

Oligomerization of the ER Ca2+ sensor, STIM1, is the key triggering step in the activation of store-operated Ca channels. We show that the cytosolic CRAC activation domain, previously known only for its role in binding to and activating Orai1 channels, is also required for stable STIM1 oligomerization.
Abstract
Oligomerization of the ER Ca2+ sensor STIM1 is an essential step in store-operated Ca2+ entry. The lumenal EF-hand and SAM domains of STIM1 are believed to initiate oligomerization after Ca2+ store depletion, but the contributions of STIM1 cytosolic domains (coiled-coil 1, CC1; coiled-coil 2, CC2; CRAC activation domain, CAD) to this process are not well understood. By applying coimmunoprecipitation and fluorescence photobleaching and energy transfer techniques to truncated and mutant STIM1 proteins, we find that STIM1 cytosolic domains play distinct roles in forming both “resting” oligomers in cells with replete Ca2+ stores and higher-order oligomers in store-depleted cells. CC1 supports the formation of resting STIM1 oligomers and appears to interact with cytosolic components to slow STIM1 diffusion. On store depletion, STIM1 lacking all cytosolic domains (STIM1-ΔC) oligomerizes through EF-SAM interactions alone, but these oligomers are unstable. Addition of CC1 + CAD, but not CC1 alone, enables the formation of stable store-dependent oligomers. Within the CAD, both CC2 and C-terminal residues contribute to oligomer formation. Our results reveal a new function for the CAD: in addition to binding and activating Orai1, it is directly involved in STIM1 oligomerization, the initial event triggering store-operated Ca2+ entry.
INTRODUCTION
Store-operated Ca2+ entry (SOCE) is a ubiquitous and essential cellular signaling pathway (Parekh and Putney, 2005). Under physiological conditions, a variety of extracellular ligands activate receptors that stimulate phospholipase C to generate inositol 1,4,5-triphosphate, releasing Ca2+ from the endoplasmic reticulum (ER). The ensuing depletion of Ca2+ from the ER triggers opening of store-operated channels (SOCs) in the plasma membrane (PM). The outline of a molecular mechanism for SOCE has emerged after the discovery of stromal interaction molecule 1 (STIM1), the ER Ca2+ sensor, and Orai1, the pore-forming subunit of the Ca2+ release-activated Ca2+ (CRAC) channel, the most extensively characterized SOC (Lewis, 2007; Cahalan, 2009; Hogan et al., 2010). Reduction of ER lumenal [Ca2+] causes STIM1 to oligomerize, triggering its accumulation in subregions of the ER closely apposed to the PM (Liou et al., 2005, 2007; Wu et al., 2006; Luik et al., 2008). These structures are referred to as “puncta” at the light microscope level (Liou et al., 2005; Zhang et al., 2005) and ER–PM junctions at the electron microscopic level (Wu et al., 2006; Orci et al., 2009). At these locations, Orai1 in the PM accumulates directly opposite STIM1 (Luik et al., 2006; Xu et al., 2006) by binding to a region known as the CRAC activation domain (CAD) or the STIM1 Orai-activating region (SOAR) (Kawasaki et al., 2009; Park et al., 2009; Yuan et al., 2009). Direct physical interaction of CAD with Orai1 opens the CRAC channel (Park et al., 2009; Zhou et al., 2009), leading to highly localized Ca2+ entry at ER–PM junctions (Luik et al., 2006). Studies of naturally occurring loss-of-function mutations as well as STIM1- and Orai1-knockout mice have shown that Ca2+ entry through CRAC channels is essential for normal function of lymphocytes, mast cells, and platelets (Feske, 2009; Hogan et al., 2010).
Oligomerization of STIM1 is the key step in coupling store depletion to SOC activation. Ikura and colleagues first proposed that oligomerization was a trigger for SOCE, based on their finding that a recombinant protein fragment containing only the lumenal EF hands and the sterile alpha motif (SAM) of STIM1 oligomerizes in response to Ca2+ removal in vitro (Stathopulos et al., 2006). Using Förster resonance energy transfer (FRET) techniques, Liou et al. (2007) later confirmed that full-length STIM1 oligomerizes in response to store depletion in cells, shortly before puncta begin to form (Muik et al., 2008). Finally, Luik et al. (2008) replaced the EF-SAM with FRB and FKBP and showed that heterodimerization with a rapamycin analog triggered puncta formation and SOCE, establishing STIM1 oligomerization as the causal link between store depletion and CRAC channel activation. Together, these studies imply that a full understanding of the SOCE mechanism requires description at the molecular level of how STIM1 oligomerization is controlled. STIM1 contains a number of protein association domains, including the EF hand and SAM domain on the lumenal side and multiple coiled-coil (CC) domains on the cytosolic side; thus, an important first step is to identify which of these regions participate in oligomerization.
STIM1 exhibits one of two oligomeric forms depending on the state of intracellular Ca2+ stores. In resting cells with replete stores, native STIM1 is believed to self-associate based on the coimmunoprecipitation of orthogonally labeled STIM1 proteins (Williams et al., 2002; Baba et al., 2006) as well as cross-linking studies of dStim expressed in Drosophila S2 cells (Penna et al., 2008). The precise stoichiometry of this resting state is not yet known (see Discussion), but self-association is prevented by internal deletion of a region encompassing the two putative cytosolic CC domains, suggesting that it is stabilized by CC interactions (Baba et al., 2006). However, this conclusion is complicated to an extent by FRET measurements suggesting that the CC domains themselves are not sufficient or necessary to promote self-association of isolated cytosolic STIM1 fragments (Muik et al., 2009). It is important to note that resting oligomers of mammalian STIM1 are inactive in that they do not spontaneously form puncta nor do they bind to or activate Orai1.
As discussed above, STIM1 is activated by the formation of higher-order oligomers in response to Ca2+ store depletion, culminating in puncta formation and SOCE. Nuclear magnetic resonance studies have shown that the resting, Ca2+ bound form of the EF-SAM fragment is monomeric because of an intramolecular interaction of the EF hands with the SAM; loss of Ca2+ disrupts this interaction, exposing hydrophobic regions that promote the generation of dimers and large aggregates (Stathopulos et al., 2008). However, it is not yet known to what extent oligomerization of the EF-SAM fragment in vitro mimics the oligomerization of full-length STIM1 in situ. In particular, does the lumenal EF-SAM domain by itself drive the formation of higher-order oligomers of STIM1, or are interactions of the cytoplasmic domains also required? Previous studies have used soluble fragments of STIM1 to identify cytosolic regions important for oligomerization (Muik et al., 2009), but these fragments are constitutively active, i.e., they activate Orai independently of store depletion (Penna et al., 2008; Muik et al., 2009; Yuan et al., 2009; Zhou et al., 2009). Hence, it is not clear that cytosolic fragments can be used to study the transition of native STIM1 from a resting to an active oligomerized state. For these reasons, it is important to determine the structural basis of oligomerization of full-length STIM1 in its native cellular environment.
To assess the roles of the STIM1 cytosolic domains in controlling oligomerization in situ, we examined a series of truncated and mutant STIM1 proteins having intact lumenal and transmembrane regions and hence normal ER localization. We applied a combination of approaches, including coimmunoprecipitation, fluorescence recovery after photobleaching (FRAP), and FRET, in addition to confocal imaging of STIM1 and Orai1 colocalization and Ca2+ imaging to detect CRAC channel activation. Our results demonstrate distinct roles for the cytoplasmic domains in resting versus depletion-activated STIM1 oligomerization. They also reveal a new role for the CAD: in addition to activating Orai1, it also enables the oligomerization of STIM1 that initiates SOCE.
MATERIALS AND METHODS
Cells and Plasmids
Human embryonic kidney (HEK) 293 cells (American Type Culture Collection, Manassas, VA) were maintained in DMEM with GlutaMax (Invitrogen, Carlsbad, CA), supplemented with 10% FBS (Hyclone, Logan, UT) and 1% penicillin/streptomycin (Mediatech, Herndon, VA), at 37°C, 6% CO2. A HEK 293 cell line stably transfected with inducible green fluorescent protein (GFP)-myc-Orai1 was generated and maintained as described (Park et al., 2009). Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Wild-type (WT) hemagglutinin (HA)-STIM1 and GFP-STIM1 plasmids have been described (Wu et al., 2006), as has GFP-myc-Orai1 (Luik et al., 2006). Cyan fluorescent protein (CFP)-STIM1 and yellow fluorescent protein (YFP)-STIM1 were gifts from J. Liou and T. Meyer (Stanford University). The intramolecular FRET probe CFP-RPTPα-SpD2-YFP2.1 (Blanchetot et al., 2002) was a gift from J. den Hertog (Netherlands Institute for Developmental Biology). mCherry-myc-Orai1, mCherry-CAD, FLAG-myc-CAD, and YFP-CAD were gifts from C. Y. Park and R. Dolmetsch (Stanford University). Truncated STIM1 mutants were made using a QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, Wilmington, DE) to introduce premature stop codons after amino acid 237 (STIM1-ΔC), 344 (STIM1-CC1), 391 (STIM1-CC2), or 448 (STIM1-CAD). Coiled-coil mutants STIM1-A369K, -L373K, -A376K, and -3K were generated by introducing the relevant mutation into GFP-, CFP-, and YFP-STIM1 or into mCherry-CAD using the QuikChange II XL kit.
Confocal Imaging
HEK 293 cells transiently transfected with 1 μg of each plasmid were plated onto sterile poly-d-lysine–coated coverslips and cultured an additional 24 h before imaging. When an inducible GFP-myc-Orai1 stable cell line was used, GFP-myc-Orai1 expression was induced with 1 μg/ml tetracycline 8 h before imaging. Cells were imaged in 2 mM Ca2+ Ringer's solution containing (in mM) 155 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 10 d-glucose, and 5 Na-HEPES, pH 7.4. For store depletion, cells were perfused with 0 mM Ca2+ Ringer's solution prepared with either 1 μM thapsigargin (TG; LC Laboratories, Woburn, MA) or 1–4 μM ionomycin (EMD Biosciences, San Diego, CA) and (in mM) 155 NaCl, 4.5 KCl, 3 MgCl2, 1 EGTA, 10 d-glucose, and 5 Na-HEPES, pH 7.4. Imaging was performed on a Leica SP2 AOBS confocal microscope system (Deerfield, IL) as described (Park et al., 2009).
FRAP
HEK 293 cells transiently transfected with 2–2.5 μg of GFP-STIM1 variants were split onto coverslips and maintained in culture until imaging 24–72 h after transfection. Cells were imaged on the confocal system described above in 2 mM Ca2+ Ringer's or 0 mM Ca2+ Ringer's + 4 μM ionomycin. Prebleach and recovery images were obtained with the 488-nm line of a 20-mW Ar laser at 50% power, 6–10% transmission. A 2–4-μm strip was photobleached at 50% power, 100% transmission for ∼2 s. Images were analyzed with ImageJ (W. S. Rasband, NIH, Bethesda, MD; http://rsb.info.nih.gov/ij/), and fluorescence recovery traces were fitted with a published equation (Ellenberg et al., 1997) modified to include an offset to account for incomplete photobleaching:
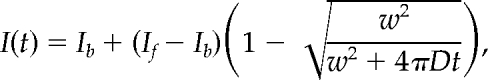 |
where I(t) is the mean intensity within the bleached region at time t, Ib is the intensity immediately after bleaching, If is the intensity after recovery, w is the width of the bleached region, and Deff is the estimated effective diffusion coefficient. Fits to this equation were made by a least-squares criterion with Igor Pro version 4.09 (WaveMetrics, Lake Oswego, OR). The effective diffusion coefficient includes contributions from projection of three-dimensional ER geometry onto a two-dimensional image and possible interactions with immobile or slowly moving proteins, both of which reduce the apparent mobility. When possible, fluorescence recoveries were measured from the same region of individual cells before and after store depletion to facilitate comparison (see example in Figure 3B).
Figure 3.

Diffusional mobility of truncated STIM1 mutants before and after store depletion. (A) FRAP in a HEK 293 cell expressing GFP-STIM1-CAD. After acquisition of prebleach images (left) a 3-μm-wide strip (indicated by the white rectangle, left) was photobleached (middle), and recovery was monitored over several minutes (right). FRAP was performed on the same cell again after store depletion with 0 mM Ca2+ Ringer + 4 μM ionomycin (bottom). Bar, 5 μm; time stamp, min:sec. (B) FRAP time course from the cell in A at rest (red) and after store depletion (black). Fits indicate a Deff of 0.23 μm2/s (rest) and 0.08 μm2/s (depleted). (C) Mean effective diffusion coefficients (±SEM) before (red) or after (black) treatment with ionomycin to deplete Ca2+ stores. Numbers of cells (rest, depleted): STIM1-ΔC (n = 8, 6); STIM1-CC1 (n = 10, 9); STIM1-CAD (n = 8, 13); STIM1-WT (n = 32, 9). Significant differences, determined by Student's t tests, between mean Deff are noted by p values.
FRET
HEK 293 cells coexpressing CFP- and YFP-STIM1 variants were imaged 48–72 h after transfection using the three-cube FRET (E-FRET) method described by Zal and Gascoigne (2004) in which FRET efficiency correlates linearly with the fraction of donor interacting with acceptor. Briefly, cells were imaged on a Zeiss 200M wide-field epifluorescence microscope (Thornwood, NY) using a 40 X F Fluar 1.3 NA objective (Zeiss) and a Polychrome II excitation source (TILL Photonics, Martinsreid, Germany). At each time point, three images were taken through the equator of the cell, using the following filter cubes: for CFP emission due to CFP excitation (IDD), 440 ± 10-nm excitation, 455 DCLP dichroic, 485 ± 20-nm emission; for YFP emission due to YFP excitation (IAA), 500 ± 10-nm excitation, 515 DCXR dichroic, 535 ± 15-nm emission; and for FRET emission due to CFP excitation (IDA), 440 ± 10-nm excitation, 455 DCLP dichroic, 535 ± 15-nm emission (all filters by Chroma Technology, Brattleboro, VT). Images were acquired every 10 s for 60 frames. After frame 15 (140 s), 2 mM Ca2+ Ringer's in the bath was replaced with 0 mM Ca2+ Ringer's + 1 μM ionomycin.
Bleedthrough factors were acquired as described by Zal and Gascoigne (2004): a = 0.057 ± 0.001 (n = 16), b = 0.015 ± 0.002 (n = 16), c = 0.009 ± 0.002 (n = 14), and d = 0.405 ± 0.001 (n = 14); values are ±SEM. After background subtraction of IDD, IAA, and IDA images, the apparent FRET efficiency, FRET-E, was calculated as follows:
where
The instrument-dependent parameter G (Zal and Gascoigne, 2004), determined using the intramolecular FRET probe CFP-RPTPα-SpD2-YFP2.1 (Blanchetot et al., 2002) expressed in HEK 293 cells, had a value of 1.34 ± 0.04 (n = 14). FRET analysis was restricted to cells with molar CFP/YFP ratios between 1:5 and 5:1 to minimize errors in FRET-E (Berney and Danuser, 2003). Ionomycin did not affect the fluorescence intensities of individually expressed CFP- or YFP-STIM1-ΔC, indicating that the signals we measured in CFP/YFP-coexpressing cells reflected changes in FRET rather than any possible effect of store depletion on individual CFP or YFP fluorescence.
All FRET-E analyses were done by averaging (±SEM) the FRET values from regions of interest (ROIs) drawn to encompass the whole cell, except where indicated in Figure 4F and Supplemental Figure 7, where the analyses were separated into central versus peripheral FRET-E (whole cell = central + peripheral). Central FRET-E was averaged from an ROI limited to the center (cytosol) of the cell. To make the central ROI, an ROI was first drawn encompassing the entire cell and then reduced in size by 10 pixels (∼1.5 μm) using the “Enlarge/Reduce” function in ImageJ. Peripheral FRET-E was determined from a 1.5-μm band from the PM; the ROI was drawn using the “Make Band” function in ImageJ, using the central ROI as the baseline and defining a band size of 10 pixels (∼1.5 μm).
Figure 4.

Mutations in the CC2 domain of CAD identify residues critical for STIM1 function and oligomerization. (A–D) Confocal images of the footprints of HEK 293 cells cotransfected with mCherry-myc-Orai1 and GFP-labeled STIM1-WT (A), STIM1-L373K (B), STIM1-A369K (C), or STIM1-A376K (D). In each panel the same cell is displayed before and after treatment with 1 μM TG to deplete stores. Bars, 5 μm. (E) Effects of STIM1 CC2 mutations on SOCE activation. Mean Fura-2 ratios (±SEM) are shown for HEK 293 cells cotransfected with mCherry-myc-Orai1 and STIM1-WT (black, n = 32), STIM1-L373K (blue, n = 29), STIM1-A376K (red, n = 21), or STIM1-A369K (green, n = 33). Stores were depleted with 1 μM TG, and extracellular Ca2+ concentration in mM is indicated above the graph. (F) Time course of mean whole cell FRET-E (±SEM) from HEK 293 cells expressing CFP- and YFP-labeled STIM1-WT (black, n = 8), STIM1-L373K (blue, n = 16), STIM1-A376K (red, n = 9), or STIM1-A369K (green; analysis of cell periphery only, green circles; n = 9). Stores were depleted with 1 μM ionomycin in Ca2+-free Ringer's solution as indicated.
Ca2+ Imaging
Transiently transfected HEK 293 cells were loaded with 1 μM Fura2 acetoxymethyl ester (Fura-2AM; Invitrogen) in medium for 25 min at 37°C and were washed three times with HEPES Physiological Saline Solution before imaging. Imaging was performed at 350- and 380-nm excitation with an Axiovert 35 inverted microscope (Zeiss) using a VideoProbe imaging system (ETM Systems, Irvine, CA) (Bautista et al., 2002). GFP-positive cells were identified with a 480 ± 15-nm excitation and a 530 ± 15-nm emission filter. mCherry-positive cells were identified using a 540 ± 12-nm excitation filter and a 580LP emission filter (Chroma).
Immunoprecipitation and Western Blotting
Six- or 10-cm dishes of HEK 293 cells were rinsed with PBS to remove media and then rinsed with TBS before lysis at 4°C in buffer containing 1% Nonidet P-40, 20 mM Tris, pH 7.4, complete protease inhibitor cocktail (Roche, Indianapolis, IN), and 150 mM (high salt) or 10 mM (low salt) NaCl. All subsequent steps were performed at 4°C. After centrifugation at 14,000 rpm for 15 min, supernatants were transferred to tubes containing washed anti-HA agarose beads (Immunology Consultants Laboratory, Newberg, OR) for 2 h. Beads were then washed four times with 0.1% Tween-20, 20 mM Tris, pH 7.4, and 500 mM (high salt) or 10 mM (low salt) NaCl before elution of protein in sample buffer. Samples were separated by SDS-PAGE on 10% polyacrylamide gels and blotted with 1:4000 anti-HA antibody (clone HA-7, Sigma, St. Louis, MO) or 1:5000 anti-GFP antibody (Millipore, Bedford, MA) followed by 1:30,000 alkaline-phosphatase–conjugated goat anti-mouse antibody (Sigma). Signal was detected with LumiPhos Western blotting buffer (Pierce, Rockford, IL).
Coiled-Coil Prediction Software
STIM1 sequences were analyzed with the COILS program (http://vit-embnet.unil.ch/software/COILS_form.html, Lupas, 1996) using a 28-residue window and the MTIDK database of two-stranded CC sequences. All residues were given equal weighting.
RESULTS
The CC1 Domain Supports Resting Self-Association of STIM1
To identify the cytosolic regions of STIM1 required to support resting oligomerization, we constructed a series of truncated STIM1 mutants and tested their ability to self-associate in cells (Figure 1A). STIM1-CAD (aa 1-448) is terminated at the C-terminal end of the CAD and contains both CC1 and CC2 cytosolic domains, STIM1-CC1 (aa 1-344) is terminated at the N-terminal boundary of CAD and contains CC1 only, and STIM1-ΔC (aa 1-237) is terminated immediately C-terminal to the transmembrane domain and lacks the entire cytoplasmic region. Orthogonally labeled STIM1 mutants were made by inserting a GFP or HA tag into the lumenal domain immediately C-terminal to the signal peptide, and their resting self-association was assessed by coimmunoprecipitation.
Figure 1.

The CC1 domain supports resting self-association of STIM1. (A) Schematic of wild-type STIM1 (STIM1-WT) and truncated STIM1 proteins used in this study, with the major known domains indicated. Premature stop codons were introduced after the indicated residues. Abbreviations: Sig, signal peptide; EF, EF-hand; SAM, sterile alpha motif; TM, transmembrane domain; CC1, coiled-coil 1; CC2, coiled-coil 2; CAD, CRAC activation domain; S/P, serine-proline–rich region; K, polybasic region. (B) Only STIM1 proteins containing CC1 + CAD remain self-associated under high salt conditions. Cells coexpressing GFP- and HA-labeled STIM1 constructs were lysed in buffer containing 150 mM NaCl, and immunoprecipitates were washed in buffer containing 500 mM NaCl. Whole cell lysates (top) and anti-HA immunoprecipitates (bottom) were fractionated by SDS-PAGE and blotted with anti-HA antibodies (left) or anti-GFP antibodies (right). Representative of four experiments. (C) STIM1-CC1 remains self-associated under low-salt conditions, but STIM1 lacking the entire C-terminus (ΔC) does not. Cells were lysed and immunoprecipitates washed in buffer containing 10 mM NaCl. Representative of five experiments. The amino acid numbering does not include the CFP, YFP, GFP, or HA tag sequences.
When HEK 293 cells were cotransfected with GFP- and HA-labeled WT STIM1 constructs, anti-HA antibody coimmunoprecipitated HA- and GFP-labeled STIM1-WT from cell lysates even under stringent (0.5 M) salt conditions (Figure 1B). These results confirm a previous report (Baba et al., 2006) that full-length STIM1 self-associates strongly in resting cells. Similarly, GFP-STIM1-CAD coimmunoprecipitated with HA-STIM1-CAD, demonstrating that it also strongly self-associates. In contrast, neither GFP-STIM1-CC1 nor GFP-STIM1-ΔC could be detected in immunoprecipitates of the corresponding HA-labeled mutant at high ionic strength. However, under low-salt conditions (10 mM) STIM1-CC1 did coimmunoprecipitate with itself, although STIM1-ΔC did not (Figure 1C). These results indicate that the CC1 domain within STIM1-CC1 supports self-association, but the resulting oligomer is somewhat labile and is strengthened by the addition of the CAD (aa 342-448) and possibly C-terminal residues further downstream. Similar results were obtained from cells pretreated with TG to deplete Ca2+ stores. Importantly, HA- and GFP-STIM1-ΔC mutants did not coimmunoprecipitate even after store depletion (Supplemental Figure 1), suggesting that when attached to the ER membrane, the lumenal EF hand-SAM domain either fails to oligomerize or does so with such low affinity that it does not survive solubilization and coimmunoprecipitation (see below).
Store-dependent Oligomerization of STIM1 Requires the CAD
After store depletion, WT STIM1 redistributes from a diffuse localization throughout the ER to form punctate clusters at ER–PM junctions. To assess the contribution of the cytoplasmic domains to STIM1 localization, we collected confocal images of cells expressing GFP-labeled STIM1 truncation mutants before and after store depletion. Each of the three mutant constructs displayed a reticular ER distribution in resting cells similar to full-length GFP-STIM1-WT (Figure 2A, left). After store depletion with ionomycin, GFP-STIM1-WT and GFP-STIM1-CAD redistributed into peripheral puncta, whereas GFP-STIM1-CC1 and GFP-STIM1-ΔC retained their reticular distribution (Figure 2A, right). These findings indicate that the CAD of STIM1 contains elements that are required for puncta formation. It was surprising to find that GFP-STIM1-CAD formed puncta in the absence of exogenous Orai1. Prior studies have shown that deletion of the C-terminal polybasic domain (aa 672-685; STIM1-ΔK) prevents STIM1 from forming puncta unless Orai1 is overexpressed (Park et al., 2009), a result that has been interpreted to suggest that the polybasic domain localizes STIM1 to ER–PM junctions by binding to negatively charged phospholipids—phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol 3,4,5-triphosphate (PIP3) in the PM (Liou et al., 2007; Walsh et al., 2009; but see also Korzeniowski et al., 2009). One possible explanation for our results is that the lysine/arginine-rich region in the CAD (aa 365-387, containing nine positive charges) may substitute for the C-terminal polybasic domain in targeting STIM1-CAD to ER–PM junctions.
Figure 2.

Store-dependent, higher-order oligomerization of STIM1 requires the CRAC activation domain (CAD). (A) Confocal images focused at the equator of HEK 293 cells expressing GFP-labeled STIM1 mutants. Resting cells were bathed in 2 mM Ca2+ Ringer's solution (left), and the same cells were store-depleted with 4 μM ionomycin in Ca2+-free Ringer's solution for 5 min (right). Arrowheads mark locations of puncta. Bar, 5 μm. (B) FRET-E images of a HEK 293 cell expressing CFP-STIM1 and YFP-STIM1 before (left) and after (right) store depletion with 1 μM ionomycin in Ca2+-free Ringer's solution for 7 min. Bar, 5 μm. (C) Time course of mean FRET-E (±SEM) from cells coexpressing CFP- and YFP-labeled STIM1-WT (black, n = 8), STIM1-CAD (blue, n = 9), STIM1-CC1 (green, n = 7), or STIM1-ΔC (red, n = 14). Extracellular solution changes are indicated above the graph.
The redistribution of STIM1-WT and STIM1-CAD into peripheral puncta after store depletion is a positive indication of oligomerization; however, the absence of puncta with other variants does not necessarily imply a failure to oligomerize. A case in point is STIM1-ΔK, which oligomerizes in response to store depletion yet fails to redistribute into puncta unless Orai1 is coexpressed (Liou et al., 2007; Park et al., 2009). To measure STIM1 oligomerization more directly in resting and store-depleted cells, we monitored FRET between CFP- and YFP-labeled STIM1 mutants (Liou et al., 2007). We adopted the E-FRET method, which approximates the FRET efficiency measured by donor recovery after acceptor photobleaching (Zal and Gascoigne, 2004). Unlike some FRET methods (e.g., FRETN and NFRET in Gordon et al., 1998; Xia and Liu, 2001, respectively), the apparent FRET efficiency (Eapp or FRET-E) is consistently proportional to the degree of molecular interaction (with respect to donor). Before the addition of ionomycin, STIM1-ΔC displayed a resting FRET value close to 0 (mean FRET-E of ∼0.025, Figure 2C, red trace), whereas STIM1-CC1, STIM1-CAD, and STIM1-WT had three- to fourfold higher resting FRET efficiencies (∼0.07–0.11; Figure 2C). The low resting FRET-E of STIM1-ΔC is consistent with its failure to coimmunoprecipitate with itself (Figure 1) and most likely indicates that STIM1-ΔC is monomeric under these store-replete conditions. Thus, the higher resting FRET-E levels for STIM1-CC1, STIM1-CAD, and STIM1-WT indicate that these proteins all form at least dimers at rest, consistent with the coimmunoprecipitation data. The resting FRET of STIM1-WT is the highest, possibly because it self-associates more strongly than the others or because cells overexpressing STIM1-WT sometimes display a few puncta at rest, suggesting a small degree of spontaneous higher-order oligomerization (Figure 2, A and B). The resting FRET of STIM1-CC1 is lower than that of STIM1-WT and STIM1-CAD, consistent with its weaker self-association inferred from coimmunoprecipitation (Figure 1). It is important to note that the donor/acceptor ratio can affect the magnitude of FRET-E (Berney and Danuser, 2003); to minimize the contribution of variable expression ratios to our results, we restricted analysis to cells with CFP/YFP fluorescence ratios between 0.2 and 5 (see Materials and Methods).
FRET studies also revealed differences in the oligomerization of STIM1 mutants after store depletion with ionomycin. Depletion rapidly increased the STIM1-ΔC FRET signal (Figure 2C, red trace) to a final level similar to the resting FRET of WT STIM1. The FRET increase of STIM1-ΔC shows that depletion of ER Ca2+ induces the monomeric STIM1-ΔC to multimerize; however, the oligomers appear to be labile, as HA- and GFP-labeled STIM1-ΔC proteins failed to coimmunoprecipitate after store depletion, even under low-salt conditions (Supplemental Figure 1). Store depletion also increased FRET between CFP-STIM1-ΔC and YFP-STIM1-WT or the reverse CFP/YFP pair (Supplemental Figure 2). In these cells, STIM1-ΔC showed only a slight redistribution into puncta, arguing against the formation of extensive oligomers of STIM1-ΔC by store depletion (see Discussion).
Surprisingly, STIM1-CC1 FRET did not increase significantly upon ionomycin treatment (Figure 2C, green trace). This result shows that the addition of CC1 to STIM1-ΔC, whereas enabling self-association at rest, prevents oligomerization of the EF-SAM domains in response to store depletion. Depletion-induced oligomerization was rescued by the addition of the CAD (Figure 2C, blue). The STIM1-CAD FRET increase was slightly smaller than that of STIM1-WT (Figure 2C, blue vs. black), suggesting a somewhat diminished level of store-dependent oligomerization consistent with this mutant's reduced capacity to form puncta and evoke SOCE relative to STIM1-WT (Park et al., 2009). Together, these results distinguish the functions of STIM1 cytoplasmic domains: although the CC1 domain is needed to form resting oligomers, the CAD is required for higher-order oligomerization in response to store depletion.
Mobility of Truncated Mutants of STIM1
To assess the role of cytoplasmic domains in STIM1 oligomerization independently, we estimated the diffusion coefficients of the truncated STIM1 mutants using FRAP. HEK 293 cells expressing a GFP-labeled STIM1 mutant (e.g., STIM1-CAD, Figure 3A) were imaged by confocal microscopy at a focal plane near the cell equator. After a series of prebleach images, a high-intensity laser scan was used to photobleach the fluorophores in a narrow strip across the ER (Ellenberg et al., 1997). We measured fluorescence recovery in the bleached region caused by diffusion of unbleached fluorophore, and used the time course to estimate the effective diffusion coefficient (Deff; see Materials and Methods). In each cell, the same region was bleached again after ionomycin treatment to determine the effects of store depletion on Deff. For the cell in Figure 3A, a comparison of the fluorescence recovery kinetics before and after ionomycin shows that store depletion greatly slowed GFP-STIM1-CAD diffusion (Figure 3B; Deff = 0.23 μm2/s at rest and 0.08 μm2/s after ionomycin treatment).
Diffusion coefficients for each truncated STIM1 mutant were estimated before and after store depletion (Figure 3C). In resting HEK 293 cells, GFP-STIM1-ΔC moved rapidly (Deff = 0.40 ± 0.08 μm2/s, n = 8 cells). This Deff is similar to that reported for other single-pass ER-membrane proteins (Ellenberg et al., 1997; Marguet et al., 1999; Nehls et al., 2000; Daigle et al., 2001), suggesting that like these other proteins, GFP-STIM1-ΔC diffuses freely within the ER membrane. After store depletion, the Deff of GFP-STIM1-ΔC remained constant, despite the evidence that under these conditions it oligomerizes and most likely forms a dimer (see Figure 2C, red trace). The lack of a detectable change in Deff is not surprising; mobility of membrane proteins in lipid membranes is only weakly dependent on size, so that a doubling of the radius of the transmembrane domain would be expected to reduce D by <10% (Saffman and Delbrück, 1975; Ramadurai et al., 2009; see Discussion). Interestingly, GFP-STIM1-CC1 diffused at less than half the rate of GFP-STIM1-ΔC in resting cells (Figure 3C). This greatly reduced mobility is not expected based on the notion that GFP-STIM1-CC1 is a dimer and GFP-STIM1-ΔC is a monomer. Rather, the result suggests that the addition of CC1 enables GFP-STIM1-CC1 to either form very large oligomers or to interact with other proteins in the cell (see Discussion). Store depletion did not alter the mobility of GFP-STIM1-CC1 (Figure 3C), consistent with the FRET evidence in Figure 2C that STIM1-CC1 fails to form higher-order oligomers in response to store depletion. Finally, although the resting mobilities of GFP-STIM1-CAD and GFP-STIM1-WT were similar to that of GFP-STIM1-CC1, their diffusion slowed by about twofold after store depletion. The diminished Deff of STIM1-WT and STIM1-CAD in response to store depletion (Figure 3C) is consistent with the formation of higher-order oligomers as inferred from the FRET experiments (Figure 2C, black and blue traces). These FRAP results complement and extend the conclusions from coimmunoprecipitation and FRET, suggesting that GFP-STIM1-ΔC is monomeric, that CC1 enables interactions between STIM1s and possibly other proteins, and that the CAD is needed in addition to CC1 to enable oligomerization in response to store depletion.
Mutations in the Predicted Coiled-Coil of the CAD Affect STIM1 Function and Oligomerization
Bioinformatic analysis of the CAD sequence with COILS (Lupas et al., 1991) predicts that residues 360-390 have a high probability of forming a CC structure (Supplemental Figure 3). In an effort to learn whether the putative CC-forming region is necessary for store-dependent oligomerization, we substituted lysine for three individual residues (A369, L373, and A376) predicted to be at position “a” or “d” in the heptad repeat and thus to lie within the hydrophobic interface of the CC (Supplemental Figure 3). Additionally, a triple mutant (STIM1-3K) was made in which the three amino acids together were mutated to lysine. Each individual mutation was predicted by COILS to have little impact, whereas the triple lysine substitution was predicted to nearly abolish the CC-forming potential of this region (Supplemental Figure 3). On the basis of these predictions, we conjectured that if store-dependent oligomerization is mediated through the CC region, the single lysine mutants of STIM1 would have little effect and only the triple mutant STIM1-3K would fail to oligomerize. We tested these constructs for their effects on STIM1 localization, Orai1 recruitment and CRAC channel activation.
As expected from previous studies, GFP-STIM1-WT, when cotransfected with mCherry-myc-Orai1, was localized diffusely throughout the ER (Figure 4A, top) and did not produce significant constitutive Ca2+ entry in resting HEK 293 cells (Figure 4E, black trace). After TG treatment to deplete stores, GFP-STIM1-WT recruited Orai1 into puncta visible at the cell footprint (Figure 4A, bottom), and readdition of extracellular Ca2+ generated a large Ca2+ response due to activated CRAC channels (Figure 4E). STIM1-L373K behaved similarly to WT STIM1; at moderate levels of expression, it exhibited diffuse ER localization at rest (Figure 4B, top) and colocalized with Orai1 in a punctate distribution after store depletion (Figure 4B, bottom). Only in very highly overexpressing cells did GFP-STIM1-L373K form constitutive puncta at rest, but these puncta were unable to recruit Orai1 (data not shown). As expected, Ca2+ signaling in cells expressing GFP-STIM1-L373K was similar to cells expressing GFP-STIM1-WT (Figure 4E, blue vs. black). Thus, L373K behaved as expected given the bioinformatics predictions.
In contrast, GFP-STIM1-A369K formed puncta at rest that colocalized with Orai1 to a moderate extent (Figure 4C, top; Supplemental Figure 4A). Basal Ca2+ influx was correspondingly elevated in these cells (Figure 4E, green trace), indicating that CRAC channels were constitutively active. The size and intensity of STIM1-A369K puncta as well as colocalization with mCherry-myc-Orai1 increased after store depletion (Figure 4C, bottom), and robust Ca2+ influx was observed upon readdition of extracellular Ca2+. Together, these data suggest that the A369K mutation partially activates STIM1 independently of store depletion.
In resting cells, GFP-STIM1-A376K formed puncta that were even more pronounced than those of GFP-STIM1-A369K. These puncta resembled WT in size, intensity and location. They were peripherally located (Supplemental Figure 4B), indicating that GFP-STIM1-A376K is capable of targeting to ER–PM junctions, yet appears to lack the ability to bind or activate Orai1. TG did not further increase the size or intensity of these puncta, nor did Orai1 colocalize with GFP-STIM1-A376K before or after store depletion (Figure 4D). Consistent with these results, GFP-STIM1-A376K did not enhance Ca2+ entry at rest or after store depletion (Figure 4E, red trace). The triple mutant, STIM1-3K (A369K/L373K/A376K), displayed a phenotype similar to that of A376K alone, with constitutive puncta that neither clustered Orai1 nor supported store-operated Ca2+ entry (Supplemental Figure 5).
Because the STIM1-A376K and STIM1-3K mutants failed to cocluster with Orai1 (Figure 4D, Supplemental Figure 5A), their inability to activate SOCE was likely due to a lack of CAD binding to Orai1. We directly tested this idea by making the same mutations in the isolated CAD fragment. mCherry-CAD-WT was recruited to the PM of cells expressing Orai1 and activated constitutive Ca2+ entry; in contrast, the mutant CAD proteins mCherry-CAD-A376K and mCherry-CAD-3K failed to colocalize with GFP-myc-Orai1 at the PM or to induce constitutive Orai1 activity (Supplemental Figure 6). Thus, the mutations inhibit CAD binding to Orai1.
To relate the functional effects of the CC2 point mutations to STIM1 oligomerization, we introduced the same mutations into CFP/YFP-labeled pairs of full length STIM1 proteins for FRET-E measurements. As expected, STIM1-L373K resembled WT STIM1, both in its basal FRET efficiency and the large increase in FRET induced by store depletion (Figure 4F, blue vs. black traces). In contrast, STIM1-A376K exhibited a high resting FRET, which did not increase further after store depletion, suggesting that STIM1-A376K is fully oligomerized in resting cells (Figure 4F, red trace). Thus, although STIM1-A376K constitutively forms high-order oligomers capable of aggregating into puncta near the PM, these oligomers are nonfunctional, presumably due to their inability to bind Orai1.
When averaged over the entire cell, the FRET signal from STIM1-A369K in resting cells was similar to that of STIM1-WT despite the fact that STIM1-A369K was partially localized to puncta and stimulated constitutive Ca2+ entry (Figure 4, C, E, and F). However, in A369K cells the FRET signal throughout the cell interior was significantly lower than the signal near the PM (Supplemental Figure 7, peripheral vs. central), suggesting that the fraction of A369K prelocalized to puncta is already oligomerized and can activate Orai1 in the absence of store depletion. Store depletion further increased FRET-E of STIM1-A369K (Figure 4F, Supplemental Figure 7), indicating that more A369K molecules adopted the higher-order oligomerized state, consistent with the increase in puncta and Ca2+ influx we observed after ionomycin (Figure 4C, E).
The C-Terminal Half of the CAD Is Required for Store-dependent STIM1 Oligomerization
To further assess the role of CC2 in STIM1 oligomerization, we examined a STIM1 protein truncated immediately C-terminal to CC2 (STIM1-CC2; Figure 5A). CFP-STIM1-CC2 was distributed throughout the ER at rest, but failed to form puncta or recruit Orai1 after store depletion (Figure 5B). Moreover, STIM1-CC2 FRET did not increase upon store depletion (Figure 5C). These results demonstrate that CC2 cannot substitute for the entire CAD sequence in enabling store-dependent oligomerization of STIM1 and suggest a requirement for residues within the CAD region C-terminal to CC2 (i.e., aa 392-448).
Figure 5.

STIM1-CC2 fails to oligomerize in response to store depletion. (A) STIM1-CC2 was constructed by introducing a premature stop codon at aa 392 as shown. (B) Confocal images of the footprint of a HEK 293 cell cotransfected with CFP-STIM1-CC2 and mCherry-myc-Orai1, before and after treatment with 1 μM TG to deplete Ca2+ stores. Bar, 5 μm. (C) Time course of mean FRET-E (±SEM) from HEK 293 cells coexpressing CFP- and YFP-labeled STIM1-CC2 (red, n = 9); the FRET-E response of STIM1-WT (black, n = 8) and STIM1-CC1 (green, n = 7) are reproduced from Figure 2C for comparison. 1 μM ionomycin was added as indicated above the graph.
DISCUSSION
Although the EF-SAM domain is thought to initiate the oligomerization of STIM1 after Ca2+ store depletion, the possible contributions of the cytoplasmic domains to this process have until now been unclear (Stathopulos et al., 2006, 2008). Our results indicate specific roles for the CC1 and CAD domains in regulating the oligomerized state of STIM1 before and after store depletion. CC1 is essential for generating resting, inactive oligomers of STIM1 in cells with replete Ca2+ stores. After store depletion, the EF-SAM domain by itself (STIM1-ΔC) oligomerizes to a limited extent, but these complexes are unstable. The cytoplasmic CAD is required to enable the formation of stable, higher-order oligomers of STIM1 in response to store depletion. These results demonstrate a new function for the CAD in initiating the SOCE cascade.
Roles for CC1 in Protein–Protein Interactions of STIM1
Resting Self-Association.
Previous studies have shown that STIM1 self-associates in resting cells with replete Ca2+ stores (Williams et al., 2002; Baba et al., 2006) and that this is prevented by internal deletion of residues 249-390, suggesting roles for CC1 and/or CC2 (Baba et al., 2006). However, in studies of cytosolic STIM1 fragments Muik et al. (2009) found that CC1 and CC2 were not able to support formation of homomers, but that longer fragments containing the CAD (233–450 or longer) acquired this ability. Using a series of truncated STIM1 mutants, we determined that CC1 or CC1 + CC2 supported resting oligomers of STIM1 that were further stabilized by the CAD region. Thus, while both studies agree that CAD stabilizes the self-association, our results differ in that we found that STIM1-CC1 and -CC2 are able to oligomerize at rest. This discrepancy highlights the different requirements for oligomerization of full-length versus cytosolic fragments of STIM1. In our study, the presence of the transmembrane and lumenal domains of STIM1 may have enhanced the self-association of STIM1-CC1/CC2, perhaps by constraining the proteins to a two-dimensional surface or by stabilizing conformations conducive to oligomerization. Moreover, because the cytosolic STIM1 fragments (aa 233-450 or longer; Muik et al., 2009) appear to mimic the active state of STIM1, they likely adopt a different conformation than resting STIM1.
Interactions with Other Proteins.
We found that although the mobility of STIM1-ΔC is quite comparable to that of other single-pass ER or Golgi membrane proteins (Cole et al., 1996; Marguet et al., 1999; Lippincott-Schwartz et al., 2001), addition of CC1 (STIM1-CC1) slows it by ∼50%. Because free diffusion of membrane proteins is determined predominantly by the size and number of transmembrane domains, this result implies that STIM1-CC1 either forms very large oligomers in the ER or that its mobility is retarded by interactions with other proteins. An estimate of the size of such an oligomer can be made from the Saffman-Delbrück equation:
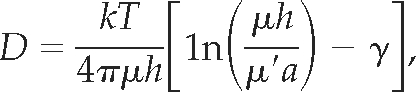 |
where D is the diffusion coefficient, k is Boltzmann's constant, T is absolute temperature, μ is the viscosity of the membrane (∼1 P), μ′ is the viscosity of the surrounding fluid (∼0.01 P), h is the width of the membrane (∼4 nm), a is the radius of the transmembrane segment, and γ is Euler's constant (0.5772; Saffman and Delbrück, 1975). This relation has recently been validated experimentally for proteins containing 1-36 transmembrane domains (radius of 0.5–4 nm) diffusing in pure lipid bilayers (Ramadurai et al., 2009). Thus, to explain the low Deff of STIM1-CC1 relative to STIM1-ΔC solely in terms of its interactions with the ER membrane, and assuming that STIM1-ΔC is monomeric with a radius of 0.5 nm, STIM1-CC1 would need to have an effective radius of 10 nm, or 20-fold larger than STIM1-ΔC. Such extensive oligomerization seems unlikely given that CC1 is the only cytosolic protein association domain in STIM1-CC1 and that CCs are generally limited to less than six strands (Grigoryan and Keating, 2008). One alternative is that diffusion of STIM1-CC1 could be slowed by binding to cytosolic, ER, or cytoskeletal proteins. One known binding partner of STIM1 is the microtubule end-binding protein EB1 (Grigoriev et al., 2008); however, STIM1-CC1 does not contain the identified S/TxIP binding site for EB1 (Honnappa et al., 2009). Diffusion could also be slowed by nonspecific interactions due to molecular crowding (Vaz et al., 1984; Zhang et al., 1991; Iino et al., 2001). This scenario seems especially plausible given the expected dimensions of CC1; an α-helix with ∼100 residues could extend as far as ∼15 nm from the ER membrane. The anomalously slow diffusion of STIM1-WT has been noted before (Liou et al., 2007); our results suggest that CC1 plays the dominant role in slowing diffusion, as the mobility of STIM1-WT with its additional 341 residues was similar to that of STIM1-CC1 (Figure 3C).
After store depletion, the mobility of STIM1-CAD and STIM1-WT declined by a further ∼50%, consistent with a previous study of STIM1-WT (Liou et al., 2007). Again, this large reduction in diffusion rate could in principle arise from the formation of very large aggregates, but may also be explained simply by moderate multimerization of STIM1 proteins that are slowed by auxiliary partners interacting with CC1. A better determination of STIM1 stoichiometry before and after store depletion will be critical for testing these possibilities.
A Role for CAD in the Store-dependent Oligomerization of STIM1
We observed oligomerization of STIM1-ΔC as a FRET-E increase after store depletion, consistent with earlier results showing that the isolated EF-SAM oligomerizes after Ca2+ removal (Stathopulos et al., 2006, 2008). From these measurements we cannot determine the precise stoichiometry of the oligomers, because FRET depends on the fraction of molecules that interact as well as the geometry of the interaction. However, it is clear that these oligomers are unstable, as shown by their failure to coimmunoprecipitate after detergent solubilization even under low-salt conditions.
Interestingly, we also found through FRET experiments that STIM1-ΔC was able to bind to STIM1-WT oligomers after store depletion (Supplemental Figure 2). Although Ikura and colleagues showed that EF-SAM formed dimers as well as higher-order aggregates upon Ca2+ removal, the aggregates were disordered (Stathopulos et al., 2008), and it was not clear whether extended arrays of full-length STIM1 form in cells through interactions of EF-SAM between neighboring STIM1 oligomers (“interoligomers”), or whether only adjacent EF-SAM domains within single oligomers interact (“intraoligomers”). Our FRET results show that interoligomeric binding does occur; however, the fact that STIM1-ΔC was not recruited in abundance to puncta by STIM1-WT illustrates that these interactions are by themselves incapable of forming large arrays. This behavior is consistent with our conclusion that the EF-SAM interaction is rather labile and that the cytosolic regions (CC1 + CAD) are needed to stabilize store-dependent higher-order oligomers.
Although STIM1-CC1 failed to oligomerize in response to store depletion, addition of the CAD (STIM1-CAD) restored oligomerization. These results clearly demonstrate that store-dependent, higher-order oligomerization of STIM1 requires not just the EF-SAM region, but also the cytoplasmic CAD. Thus, in addition to its known role in binding and activating Orai1 (Park et al., 2009; Yuan et al., 2009) the CAD is also critically involved in initiating the SOCE process. This additional role for the CAD may explain why in a previous study a STIM1-ΔCAD deletion mutant failed to form puncta in the absence of Orai1, even though it retained an intact polybasic tail region (Park et al., 2009).
Our results demonstrate that both CC2 and a C-terminal domain within the CAD influence store-dependent oligomerization. Mutations within the CC2 in CAD had diverse effects on STIM1 oligomerization, targeting and activity. STIM1-A376K at rest displayed a very high resting FRET efficiency, indicative of constitutive oligomerization, as well as constitutive targeting to ER–PM junctions. However, A376K puncta did not recruit Orai1, suggesting that this mutant retained Orai1-independent PM binding but lost its ability to bind to Orai1. In this way, STIM1-A376K is the converse of the STIM1-ΔK mutant that retains Orai1-dependent but not -independent targeting (Liou et al., 2007; Park et al., 2009). One possibility is that by promoting oligomer formation, the A376K mutation increases the avidity of the polybasic region for PIP2 and PIP3 in the PM (Ercan et al., 2009; Walsh et al., 2009), but that an altered oligomer conformation or K376 itself may prevent binding to Orai1. In either case, these results, together with the effects of artificially cross-linking STIM1 with a heterodimerizer system (Luik et al., 2008), underscore the causal role of higher-order STIM1 oligomerization for targeting to ER–PM junctions and provide further evidence that CAD-Orai1 binding is required for CRAC channel opening. Recent studies by Frischauf et al. (2009) have shown the importance of A373 and A376 in physical interactions with a putative CC domain in the C-terminus of Orai; although those authors found that A373S inhibited interactions with Orai1, we observed no effect of A373K on STIM1 targeting or Orai1 activation, perhaps because the lysine substitution, unlike serine, was not predicted to disrupt CC formation.
Despite the effects of CC2 mutations on STIM1 oligomerization, STIM1-CC2 was not sufficient to support store-dependent oligomerization as assayed by FRET, implying that C-terminal residues between 391 and 448 are also required. This result may explain the inability of a STIM1ΔST mutant (aa 1-390) to form puncta or activate Orai1 in a previous study (Baba et al., 2006). Interestingly, on the basis of studies of cytosolic STIM1 fragments, Muik et al. (2009) have proposed that residues 420-450 of STIM1 constitute an assembly domain. Their results are consistent with our findings that CAD residues downstream of CC2 are required for the formation of STIM1 oligomers after store depletion.
Toward a Mechanism for STIM1 Oligomerization
In this article we have described essential roles for cytosolic domains in regulating the oligomerization of STIM1. Our results provide an important link between structural work on lumenal EF-SAM fragments in vitro (Stathopulos et al., 2006, 2008) and FRET studies of soluble STIM1 fragments in the cytosol (Muik et al., 2009). We can now combine our results with those of previous studies into a working model for STIM1 oligomerization. In resting cells, STIM1 may exist mostly as dimers (Penna et al., 2008) formed by homotypic intermolecular interactions between CC1 regions that are additionally stabilized by the CAD; in this resting state, the Ca2+-bound EF-SAM region of each STIM1 monomer is engaged in intramolecular binding. Ca2+ is released from the EF hand upon store depletion, allowing the EF-SAM region to bind to its neighbor within the dimer and/or to EF-SAM regions on adjacent dimers. This higher-order oligomerization requires the cytoplasmic CAD and results in tetrameric or greater stoichiometry. It is tempting to think that these oligomers may in fact be tetramers formed from the dimerization of STIM1 dimers, based on the propensity of CAD fragments to form tetramers in solution (Park et al., 2009). The formation of tetramers is expected to increase the avidity of the polybasic domain for membrane phospholipids, thus trapping it at ER–PM junctions. Tetramers of the CAD are then in place to bind, trap, and activate passing Orai1 channels in the overlying PM (Kawasaki et al., 2009; Park et al., 2009; Yuan et al., 2009; Zhou et al., 2009). In this way the CAD of STIM1 plays at least two distinct roles in SOCE: it promotes the transition of STIM1 into an oligomerized, active conformation and directly binds to and activates its target, Orai1. Several critical assumptions in this model remain to be addressed: in particular, the stoichiometry of the resting and activated forms of STIM1 and the question of whether a conformational change occurs to expose the CAD or assemble it into oligomers.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank L. Lokteva for making the CFP- and YFP-STIM1 mutant constructs and P. Hoover (Stanford University), C. Y. Park, J. den Hertog, J. Liou, and T. Meyer for plasmids. We also thank M. Yen for help with FRET experiments and data analysis, A. Brunger and the Lewis Lab for helpful discussions, and R. Dolmetsch and members of the Lewis Lab for comments on the manuscript. This work was supported by a National Science Foundation Graduate Research Fellowship (E.D.C.), a Gabilan Stanford Graduate Fellowship (E.D.C.), and the Mathers Charitable Foundation and an R01 grant from the National Institute of General Medical Sciences (R.S.L.).
Abbreviations used:
- CAD
CRAC activation domain
- CC
coiled-coil
- CRAC
Ca2+ release-activated Ca2+
- SAM
sterile alpha motif
- SOC
store-operated channel
- SOCE
store-operated Ca2+ entry
- STIM
stromal interaction molecule.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-02-0145) on April 7, 2010.
REFERENCES
- Baba Y., et al. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc. Natl. Acad. Sci .USA. 2006;103:16704–16709. doi: 10.1073/pnas.0608358103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista D. M., Hoth M., Lewis R. S. Enhancement of calcium signalling dynamics and stability by delayed modulation of the plasma-membrane calcium-ATPase in human T cells. J. Physiol. 2002;541:877–894. doi: 10.1113/jphysiol.2001.016154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berney C., Danuser G. FRET or no FRET: a quantitative comparison. Biophys. J. 2003;84:3992–4010. doi: 10.1016/S0006-3495(03)75126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchetot C., Tertoolen L. G., den Hertog J. Regulation of receptor protein-tyrosine phosphatase alpha by oxidative stress. EMBO J. 2002;21:493–503. doi: 10.1093/emboj/21.4.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan M. D. STIMulating store-operated Ca2+ entry. Nat. Cell Biol. 2009;11:669–677. doi: 10.1038/ncb0609-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole N. B., Smith C. L., Sciaky N., Terasaki M., Edidin M., Lippincott-Schwartz J. Diffusional mobility of Golgi proteins in membranes of living cells. Science. 1996;273:797–801. doi: 10.1126/science.273.5276.797. [DOI] [PubMed] [Google Scholar]
- Daigle N., Beaudouin J., Hartnell L., Imreh G., Hallberg E., Lippincott-Schwartz J., Ellenberg J. Nuclear pore complexes form immobile networks and have a very low turnover in live mammalian cells. J. Cell Biol. 2001;154:71–84. doi: 10.1083/jcb.200101089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenberg J., Siggia E. D., Moreira J. E., Smith C. L., Presley J. F., Worman H. J., Lippincott-Schwartz J. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan E., Momburg F., Engel U., Temmerman K., Nickel W., Seedorf M. A conserved, lipid-mediated sorting mechanism of yeast Ist2 and mammalian STIM proteins to the peripheral ER. Traffic. 2009;10:1802–1818. doi: 10.1111/j.1600-0854.2009.00995.x. [DOI] [PubMed] [Google Scholar]
- Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol. Rev. 2009;231:189–209. doi: 10.1111/j.1600-065X.2009.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischauf I., Muik M., Derler I., Bergsmann J., Fahrner M., Schindl R., Groschner K., Romanin C. Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1,2,3 channels by a STIM1 coiled-coil mutant. J. Biol. Chem. 2009;284:21696–21706. doi: 10.1074/jbc.M109.018408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon G. W., Berry G., Liang X. H., Levine B., Herman B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 1998;74:2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev I., et al. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr. Biol. 2008;18:177–182. doi: 10.1016/j.cub.2007.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan G., Keating A. E. Structural specificity in coiled-coil interactions. Curr. Opin. Struct. Biol. 2008;18:477–483. doi: 10.1016/j.sbi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan P. G., Lewis R. S., Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honnappa S., et al. An EB1-binding motif acts as a microtubule tip localization signal. Cell. 2009;138:366–376. doi: 10.1016/j.cell.2009.04.065. [DOI] [PubMed] [Google Scholar]
- Iino R., Koyama I., Kusumi A. Single molecule imaging of green fluorescent proteins in living cells: E-cadherin forms oligomers on the free cell surface. Biophys. J. 2001;80:2667–2677. doi: 10.1016/S0006-3495(01)76236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T., Lange I., Feske S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem. Biophys. Res. Commun. 2009;385:49–54. doi: 10.1016/j.bbrc.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniowski M. K., Popovic M. A., Szentpetery Z., Varnai P., Stojilkovic S. S., Balla T. Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J. Biol. Chem. 2009;284:21027–21035. doi: 10.1074/jbc.M109.012252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis R. S. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–287. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- Liou J., Fivaz M., Inoue T., Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA. 2007;104:9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J., Kim M. L., Heo W. D., Jones J. T., Myers J. W., Ferrell J. E., Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., Snapp E., Kenworthy A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2001;2:444–456. doi: 10.1038/35073068. [DOI] [PubMed] [Google Scholar]
- Luik R., Wang B., Prakriya M., Wu M., Lewis R. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538–542. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luik R. M., Wu M. M., Buchanan J., Lewis R. S. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006;174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A. Prediction and analysis of coiled-coil structures. Methods Enzymol. 1996;266:513–525. doi: 10.1016/s0076-6879(96)66032-7. [DOI] [PubMed] [Google Scholar]
- Lupas A., Van Dyke M., Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Marguet D., Spiliotis E. T., Pentcheva T., Lebowitz M., Schneck J., Edidin M. Lateral diffusion of GFP-tagged H2Ld molecules and of GFP-TAP1 reports on the assembly and retention of these molecules in the endoplasmic reticulum. Immunity. 1999;11:231–240. doi: 10.1016/s1074-7613(00)80098-9. [DOI] [PubMed] [Google Scholar]
- Muik M., Fahrner M., Derler I., Schindl R., Bergsmann J., Frischauf I., Groschner K., Romanin C. A cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J. Biol. Chem. 2009;284:8421–8426. doi: 10.1074/jbc.C800229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muik M., et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J. Biol. Chem. 2008;283:8014–8022. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- Nehls S., Snapp E. L., Cole N. B., Zaal K. J., Kenworthy A. K., Roberts T. H., Ellenberg J., Presley J. F., Siggia E., Lippincott-Schwartz J. Dynamics and retention of misfolded proteins in native ER membranes. Nat. Cell Biol. 2000;2:288–295. doi: 10.1038/35010558. [DOI] [PubMed] [Google Scholar]
- Orci L., Ravazzola M., Le Coadic M., Shen W., Demaurex N., Cosson P. STIM1-induced precortical and cortical subdomains of the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 2009;106:19358–19362. doi: 10.1073/pnas.0911280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh A. B., Putney J. W., Jr Store-operated calcium channels. Physiol. Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Park C. Y., Hoover P. J., Mullins F. M., Bachhawat P., Covington E. D., Raunser S., Walz T., Garcia K. C., Dolmetsch R. E., Lewis R. S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna A., Demuro A., Yeromin A. V., Zhang S. L., Safrina O., Parker I., Cahalan M. D. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456:116–120. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadurai S., Holt A., Krasnikov V., van den Bogaart G., Killian J. A., Poolman B. Lateral diffusion of membrane proteins. J. Am. Chem. Soc. 2009;131:12650–12656. doi: 10.1021/ja902853g. [DOI] [PubMed] [Google Scholar]
- Saffman P. G., Delbrück M. Brownian motion in biological membranes. Proc. Natl. Acad. Sci. USA. 1975;72:3111–3113. doi: 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopulos P., Li G., Plevin M., Ames J., Ikura M. Stored Ca2+ depletion-induced oligomerization of STIM1 via the EF-SAM region: an initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 2006;281:35855–35862. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- Stathopulos P. B., Zheng L., Li G.-Y., Plevin M. J., Ikura M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. 2008;135:110–122. doi: 10.1016/j.cell.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Vaz W.L.C., Goodsaidzalduondo F., Jacobson K. Lateral diffusion of lipids and proteins in bilayer-membranes. FEBS Lett. 1984;174:199–207. [Google Scholar]
- Walsh C. M., Chvanov M., Haynes L. P., Petersen O. H., Tepikin A. V., Burgoyne R. D. Role of phosphoinositides in STIM1 dynamics and store-operated calcium entry. Biochem. J. 2009;425:159–168. doi: 10.1042/BJ20090884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R., Senior P., Van Stekelenburg L., Layton J., Smith P., Dziadek M. Stromal interaction molecule 1 (STIM1), a transmembrane protein with growth suppressor activity, contains an extracellular SAM domain modified by N-linked glycosylation. Biochim. Biophys. Acta. 2002;1596:131–137. doi: 10.1016/s0167-4838(02)00211-x. [DOI] [PubMed] [Google Scholar]
- Wu M. M., Buchanan J., Luik R. M., Lewis R. S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z., Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 2001;81:2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P., Lu J., Li Z., Yu X., Chen L., Xu T. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem. Biophys. Res. Commun. 2006;350:969–976. doi: 10.1016/j.bbrc.2006.09.134. [DOI] [PubMed] [Google Scholar]
- Yuan J. P., Zeng W., Dorwart M. R., Choi Y. J., Worley P. F., Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009;11:337–343. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zal T., Gascoigne N. R. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys. J. 2004;86:3923–3939. doi: 10.1529/biophysj.103.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Crise B., Su B., Hou Y., Rose J. K., Bothwell A., Jacobson K. Lateral diffusion of membrane-spanning and glycosylphosphatidylinositol-linked proteins: toward establishing rules governing the lateral mobility of membrane proteins. J. Cell Biol. 1991;115:75–84. doi: 10.1083/jcb.115.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. L., Yu Y., Roos J., Kozak J. A., Deerinck T. J., Ellisman M. H., Stauderman K. A., Cahalan M. D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Meraner P., Kwon H. T., Machnes D., Masatsugu O. H., Zimmer J., Huang Y., Stura A., Rao A., Hogan P. G. STIM1 gates the store-operated calcium channel ORAI1 in vitro. Nat. Struct. Mol. Biol. 2009;17:112–116. doi: 10.1038/nsmb.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.