distribution of GDT scores is strongly bimodal. As illustrated in Figure 3, models therefore fall under one of two clearly separable peaks in GDT-HA or GDT-TS, separated by a valley at 33 for GDT-HA or at 50 for GDT-TS. These distributions are discussed and used in the Results sections on full-model measures and on robust “right fold” identification. This basic bimodal division also holds within most individual target domains (though there is much variability between targets in the positions and shapes of the peaks), implying that the TBM-wide bimodality is not caused by bimodality of target difficulty. This property of the distributions suggests a possible cutoff for models that have an approximately correct fold and are therefore appropriate for the more detailed, local quality assessment our new metrics provide. Accordingly, we only considered the following: (1) models with GDT-HA ≥ 33 for our domain-based metrics and (2) models with at least one domain with GDT-HA ≥ 33 for our whole target-based metrics. (Note that each target has at most three domains except for T0487 with five domains, so we increased its model requirement to two domains with GDT-HA ≥ 33.) For full-model measures, this model-based GDT-HA cutoff was judged preferable to the target-based system used for GDT-TS (server groups evaluated in all targets and all groups on human targets2,6), because restricting assessment to the small number of high-accuracy targets in the human category would yield only 24/88 human groups with a statistically reasonable number of targets, whereas filtering by model can more than double that number to 51/88.
Figure 3.
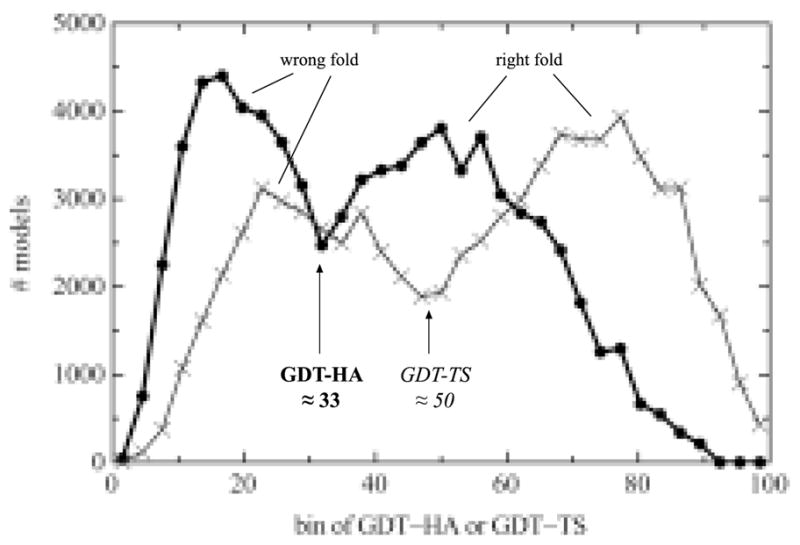
Bimodal distributions of GDT-HA and GDT-TS scores. All CASP8 TBM models were placed into 33 equally spaced bins, separately for GDT-HA and for GDT-TS. The division between “right fold” and “wrong fold” occurs at approximately GDT-HA of 33 (which we used for our later analysis) and GDT-TS of 50. Note that bimodal distributions were also observed within most individual targets (data not shown).
Since NMR targets are ensembles of multiple models and are derived primarily from local inter-atomic distance measurements, they require different treatment than crystal structures for some purposes. Modifications adopted for the rotamer-match metric are described above. In defining domain targets for the official GDT evaluations9, NMR targets were trimmed according to the same 3.5Å cutoff on differences in superimposed Cα coordinates that was used for multiple chains in x-ray structures. Although model 1 of the NMR ensemble was usually used as the reference, in some cases another model was chosen as staying closer to the ensemble center throughout the relevant parts of the entire target structure. The outer edges of NMR ensembles typically diverge somewhat even when the local conformation is well defined by experimental data. Except for GDC-sc, the full-model metrics are still meaningful despite gradual divergence in coordinate space. Therefore we specified alternative “D9” target definitions for many of the NMR targets, which were trimmed only where local conformation became poorly correlated within the ensemble. This was manually judged using the translational “co-centering” tool in KiNG graphics29. The resulting residue ranges were also used for CASP8 disorder assessment30. A D9 alternative target was defined for the T0409 domain-swap dimer target (see Results), by constructing a reconnected, compact monomer version.
Results
Information Content of Full Model Measures
Structure prediction is progressing to a level of accuracy where models can be routinely used to generate detailed biological hypotheses. To track this maturation, we have added new metrics to TBM assessment to probe the fine-grained structure quality we think homology models can ultimately achieve. In evaluating the suitability of these full-model metrics for CASP8 assessment, it is important to understand their relationship to traditional superposition-based metrics. Any appropriate new metric of model quality should show an overall positive correlation to GDT scores, but should also provide additional, orthogonal information with a significant spread and some models scoring quite well.
Figure 4 plots each of the six full-model measures against either GDT-TS or GDT-HA, showing strong positive correlation in all cases. (Note that the correlation is technically negative for MPscore, but lower MPscore is better.) Plots 4a and 4b, including all models across the full GDT range, show that detail is relatively uncoupled for the lower half of the GDT range but well correlated for the upper half, in correspondence with the bimodal GDT distributions in Figure 3 above. Therefore parts c-f of Figure 4 plot only the best models with GDT-HA ≥ 33. Tables with the detailed score data on all the full-model measures, by target and group, are available on our web site (http://kinemage.biochem.duke.edu) and at the Prediction Center.
Figure 4.

Distributions of the new full-model scores for individual models. Panels a–b include all models regardless of GDT, whereas panels c-f include only best models with GDT-HA ≥ 33. Dual linear fits are on models with GDT-TS < 55 vs. ≥ 55 in panels a-b and on models with GDT-HA < 60 vs. ≥ 60 in panel e; these divisions were chosen manually to highlight visible inflection points. Larger dots in panels c–f are median values for bins of 3 GDT-HA units; bins at high GDT-HA include many fewer models, producing high variability for some measures (e.g. corRot). The fit lines are well below the median points in panel e, because many points lie at zero MCRS. Note that the y-axis for MPscore in panel f has been reversed relative to other panels, because lower MPscores are better.
The slope, linearity, and scatter vary: correlation coefficients for fits of models with the “right fold” (see section below) to GDT-HA range from 0.24 for MPscore to 0.87 for GDC-sc. Large dots plot median values of each measure within bins spaced by 3 GDT-HA units, to improve visibility of the trends, although with high variability at the tails due to less occupied bins. Taken together, these results show that as a general rule all aspects improve together, but different detailed parameters couple in different ways to getting the backbone Cα atoms into roughly the right place, as evidenced by the varying levels of saturation and scatter.
Not too surprisingly, GDC-sc has the tightest correlation to GDT-HA. It measures match of sidechain end positions between model and target, for which match of Cα positions is a prerequisite. The vertical spread of scores indicates some independent information, but less than for the other full-model scores. However, GDC-sc shows the most pronounced upturn at high GDT-HA, an effect detectable for most of the six plots. It will require further investigation to decide to what extent this is caused by copying from more complete templates and to what extent there is a threshold of backbone accuracy beyond which it becomes much more feasible to achieve full-model accuracy. Taken together, the GDC-sc, corRot, and HBsc measures assess the challenging optimization problem of sidechain placement in distinct ways, and they can provide tools to push future CASP assessments in the direction of higher-resolution, closer-to-atomic detail.
Interestingly, the model-only “quality” measures – i.e. MCRS and MPscore – also correlate with correct backbone superposition scores (Fig. 3e–f). Seemingly, proteins must relax (in terms of sterics and covalent geometry) into the proper backbone conformation, but details of the relationships differ in revealing ways. MolProbity score has high scatter and relatively low slope, but is linear over the entire range; it includes the clashscore for all atoms, an extremely demanding criterion which improves at higher GDT-HA but still leaves much scope for further gains. In contrast, mainchain reality score, which measures Ramachandran, steric, and geometric ideality along the backbone, is often quite dire in poor models (e.g. over half of the residues with geometry outliers, sometimes by > 50σ), but it saturates to quite good values on the upper end. The dearth of any really bad MCRS models for good GDT-HA suggests that modeling physically realistic mainchain may be essential for achieving really accurate predictions; however, as noted for GDC-sc, this relationship needs further study.
The H-bond recapitulation measures, developed from ideas introduced in CASP76, seem clearly to be informative. The new separation of mainchain and sidechain H-bonds appears to be helpful, since they show strongly correlated but distinctly different 2D distributions which would be less informative if combined. In both cases, the diagnostic range is for models with better than average GDT scores (Fig. 4a and b), and that range is therefore used in assessment. At low GDT, almost no sidechain H-bonds are matched, whereas mainchain H-bonds show an artificial peak due to secondary-structure prediction of α-helices without correct tertiary structure. To correct this overemphasis, future versions of HBmc could somewhat down-weight either specifically helical H-bonds or perhaps all short-range backbone H-bonds (i to i+4 or less). The upper half of both H-bond measures shows the desirable behavior of a very strong correlation and high slope relative to GDT, but with a large spread indicative of a significant contribution from independent information.
Group Rankings on Full-Model Measures
Traditionally, CASP assessment has involved a single ranking of groups relative to each other, to determine which approaches represent the current state of the art. A group’s official ranking is arrived at by (1) determining the top 25 groups in terms of average GDT-TS (or GDT-HA) Z-score on all first models with Z-score ≥ 0, then (2) performing a paired t-test for each of those 25 groups against every other on common targets to determine the statistical significance of the pairwise difference7,5,15,6,2.
The full-model assessment presented here is analogous to previous rankings in that we compute group average Z-scores on models above GDT-HA raw score of 33 for the top 20 groups. It differs in using the best model (by GDT-TS) rather than model 1, in using raw GDT rather than Z-score for the model cutoff, and in evaluating the full model. A further difference from recent versions is consideration of multiple dimensions of performance: the two model-only and the four match-to-target full-model scores as well as GDT-TS or HA. Those 6 full-model scores are combined with each other and the result averaged with GDT-HA Z for our final ranking of high-accuracy performance. Table I lists the top 20 prediction groups on each of the full-model measures, on the overall full-model average Z-score among groups in the top half of GDT rank, and on the average of the full-model and the GDT-HA Z scores. A more complete version of Table I, with specific scores for all qualifying groups, is available as supplementary information. Figure 5 shows the combined performance on GDT and full-model scores more explicitly by a two-dimensional plot of group-average full-model Z-score versus group-average GDT-HA Z-score, with diagonal lines to follow the final ranking that combines those two axes.
Table I.
Predictor Group Rankings on Combined Full-Model, High-Accuracy Scores
| model-only measures | match-to-target measures | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| group ID | group name | 6full+GDT HA rank | 6full rank | MCRS avg Z | MPscore avg Z | HBmc avg Z | HBsc avg Z | GDC-sc avg Z | corRot avg Z |
| 489 | DBaker | 1 | 2 | Yasara | Yasara | LevittGroup | Lee-s | Lee | Lee-s |
| 293-s | Lee-s | 2 | 5 | Lee | Ozkan-Shell | Sam-T08-h | DBaker | Lee-s | Lee |
| 453 | Multicom | 3 | 3 | DBaker | DBaker | DBaker | Lee | IBT_LT | Bates_BMM |
| 407 | Lee | 4 | 4 | Lee-s | A-Tasser | Keasar | Keasar-s | Multicom | Multicom |
| 046 | Sam-T08-h | 5 | 9 | Bates_BMM | Robetta | Mufold | Yasara | McGuffin | ChickenGeo |
| 379 | McGuffin | 6 | 16 | MuProt | Bates_BMM | Multicom | LevittGroup | Zhang | Robetta |
| 196 | ZicofSTP | 7 | 23 | Robetta | Lee-s | Zhang | Sam-T08-h | LevittGroup | Sam-T08-s |
| 138 | ZicofSTPfData | 8 | 22 | Multicom | Keasar | PoemQA | Robetta | Zhang-s | DBaker |
| 299 | Zico | 9 | 8 | MulticomRef | Lee | Bates_BMM | McGuffin | ChickenGeo | Fais@hgc |
| 310 | Mufold | 10 | 21 | PoemQA | Multicom | Sam-T08-s | Multicom | Sam-T08-s | Pcons_multi |
| 283 | IBT_LT | 11 | 15 | Elofsson | Pcons_dot_net | Keasar-s | Sam-T08-s | Sam-T08-h | LevittGroup |
| 178 | Bates_BMM | 12 | 32 | Pcons.net | ChickenGeo | Lee | Ozkan-Shell | ZicofSTPfData | Zhang |
| 147s | Yasara | 13 | 26 | HHpred5 | Sam-T08-h | McGuffin | MulticomClust | Mufold | Zhang-s |
| 071 | Zhang | 14 | 1 | Fais-s | Mufold | Lee-s | GeneSilico | Bates_BMM | Yasara |
| 081 | ChickenGeo | 15 | 20 | GSKudlatyPred | Sam-T08-s | Yasara | Keasar | DBaker | Pcons_dot_net |
| 485 | Ozkan-Shell | 16 | 10 | Hao_Kihara | Pcons_multi | ZicofSTP | ZicofSTP | ZicofSTP | IBT_LT |
| 034 | Samudrala | 17 | 29 | MulticomRank | Samudrala | ZicofSTPfData | MulticomRank | Zico | Keasar |
| 425s | Robetta | 18 | 7 | MulticomClust | MulticomCMFR | Zico | Fams-multi | Fams-multi | MulticomCMFR |
| 426s | Zhang-s | 19 | 41 | MulticomCMFR | Hao_Kihara | IBT_LT | ZicofSTPfData | Samudrala | Sam-T08-h |
| 434 | Fams-ace2 | 20 | 30 | PS2-s | IBT_LT | Zhang-s | IBT_LT | Fams-ace2 | Phyredenovo |
Groups in boldface appear in the top 4 at least once and in the top 20 for 5 out of the 6 full-model metrics.
MCRS = mainchain “reality” score: all-atom clashes, Ramachandran outliers, bond length or angle outliers for backbone
MPscore = MolProbity score: all-atom clashes, Ramachandran and rotamer outliers (scaled) for whole model
HBmc = fraction of target mainchain Hbonds matched in model
HBsc = fraction of target sidechain Hbonds matched in model
GDC-sc = GDT-style score for atom at end of each sidechain except Gly or Ala, 0.5 to 5Å limits (by LGA program)
corRot = fraction of target sidechain rotamers matched by model (all chi angles)
6full rank: group ranking based on the average of all 6 full-model-measure Z-scores; over all best models with GDT-HA > 33
6full+GDT HA rank: group ranking based on the sum of (1) by-domain, best-model GDT-HA Z-score and (2) average of 6 full-model-measure Z-scores
Figure 5.

a) Group-average Z-score for the 6 full-model scores, plotted vs. group-average Z-score for GDT-HA. b) Close-up of the upper-right quadrant from panel a, with the groups highlighted that did well on the combined score from both axes (emphasized by the diagonal lines). Group Z-scores are averaged over best models with GDT-HA ≥ 33; groups with a qualifying model for < 20 targets are excluded.
A small set of top-tier groups scored outstandingly well on most of the six model-only and model-to-target metrics (Table I). Yasara is highest on model-only criteria and LevittGroup on mainchain H-bonds, while Lee and Lee-server sweep the sidechain scores. Most of the same top groups also excelled in Cα positioning (Fig. 5). DBaker is the clear overall winner on this combined evaluation of Cα superposition and structure quality/all-atom correctness. Lee, Lee-server, MultiCom, Sam-T08-h, and McGuffin are in the next rank on the combined measure (Fig. 5), while Bates-BMM, IBT-LT, and Yasara are also notable for each scoring in the top 20 on 5 of the 6 full-model measures and once in the top 3 (Table I). An accompanying paper31 discusses aspects of TBM methodology that can contribute to the differences in detailed performance on this two-dimensional measure.
To examine these relationships further, group-average Z-scores were plotted for the six new quality and match-to-target measures individually against group-average Z-scores for GDT-HA. In addition to trends seen in the all-model plots of Figure 4, group-average scores for sidechain rotamer match-to-target (corRot) show two strong clusters, one at high and one at low values (Figure 6). Through the range of −1 to +0.5 GDT-HA, corRot is nearly independent of GDT-HA in both clusters. This suggests that many intermediate groups don’t pay attention to sidechain placement and/or use poor rotamer libraries, leaving sidechain and backbone modeling uncoupled. For the very best GDT-HA groups at the extreme right of the plot, however, corRot is also excellent, which implies that proper sidechain modeling may in fact be necessary for reliably achieving highly accurate backbone placement. There is no evidence that excellence in any of the full-model metrics is achieved by a tradeoff with GDT scores – rather they tend to improve together.
Figure 6.

Group-average Z-score for rotamer correctness, plotted vs. group-average Z-score for GDT-HA. The horizontal line at corRot Z-score of 0 was drawn manually to visually highlight the gap between group clusters on sidechain performance. Group-average Z-scores are for best models with GDT-HA≥ 33; groups attempting < 20 targets are excluded.
Robust “Right Fold” Identification
We also sought to assess which groups excelled at template or fold identification, to help delineate the state of the art for that stage of homology modeling. To do so, we computed the percentage of all of a group’s models with approximately the “right fold”, defined as GDT-HA ≥ 33 (Fig. 3) as per our threshold for reasonably accurate models used above. However, success rates on this metric are also dependent on average difficulty of attempted targets. Therefore Figure 7 plots “right fold” percentage as a function of average target difficulty. Prediction groups fall into three loose areas of target difficulty: those who predicted the harder human targets (at left in Fig. 7), those who predicted all targets (center), and those who predicted only the easier server targets (right). Table II lists the top groups in each of these three divisions.
Figure 7.
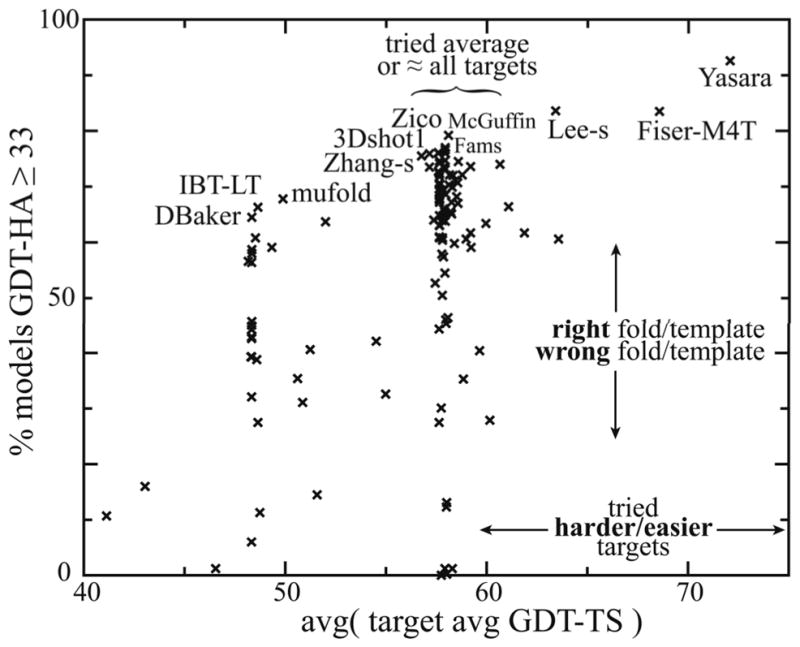
Percentage of models with roughly the “right fold”, plotted vs. difficulty of targets attempted. The percentage of all of a group’s models with GDT-HA≥ 33 (“right fold”) is on the y-axis. The average across a group’s attempted targets of all-model, all-group average GDT-TS (a measure of target difficulty) is on the x-axis. All groups attempting at least 20 targets are included. Names of several groups along the “outstanding edge” are labeled.
Table II.
Groups Robustly in Top Half of GDT-HA
| target choice | group ID | group name | # targets attempted | avg of (target avg GDT-TS) | % models GDT-HA ≥ 33 |
|---|---|---|---|---|---|
| easier targets | 147s | Yasara | 60 | 72.06 | 92.6 |
| 293s | Lee-server | 78 | 63.40 | 83.6 | |
| 394s | Fiser-M4T | 76 | 68.56 | 83.5 | |
| average targets or ≈ all targets | 379 | McGuffin | 119 | 58.09 | 79.2 |
| 299 | Zico | 119 | 57.93 | 77.0 | |
| 138 | ZicoFullSTPFullData | 119 | 57.93 | 76.6 | |
| 266 | FAMS-multi | 120 | 57.59 | 76.1 | |
| 434 | Fams-ace2 | 120 | 57.59 | 76.0 | |
| 196 | ZicoFullSTP | 119 | 57.93 | 76.0 | |
| 282 | 3DShot1 | 113 | 57.14 | 75.9 | |
| 485 | Ozkan-Shell | 27 | 56.75 | 75.5 | |
| 453 | Multicom | 119 | 57.93 | 74.7 | |
| 426s | Zhang-server | 121 | 57.64 | 74.6 | |
| 007s | FFASstandard | 121 | 58.58 | 74.5 | |
| 425s | Robetta | 121 | 57.64 | 74.2 | |
| 475 | AMU-Biology | 98 | 60.64 | 74.0 | |
| 193s | CpHModels | 120 | 59.19 | 73.6 | |
| 419 | 3DShotMQ | 113 | 57.14 | 73.5 | |
| 340 | ABIpro | 119 | 57.86 | 73.5 | |
| 149 | A-Tasser | 119 | 57.93 | 73.1 | |
| 407 | Lee | 120 | 57.59 | 72.7 | |
| 409s | Pro-sp3-Tasser | 121 | 57.64 | 72.4 | |
| 154s | HHpred2 | 121 | 57.64 | 72.3 | |
| 436s | Pcons_dot_net | 117 | 58.14 | 72.1 | |
| 142s | FFASsuboptimal | 121 | 58.77 | 72.1 | |
| 135s | Pipe_int | 111 | 58.31 | 72.0 | |
| 122s | HHpred4 | 121 | 57.64 | 71.8 | |
| 297s | GeneSilicoMetaServer | 119 | 58.43 | 71.1 | |
| 247s | FFASflextemplate | 120 | 58.54 | 70.7 | |
| 443s | MUProt | 121 | 57.64 | 70.6 | |
| 182s | MetaTasser | 121 | 57.64 | 70.4 | |
| 438s | Raptor | 121 | 57.64 | 70.3 | |
| 429s | Pcons_multi | 121 | 58.11 | 70.3 | |
| harder targets | 310 | Mufold | 51 | 49.88 | 67.8 |
| 283 | IBT_LT | 52 | 48.63 | 66.3 | |
| 489 | DBaker | 52 | 48.33 | 64.5 | |
| 353 | CBSU | 29 | 51.99 | 63.7 | |
| 200 | Elofsson | 53 | 48.51 | 60.8 | |
| 198 | Fais@hgc | 43 | 49.32 | 59.1 | |
| 178 | Bates_BMM | 52 | 48.33 | 58.7 | |
| 371 | GeneSilico | 52 | 48.33 | 58.0 | |
| 208 | MidwayFolding | 51 | 48.15 | 56.6 | |
| 442 | LevittGroup | 51 | 48.32 | 56.4 | |
Group names in boldface indicate servers.
Despite this clustering, the top of Figure 7 is roughly linear with an upward slope; groups along this “outstanding edge” can be considered exemplary given their target choice. This distribution suggests that groups play to their strengths by focusing on targets for which their specialties will be most useful. In particular, note that server groups dominate for easier targets but human groups comprise the top groups for average and more difficult targets (Table II). Within each of the three areas of target difficulty, these relative rankings provide a meaningful measure of reproducible success at correct template/fold identification. This score for the central set of groups attempting essentially all targets, especially for the automated servers, can act as a suitable accompaniment to the full-model, high-accuracy score shown in Table I and Figure 5.
Self-Scoring: Model 1 vs. Best Model
To complement our use of best models for the new assessment metrics, it is important to measure separately the success of prediction groups in identifying which of their (up to five) submitted models is the best match to the target. That ability is very important to end users of predictions who want a single definitive answer, especially from publicly available automated servers. This self-scoring aspect was assessed by first calculating for each group the randomly expected number of targets for which their model 1 would be also their best model on the traditional GDT-TS metric, nM1best,exp, accounting for different groups submitting different numbers of models (including only groups that submitted at least two models per target on average):
where n̄models is the average number of models per target by the group in question. The actual number of targets for which a group’s model 1 was also their best model can then be calculated and converted to the number of standard deviations from that expected from random chance:
where “act” and “exp” subscripts denote actual and expected quantities.
Figure 8 plots this self-scoring metric for each group versus the average difference in GDT among their sets of models. Most prediction groups are at least 3σ better than random at picking their best model as model 1, but few are right more than 50% of the time. As seen in Figure 8, servers turn out overwhelmingly to dominate the top tier of this metric, making up all of the 8 top-scoring groups and all but one of the top 20. Not surprisingly, groups do somewhat better if their 5 models are quite different, but the correlation coefficient is only 0.3 and accounts for only a small part of the total variance. Unfortunately, success at self-scoring is essentially uncorrelated with high average GDT-TS score (correlation coefficient 0.048). It seems plausible that the best self-scorers are the groups whose prediction procedure is fairly simple and clearly defined, so that they can cleanly judge the probable success of that specific procedure. Although we applaud the self-scoring abilities of these servers, we do not think that these statistics convincingly uphold the traditional CASP practice of combining successful prediction and successful self-scoring together into a single metric. Both aspects are very important to further development of the field, but they seem currently to remain quite unrelated, and we believe they should therefore be assessed and encouraged separately.
Figure 8.
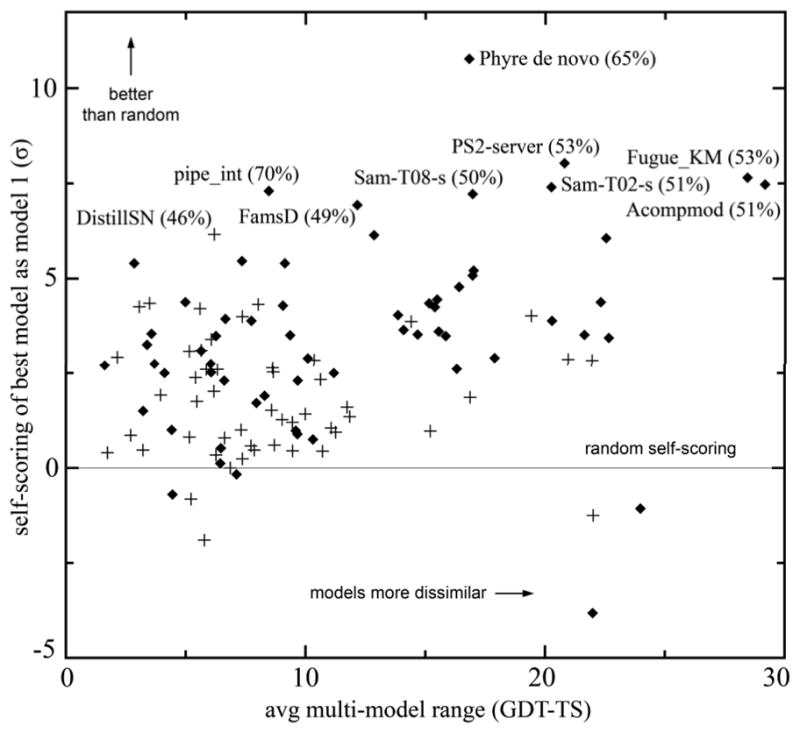
Ability of groups to self-select their best model as model 1. The difference from the percentage expected based on random chance (correcting for different average numbers of models) is plotted vertically (in units of standard deviations); range of scores within a group’s model sets is plotted horizontally. For the best self-scorers, the group name and the percentage of “model 1s” that were actually “best models” are shown. Diamonds indicate server groups, which dominate the top self-scorers; pluses indicate human groups.
Model Compaction or Stretching
Large geometrical outliers on main-chain bond lengths and angles can result from difficulties stitching together model fragments or from inconsistencies in building a local region, while small but consistent non-ideal values can indicate overall scaling problems. Previous CASP assessors have found that a few predictor groups built models with quite extreme compaction across large regions5, which had the side-effect of achieving artificially high GDT scores. As assessors we felt the need to check for such unrealistic distortions on a per-group basis by measuring the average of signed bond length and angle non-idealities over all models submitted; these deviations should average out to zero if there is no systematic directionality. Among groups with poor values on the geometry components of the mainchain reality score, the most skewed bond lengths found for any group had an average difference less than 1 standard deviation short. This represents less than a 1% compaction in the models, which seems unlikely to produce any significant effect on overall GDT scores. Such small systematic properties are unlikely to be intentional, although this phenomenon does highlight the unintended consequences of focusing assessment too strongly on a single measure: prediction methods can inadvertently become “trained” to optimize that metric at the expense of other factors.
Local compaction or stretching is much more common, and in some cases could be an informative diagnostic. The most interesting cases occur along individual β-strands, occasionally compacted but more frequently stretched, to an extent that would match compensation for a single-residue deletion. Trying to span what should be 7 residues with only 6, as in the example shown in Figure 9, produces a string of bond-length outliers at 10σ or more, marked as stretched red springs. This response to avoiding prediction of the specific deletion location keeps all Cα differences under 4Å, but gets the alternation of sidechain direction wrong for half the residues on average. This is not an entirely unreasonable strategy, but would not be part of an optimal predicted model and could not easily be improved by refinement. It would be preferable to assume the structural deletion occurs at one of the strand ends, and to choose the better model of those two alternatives.
Figure 9.

An over-extended β-strand, with main-chain bond-length outliers up to 40σ, marked as stretched-out red springs. T0487-D1, PDB code: 3DLB, argonaute complex37.
Modeling Insertions
One of the classic difficulties in template-based modeling is dealing with regions of inserted sequence relative to any available template. Methods for modeling insertions have become much more powerful in recent years, especially the flexible treatment of information from many partial templates. That otherwise salutary fact made a systematic analysis of this problem too complex for the time scale of this assessment. However, several individual examples were studied.
A very large insertion usually amounts to free modeling of a new domain, such as the FM domain 2 of T041632. Insertion or deletion of only one or two residues within a helix or strand is presumably best treated by comparing relevant short fragments such as strands with β-bulges, with attention to hydrophobicity patterns and to location of key sequence changes such as Gly, Pro, and local sidechain-mainchain H-bonds. Anecdotally it seems there is still room for improvement, with the greatly stretched β-strand of Figure 9 as one example.
The most obvious insertion modeling problems come from an intermediate number (~3–20) of extra residues, which nearly always means insertion of a new loop or lengthening an existing one. The problem of modeling new loops has two distinct parts: first is the alignment problem of figuring out where in the sequence the extra residues will choose to pop out away from the template structure, and second is the modeling of new structure for the part that loops out. Evolutionary comparisons have taught us that the structural changes from insertions are almost always quite localized and that they seldom occur within secondary structure33. Therefore the alignment problem needs to compromise suitably between optimal sequence alignment and the structural need to shift the extra piece of structure toward loops and toward the surface.
As an example, in T0438 loop 255-266 is an insertion relative to both sequence and structure of 2G39, a good template declared as the parent for 9 distinct models from 7 different server groups. Sequence alignment is somewhat ambiguous across a stretch of over 30 template residues, and the 9 models place the insertion in 5 different locations: Δ0, Δ-4, Δ+2, Δ+4, and Δ+10. Figure 10a shows the T0438 loop insertion (green) and the 9 different models (magenta). Three models insert the loop in exactly the right place: one from AcompMod (002_1) and two different pairs of identical models (each pair has all coordinates the same: 220_2 = 351_2 and 220_4 = 351_4) from related Falcon servers. However, none get the loop conformation quite right.
Figure 10.

Evaluating loop insertion models for residues 255-266 of T0438. PDB code: 2G39 (MCSG unpublished). a) Loop insertions (magenta) for the 9 distinct server models (backbone in brown) that declared the template 2G39 (blue), as compared to the actual insertion in T0438 (green) relative to 2G39. b) Correctly aligned insertion for model 002_1 with few geometry problems. c) Incorrectly aligned insertion with significant geometry problems. Red spikes are steric clashes with ≥ 0.5Å overlap of van der Waals radii, green kinks are Ramachandran outliers, gold sidechains are rotamer outliers, pink balls indicate Cβ atoms with excessive deviations from their ideal positions12, blue and red springs are too-short and too-long bond lengths, and blue and red fans are too-tight and too-wide bond angles.
During prediction, by definition no match-to-target measures are available, but model-only measures could be used. To test this, the above 9 models were run through MolProbity13 and local density of validation outliers was examined around the new loops. To increase signal-to-noise, the cutoff for serious clashes was loosened from 0.4 to 0.5Å overlap. Nearly all models have a steric clash at the loop base, between the backbone of the two residues flanking the loop; those therefore do not distinguish between correct and incorrect placement, but show that the ends of insertions are usually kept a bit too close together. The three correctly placed loops, and one offset but entirely solvent-exposed insertion, have only 1 to 3 other outliers (backbone clashes, Ramachandran outliers, bad sidechain rotamers, bond-length and bond-angle outliers, or large Cβ deviations12) and are not notably different from the rest of the model. However, the other 5 incorrectly placed insertions have between 16 and 28 other outliers and can easily be spotted as among the one or two worst local regions in their models. Figure 10b shows outliers for a correctly placed loop, and 10c shows outliers for an incorrectly placed one. For this target, at least, it would clearly be possible during the prediction process to use local model-validation measures to distinguish between plausible and clearly incorrect predicted loop insertions.
Outstanding Individual Models
To complement the group-average statistics, we have also compiled information on outstanding individual models for specific targets. As represented by the three divisions in Table III, outstanding models for a given target were identified in three rather different ways: (1) if their trace stood out from the crowd, to the lower right on the cumulative GDT-TS plot21; (2) if they involved correct identification of a tricky aspect such as domain orientation; or (3) if they had outstanding full-model statistics within a set of models with high and very similar GDT scores.
Table III.
Outstanding Individual Models on a Specific Target
| target | group_model | |||
|---|---|---|---|---|
| Outstanding on cumulative GDT-TS plot: | ||||
| T0395 | DBaker_1 | IBT-LT_1 | ||
| T0407-D2 | IBT-LT_1 | DBaker_3 | ||
| T0409-D9 | Ozkan-Shell_3 | |||
| T0414-D1 | Phyre_de_novo-s_1 | Fams-ace2_3 | ||
| T0419-D2 | Tasser_2 | Zhang_4 | ||
| T0430-D2 | Pcons_multi-s_4 | Falcon-s_1 | ||
| T0460-D1 | DBaker_3 | Jones-UCL_1 | ||
| T0464 | DBaker_5 | |||
| T0467-D9 | DBaker_1 | A-Tasser_2 | ||
| T0476 | DBaker_1 | Mufold-MD-s_2 | ||
| T0478-D2 | Falcon-s_1 | |||
| T0482-D9 | DBaker_3 | Chicken-George_3 | ||
| T0487-D4 | DBaker_1 | IBT-LT_1 | ||
| T0495 | Sam-T08-h_1 | |||
| Outstanding on combining domains or related targets: | ||||
| T0393-D1,D2 | IBT-LT_1 | |||
| T0398-D1,D2 | Muster_1 | Fams-ace2_5 | ||
| T0429-D1,D2 | Tasser_1,3 | Raptor-s_3 | DBaker_5 | |
| T0472-D1,D2 | Pipe_int-s_1 | Pro-sp3-Tasser_1 | Raptor-s_1 | |
| T0498 & T0499 | Softberry | Feig | IBT-LT | DBaker |
| Outstanding on full-model metrics, among top GDT-HA: | ||||
| T0390-D1 | McGuffin | Pcons-multi-s | ||
| T0392-D1 | Pcons-multi-s | MultiCom | ||
| T0396-D1 | CpHModels-s | IBT-LT | ||
| T0450-D1 | FFASsubopt-s, flex-s | Robetta-s | ||
| T0458-D1 | FFASsubopt-s | MultiCom-Cluster-s | ||
| T0490-D1 | Lee, Lee-s | McGuffin | PoemQA | |
| T0494-D1 | McGuffin | Lee, Lee-s | ||
| T0502-D1 | Shortle | |||
| T0508-D1 | Lee, Lee-s | |||
| T0511-D1 | Lee, Lee-s | |||
Notes: Server groups have “-s” appended to their names;
“-D9” targets were evaluated with alternative domain definitions
Figure 11b illustrates the most dramatic cumulative GDT-TS plot, for T0460, with two individual models very much better than all others: 489_3 (DBaker; green backbone in Fig. 11a) and 387_1 (Jones-UCL). The target is an NMR ensemble (2K4N), shown (black in Fig. 11a) trimmed of the disordered section of a long β-hairpin loop. This is an FM/TBM target, because although there are quite a few reasonably close templates, they each differ substantially from the target for one or more of the secondary-structure elements. Only the two best models achieved a fairly close match throughout the target (GDT-TS of 63 and 54, vs. the next group at 40–44); each presumably either made an especially insightful combination among the templates or else did successful free modeling of parts not included in one or more of the better templates.
Figure 11.

Two outstanding predictions for the TBM/FM target T0460-D1. a) Cα traces are shown for the target in black, for the 134/521 predicted models with LGA-S3 from 30 to 60 in peach, and for the particularly exceptional model 489_3 (DBaker) in green. PDB code: 2K4N (NESG, unpublished). b) Cumulative superposition correctness plot 21 from the Prediction Center website. The percentage of model Cα atoms positioned within a distance cutoff of the corresponding target Cα atom after optimal LGA superposition is shown (x-axis) for a range of such distance cutoffs (y-axis); all models for T0460-D1 are shown in peach. Thus lines lower and further to the right indicate predictions that better coincide with the target. The rightmost lines are models 489_3 (DBaker, green) and 387_1 (Jones-UCL, blue).
T0395 has a long, meandering C-terminal extension relative to any of the evident templates, and its backbone forms a knot (it is related to a set of still undeposited knotted targets from CASP74); that extension was trimmed from the official T0395-D1 target9. However, two models – 283_1 (IBT-LT) and 489_1 (DBaker) – placed the small C-terminal helix quite closely and residues 236-292 fairly well, although neither predicted the knot. No other models came anywhere close.
T0409 (3D0F) is a domain-swap dimer, so that the single chain is non-compact. An alternative assessment was done using a reconnected model for a hypothetical unswapped compact monomer, on which 485_3 (Ozkan-Shell) was the outstanding model.
As an additional note, T0467 was canceled because the ensemble submitted to the Prediction Center was very loose; it is therefore not included in Table III. However, the PDB-deposited ensemble (2K5Q) was suitably superimposed, and two outstanding models were identified: 489_1 (DBaker) and 149_2 (A-Tasser).
Figure 12 shows one of the cases where a few prediction groups assigned the correct orientation between two target domains. T0472 (2K49) is a tightly packed gene-duplication of an α-βββ sub-domain (ribbons in Fig. 12a). There are single-chain templates only for one repeat, and template dimers show a variety of relationships. As can be seen in the alignment plot of Figure 12b, the top three models placed both halves correctly – 409_1 (Pipe_int-s), 135_1 (Pro-sp3-Tasser), and 438_1 (Raptor-s) – while all other models align only onto one half or the other. These three models have the best GDT-TS scores for the whole target and for domain 1 (which requires placing the C-terminal helix against the first 3 β-strands), but are not the top scorers for the TBM-HA domain 2.
Figure 12.
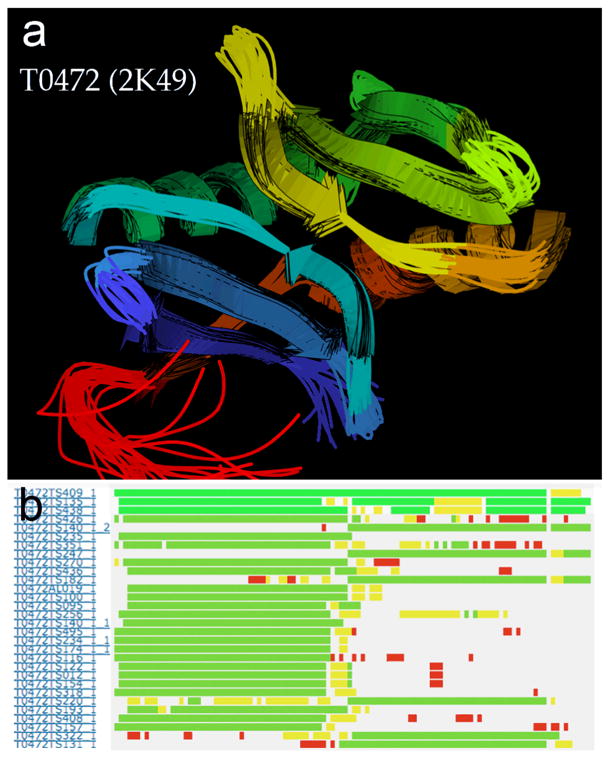
Evaluation of relative sub-domain orientation for T0472 (PDB code: 2K49, NESG, unpublished). a) Ribbon representation of the NMR ensemble for T0472. Note the twofold pseudo-symmetry between the similar compact, sheet-to-helix bundles in the top-right and bottom-left. b) Position-specific alignment plot for the whole target T0472, from the Prediction Center website. Domain 1 is on the left, domain 2 on the right. Residues along the sequence (x-axis) are colored white, red, yellow, and green for increasingly accurate alignment. Note the top three models (y-axis), which are the only ones with good alignment in both sub-domains.
It would be expected that a group especially good at modeling relative domain orientations should have an outstanding GDT-TS Z-score for whole targets (as opposed to by-domain targets). The top-scoring group on whole targets is DBaker, with an average Z of 1.001 vs. the next-highest at 0.828 (Zhang). However, those high whole-target Z-scores are earned primarily on single-domain rather than two-domain targets, by unusually good modeling of difficult loops or ends that were trimmed off the domain targets.
Another case of recognizing a non-obvious relationship is the four groups whose predictions matched both T0498 and T0499. These are the nearest thing to a “trick question” in CASP8, since they represent a pair of structures designed and evolved to have nearly identical sequences (only 3 residues different) but very distinct folds: T0498 resembles the 3-helix bundle of Staphylococcal protein A and T0499 resembles the ββ-α-ββ structure of the B-domain of Staphylococcal protein G. Both sequences are confusingly close to that of protein G, but there are possible templates (1ZXG and 1ZXH) from an earlier pair of less-similar designs34. The four prediction groups that correctly matched both targets – Softberry, Feig, IBT-LT, and DBaker – may well have done so by identifying that earlier work; however, we feel that making effective use of outside information is an important and positive asset in template-based modeling.
The final section in Table III includes only easier targets (mostly TBM-HA, server-only), where many models have high and very similar GDT scores. Among those, there can be a wide spread of full-model scores, and the listed examples were selected as clearly outstanding on combined scores. Figure 13a shows such a plot for T0494, and parts b and c compare the conformational outliers for one of these outstanding models (from Lee, McGuffin, and Lee-s) versus a model with equivalent GDT score but poor full-model scores of both model-only and match-to-target types. Such cases provide examples of “value added” beyond the Cαs to produce a predicted model of much greater utility for many end uses.
Figure 13.

Differentiating models with equally good GDT scores, based on full-model performance for both physical realism and match to target. a) Average full-model Z-score, plotted against raw GDT-HA, on individual best models for target T0494-D1 (PDB code: 2VX3, SGC, unpublished). b) Model 407_3 (Lee) has a GDT-HA of 65.9 and the best average full-model Z-score on this target. c) Another model with essentially the same GDT-HA (65.2) has a much lower full-model Z-score, including poorer match to target sidechains and H-bonds; the six individual scores are listed. Mainchain-mainchain steric clashes, rotamer and Ramachandran outliers, and Cβ deviations are flagged in color for parts b and c, which show a representative portion of the model structures.
Discussion
It has been a fascinating privilege to become deeply immersed in the complex and diverse world of current protein structure prediction. The best accomplishments in CASP8 are truly remarkable in ways that were only vague and optimistic hopes 15 years ago. Groups whose work is centrally informed by the process of evolution can now often pull out from the vast and noisy sequence universe the relevant parts of extremely distant homologs, and assemble them to successfully cover a target. On the other hand, methods centrally informed by the process of protein folding can often build up from the properties of amino acids and their preferred modes of structural fragment combination, to model the correct answer for a specific target.
Not surprisingly, however, such outstanding successes are not yet being achieved by most groups, and not yet on most targets by anyone. The prediction process has many stages and aspects that demand quite different methods and talents. Our assessments have striven to separate out various of those aspects and to recognize and reward excellence in them. Indeed, there is a new breadth in the groups singled out by the various new measures: in some cases the same prediction methods that succeed best at the fundamental GDT Cα measures also succeed well on other aspects, but in other cases new players are spotlighted who have specific strengths that could become part of a further synthesis.
Full-model measures
We chose to emphasize local, full-model quality and correctness in this set of assessments in the service of two long-range aspirations. One is that such quality is fundamental to many of the biological uses of homology modeling; the second is that full-model quality will be an essential attribute of the fully successful predictions this field will eventually achieve. The results reported above show that the six new full-model measures exhibit the right behavior for potentially useful assessments: (1) they each correlate robustly with GDT scores if measured for models in the upper part of the bimodal GDT distribution, but their spread of scores indicates that they contribute independent information (Fig. 4); (2) a substantial number of models, and of predictor groups, score well on them, but they are not trivially achievable; and (3) for individual targets, examination of predicted models with high versus low combined full-model scores reveals features convincingly diagnostic of better versus worse predictions of the target (e.g., Fig. 13).
Therefore we conclude that the general approach of full-model assessment is suitable for evaluating CASP template-based models. These new metrics have only had the benefit of one cycle of intensive development and should continue to be improved; some suggestions for desirable modifications are noted below. However, we feel strongly that template-based modeling is ready for full-model assessment, by these or similar measures.
An especially salient point is that excellent scores on the model-only measures (MolProbity and mainchain reality scores), as well as on the match-to-target full-model measures, correspond with the best backbone predictions, both at the global and the local level within a model. For the easiest targets this could result from copying very good templates, but not for hard targets. It would be valuable in the future to study this relationship quantitatively and in a method-specific manner, but current evidence strongly suggests the practical utility of using physical realism to help guide modeling toward more correct answers.
Assessing components of the TBM process
High-accuracy assessment for CASP8 was carried out here over a scope defined by predicted models with GDT-HA ≥ 33, rather than over a scope defined by targets designated as TBM-HA; this general approach was suggested after CASP78. Three types of evaluations were done: (1) “right fold” or right template identification for the initial step (Table II); (2) full-model quality and correctness for the modeling step, in 6 components and overall (Table I); and (3) individual outstanding high-accuracy models (listed in the last section of Table III). It is important to note that each of these evaluations is inherently two-dimensional, in the sense of needing to be considered jointly with another reference metric such as GDT-TS (Fig. 5), GDT-HA (Fig. 6 and 13), or target difficulty (Fig. 7).
Some overall aspects of prediction can be studied for all models (such as Fig. 4a,b), but any assessment of predictor-group performance must use one model per target (out of up to 5 possible submissions). The two reasonable choices are model 1 (as designated by the predictor) or the best model (the most accurate by GDT-TS); this is an extremely contentious issue with strong opinions on both sides. The official TBM group assessment by GDT-TS has always used model 1 and continues to do so for CASP82; some groups have specifically molded their practices to that expectation. FM assessment always looks for the best among all models, since excellent free models are too rare to accept missing one. It is completely clear that having a prediction define a single optimal model would be extremely valuable for end users, and also that it will eventually be true for a mature prediction technology. Therefore self-scoring skill should definitely be assessed and rewarded, but currently it seems surprisingly difficult.
To provide a counterpoint to the model 1 GDT evaluation, to seek out excellence wherever feasible, and perhaps also because we find it difficult to ignore 80% of the available data, we chose to use the best model in all of our full-model scores. Then, separately, we assessed the ability of groups to pick their best model as model 1, measured across the entire range of targets. Those results (Table II and Figure 7) show that self-scoring is very much better than random, especially for some server groups, but that it is seldom correct more than 50% of the time and is completely uncorrelated with average prediction quality. This is a considerably more optimistic evaluation than found for refinement35 and less optimistic than found for high-accuracy targets in CASP78. Overall, however, none of these studies show self-scoring to be at all reliable. We would strongly suggest that it be assessed prominently but separately from other aspects of prediction.
As we gather has often been true for past assessors, some of our new ideas did not work as well as expected. For instance, we expected that using Cβ rather than Cα atoms for a GDT measure would be sensitive to alignment and orientation as well as placement, especially for β-strands. However, the correlation with GDT-TS was far too tight to be useful, and we then developed the more satisfactory GDC-sc measure using sidechain ends.
Many CASP score distributions are bimodal (e.g., Fig. 3) or otherwise highly non-normal, and their shapes vary between targets. This problem is one reason why GDT Z-scores are usually truncated at zero7,2 and one reason why our full-model measures omit models with GDT-HA < 33. We experimented with “robust” statistics36 that use medians in place of averages and median absolute deviation (MAD) scores in place of Z-scores, but the CASP distributions are so far from being unimodal and Gaussian that the median/MAD statistics gave no noticeable improvement and were not adopted. The full-model scores with model-level GDT-HA ≥ 33 filtering showed skewed but unimodal distributions and could acceptably be averaged into an overall full-model Z-score.
On the other hand, several of the new TBM assessments have already shown broader applicability by being incorporated into other aspects of CASP assessment. Our alternative domain definitions for NMR targets were used in disorder assessment30, and the assessment by the six full-model criteria turned out to demonstrate useful improvements obtained by predictors in the model refinement section35.
Future suggestions – procedural
As former outsiders to the CASP process, we undoubtedly miss some of the underlying history and subtleties, but hope that a fresh perspective can identify new trends and possibilities.
The need to diagnose and correct the many problems with model file format and content made the process of evaluating submitted predictions more difficult, as well as potentially producing incorrect assessment scores (see Methods: Model file preprocessing). For future CASP experiments, we would urge more complete and explicit format and content specifications and a much more thorough checking procedure at submission. Ultimately, this is in the predictor groups’ best interests, both for an accurate evaluation within CASP and, more importantly, for broader use in the scientific community. If a model file generated by structure prediction does not follow normal format standards, end users cannot take advantage of general-purpose molecular visualization, modeling, and analysis software to study that predicted model.
Many prediction assessment tools both internal and external to CASP are available and routinely run by the Prediction Center, but there were several tools developed by previous assessors that we either were not equipped to run (such as molecular replacement tests8) or redeveloped for CASP8 use (such as H-bond match to target6). Of the newly developed full-model measures, only the MolProbity score is publicly available in the form needed for prediction assessment (at http://molprobity.biochem.duke.edu). We would encourage an effort to provide all promising evaluation software in a form suitable for use by the Prediction Center, by future assessors, and by individual predictors or users of models. We plan to contribute to such an effort, for both format correction tools and full-model assessment measures.
As explained in Methods: Model Selection and Filtering, we found the standard rules for trimming targets9 too strict in the case of NMR ensembles, especially for measures that are more sensitive to local conformation and less to absolute coordinates. Even for superposition-based metrics, more of the NMR ensemble could be meaningfully included if the 3.5Å cutoff were measured from a model chosen as the most centrally positioned representative rather than between all pairs of models, and one or two outlier NMR models could be allowed where the rest clustered satisfactorily. We feel also that a general decision should be made by CASP predictors, assessors, and organizers as to whether TBM prediction has advanced to a level where all well-ordered, compact, natural parts of a domain sequence should be assessed even if no template covers that specific portion.
In order to support better assessment of separate aspects of template-based modeling, it would be desirable to expand the methodological information the “parent” record is meant to provide. As an important start, when server models are used they should be considered and declared as templates. Prediction is now often done from complex combination of fragments, in which case naming one or a few parent templates may be inappropriate. Additional keywords could be defined to allow generic description of the methodologies and sources used, and gradually after an adjustment period it could be required that something be entered for each submitted model. The keywords should be neutral to group identities, and checks put in place to test for incorrect claims; some automated comparisons between models and templates were done in CASP57, and expansion of such a system to model-model comparisons would provide a good check. With this carefully limited but crucial extra information, assessors would be in a position to make much more focused and useful comparative evaluations.
It appears to us that the boundaries between FM, TBM, and HA target types are becoming increasingly blurred, while distinctive styles and aspects of methodology are more evident than ever. Pure FM targets with no structural templates whatsoever have nearly disappeared9,32, but it is still of central scientific value to develop and test de novo prediction. In evaluating high-accuracy details, we started out using target distinctions of TBM vs. TBM-HA and human vs. server categories, but discovered we could achieve much better coverage and statistics by separating on model characteristics than on target characteristics. (As explained at the end of Methods, our high-accuracy assessments could include more than twice as many human groups if defined by >20 good models than if defined by >20 easy targets.) The modest amount of additional model-file information suggested above would further enable meaningful assessments to be made both within and between methodologies.
Future suggestions – content
Several potential improvements in the full-model criteria are evident now, after their use in CASP8. For mainchain H-bonds, the plot in Figure 4a makes it clear that H-bonds short-range in sequence (≤ i to i+4) should be down-weighted somewhat. In general, many measures including GDT scores could profit from investigating differential weighting by secondary-structure type, since an overall fold is influenced about as much by a single β-strand as by a single α-helix but the latter has about twice as many residues per unit length; relative weights of 1:2:2 for helix:beta:coil would be reasonable default values to start from. For sidechain-specific measures, it would be preferable to compromise between using all (as here and 8) and omitting all6 surface sidechains. Our vote would be for down-weighting or omitting the subset of sidechains that are fully exposed without good contacts to other structure within their own domain.
More generally, some form of compactness measure would be desirable, although finding a suitable one is more difficult than it sounds. As in CASP76, we limited contact analysis to hydrogen bonding; however, more general forms should probably be explored again in the future.
Finally, it is clear that our answer to the question posed in the Introduction is “Yes!” Template-based modeling is indeed ready to benefit from full-model assessment, and so full-model measures of some sort should definitely be continued in future CASPs.
Supplementary Material
Acknowledgments
We would like to thank the Prediction Center for their capable and timely support, Andriy Kryshtafovych in particular for special runs such as for “D9” alternative target definitions, Scott Schmidler for advice on statistics, the organizers and previous assessors for their work and insights, the experimentalists for providing targets, and the predictors for helpful discussions at the CASP8 meeting. This work was supported in part by NIH grants GM073930 and GM073919.
Footnotes
The work presented in this paper was conducted at Duke University in Durham, NC.
References
- 1.Kryshtafovych A, Fidelis K, Moult J. Progress from CASP6 to CASP7. Proteins. 2007;69 (Suppl 8):194–207. doi: 10.1002/prot.21769. [DOI] [PubMed] [Google Scholar]
- 2.Cozzetto D, Kryshtafovych AFK, Moult J, Rost B, Tramontano A. Evaluation of template-based models in CASP8 with standard measures. Proteins. 2009;(Suppl 9) doi: 10.1002/prot.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zemla A. LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31(13):3370–4. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kinch LN, Wrabl JO, Krishna SS, Majumdar I, Sadreyev RI, Qi Y, Pei J, Cheng H, Grishin NV. CASP5 assessment of fold recognition target predictions. Proteins. 2003;53 (Suppl 6):395–409. doi: 10.1002/prot.10557. [DOI] [PubMed] [Google Scholar]
- 5.Tress M, Ezkurdia I, Grana O, Lopez G, Valencia A. Assessment of predictions submitted for the CASP6 comparative modeling category. Proteins. 2005;61 (Suppl 7):27–45. doi: 10.1002/prot.20720. [DOI] [PubMed] [Google Scholar]
- 6.Kopp J, Bordoli L, Battey JN, Kiefer F, Schwede T. Assessment of CASP7 predictions for template-based modeling targets. Proteins. 2007;69 (Suppl 8):38–56. doi: 10.1002/prot.21753. [DOI] [PubMed] [Google Scholar]
- 7.Tramontano A, Morea V. Assessment of homology-based predictions in CASP5. Proteins. 2003;53 (Suppl 6):352–68. doi: 10.1002/prot.10543. [DOI] [PubMed] [Google Scholar]
- 8.Read RJ, Chavali G. Assessment of CASP7 predictions in the high accuracy template-based modeling category. Proteins. 2007;69 (Suppl 8):27–37. doi: 10.1002/prot.21662. [DOI] [PubMed] [Google Scholar]
- 9.Tress ML, Ezkurdia I, Richardson JS. Domain definition and target classification for CASP8. Proteins. 2009;(Suppl 9) doi: 10.1002/prot.22497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Word JM, Lovell SC, LaBean TH, Taylor HC, Zalis ME, Presley BK, Richardson JS, Richardson DC. Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. J Mol Biol. 1999;285(4):1711–33. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
- 11.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000;40(3):389–408. [PubMed] [Google Scholar]
- 12.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50(3):437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 13.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35(Web Server issue):W375–83. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berman HM, Bhat TN, Bourne PE, Feng Z, Gilliland G, Weissig H, Westbrook J. The Protein Data Bank and the challenge of structural genomics. Nat Struct Biol. 2000;7 (Suppl):957–9. doi: 10.1038/80734. [DOI] [PubMed] [Google Scholar]
- 15.Wang G, Jin Y, Dunbrack RL., Jr Assessment of fold recognition predictions in CASP6. Proteins. 2005;61 (Suppl 7):46–66. doi: 10.1002/prot.20721. [DOI] [PubMed] [Google Scholar]
- 16.Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol. 1999;285(4):1735–47. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 17.Arendall WB, 3rd, Tempel W, Richardson JS, Zhou W, Wang S, Davis IW, Liu ZJ, Rose JP, Carson WM, Luo M, et al. A test of enhancing model accuracy in high-throughput crystallography. J Struct Funct Genomics. 2005;6(1):1–11. doi: 10.1007/s10969-005-3138-4. [DOI] [PubMed] [Google Scholar]
- 18.Engh RA, Huber R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogrp. 1991;A47:392–400. [Google Scholar]
- 19.Dill KA, Bromberg S. Molecular Driving Forces: Statistical Thermodynamics in Chemistry and Biology. New York: Garland Science; 2002. p. 704. [Google Scholar]
- 20.Canutescu AA, Shelenkov AA, Dunbrack RL., Jr A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003;12(9):2001–14. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kryshtafovych A, Prlic A, Dmytriv Z, Daniluk P, Milostan M, Eyrich V, Hubbard T, Fidelis K. New tools and expanded data analysis capabilities at the Protein Structure Prediction Center. Proteins. 2007;69 (Suppl 8):19–26. doi: 10.1002/prot.21653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MySQL AB. MySQL Administrator’s Guide and Language Reference. 2. MySQL Press; 2006. [Google Scholar]
- 23.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24(23):2780–1. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Team RDC. R. A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2005. [Google Scholar]
- 25.Richardson DC, Richardson JS. The kinemage: a tool for scientific communication. Protein Sci. 1992;1(1):3–9. doi: 10.1002/pro.5560010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richardson JS, Richardson DC. MAGE, PROBE, and Kinemages. In: Rossmann MG, Arnold E, editors. International tables for crystallography. Vol. F. Crystallography of biological macromolecules. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2001. pp. 727–730. [Google Scholar]
- 27.Kleywegt GJ, Harris MR, Zou JY, Taylor TC, Wahlby A, Jones TA. The Uppsala Electron-Density Server. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2240–9. doi: 10.1107/S0907444904013253. [DOI] [PubMed] [Google Scholar]
- 28.Wallner B, Elofsson A. Prediction of global and local model quality in CASP7 using Pcons and ProQ. Proteins. 2007;69 (Suppl 8):184–93. doi: 10.1002/prot.21774. [DOI] [PubMed] [Google Scholar]
- 29.Block JN, Zielinski DJ, Chen VB, Davis IW, Vinson EC, Brady R, Richardson JS, Richardson DC. KinImmerse: Macromolecular VR for NMR ensembles. Source Code Biol Med. 2009;4:3. doi: 10.1186/1751-0473-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noivirt O, Prilusky J, Sussman JL. Assessment of disorder predictions in CASP8. Proteins. 2009;(Suppl 9) doi: 10.1002/prot.22586. [DOI] [PubMed] [Google Scholar]
- 31.Krieger E, Joo K, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K. Improving physical realism, stereochemistry and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;(Suppl 9) doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ben-David M, Noivirt O, Paz A, Prilusky J, Sussman JL, Levy K. Assessment of CASP8 structure predictions for template free targets. Proteins. 2009;(Suppl 9) doi: 10.1002/prot.22591. [DOI] [PubMed] [Google Scholar]
- 33.Heinz DW, Baase WA, Dahlquist FW, Matthews BW. How amino-acid insertions are allowed in an alpha-helix of T4 lysozyme. Nature. 1993;361(6412):561–4. doi: 10.1038/361561a0. [DOI] [PubMed] [Google Scholar]
- 34.He Y, Yeh DC, Alexander P, Bryan PN, Orban J. Solution NMR structures of IgG binding domains with artificially evolved high levels of sequence identity but different folds. Biochemistry. 2005;44(43):14055–61. doi: 10.1021/bi051232j. [DOI] [PubMed] [Google Scholar]
- 35.MacCallum JL, Hua L, Schnieders MJ, Pande VS, Jacobson MP, Dill KA. Assessment of the protein-structure refinement category in CASP8. Proteins. 2009;(Suppl 9) doi: 10.1002/prot.22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huber PJ. Robust Statistics. New York: John Wiley & Sons; 1981. [Google Scholar]
- 37.Wang Y, Sheng G, Juranek S, Tuschl T, Patel DJ. Structure of the guide-strand-containing argonaute silencing complex. Nature. 2008;456(7219):209–13. doi: 10.1038/nature07315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.