

Abstract
Objective
In dyslipidemic states, platelets become hyperreactive, secreting molecules that promote atherosclerosis. We have shown that the semaphorin family member, sema4D (CD100) is expressed on the surface of platelets and proposed that its role includes promoting thrombus growth by binding to nearby platelets and endothelial cells, both of which express sema4D receptors. Here we tested the hypothesis that deleting sema4D will attenuate the adverse consequences of dyslipidemia on platelets and the vessel wall.
Methods
Platelet function and atherosclerotic lesion formation were measured in LDLR(-/-) and sema4D(-/-)LDLR(-/-) mice after 6 months on a high fat diet.
Results
All of the mice developed the dyslipidemia expected on this diet in the absence of functional LDL receptors. However, when compared to LDLR(-/-) mice, sema4D(-/-)LDLR(-/-) mice had reduced lipid deposition in the descending aorta, a 6-fold decrease in the frequency of arterial occlusion and a reduction to near wild type levels in the accumulation of platelets following injury. These differences were retained ex vivo, with a marked decrease in platelet accumulation on collagen under flow and in platelet aggregation.
Conclusions
These results show that loss of sema4D expression reduces the platelet hyperactivity otherwise found in dyslipidemia, and confers protection against the development of atherosclerosis.
Keywords: semaphorin 4D, atherosclerosis, platelet, dyslipidemia
Introduction
Although the best known role of platelets is in hemostasis, the interaction between platelets and the vascular wall is not limited to the setting of acute injury and can have harmful as well as beneficial effects. Platelets help to maintain the integrity of the endothelial monolayer 1, but they are also believed to contribute to the development of atherosclerosis especially in dyslipidemic states, where platelet reactivity to agonists increases 2. The mechanisms by which platelets contribute to atherogenesis are only partly understood. In rabbits 3 and mice 4, hypercholesterolemia is associated with increased adhesion of platelets to intact endothelium even before lesions are clearly established. The chemokines and cytokines released by activated platelets feed the inflammatory response, attracting monocytes and neutrophils, and then facilitating their migration into adjacent tissues 5, 6. Dyslipidemia makes this more likely to happen. Genetic deletion of some of the molecules needed for a normal platelet response to injury has been shown to confer protection in mouse models of atherosclerosis 7, 8 9 10, as have inhibitors of platelet function 11 12, 13. Conversely, deletion of the platelet PGI2 receptor, which removes a normal brake on platelet activation, accelerates atherosclerosis 12. Therefore, one key to understanding how platelets affect the vessel wall is to understand the relationship between platelet activation and the evolution of vascular disease, and how the suppression of the former can retard the latter.
In addition to receptors for agonists and integrins such as αIIbβ3, the platelet surface expresses signaling molecules that can mediate interactions between cells 14. We have previously shown that ephrins and Eph kinases are a ligand/receptor pair that works in trans between adjacent platelets 15. More recently, we proposed that the semaphorin family member, sema4D (CD100), and its receptors do so as well, modulating thrombus growth and stability in the setting of vascular injury 16. Sema4D is a type I transmembrane molecule expressed as a disulfide-linked homodimer that was originally reported on T-cells 17-19. Its extracellular domain has been shown to be a ligand for CD72, plexin-B1 and possibly plexins-B2 and -C1 as well. Cell activation causes the sema4D exodomain to be cleaved and shed, producing a large soluble fragment that can activate plexin-B1 on endothelial cells20 and inhibit the migration of monocytes 21 and dendritic cells 22, both of which have been implicated in the progression of atherosclerosis. In previous studies, we have shown that sema4D is expressed on platelets and that sema4D(-/-) mice have defects in thrombus formation in vivo and in collagen-induced platelet aggregation in vitro 16. Those studies also show that activated platelets, like activated T-cells, shed the sema4D exodomain. The rate of shedding is slow compared to platelet aggregation, but the soluble fragment retains biological activity when added to platelets ex vivo.
The presence of sema4D on platelets and T-cells, along with the presence of sema4D receptors on endothelial cells, monocytes and dendritic cells, puts these molecules in locations that are intimately involved in atherothrombosis. In the present studies, we have examined the contribution of sema4D to platelet function in the setting of dyslipidemia, and asked whether loss of sema4D expression is protective against the development of atherosclerosis. The studies take advantage of the sema4D(-/-) mouse line, which we have crossed with proatherogenic LDLR(-/-) mice. The results show that deleting sema4D attenuates much of the increase in platelet responsiveness that is otherwise observed in dyslipidemic states. It also impairs atherogenesis. Although expression of sema4D is not limited to platelets, the effects that we observed are at least in part platelet autonomous and, therefore, provide an additional link between platelets and the development of atherosclerosis.
Methods
Mice
Sema4D-/- mice on a C57Bl/6 genetic background 23 were crossed with C57Bl/6 LDLR(-/-) mice obtained from Jackson Laboratory (Bar Harbor, ME). Their progeny were bred to generate sema4D(-/-)LDLR(-/-) double knockouts. Mice were started on a high fat diet (0.2% cholesterol 21% saturated fat; formula TD88137, Harlan Teklad, Indianapolis, IN) at 8 weeks of age and maintained on the diet for up to 6 months.
Examination of the aorta
The intact aorta was perfused with PBS, fixed in formalin (Fisher Scientific, Hampton, NH), cleaned, opened and stained with Sudan IV (Sigma-Aldrich, St. Louis, MO). Lipid deposits were quantified using Image-Pro (Media Cybernetics, Inc., Bethesda, MD).
Immunohistochemistry
Mouse hearts were embedded in Tissue Tek O.C.T (Sakura-Finetek, Inc., Torrance, CA) and 8 μm serial sections of the aortic root mounted on masked slides (Carlson Scientific, Peotone, IL). Acetone fixed and peroxidase quenched sections were incubated with biotinylated rat anti-mouse CD4 and rat anti-mouse CD8 (made in-house), rat anti-mouse CD22 (Southern Biotech, Birmingham, AL) and rat anti-mouse CD11b (BD Biosciences, Pasadena, CA) and FITC-conjugated mouse anti-α-smooth muscle actin (SMA) clone 14A (Sigma, St. Louis, MO). Sections stained for SMA were subjected to an intermediate step with biotinylated goat anti-FITC (Vector Laboratories, Burlingame, CA) antibody. Antibody binding was detected following amplification with Vectastain ABC avidin-biotin (Vector Laboratories, Burlingame, CA), and developed with diaminobenzidine (Dako, Carpinteria, CA) as substrate. All sections were counterstained with Gill's Formulation No. 1 hematoxylin (Fisher Scientific, Pittsburgh, PA). Isotype matched controls were run in parallel and showed negligible staining in all cases.
Plasma lipid analysis
Heparinized blood (150 U/ml, 1:9 dilution with blood) was obtained from the inferior vena cava of anesthetized mice that had been fasted for 4 hours. Plasma total cholesterol and triglyceride levels were measured using a Cobas Fara II autoanalyzer (Roche Diagnostic Systems, Nutley, NJ).
Vascular injury
Thrombus formation was visualized in the cremaster muscle microcirculation 24. Following anesthesia (90 mg/kg pentobarbital, i.p.), the cremaster muscle was exteriorized, spread on the pedestal of a custom built chamber while being superfused with bicarbonate buffer (37°C) and bubbled with 95%N2/5%CO2. After 10 minutes, Alexa-488-labeled anti-CD41 antibody F(ab)2 fragments (MWReg30, 240 μg/kg, BD Bioscience) were administered via the jugular vein. After an additional 5 minutes, an arteriole (30-50 μm diameter) was injured using a pulsed nitrogen dye laser fired through the microscope objective. Images were captured using a CCD camera (SensiCam, Cooke, Auburn Hills, MI) coupled to Slidebook 4.2 image acquisition software (Intelligent Imaging Innovations, Denver, CO). Platelet accumulation is reported as the background-subtracted median fluorescence intensity. Thrombus area calculations reflect the number of pixels exceeding background.
Microfluidics chamber
The method is described in detail elsewhere 25. Glass slides were coated with 0.3 mg/ml Type I collagen (PureCol™, Inamed Biomaterials) by microfluidics patterning. Channels that were 100 μm high and wide were placed perpendicular to a 100 μm strip of collagen. Whole mouse blood anti-coagulated with 93 μM PPACK (final) and labeled with 1:100 (vol/vol) Alexa-488-conjugated CD41 monoclonal antibody was perfused at 1000 s-1 followed by an increase to 10,000 s-1 during which platelet poor mouse plasma was substituted for whole blood. Platelet accumulation was observed using a Nikon Eclipse TE2000-U inverted microscope equipped with a Hamamatsu Digital Camera C9300.
Platelet aggregation
Heparinized blood was diluted 1:1 with HEPES-Tyrode's buffer (137 mM NaCl, 20 mM HEPES, 5.6 mM glucose, 1 mg/ml BSA, 1 mM MgCl2, 2.7 mM KCl, 3.3 mM and NaH2PO4, pH 7.4), and centrifuged at 129×g for 7 min to prepare platelet-rich plasma (PRP). Platelet counts (Beckman-Coulter Z1) were adjusted to 2.5×108/ml with autologous platelet poor plasma. Platelet aggregation was measured in a Chrono-log lumi-aggregometer (Havertown, PA).
Results
Loss of sema4D expression ameliorates the effects of dyslipidemia on platelet function in vivo
Dyslipidemia affects platelet function, making them more reactive to agonists and, arguably, more likely to become activated in response to vascular injury or plaque rupture 2, 26. Having shown previously that loss of sema4D expression impairs platelet responses in vitro and in vivo in otherwise normal C57Bl/6 mice fed a standard chow diet 16, we began the present studies by asking whether the eliminating sema4D-dependent events could provide a means to reduce platelet hyperactivity in the setting of dyslipidemia. To accomplish this, sema4D(-/-) mice were crossed with LDLR(-/-) mice, all in the C57Bl/6 background, and then placed on a high fat, high cholesterol diet for 3 or 6 months beginning at 8 weeks of age, as were male wild type C57Bl/6 mice for comparison. Lipid profiles confirmed the dyslipidemia expected on this diet in mice that lack LDLR, with similar levels achieved in both knockout strains, but not the WT mice (Table 1).
Table 1.
Plasma lipid profiles in male mice that had been placed on a high fat diet for 6 months starting at 8 weeks of age (mean ± SEM).
| Total cholesterol (mg/dl) |
HDL cholesterol (mg/dl) |
Total triglycerides (mg/dl) |
|
|---|---|---|---|
|
sema4D(-/-)LDLR(-/-) n=5 |
2055±778 | 270±109 | 297±185 |
|
LDLR(-/-) n=5 |
2188±476 | 275±64 | 476±151 |
|
Wild type n=5 |
293±61 | 212±32 | 23±5 |
Thrombus formation in vivo was measured using a laser to produce focal endothelial injuries in cremaster muscle arterioles 24. In this model, platelet accumulation is detected in real time using fluorescently-tagged F(ab)2 fragments of a nonblocking anti-CD41 (αIIb) antibody. Two sets of studies were performed. In the first, sema4D(-/-) and matched sema4D(+/+) (referred to as WT) mice were compared while receiving a normal chow diet. Platelet accumulation following injury occurred in both strains of mice, but the initial rate and maximum extent of accumulation were reduced by 83% and 60% respectively in the sema4D(-/-) mice compared to the wild type mice (Figure 1A&C). The second set of studies compared sema4D(-/-)LDLR(-/-) and LDLR(-/-) mice that had been on the high fat diet for 6 months. Again large differences were observed, with the initial rate and maximum extent of platelet accumulation reduced by 48% and 58% in the sema4D(-/-)LDLR(-/-) mice compared to the LDLR(-/-) mice (Figure 1B&D).
Figure 1. Platelet response to injury in vivo.

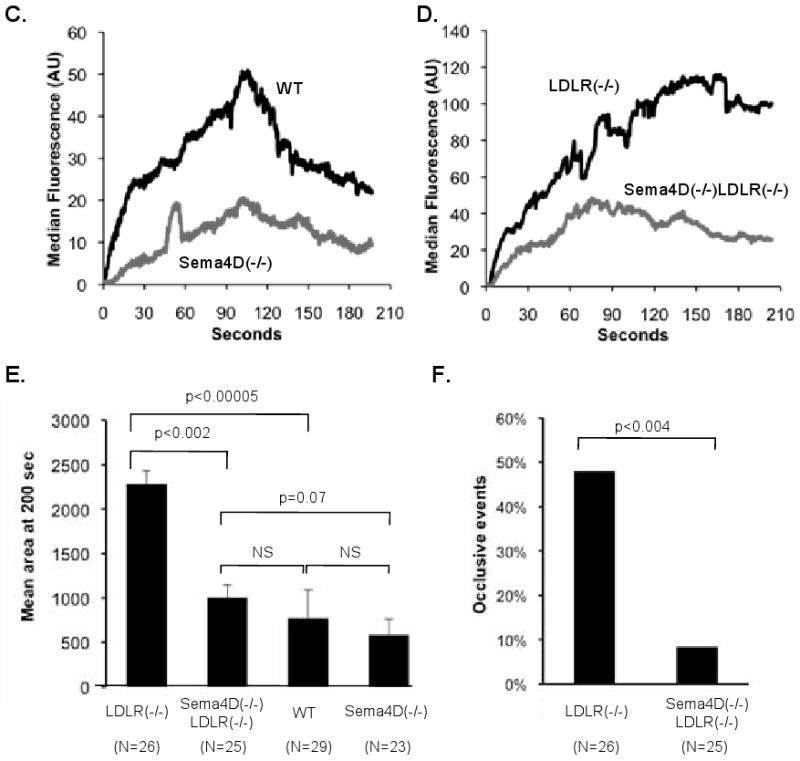
Platelet accumulation (green) in cremaster muscle arterioles damaged with a pulsed laser. (A&B) Video captures 2 min after injury. The WT and sema4D(-/-) mice were on a normal diet. LDLR(-/-) and sema4D(-/-)LDLR(-/-) mice were studied after 6 months on a high fat diet. (C&D) Median fluorescence intensity. WT: 29 injuries in 4 mice. Sema4D(-/-): 23 injuries in 3 mice. LDLR(-/-): 26 injuries in 3 mice. Sema4D(-/-)LDLR(-/-): 25 injuries in 3 mice. (E) Mean thrombus area. (F) Frequency of complete occlusion.
A comparison of these two sets of observations highlights both the consequences of dyslipidemia and the effects of deleting sema4D. Because the two sets of observations were performed with different batches of fluorescent antibody, mean thrombus area (i.e. all pixels exceeding background), rather than median fluorescence intensity (a measure that more closely approximates total platelet accumulation over time) was used for the comparison. Consistent with previous studies on the effects of dyslipidemia, the calculated mean thrombus area near the end of the observation period after injury was 3 times greater in the LDLR(-/-) mice fed a high fat diet than in matched wild type C57Bl/6 mice fed a regular chow diet (Figure 1E, p<0.00005). Thrombus area in the sema4D(-/-)LDLR(-/-) mice was also substantially less than in the LDLR(-/-) mice (p<0.002). In fact at that time point, mean thrombus area in the sema4D(-/-)LDLR(-/-) mice fed the high fat diet approached that observed in the wild type mice fed the chow diet, reflecting a large effect of the sema4D knockout in this setting (Figure 1E). Note that in terms of thrombus area, the sema4D(-/-) mice on the chow diet were not significantly different from the WT mice fed the same diet. In part this reflects the narrowing of the differences between WT and sema4D(-/-) mice late in the observation period when thrombus size tends to decrease (Figure 1C), but it also suggests that, as might be expected, total platelet accumulation over time is a more sensitive measure of the effects of sema4D deficiency on thrombus formation in response to laser injury under normolipidemic conditions than is mean thrombus area.
A second metric of platelet hyperactivity in vivo was also affected by the deletion of sema4D. In normolipidemic conditions, the laser injury in this model rarely results in complete occlusion of the arteriole in wild-type mice (data not shown). However, complete occlusion of the arteriole following laser injury occurred in nearly half (48%) of the injuries in dyslipidemic LDLR(-/-) mice, but only 8% of injuries in dyslipidemic sema4D(-/-)LDLR(-/-) mice (p=0.004; Figure 1F). Thus, loss of sema4D expression results in a significant reduction in the response of platelets to vascular injury. This effect of sema4D deficiency on platelet responsiveness is preserved in the context of dyslipidemia, conditions under which (as will be shown below) sema4D deficiency also significantly reduces atherosclerosis.
Loss of sema4D expression also reduces the effects of dyslipidemia on platelet function ex vivo
Since the expression of sema4D and its receptors is not limited to platelets, the results obtained in the in vivo models are not necessarily due solely to the loss of expression of sema4D from platelets. Therefore, we next asked whether loss of sema4D expression diminishes the effects of dyslipidemia when platelets are studied ex vivo. In the first of these studies, platelet accumulation on a collagen-coated surface was measured in a microfluidics flow chamber 25. Whole blood obtained from sema4D(-/-)LDLR(-/-) and LDLR(-/-) mice that had been fed the high fat diet for 6 months, was anticoagulated with the thrombin inhibitor, PPACK, and perfused through the chamber for 5 minutes at 1000 sec-1. The platelets were labeled with fluorescent anti-CD41 and observed in real time. Platelets from both mouse lines accumulated on the collagen surface as multicellular aggregates. However, the maximal extent of accumulation was reduced by 41% in the sema4D(-/-)LDLR(-/-) mice compared to the LDLR(-/-) mice (Figure 2A, p=0.034). A similar attenuation of thrombus growth is observed when Sema4D knockout platelets from eulipemic mice (LDLR(+/+)) are exposed to collagen under flow in this system (Zhu et al, manuscript in preparation). After 5 minutes at 1000 sec-1, the flow rate was increased 10-fold to 10,000 sec-1 to challenge the stability of pre-existing aggregates (Figure 2B). Dispersal of the sema4D(-/-)LDLR(-/-) platelets occurred more rapidly than the LDLR(-/-) platelets, indicating that the sema4D knockout affects thrombus stability as well as thrombus growth in the setting of dyslipidemia.
Figure 2. Platelet accumulation on collagen in a microfluidics flow chamber.
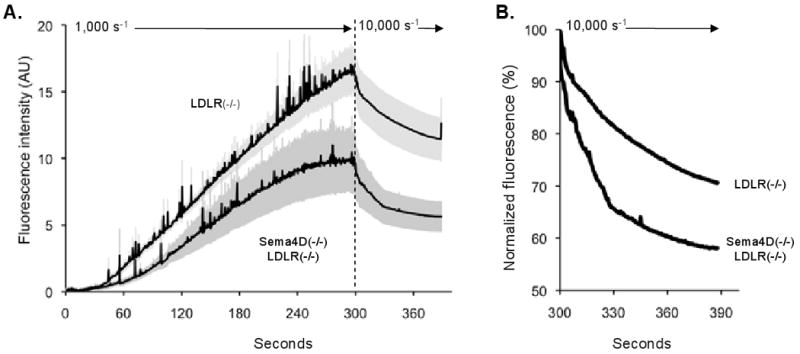
(A) Whole blood anticoagulated with PPACK was perfused through the chamber at 1000 s-1 for 300 seconds, then platelet poor plasma was substituted and shear was increased 10-fold. Mean ± SEM (N=7). (B) Data at 10,000 s-1 were normalized by dividing by the fluorescence intensity achieved just before the shear increase.
Finally, we have previously shown that collagen-induced platelet aggregation is inhibited in sema4D(-/-) mice, shifting the collagen dose/response curve to the right 16. A similar study using platelet rich plasma obtained from LDLR(-/-) and sema4D(-/-)LDLR(-/-) mice shows that the consequences of deleting sema4D persist in the face of dyslipidemia. At 2 μg/ml collagen shape change occurred normally, but the maximum extent of aggregation was approximately 50% less with the platelets from the sema4D(-/-)LDLR(-/-) mice than with LDLR(-/-) platelets (p=0.02, Figure 3).
Figure 3. Loss of sema4D expression impairs platelet aggregation.
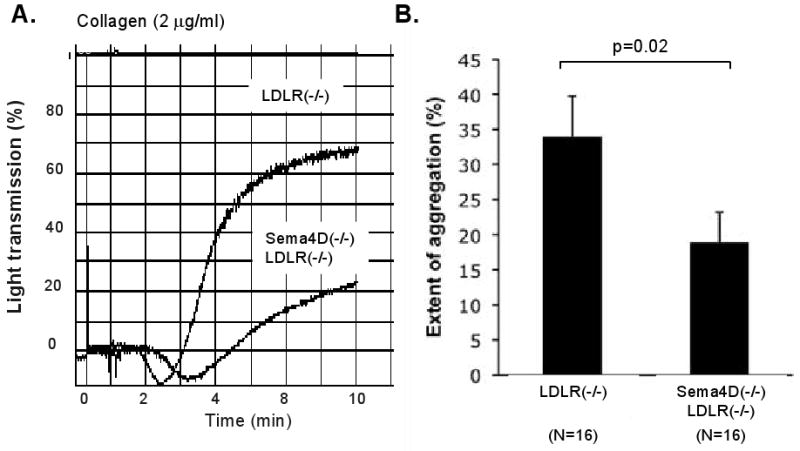
(A) Collagen-induced platelet aggregation from mice maintained on a high fat diet for 6 months. (B) Summary of 16 studies performed with platelets from 5 mice of each genotype (mean ± SEM).
Atherosclerosis is reduced in mice that lack sema4D
Since the absence of sema4D reduces the platelet hyperactivity otherwise seen in the setting of dyslipidemia, we next asked whether it would alter the progression of atherosclerosis usually observed when mice lacking LDL receptors are placed on a high fat diet. Histological examination revealed extensive lipid deposits in the aortas of LDLR(-/-) mice, especially after 6 months on the high fat diet. The deposits were visibly smaller in mice that lacked both LDLR and sema4D, and negligible in the wild type mice receiving the same diet (Figure 4A).
Figure 4. Loss of sema4D expression reduces atherosclerotic lesion size.
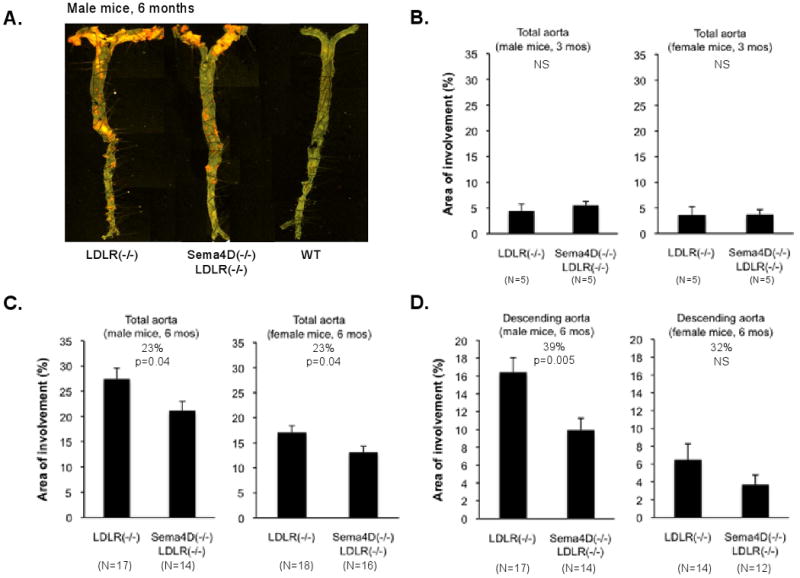
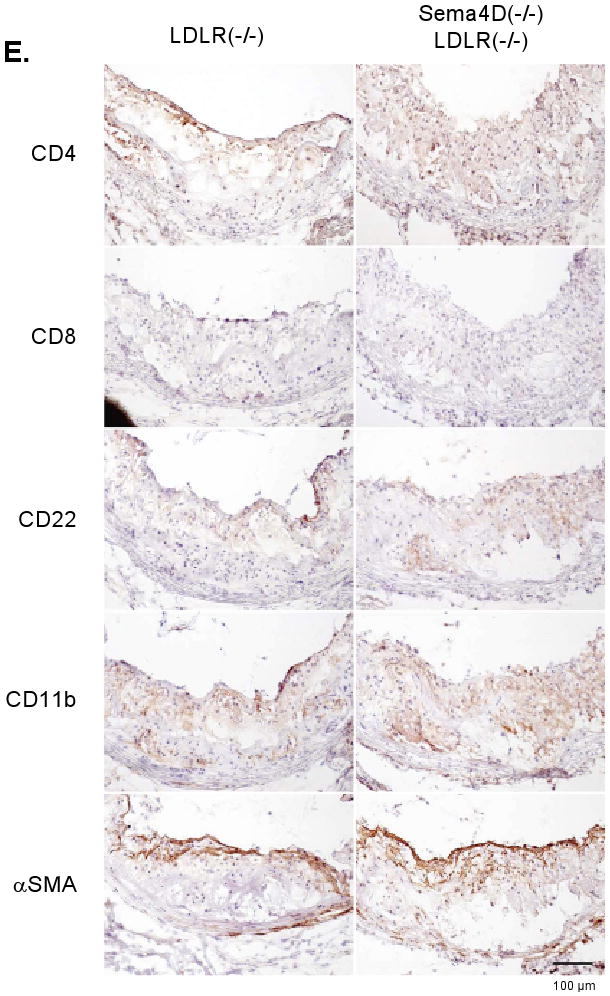
(A) Aortas after 6 months on the high fat diet showing lipid-rich deposits. (B,C,D) Analysis of lesion size. (E) Sections from the aortic root. Markers: CD4 and CD8, T-cells; CD22, B-cells; CD11b, monocytes, macrophage and neutrophils; smooth muscle actin (αSMA), smooth muscle cells and fibrotic caps. No differences were observed.
Lesion size was quantified after 3 and 6 months on the high fat diet, with most of the studies being performed with mice that had received the high fat diet for 6 months. At the 3 month time point, lipid deposits were small in both the male and female mice, and there was no evident difference between the LDLR(-/-) mice and the sema4D(-/-)LDLR(-/-) mice (Figure 4B). Note, however, that the number of mice studied at the 3 month time point was small (5 in each arm of the experiment), so small differences might have been missed. By the 6 month time point, lesion size had grown substantially, especially in the male mice. A comparison between the LDLR(-/-) and the sema4D(-/-)LDLR(-/-) mice at 6 months showed that loss of sema4D expression was associated with a 23% decrease in total lesion size in the entire aorta (p=0.04) in both the male and the female mice (Figure 4C). An even greater difference (39%) between the LDLR(-/-) and the sema4D(-/-)LDLR(-/-) mice was observed regionally in the descending aorta of the male mice at 6 months (p=0.005, Figure 4D). The female mice showed the same trend, but it did not reach statistical significance.
Finally, as noted in the introduction, sema4D is expressed on T-cells, as well as platelets, and B-cells, monocytes and endothelial cells express sema4D receptors. T-cells in particular have been shown to be present in atherosclerotic lesions in humans and mice 27, 28 and would be expected to be affected by the global absence of sema4D in the mouse lines that we studied. Since a megakaryocyte-selective deletion of sema4D is not available for comparison, we performed immunohistochemistry of aortic lesions using markers for T-cells, B-cells, monocytes and macrophage. No consistent differences were observed (Figure 4E).
Discussion
The idea that platelets contribute to atherogenesis is not a new one, but increasing evidence has been found to support it. Here we have tested the hypothesis that loss of the semaphorin family member, sema4D, would be protective against both the gain of platelet function normally seen in dyslipidemia and the subsequent evolution of atherosclerotic disease. Sema4D is of interest in this context for several reasons. First, sema4D supports platelet activation by collagen, which is impaired in mice lacking sema4D and in human platelets preincubated with anti-sema4D 16, 29. Second, sema4D receptors are expressed by many of the cells believed to be involved in atherogenesis, including endothelial cells, monocytes, activated macrophage, B-cells, and dendritic cells in addition to platelets. Finally, activated platelets shed sema4D as a single large exodomain fragment that retains its ability to activate sema4D receptors. Soluble sema4D shed in the context of atherosclerotic lesions, like surface-expressed sema4D on platelets, can potentially affect the behavior of nearby cells expressing its receptors. The results that we obtained show that loss of sema4D expression attenuates the progression of atherosclerosis in LDLR(-/-) mice despite the presence of profound dyslipidemia. It also greatly reduces the increase in platelet reactivity otherwise found in this setting.
Studies from a number of laboratories have demonstrated that platelets become hypersensitive to agonists when studied in the presence of dyslipidemia in humans or in mouse models 2. The basis for the increase in platelet function observed in dyslipidemia is only partially understood, but a recent study shows that oxidized LDL binds to CD36 (GP IV) on platelets and that deletion of CD36 reduces the effects of dyslipidemia on platelets 26. Consistent with these observations, we found that LDLR(-/-) mice on a high fat diet formed larger thrombi after vascular injury than did wild type mice and were much more likely to develop occlusive thrombi. Loss of sema4D reduced thrombus size and the frequency of occlusion. It should be noted that while these studies were performed in the microvasculature, our previous study demonstrated thrombus formation was attenuated in both the microvasculature and the carotid artery of sema4D knockout mice following oxidative injuries produced by, respectively, excited rose Bengal dye and FeCl3 16. It is likely, therefore, that our findings in the microvasculature of dyslipidemic mice would translate to the macrovasculature where atherosclerotic lesions develop. Further, when studied ex vivo we found that loss of sema4D expression limited platelet accumulation on collagen under flow and destabilized the thrombi that formed.
Collectively, these results not only confirm earlier observations that platelets become hyperresponsive in dyslipidemia, but also show that removing sema4D attenuates much of the increase. Mechanistically, we show elsewhere that loss of sema4D leads to impaired activation of the tyrosine kinase, Syk, which, in turn, decreases activation of phospholipase Cγ2, and reduces the increase in cytosolic Ca++ that otherwise occurs when platelets are activated by collagen 29. Although not directly examined in the present study, a defect in this signaling pathway could explain the attenuation of platelet responses in the dyslipidemic setting.
This study is not the first in which the knockout of a protein expressed on platelets that has been shown to affect the progression of atherosclerosis. What general lessons can be learned by comparing these mice? First, not all of deletions that affect platelet interactions with collagen have the same effect on atherogenesis. Platelets have at least three collagen receptors: the GP VI/FcRγ complex, integrin α2β1, and GP Ibα, which binds via von Willebrand factor. Although deletion of either FcRγ 8 or von Willebrand factor 30, is protective against atherosclerosis, loss of α2β1 is not 31. Second, there does not seem to be a linear relationship between the extent to which a particular gene deletion impairs platelet function and the extent to which it ameliorates atherosclerosis. Eliminating αIIbβ3 on platelets has a much greater effect on platelet function than does the sema4D knockout, but the effects of the two knockouts on atherogenesis are similar (reference 10 and the present study). Among other possibilities, this may mean that platelet signaling defects, particularly those that affect secretion, have a greater impact on atherogenesis than do adhesion or cohesion defects caused by deletion of integrins.
The sema4D knockout mice used in the present studies have a global defect in sema4D expression. Although our studies show that the sema4D knockout has cell-autonomous effects on platelet function, sema4D is also expressed on T-cells and sema4D receptors are on activated macrophage, dendritic cells and endothelial cells. Defects in these cells would not be expected to contribute to the defects in platelet function that we observed ex vivo, but might well affect the development of atherosclerosis. It is reasonable, therefore, to ask whether the global loss of sema4D expression affects atherogenesis because of its absence from platelets or from other types of cells. Tissue-specific knockouts would be helpful in answering this question, but are not yet available. As an alternative, we sought evidence that other hematopoietic cells are involved by looking for differences in T-cell, B-cell and macrophage infiltration into the atherosclerotic lesions in LDLR(-/-) and sema4D(-/-)LDLR(-/-) mice. No differences were observed, but this deserves to be re-visited. Therefore, our results support a role for sema4D in the enhancement of both atherogenesis and platelet activation, but further study is required to determine whether the role of sema4D in platelet function is directly related to its ability to enhance atherogenesis. However, since other studies have demonstrated that inhibition of platelet function can attenuate atherosclerosis 7-11, 13, 30, we would like to speculate that the attenuation in the sema4D(-/-) mice is due at least in part to reduced platelet function.
Supplementary Material
Acknowledgments
Funding. National Institutes of Health P50-HL81012 (LFB), R33-HL087385 (LFB and SLD), P01-HL62250 (EP) and F32-HL090304 (KBN). American Heart Association post-doctoral fellowship 0525630U (TJS). EP also acknowledges support from the Pennsylvania Department of Health and the Ludwig Institute for Cancer Research. Acknowledgements. We thank Dr. Daniel Rader (University of Pennsylvania) for assistance with the mouse lipid profiles, and Min Wang and Hua Li for their technical assistance.
Footnotes
Disclosures. None.
Literature cited
- 1.Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N Engl J Med. 2008;359:1261–1270. doi: 10.1056/NEJMra0800887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akkerman JW. From low-density lipoprotein to platelet activation. Int J Biochem Cell Biol. 2008;40:2374–2378. doi: 10.1016/j.biocel.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Theilmeier G, Michiels C, Spaepen E, Vreys I, Collen D, Vermylen J, Hoylaerts MF. Endothelial von Willebrand factor recruits platelets to atherosclerosis-prone sites in response to hypercholesterolemia. Blood. 2002;99:4486–4493. doi: 10.1182/blood.v99.12.4486. [DOI] [PubMed] [Google Scholar]
- 4.Massberg S, Brand K, Gruner S, Page S, Muller E, Muller I, Bergmeier W, Richter T, Lorenz M, Konrad I, Nieswandt B, Gawaz M. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002;196:887–896. doi: 10.1084/jem.20012044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huo Y, Ley KF. Role of platelets in the development of atherosclerosis. Trends Cardiovasc Med. 2004;14:18–22. doi: 10.1016/j.tcm.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 6.Croce K, Libby P. Intertwining of thrombosis and inflammation in atherosclerosis. Curr Opin Hematol. 2007;14:55–61. doi: 10.1097/00062752-200701000-00011. [DOI] [PubMed] [Google Scholar]
- 7.Burger PC, Wagner DD. Platelet P-selectin facilitates atherosclerotic lesion development. Blood. 2003;101:2661–2666. doi: 10.1182/blood-2002-07-2209. [DOI] [PubMed] [Google Scholar]
- 8.Hernandez-Vargas P, Ortiz-Munoz G, Lopez-Franco O, Suzuki Y, Gallego-Delgado J, Sanjuan G, Lazaro A, Lopez-Parra V, Ortega L, Egido J, Gomez-Guerrero C. Fcgamma receptor deficiency confers protection against atherosclerosis in apolipoprotein E knockout mice. Circ Res. 2006;99:1188–1196. doi: 10.1161/01.RES.0000250556.07796.6c. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi T, Tahara Y, Matsumoto M, Iguchi M, Sano H, Murayama T, Arai H, Oida H, Yurugi-Kobayashi T, Yamashita JK, Katagiri H, Majima M, Yokode M, Kita T, Narumiya S. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J Clin Invest. 2004;114:784–794. doi: 10.1172/JCI21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massberg S, Schurzinger K, Lorenz M, Konrad I, Schulz C, Plesnila N, Kennerknecht E, Rudelius M, Sauer S, Braun S, Kremmer E, Emambokus NR, Frampton J, Gawaz M. Platelet adhesion via glycoprotein IIb integrin is critical for atheroprogression and focal cerebral ischemia: an in vivo study in mice lacking glycoprotein IIb. Circulation. 2005;112:1180–1188. doi: 10.1161/CIRCULATIONAHA.105.539221. [DOI] [PubMed] [Google Scholar]
- 11.Cyrus T, Sung S, Zhao L, Funk CD, Tang S, Pratico D. Effect of low-dose aspirin on vascular inflammation, plaque stability, and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation. 2002;106:1282–1287. doi: 10.1161/01.cir.0000027816.54430.96. [DOI] [PubMed] [Google Scholar]
- 12.Cayatte AJ, Du Y, Oliver-Krasinski J, Lavielle G, Verbeuren TJ, Cohen RA. The thromboxane receptor antagonist S18886 but not aspirin inhibits atherogenesis in apo E-deficient mice: evidence that eicosanoids other than thromboxane contribute to atherosclerosis. Arterioscler Thromb Vasc Biol. 2000;20:1724–1728. doi: 10.1161/01.atv.20.7.1724. [DOI] [PubMed] [Google Scholar]
- 13.Afek A, Kogan E, Maysel-Auslender S, Mor A, Regev E, Rubinstein A, Keren G, George J. Clopidogrel attenuates atheroma formation and induces a stable plaque phenotype in apolipoprotein E knockout mice. Microvasc Res. 2009 doi: 10.1016/j.mvr.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Brass LF, Zhu L, Stalker TJ. Minding the gaps to promote thrombus growth and stability. J Clin Invest. 2005;115:3385–3392. doi: 10.1172/JCI26869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prevost N, Woulfe DS, Jiang H, Stalker TJ, Marchese P, Ruggeri Z, Brass LF. Eph kinases and ephrins support thrombus growth and stability by regulating integrin outside-in signaling in platelets. Proc Natl Acad Sci U S A. 2005;102:9820–9825. doi: 10.1073/pnas.0404065102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu L, Bergmeier W, Wu J, Jiang H, Stalker TJ, Cieslak M, Fan R, Boumsell L, Kumanogoh A, Kikutani H, Tamagnone L, Wagner DD, Milla ME, Brass LF. Regulated surface expression and shedding support a dual role for semaphorin 4D in platelet responses to vascular injury. Proc Natl Acad Sci U S A. 2007;104:1621–1626. doi: 10.1073/pnas.0606344104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bougeret C, Mansur IG, Dastot H, Schmid M, Mahouy G, Bensussan A, Boumsell L. Increased surface expression of a newly identified 150-kDa dimer early after human T lymphocyte activation. J Immunol. 1992;148:318–323. [PubMed] [Google Scholar]
- 18.Hall KT, Boumsell L, Schultze JL, Boussiotis VA, Dorfman DM, Cardoso AA, Bensussan A, Nadler LM, Freeman GJ. Human CD100, a novel leukocyte semaphorin that promotes B-cell aggregation and differentiation. Proc Natl Acad Sci U S A. 1996;93:11780–11785. doi: 10.1073/pnas.93.21.11780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furuyama T, Inagaki S, Kosugi A, Noda S, Saitoh S, Ogata M, Iwahashi Y, Miyazaki N, Hamaoka T, Tohyama M. Identification of a novel transmembrane semaphorin expressed on lymphocytes. J Biol Chem. 1996;271:33376–33381. doi: 10.1074/jbc.271.52.33376. [DOI] [PubMed] [Google Scholar]
- 20.Basile JR, Afkhami T, Gutkind JS. Semaphorin 4D/plexin-B1 induces endothelial cell migration through the activation of PYK2, Src, and the phosphatidylinositol 3-kinase-Akt pathway. Mol Cell Biol. 2005;25:6889–6898. doi: 10.1128/MCB.25.16.6889-6898.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delaire S, Billard C, Tordjman R, Chedotal A, Elhabazi A, Bensussan A, Boumsell L. Biological activity of soluble CD100. II. Soluble CD100, similarly to H-SemaIII, inhibits immune cell migration. J Immunol. 2001;166:4348–4354. doi: 10.4049/jimmunol.166.7.4348. [DOI] [PubMed] [Google Scholar]
- 22.Chabbert-de Ponnat I, Marie-Cardine A, Pasterkamp RJ, Schiavon V, Tamagnone L, Thomasset N, Bensussan A, Boumsell L. Soluble CD100 functions on human monocytes and immature dendritic cells require plexin C1 and plexin B1, respectively. Int Immunol. 2005;17:439–447. doi: 10.1093/intimm/dxh224. [DOI] [PubMed] [Google Scholar]
- 23.Shi W, Kumanogoh A, Watanabe C, Uchida J, Wang X, Yasui T, Yukawa K, Ikawa M, Okabe M, Parnes JR, Yoshida K, Kikutani H. The class IV semaphorin CD100 plays nonredundant roles in the immune system: defective B and T cell activation in CD100-deficient mice. Immunity. 2000;13:633–642. doi: 10.1016/s1074-7613(00)00063-7. [DOI] [PubMed] [Google Scholar]
- 24.Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. 2002;8:1175–1180. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 25.Neeves KB, Maloney SF, Fong KP, Schmaier AA, Kahn ML, Brass LF, Diamond SL. Microfluidic focal thrombosis model for measuring murine platelet deposition and stability: PAR4 signaling enhances shear-resistance of platelet aggregates. J Thromb Haemost. 2008;6:2193–2201. doi: 10.1111/j.1538-7836.2008.03188.x. [DOI] [PubMed] [Google Scholar]
- 26.Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, Chen J, Zhang R, Silverstein RL, Hazen SL. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roselaar SE, Kakkanathu PX, Daugherty A. Lymphocyte populations in atherosclerotic lesions of apoE -/- and LDL receptor -/- mice. Decreasing density with disease progression. Arterioscler Thromb Vasc Biol. 1996;16:1013–1018. doi: 10.1161/01.atv.16.8.1013. [DOI] [PubMed] [Google Scholar]
- 28.Hansson GK, Holm J, Jonasson L. Detection of activated T lymphocytes in the human atherosclerotic plaque. Am J Pathol. 1989;135:169–175. [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu L, Jiang H, Kumanogoh A, Kikutani H, Brass LF. Defective activation of phospholipase Cγ2 by collagen in platelets lacking the semaphorin family member, sema4D. Blood. 2007;110:3631a. [Google Scholar]
- 30.Methia N, Andre P, Denis CV, Economopoulos M, Wagner DD. Localized reduction of atherosclerosis in von Willebrand factor-deficient mice. Blood. 2001;98:1424–1428. doi: 10.1182/blood.v98.5.1424. [DOI] [PubMed] [Google Scholar]
- 31.Grenache DG, Coleman T, Semenkovich CF, Santoro SA, Zutter MM. Alpha2beta1 integrin and development of atherosclerosis in a mouse model: assessment of risk. Arterioscler Thromb Vasc Biol. 2003;23:2104–2109. doi: 10.1161/01.ATV.0000097282.22923.EF. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.