

Abstract
Pancreatic insulin-producing β-cells have a long lifespan, such that in healthy conditions they replicate little during a lifetime. Nevertheless, they show increased self-duplication upon increased metabolic demand or after injury (i.e. β-cell loss). It is unknown if adult mammals can differentiate (regenerate) new β-cells after extreme, total β-cell loss, as in diabetes. This would imply differentiation from precursors or other heterologous (non β-cell) source. Here we show β-cell regeneration in a transgenic model of diphtheria toxin (DT)-induced acute selective near-total β-cell ablation. If given insulin, the mice survived and displayed β-cell mass augmentation with time. Lineage-tracing to label the glucagon-producing α-cells before β-cell ablation tracked large fractions of regenerated β-cells as deriving from α-cells, revealing a previously disregarded degree of pancreatic cell plasticity. Such inter-endocrine spontaneous adult cell conversion could be harnessed towards methods of producing β-cells for diabetes therapies, either in differentiation settings in vitro or in induced regeneration.
Keywords: transgenic, mouse, pancreas, islet, insulin, glucagon, diabetes, regeneration, transdifferentiation, reprogramming, cell plasticity, cell lineage tracing, cell ablation, diphtheria toxin, precursor cell, β , α
In vivo adult lineage reprogramming (transdifferentiation), i. e. the notion that adult differentiated cells can change fates from one cell type to another, has little experimental support with mouse models 1,2. In pancreas, nevertheless, there is evidence of induced exocrine acinar cell reprogramming: ectopic expression of pro-endocrine factors resulted in conversion of acinar cells into insulin-producing β-cells 3. In another study, loss of c-Myc activity in pancreatic progenitor cells lead to a progressive transdifferentiation of adult acinar cells into adipocytes 4.
The adult endocrine pancreas, i.e. the islets of Langerhans, is made of four different hormone-producing cell types: β-cells produce insulin, α-cells glucagon, δ-cells somatostatin and PP cells pancreatic polypeptide. During development, a fifth cell type, ε, contains ghrelin. In normal conditions, β-cell maintenance relies on their long lifespan, as they proliferate little 5,6. In response to increased physiological demand, there is increased β-cell proliferation 7,8. If the β-cell mass decreases below certain critical levels (around 10% of the normal values 9), there is diabetes onset; this is very clear for the juvenile form of the disease, called Type 1 diabetes (T1D), usually of autoimmune etiology. There is evidence of β-cell regeneration in children with T1D and in young diabetic rats 10-12, as well as after experimental surgical or chemical pancreatic injury 13-16. In these conditions, β-cell replication accounts for β-cell regeneration 13-16, although in the absence of appropriate cell lineage tracing studies other processes cannot be excluded 17-19. In this respect, the formation of new β-cells from precursors expressing Neurogenin3, thus mimicking embryonic islet development, was reported in adult mice with acute pancreatitis induced by ductal ligation 20.
β-cell loss in all available experimental models of diabetes is partial, uncontrolled, or associated with inflammation or autoimmunity. Here, we studied the inherent regenerative capacity of the adult pancreas to produce new β-cells after their near-total loss, a condition close to T1D, but without autoimmunity. Such an extreme situation allowed us to explore whether new insulin-producing cells can emerge from other sources than pre-existing β-cells, since these were almost totally depleted. For this purpose, we used two in vivo genetic approaches: cell ablation combined with cell lineage tracing 21,22. We created a model of inducible, rapid β-cell removal (>99%) by administration of diphtheria toxin (DT) 22,23. In mice, the transgenic expression of the DT receptor (DTR) followed by systemic administration of DT permits an exquisite, specific cell ablation by apoptosis 24,25. We thus generated mice in which β-cells bore DTR. In this model, β-cell regeneration was monitored in combination with cell lineage tracing devised to investigate the origin of newly formed β-cells. We found that the adult pancreas can generate new β-cells after their near total loss, mainly by the spontaneous reprogramming of α-cells.
Ablation of β-cells
We generated mice bearing a transgene containing an insulin promoter and the diphtheria toxin receptor coding sequence (RIP-DTR). The transgene was targeted to the Hprt locus of the X chromosome. The aim was to ablate either 50% or 100% of the β-cell mass using hemizygous females (in which there is random X inactivation) or males, respectively (Fig. 1a). DTR expression per se did not cause any distinguishable phenotype. Administration of DT to hemizygous RIP-DTR females did not affect their basal glycemia or life expectancy, whereas males and homozygous females became rapidly hyperglycemic (Supplementary Fig. 1a).
Figure 1. β-cell ablation and regeneration.

a, RIP-DTR mice express DTR on 100% or 50% of β-cells (blue cells in the cartoon: DTR-bearing β-cells). b-c, Measurement of β-cell mass after ablation and regeneration. Ablation of 99.6% of the β-cell mass is followed by a 3-fold increase between 15 and 30 days (from 5.9μg to 18.5μg), and up to 10-44-fold 10 months later. One-way ANOVA (p=0.0009) and Mann-Whitney tests (* p<0.05). d, Representative islets at various moments after DT. Bars: 20μm.
All subsequent experiments were performed using 2-month-old male mice. DT treatment triggered full-blown diabetes, with polyuria, polydipsia, polyphagia, ketoacidosis, and weight loss and, in absence of insulin treatment, death (Supplementary Fig. 1b-d, and not shown). Two weeks after DT, the pancreatic insulin content (Supplementary Fig. 1e) and the insulin transcription level (Supplementary Fig. 1f), had dropped to 0.3% and 0.01% of the control value, respectively. β-cell loss was confirmed histologically (Fig. 1a and Supplementary Fig. 2a-c): the β-cell mass decreased from 1,594μg to 6μg 15 days post-DT (Fig. 1b-c), which corresponds to a disappearance of 99.6% of the β-cells. Apoptotic β-cells and mild islet fibrosis were apparent in the days following DT injections, but inflammation, insulitis or extra-insular cell death were not observed (Supplementary Fig. 2d, and not shown).
β-cell regeneration
To explore the possibility of β-cell regeneration and its kinetics, mice were sacrificed at different time points after β-cell ablation, for a period of up to 10 months. Between 15 days and one month, β-cell mass and total pancreatic insulin content increased by a factor of 3 (from 5.9±1.9μg to 18.5±4.6μg; see below Fig.1b-d, and not shown). During this initial period, transcription of the 2 insulin genes increased by a factor of 10 (Supplementary Fig. 1f; also Supplementary Fig. 10a).
In long-term experiments, mice were kept alive for up to 10 months after ablation. During the initial 5 months, animals were regularly given subcutaneous insulin implants whenever their glycemia was above 20mM (16 mice were studied in total). From the 6th month on, all mice survived without further insulin treatment, thus showing clear signs of recovery (Supplementary Fig. 3a). The β-cell mass was found increased in all animals: 10-fold in mice that remained diabetic, and up to 44-fold in animals that displayed improved glycemic control. This increment corresponds on average to 10% of the normal β-cell mass (between 4% and 17%, respectively; Fig. 1b-d). About 10% of a normal β-cell mass is found in patients with recent-onset T1D, and represents the lowest amount of β-cells able to ensure a near normal basal glycemia 9.
Almost all medium and large islets showed signs of β-cell regeneration. In fact, 60% of islets contained no or up to 2 β-cells per islet section 15 days after ablation, whereas 10 months later 96% of islet sections contained more than 2 β-cells (Supplementary Fig. 3b,c). This suggests that all islets in adult pancreas can regenerate β-cells. No β-cells were found in extra-insular locations.
Spared β-cells do not increase replication
The first month after β-cell ablation is a period of intense regeneration in RIP-DTR mice: β-cell mass triplicates between 15 and 30 days after β-cell destruction (Fig. 1b,c). Therefore, the origin of β-cells that are found 1 month after β-cell killing was studied by lineage tracing. We used the tamoxifen-dependent Cre/loxP system (“pulse-chase” rationale) to label pre-existing β-cells and measure the contribution to regeneration of the rare β-cells spared by DT. We generated transgenic mice bearing the transgenes RIP-CreERT (inducible tagger) 7, R26-YFP as reporter 26 and RIP-DTR (toxigene) (Fig. 2a and Supplementary Fig. 4a). Administration of tamoxifen induces the expression of the reporter protein YFP from the Rosa26 locus exclusively in β-cells: roughly all of them (95.4±0.5%) were YFP+ (Fig. 2c-e). We measured the contribution of surviving β-cells or their progeny to regeneration as follows: animals were given tamoxifen and 7-days later DT, and the proportion of YFP-tagged cells was determined after 15 and 30 days (Supplementary Fig. 4a). Fifteen days after ablation, only 80±2.9% of the escaping remaining β-cells were YFP+; this proportion further dropped to 7.6±1.8% of the β-cells found 30 days after DT treatment (Fig. 2b-k). These results suggest that: i) there is formation of new β-cells from non-β cell origins very rapidly after ablation (about 20% and 90% of them were not labeled 2 weeks and 1 month after DT treatment, respectively), and ii) escaping β-cells likely do not contribute to the expansion of the β-cell mass, since the proportion of labeled β-cells decreased by 10-fold, from 80% to 7.6%, while β-cell mass tripled.
Figure 2. Conditional β-cell lineage tracing.
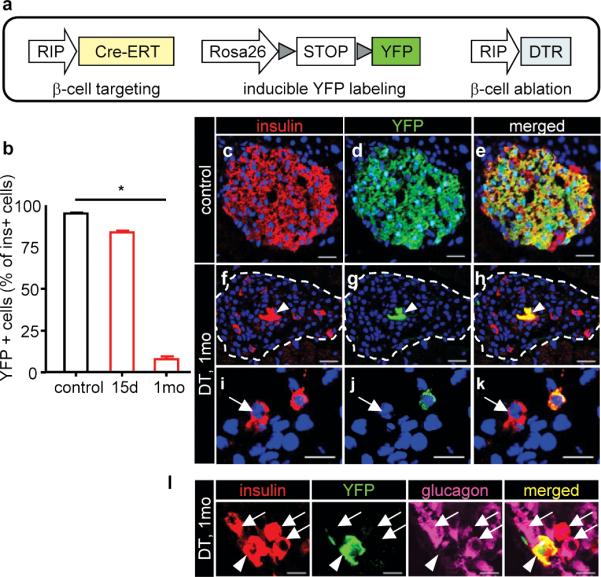
a, Transgenes. b, Proportion of YFP+ β-cells. Controls: 95.4±0.5% (n=4; 159-499 β-cells scored/mouse; 5-12 islets/individual). Two weeks and 1 month after DT, 80.6±2.9% and 7.6±1.8% β-cells were labeled, respectively (15 days: n=3 mice, 80-174 β-cells from 15-25 islets/mouse; 1 month: n=3 mice, 54-73 β-cells from 15-24 islets/mouse). *P<0.01. One-way ANOVA (p=0.0181) and Dunn's multiple comparison test (* p<0.05). c-e, Most β-cells express YFP in controls. f-k, Few β-cells are YFP+ after one month (arrowhead) (f-h). In i-k, 2 β-cells are shown (arrow: YFP-negative β-cell). l, glucagon+/insulin+ cells are YFP-negative (arrows); YFP+/insulin+ cells are glucagon-negative (arrowhead). Bars: 20 μm (c-h); 10μm (i-l).
Contrary to previous models of β-cell ablation/regeneration 16,27,28, we noticed that as compared with DT-untreated controls, β-cell proliferation in RIP-DTR mice was not increased during this period (15 and 30 days after DT), independently of whether β-cells were labeled or not (0.96% β-cells were Ki67+ vs. 0.7% in healthy mice of the same age; 207 β-cells were scored out of 329 islets from 119 sections of 5 DT-treated mice, and 4,367 β-cells from 5 mice in controls. P=0.9, NS; Fischer's test). The same pattern of low proliferation was observed long time after ablation (at 10 months: 0.3% β-cells were Ki67+; 869 β-cells scored from 8 mice); this suggests that long-term regeneration (Fig. 1) does not rely on increased β-cell replication.
Together, dilution of labeled β-cells while β-cell mass increases, and without increased β-cell proliferation, is most consistent with a model of regeneration from heterologous, i.e. non β-cell, origins.
Bihormonal cells arise after β-cell loss
Increased glucagon production and secretion are often associated with insulin deficiency in T1D and T2D humans 29. In RIP-DTR mice, the total pancreatic glucagon content, glucagonemia, and glucagon gene expression were increased by 2-fold after β-cell loss (Supplementary Fig. 5a-c). Islets became prominently composed of α-cells, yet α-cell proliferation and mass remained unchanged during the whole period of analysis (up to 10 months; Supplementary Fig. 5d,e). In the days following β-cell destruction, cells co-expressing glucagon and insulin became frequent: about 1/3 of the rare cells containing insulin (Supplementary Fig. 5f,g) or conversely, 1-3% of the glucagon-expressing cell population (not shown). Interestingly, the glucagon+/insulin+ cells remained detectable at all times after ablation, with similar relative proportions 10 months after ablation (Supplementary Fig. 5f). In the RIP-CreERT-mediated lineage tracing described above, these bihormonal cells were never YFP-labeled, thus revealing that they were not β-cells that escaped ablation and became glucagon-expressers (Fig. 2l). They should therefore be either pre-existing α-cells that start producing insulin, or undefined precursors that start producing and storing the 2 hormones, or both. Interestingly, other β-cell markers, such as the transcription factors Pdx1 and Nkx6.1 were also found in a fraction of glucagon-expressing cells very rapidly after DT treatment, and subsequently throughout all the period of analysis, up to 10 months (see below Fig. 4c and Supplementary Fig. 6a,b).
Figure 4. β-cell marker expression.

a, Control mice: Nkx6.1 is expressed in β-cells. Bars: 20 μm. b, One month post-ablation, 5.05±0.8% of glucagon-expressing cells are Nkx6.1+ (2,855 cells in 277 islets, 5 mice, vs. 0.37±0.3% in controls; 674 α-cells in 39 islets, 3 mice; P=0.035). c, Nkx6.1 expression in glucagon+ cells (top). Some cells also express insulin (middle). YFP+/Insulin+/Nkx6.1+/Glucagon-negative cell (bottom). Bars: 10 μm. d, Proposed reprogramming sequence.
Although marker colocalization per se is not a proof of ontogenetic relationships between different cell types 12, we reasoned that cells co-expressing glucagon and β-cell-specific markers, in particular insulin, might represent emergent β-cells.
α-cells transdifferentiate to β-cells
We devised a conditional α-cell lineage analysis to determine whether pre-existing α-cells are at the origin of glucagon+/insulin+ co-expressing cells and new β cells. We generated a transgenic strain in which only α-cells are selectively and irreversibly labeled before β-cell ablation (tetracycline-dependent Cre/loxP system). These mice were termed Glucagon-rtTA. They bore in addition the TetO-Cre responder transgene 30, the reporter R26-YFP 26, and the RIP-DTR transgene (Fig. 3a; Supplementary Fig. 4b). TetO (tetracycline-responsive promoter/operator) drives the expression of Cre recombinase after rtTA activation with the tetracycline analog doxycycline (DOX, “the pulse”). Almost no α-, β- or δ-cells were YFP-labeled without DOX in 2-month-old males (some 0.2%), whereas almost 90% of α-cells were irreversibly tagged when DOX was given during 15 days after weaning (Supplementary Fig. 7). The Glucagon-rtTA inducible system is thus efficient. Two weeks after ending DOX administration, five 2-month-old males received DT and were sacrificed after one additional month, to assess for the possible direct contribution of adult α-cells to β-cell regeneration: any α-to-β-cell reprogramming would result in the presence of YFP-labeled β-cells (Supplementary Fig. 4b). Shortly after ablation, islets were composed almost exclusively of YFP+ α-cells (Fig. 3g-j; see also Supplementary Fig. 10b). We found that nearly 90% glucagon+/insulin+ co-expressing cells were YFP-labeled (Fig. 3l-o), indicating that they were pre-existing α-cells that started expressing insulin.
Figure 3. α-to-β reprogramming.

a, Transgenes used for the conditional α-cell lineage tracing. b-e, DOX-treated mice, without DT. f, Most α-cells are YFP+ in controls (88.1±4.42%; n=3 mice; 2,258 α-cells scored, 108 islets). g-j, One month after DT, islets are mostly composed of YFP+ α-cells. k, Proportion of YFP+/insulin+ cells in DOX-treated mice. DT-treated group: 63.6±8.6% (511 β-cells from 239 islets; 5 mice). l-o, YFP+/insulin+/glucagon+ cell (arrowhead; 89.87±3.04% of insulin+ cells). p-s, YFP+/insulin+ cells, not expressing glucagon (arrowheads). Bars: 20 μm (b-j); 10 μm (l-s).
On average, 65% of the cells expressing insulin one month after β-cell ablation were YFP+ (Fig. 3k, p-s); almost 90% of them (YFP+/insulin+) still contained glucagon as well (Fig. 3l-s). This lineage tracing revealed their direct origin from adult α-cells, which were irreversibly tagged before injury: they were reprogrammed α-cells.
This result was confirmed with two independent different experiments. In the first study, an important proportion of tagged insulin+ cells appeared following the massive β-cell ablation using a constitutive lineage-tracing with the Glucagon-Cre strain 21,31; this further supports the concept of an adult α-cell origin for regenerated β-cells in RIP-DTR mice (Supplementary Fig. 8).
The second confirmatory experiment revealed the absolute requirement of α-cells for the formation of glucagon+/insulin+ co-expressing cells after near-total β-cell loss: the bihormonal cells were absent when α-cells were co-ablated with β-cells in mice bearing an additional transgene, termed Glucagon-DTR, engineered to ablate α-cells (Supplementary Fig. 9).
We explored the proliferation rate of reprogrammed α-cells (i.e. YFP+/insulin+ in Glucagon-rtTA mice) and, again, we found no significantly increased insulin+ cell replication one month after ablation (1.3%; 2 β-cells out of 149 scored were Ki67+, one YFP+ and the other YFP-; 456 islets from 163 sections; 5 mice were analyzed) as compared with healthy mice of the same age (0.9%; 19 β-cells Ki67+ out of 2,192 from 68 islets of 5 mice. P=0.70, NS; Fischer's test).
Gene expression quantification revealed that several β-cell markers were severely downregulated after β-cell loss, as expected. Interestingly, expression of these very genes started to increase between one and two weeks after ablation, coincident with the beginning of regeneration (Supplementary Fig. 10a). This up-regulation of β-cell-specific genes occurred selectively within islets, which are mainly composed of α-cells at this stage (Supplementary Fig. 10b-d). We confirmed the expression of some of these β-cell markers at the protein level in glucagon-expressing cells, in bihormonal cells (YFP+/glucagon+/insulin+) and in the scarcer YFP+/insulin+ cells having lost glucagon expression: Nkx6.1 (Fig. 4a-c), Pdx1 (Supplementary Fig. 6) or the β-cell-specific glucose transporter type 2 (GLUT2; Supplementary Fig. 11).
Together, these observations are compatible with a model in which α-cells become β-cells (Fig. 4d).
Discussion
We have observed that the adult pancreas has the ability of making β-cells from heterologous origins in a pathological situation whereby β-cells have been completely lost, or almost. Regeneration in RIP-DTR mice is weaker than in other mouse models of less severe β-cell destruction 16,27,28 probably because there is almost no β-cells left after DT treatment; but why β-cell replication is not increased after the massive injury remains unclear, and is in contrast with observations reported after partial β-cell loss 16,27,28. This fact alone reveals the biological significance of studying regeneration and tissue responses under various contexts, such as the degree of injury or the age of disease onset.
Expression of Pdx1 may be part of the α-cell conversion mechanism: ectopic Pdx1 activity, alone or combined with other factors, drives hepatocytes or acinar cells into insulin production 3,32,33. Pdx1 binds directly to insulin and glucagon promoters 34, thus inhibiting glucagon expression and inducing insulin transcription 35. Other β-cell factors may determine the α-to-β reprogramming, such as Nkx6.1, which may also contribute to glucagon gene inhibition and activation of β-cell-specific genes 36, or Pax4, which regulates the balance between α- and β-cells by antagonizing Arx in endocrine progenitors 37. In this respect, it was recently reported that expression of Pax4 in embryonic α-cells using the Glucagon-Cre transgenics 21 induces their conversion into β-cells 38. Because mature α- and β-cells share a number of transcription factors (such as Isl1 and Pax6) 39 and a common ancestor 21,39, the α-cell represents an appropriate candidate for reprogramming to β-cell phenotype. Moreover, α- and β-cells are functionally very close, with a similar machinery to metabolize glucose and secrete hormones 40 : both cell types express glucokinase and ATP-regulated K+-channels, suggesting that they differ in glucose transport but not in glucose utilization 41,42. Expression of GLUT2 in insulin-producing reprogrammed α-cells, in addition to Nkx6.1 and Pdx1, should allow them to secrete insulin upon glucose stimulation, like functional β-cells.
Previous models of β-cell injury do not report heterologous regeneration of β-cells, yet this was not explored with lineage tracing analyses 16,27,28. In these models, remaining β-cells were abundant (at least 20% of the initial β-cell mass), which suggests that β-cell loss must be near total for triggering heterologous β-cell formation. In this regard, milder β-cell ablation in RIP-DTR mice, by using hemizygous females (50% ablation) or in males treated with lower doses of DT (95-98% ablation), has a different outcome: in these situations, either there is no measurable regeneration (after 50% ablation; Supplementary Fig. 12), or induction of α-cell reprogramming is decreased, with a more important contribution of spared β-cells (not shown). The amount of β-cell loss thus determines whether there is regeneration (Supplementary Fig. 12) and, together with the type of injury, it influences the degree of cell plasticity and regenerative resources of the adult pancreas (Supplementary Fig. 12).
These observations raise issues with respect to cell plasticity and regenerative recovery from lesion: α-cells were never considered previously as a potential source of cells for β-cell therapy in diabetics. Our results argue that a deep lesion (total or near-total β-cell ablation, as in T1D) causes the release of some form of signal that allows prolonged and substantial β-cell regeneration. The presence of bihormonal cells (glucagon+/insulin+) long time after lesion induction is compatible with α-cell reprogramming not being limited by temporal restrictions, and thus with regeneration in aged individuals. Alternatively or in addition, persistence of glucagon staining in some reprogrammed α-cells may reflect impaired or inhibited glucagon granule exocytosis: in cells possessing multiple types of regulated secretory granules, exocytosis can be activated independently for each of them 43.
We found that the proportion of β-cells derived from reprogrammed α-cells is very variable among individuals having the same degree (>99%) of β-cell destruction: between 32% and 81% (Fig. 3k). This further stresses the adaptability of adult pancreas and reveals high versatility in response to injury. This plasticity is reminiscent of the various mechanisms of liver regeneration 44.
In long-term human T1D patients, occasional β-cells are found scattered in the pancreas, as well as circulating C-peptide, an indicator of proinsulin processing and insulin secretion 11,45,46. Also, complete β-cell function recovery has been reported in young T1D patients 47,48. Whether this is the consequence of a continuous regeneration of new β-cells, as we have seen in mice, or persistence of few β-cells, which would escape autoimmunity, is not known 45. Nevertheless, our observations in mice should encourage attempts of treatment by inducing and enhancing regeneration after controlling the autoimmune aggression.
Finally, these findings suggest that the production of new models for selective and total cell ablation could lead to discoveries regarding regeneration induction and cell plasticity in other organs, including pathological conditions such as dysplasia or cancer.
METHODS SUMMARY
Generated mice
RIP-DTR transgene was prepared by sub-cloning the human HB-EGF cDNA 25 into a plasmid containing a 0.7kb-long fragment of the rat insulin II promoter, and a 1.6kb-long sequence containing an intron and the polyA signal of the rabbit β-globin gene, as described 21,23. The transgene was introduced into a pDEST vector for homologous recombination at the HPRT locus in BPES cells (C57Bl/6-129 background, Speedy Mouse®, Nucleis). Recombinant BPES cells were used to generate chimeras. The Glucagon-DTR construct was arranged by replacing the Cre cDNA of Glucagon-Cre plasmid 21 with the human HB-EGF cDNA. We generated 7 independent F0 founders by pronuclear injection 49, 2 of which had optimal expression of DTR. Glucagon-rtTA construct was generated by replacing the Cre sequence of Glucagon-Cre plasmid 21 by the rtTA-Advanced cDNA sequence of pTet-On Advanced vector (Clontech, cat n°630930). Thirteen F0 founders were obtained by pronuclear injection. Two of the 13 strains showed equivalent efficiency of glucagon cell labeling after DOX treatment. Diphtheria toxin, tamoxifen, doxycycline and insulin treatments. Diphtheria toxin (DT) (D0564, Sigma) was given to 2 month-old mice in 3 i.p. injections (126 ng of DT per injection, on days 0, 3 and 4). Tamoxifen was freshly prepared (50 mg/ml; TAM; Sigma T5648) and administered with a gastric catheter (5 doses of 10 mg, every 2 days). TAM (10mg) was diluted in 10μl 100% EtOH, completed to 200μl with 0.9% NaCl and sonicated for 60 seconds at minimum intensity. Doxycycline (DOX; 1 mg/ml) (D9891, Sigma) was added to drinking water for 2 weeks. After DOX removal, mice were kept during 15 additional days without treatment before DT administration; this period is sufficient for DOX clearance 50. Mice received subcutaneous implants of insulin (Linbit, Canada) when hyperglycemic (>20 mM) in the long-term regeneration experiments.
Supplementary Material
Acknowledgements
We are grateful to Pierre Vassalli and Christopher V. E. Wright for their insightful comments and support. We also thank R. Stein, S. Kim and A. Ruiz i Altaba for discussions, and G. Flores, C. Gysler, O. Fazio, G. Philippin and C. Vesin for their technical help. We thank D. Melton for the RIP-CreER transgenic strain as well as B. Thorens and C.V.E. Wright for anti-Glut2 and anti-Pdx1 antibodies, respectively. Work was funded with grants from the NIH/NIDDK (“Beta Cell Biology Consortium”), JDRF, Swiss National Science Foundation (and the NCCR “Frontiers in Genetics”), and 6FP EU Integrated Project “Beta Cell Therapy for Diabetes” to P.L.H.
Footnotes
The authors declare no competing financial interests.
‘Supplementary Information accompanies the paper on www.nature.com/nature.’
References
- 1.Zhou Q, Melton DA. Extreme makeover: converting one cell into another. Cell Stem Cell. 2008;3(4):382–388. doi: 10.1016/j.stem.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 2.Uhlenhaut NH, et al. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell. 2009;139(6):1130–1142. doi: 10.1016/j.cell.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 3.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455(7213):627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonal C, et al. Pancreatic Inactivation of c-Myc Decreases Acinar Mass and Transdifferentiates Acinar Cells Into Adipocytes in Mice. Gastroenterology. 2009;136:309–319. doi: 10.1053/j.gastro.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 5.Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54(9):2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- 6.Desgraz R, Herrera PL. Pancreatic neurogenin 3-expressing cells are unipotent islet precursors. Development. 2009;136(21):3567–3574. doi: 10.1242/dev.039214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429(6987):41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 8.Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes. 2009;58(6):1365–1372. doi: 10.2337/db08-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matveyenko AV, Butler PC. Relationship between beta-cell mass and diabetes onset. Diabetes Obes Metab. 2008;10(Suppl 4):23–31. doi: 10.1111/j.1463-1326.2008.00939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang RN, Bouwens L, Kloppel G. Beta-cell growth in adolescent and adult rats treated with streptozotocin during the neonatal period. Diabetologia. 1996;39(5):548–557. doi: 10.1007/BF00403301. [DOI] [PubMed] [Google Scholar]
- 11.Herold KC, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346(22):1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 12.Thyssen S, Arany E, Hill DJ. Ontogeny of regeneration of beta-cells in the neonatal rat after treatment with streptozotocin. Endocrinology. 2006;147(5):2346–2356. doi: 10.1210/en.2005-0396. [DOI] [PubMed] [Google Scholar]
- 13.Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes. 1993;42(12):1715–1720. doi: 10.2337/diab.42.12.1715. [DOI] [PubMed] [Google Scholar]
- 14.Wang RN, Kloppel G, Bouwens L. Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia. 1995;38(12):1405–1411. doi: 10.1007/BF00400600. [DOI] [PubMed] [Google Scholar]
- 15.Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48(12):2270–2276. doi: 10.2337/diabetes.48.12.2270. [DOI] [PubMed] [Google Scholar]
- 16.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest. 2007;117(9):2553–2561. doi: 10.1172/JCI32959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baeyens L, et al. In vitro generation of insulin-producing beta cells from adult exocrine pancreatic cells. Diabetologia. 2005;48(1):49–57. doi: 10.1007/s00125-004-1606-1. [DOI] [PubMed] [Google Scholar]
- 18.Bonner-Weir S, Weir GC. New sources of pancreatic beta-cells. Nat Biotechnol. 2005;23(7):857–861. doi: 10.1038/nbt1115. [DOI] [PubMed] [Google Scholar]
- 19.Trucco M. Regeneration of the pancreatic beta cell. J Clin Invest. 2005;115(1):5–12. doi: 10.1172/JCI23935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu X, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132(2):197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 21.Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. 2000;127(11):2317–2322. doi: 10.1242/dev.127.11.2317. [DOI] [PubMed] [Google Scholar]
- 22.Herrera PL, Nepote V, Delacour A. Pancreatic Cell Lineage Analyses in Mice. Endocrine. 2002;19(3):267–278. doi: 10.1385/ENDO:19:3:267. [DOI] [PubMed] [Google Scholar]
- 23.Herrera PL, et al. Ablation of islet endocrine cells by targeted expression of hormone- promoter-driven toxigenes. Proc Natl Acad Sci U S A. 1994;91(26):12999–13003. doi: 10.1073/pnas.91.26.12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naglich JG, Metherall JE, Russell DW, Eidels L. Expression cloning of a diphtheria toxin receptor: identity with a heparin-binding EGF-like growth factor precursor. Cell. 1992;69(6):1051–1061. doi: 10.1016/0092-8674(92)90623-k. [DOI] [PubMed] [Google Scholar]
- 25.Saito M, et al. Diphtheria toxin receptor-mediated conditional and targeted cell ablation in transgenic mice. Nat Biotechnol. 2001;19(8):746–750. doi: 10.1038/90795. [DOI] [PubMed] [Google Scholar]
- 26.Srinivas S, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1(1):4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cano DA, et al. Regulated beta-cell regeneration in the adult mouse pancreas. Diabetes. 2008;57(4):958–966. doi: 10.2337/db07-0913. [DOI] [PubMed] [Google Scholar]
- 28.Wang ZV, et al. PANIC-ATTAC: a mouse model for inducible and reversible beta-cell ablation. Diabetes. 2008;57(8):2137–2148. doi: 10.2337/db07-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerich JE, Lorenzi M, Karam JH, Schneider V, Forsham PH. Abnormal pancreatic glucagon secretion and postprandial hyperglycemia in diabetes mellitus. JAMA. 1975;234(2):159–155. [PubMed] [Google Scholar]
- 30.Perl AK, Wert SE, Nagy A, Lobe CG, Whitsett JA. Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci U S A. 2002;99(16):10482–10487. doi: 10.1073/pnas.152238499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quoix N, et al. The GluCre-ROSA26EYFP mouse: a new model for easy identification of living pancreatic alpha-cells. FEBS Lett. 2007;581(22):4235–4240. doi: 10.1016/j.febslet.2007.07.068. [DOI] [PubMed] [Google Scholar]
- 32.Li WC, Horb ME, Tosh D, Slack JM. In vitro transdifferentiation of hepatoma cells into functional pancreatic cells. Mech Dev. 2005;122(6):835–847. doi: 10.1016/j.mod.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Fodor A, et al. Adult rat liver cells transdifferentiated with lentiviral IPF1 vectors reverse diabetes in mice: an ex vivo gene therapy approach. Diabetologia. 2007;50(1):121–130. doi: 10.1007/s00125-006-0509-8. [DOI] [PubMed] [Google Scholar]
- 34.Chakrabarti SK, James JC, Mirmira RG. Quantitative assessment of gene targeting in vitro and in vivo by the pancreatic transcription factor, Pdx1. Importance of chromatin structure in directing promoter binding. J Biol Chem. 2002;277(15):13286–13293. doi: 10.1074/jbc.M111857200. [DOI] [PubMed] [Google Scholar]
- 35.Ritz-Laser B, et al. Ectopic expression of the beta-cell specific transcription factor Pdx1 inhibits glucagon gene transcription. Diabetologia. 2003;46(6):810–821. doi: 10.1007/s00125-003-1115-7. [DOI] [PubMed] [Google Scholar]
- 36.Schisler JC, et al. The Nkx6.1 homeodomain transcription factor suppresses glucagon expression and regulates glucose-stimulated insulin secretion in islet beta cells. Proc Natl Acad Sci U S A. 2005;102(20):7297–7302. doi: 10.1073/pnas.0502168102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collombat P, et al. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003;17(20):2591–2603. doi: 10.1101/gad.269003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collombat P, et al. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell. 2009;138(3):449–462. doi: 10.1016/j.cell.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonal C, Herrera PL. Genes controlling pancreas ontogeny. Int J Dev Biol. 2008;52(7):823–835. doi: 10.1387/ijdb.072444cb. [DOI] [PubMed] [Google Scholar]
- 40.Rorsman P, Salehi SA, Abdulkader F, Braun M, MacDonald PE. K(ATP)-channels and glucose-regulated glucagon secretion. Trends Endocrinol Metab. 2008;19(8):277–284. doi: 10.1016/j.tem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 41.Heimberg H, De Vos A, Pipeleers D, Thorens B, Schuit F. Differences in glucose transporter gene expression between rat pancreatic alpha- and beta-cells are correlated to differences in glucose transport but not in glucose utilization. J Biol Chem. 1995;270(15):8971–8975. doi: 10.1074/jbc.270.15.8971. [DOI] [PubMed] [Google Scholar]
- 42.Heimberg H, et al. The glucose sensor protein glucokinase is expressed in glucagon-producing alpha-cells. Proc Natl Acad Sci U S A. 1996;93(14):7036–7041. doi: 10.1073/pnas.93.14.7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burgoyne RD, Morgan A. Secretory granule exocytosis. Physiol Rev. 2003;83(2):581–632. doi: 10.1152/physrev.00031.2002. [DOI] [PubMed] [Google Scholar]
- 44.Fausto N. Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology. 2004;39(6):1477–1487. doi: 10.1002/hep.20214. [DOI] [PubMed] [Google Scholar]
- 45.Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48(11):2221–2228. doi: 10.1007/s00125-005-1949-2. [DOI] [PubMed] [Google Scholar]
- 46.Meier JJ, et al. Direct evidence of attempted beta cell regeneration in an 89-year-old patient with recent-onset type 1 diabetes. Diabetologia. 2006;49(8):1838–1844. doi: 10.1007/s00125-006-0308-2. [DOI] [PubMed] [Google Scholar]
- 47.Karges B, et al. Complete long-term recovery of beta-cell function in autoimmune type 1 diabetes after insulin treatment. Diabetes Care. 2004;27(5):1207–1208. doi: 10.2337/diacare.27.5.1207. [DOI] [PubMed] [Google Scholar]
- 48.Karges B, et al. Immunological mechanisms associated with long-term remission of human type 1 diabetes. Diabetes Metab Res Rev. 2006;22(3):184–189. doi: 10.1002/dmrr.600. [DOI] [PubMed] [Google Scholar]
- 49.Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the Mouse Embryo. A Laboratory Manual. Second ed. Cold Spring Harbor Laboratoy Press; 1994. [Google Scholar]
- 50.Nguyen VX, Nix DE, Gillikin S, Schentag JJ. Effect of oral antacid administration on the pharmacokinetics of intravenous doxycycline. Antimicrob Agents Chemother. 1989;33(4):434–436. doi: 10.1128/aac.33.4.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.