

Abstract
Purpose of review
the purpose of this review is to discuss recent findings as they pertain to anabolic and catabolic signaling pathways involved in the regulation of adult skeletal muscle mass.
Recent findings
research conducted over the past few years has continued to refine our understanding of the pathways that govern skeletal muscle mass, in particular the mTOR, FoxO and NF-κB pathways. Alternative signaling pathways have also emerged as important regulators of muscle mass such as the β-catenin pathway.
Summary
a better understanding of the anabolic and catabolic processes which regulate skeletal muscle mass is critical for the development of more effective therapeutics to prevent the loss of muscle with disuse, aging and disease.
Keywords: anabolic, catabolic, skeletal muscle, mTOR, NF-κB
Introduction
Beyond skeletal muscle's primary function as a force generator for locomotion, there is a growing recognition of the important role skeletal muscle plays in overall health through its impact on whole-body metabolism as well as directly influencing quality of life issues with chronic disease and aging. The acknowledgement of skeletal muscle's broad influence on general health has heightened interest in trying to understand the mechanisms responsible for the maintenance of muscle mass. The maintenance of skeletal muscle mass in the mature individual is primarily dictated by the balance between the rates of protein synthesis and protein degradation. Research over the previous decade has revealed that growth factors, hormones, cytokines, nutrients and mechanical loading are all environmental triggers capable of activating cellular signaling pathways which can push the balance in favor of protein synthesis or degradation leading to a gain or loss, respectively, of skeletal muscle mass. The primary purpose of this brief review is to highlight the advances that have been made over the past few years in our understanding of the signaling pathways controlling the rate of protein synthesis and degradation.
Anabolic pathways
In general, the cellular protein synthetic rate is considered to be determined by two factors, translational efficiency and translational capacity. Translational efficiency is defined as protein synthesis per unit amount of RNA where as translational capacity is defined as the total ribosomal content per unit tissue (1). The vast majority of research on muscle anabolism has implicitly focused on defining those signaling pathways which lead to enhanced translational efficiency, though recent studies have begun to investigate the signaling events that regulate ribosomal biogenesis i.e. translational capacity (2-4). As shown in Figure 1, mTOR (mammalian target of rapamycin: TORC1 complex) is currently thought to be the major hub for the integration of an array of upstream signaling pathways which, when activated, ultimately result in increased translational efficiency. Enhanced translational efficiency occurs over a relatively short time frame and is typically viewed independent of any change in the translational capacity of the cell. While it is almost certain that the processes which regulate translational efficiency and translational capacity are coordinated, the underlying mechanism responsible for such coordination remains to be fully elucidated. Nader and colleagues reported rapamycin prevented the increase in ribosomal RNA during muscle hypertrophy suggesting mTOR/TORC1 may in fact be responsible for coordinating changes in translational efficiency and capacity (3). The first part of the anabolic section will discuss recent findings on those pathways which increase translational efficiency in response to a stimulus followed by a brief discussion on some initial studies examining the control of ribosomal biogenesis during skeletal muscle hypertrophy.
Figure 1.
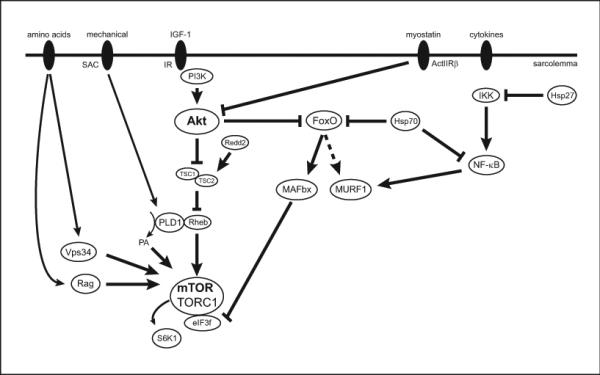
Anabolic and catabolic pathways regulating skeletal muscle mass. The major anabolic pathway regulating protein synthesis in skeletal muscle is mTOR/TORC1 signaling. Upstream triggers (IGF-1, mechanical, amino acids) activate mTOR signaling through a number of different intermediary proteins such as Rheb (Ras homolog enriched in brain), PLD1 (phospholipase D 1) and its metabolite PA (phosphatidic acid), Vps34 (vacuolar protein sorting 34) and Rag GTPases (Rag). The activity of mTOR can be inhibited by Redd2 through a TSC2-dependent mechanism or enhanced by eIF3f acting as a scaffolding protein for mTOR and its downstream target S6K1. The activity of the mTOR pathway can also be modulated through crosstalk with the TGF-β pathway through myostatin inhibition of Akt activation or FoxO up-regulation of MaFbx resulting in eIF3f degradation. Catabolic pathways discussed include FoxO and NF-κB. The FoxO pathway is inhibited by Akt phosphorylation and regulates the expression of the muscle-specific ubiquitin ligases MAFbx and MURF1, though the evidence for MURF1 is currently unclear. NF-κB pathway can be activated by different cytokines (TNF-α and IL-6) and has been shown to regulate MURF1. The activities of both FoxO and NF-kB signaling pathways can be modulated through interactions with the heat shock proteins Hsp 70 and Hsp27.
mTOR signaling
In adult skeletal muscle the mTOR/TORC1 signaling pathway is currently recognized as the major pathway regulating protein synthesis leading to increased translational efficiency (see Figure 1). Research over the past few years has continued to refine our understanding of how this pathway is modulated and its cross-talk with other pathways such as the TGF-β pathway. For an in-depth review of the mTOR signaling pathway the reader is referred to more comprehensive reviews (5, 6).
One of the most well characterized upstream triggers of mTOR signaling in skeletal muscle is IGF-1. Numerous studies have documented the ability of IGF-1 to induce muscle hypertrophy by Akt activation of mTOR signaling. What remained on open question was whether or not IGF-1 activation of mTOR was absolutely necessary for muscle hypertrophy. To address this question, Spangenburg and coworkers examined whether or not mice which over-express a dominant negative form of the IGF-1 receptor (MKR mouse) were capable of mounting a hypertrophic response following synergist ablation (7). Somewhat surprising, these investigators found no difference in the magnitude of hypertrophy between wild-type and MKR mice in response to increased mechanical loading induced by synergist ablation. Furthermore, both strains of mice showed equivalent levels of Akt activation and the downstream target of mTOR, p70S6K. These findings are significant for two reasons; first, it clearly demonstrates that IGF-1 receptor signaling is not critical for the induction of muscle hypertrophy and, secondly, it points to the involvement of some other upstream mediator of activation of mTOR signaling besides IGF-1.
Previous work by Hornberger and colleagues described mechanical activation of mTOR by an IGF-1 independent pathway involving phospholipase D (PLD) via its metabolite phosphatidic acid (PA) (8). More recently, work from the Hornberger laboratory extend these initial findings by reporting that mTOR activation following eccentric contractions required PLD synthesis of PA which was independent of PI3K-Akt activity (9). The role of PLD in mTOR signaling was further clarified when it was shown that PLD1, but not PLD2, was a downstream effector of Rheb (Ras homolog enriched in brain) activation of mTOR by physically interacting with Rheb in a GTP-dependent manner (10). Furthermore, Rheb regulation of PLD1 activity was shown to be sensitive to amino acid availability through an unknown mechanism that likely involves Rheb binding or, more speculative, through interaction with the amino acid sensitive PI3K, Vps34 (10).
Vps34 (vacuolar protein sorting 34) is a class III, PI3K previously shown to mediate amino acid activation of p70S6K by mTOR (11, 12). In skeletal muscle, MacKenzie et al., reported high-resistance contractions significantly increased Vps34 activity possibly in response to increased intramuscular leucine levels (13). Based on the temporal response of p70S6K activation, these authors proposed an interesting model in which the initial activation of mTOR was through the PLD/PA pathway via mechanical stimuli, followed by amino acid stimulation of Vps34 activity, and a final phase of mTOR activation by growth factors. Of particular interest to skeletal muscle, is the finding that amino acids cause an increase in intracellular Ca2+ resulting in calmodulin activation of Vps34, though this finding remains controversial (14, 15). In addition to Vps34, two groups reported the exciting discovery that the Rag family of GTPases was necessary and sufficient for amino acid activation of the mTOR pathway (16, 17). Determining the exact role of Vps34 and the Rag GTPases in regulating protein synthesis in adult skeletal muscle represents an exciting avenue for future study.
Despite the growing complexity of the mTOR pathway (18), and the relevancy of these new finding to muscle growth, a study by Miyazaki and Esser provided evidence suggesting the mTOR signaling pathway may be regulated in a muscle-specific fashion (19). The stress response gene Redd2 (regulated in development and DNA damage response 2) was found to be highly enriched in skeletal muscle and was capable of inhibiting, by a TSC2-dependent mechanism, basal mTOR activity as well as in response to mechanical stretch and leucine (19). Consistent with this finding, Redd2 mRNA expression was down-regulated 50% in both young and old human skeletal muscle in response to an anabolic stimulus and by 90% following mechanical overload of the mouse plantaris muscle by synergist ablation (20) (unpublished observation MM & KE). It will be of great interest to determine if the muscle-specific inactivation of Redd2 will help to maintain skeletal muscle mass during periods of disuse.
β-catenin/c-Myc signaling
In addition to translational efficiency, the rate of protein synthesis is also determined by the translational capacity of the cell as reflected by the ribosomal content. As mentioned earlier, the mTOR pathway has been shown to regulate ribosomal biogenesis in muscle by a UBF-mediated increase in rRNA transcription (3). Studies have begun to provide evidence, however, for the importance of a β-catenin/c-Myc signaling pathway which operates independent of the mTOR pathway in regulating ribosomal biogenesis. Collectively, it has been shown that after seven days of mechanical overload, nuclear β-catenin levels were increased by over 4-fold with a corresponding increase in myonuclear c-Myc expression and a ~ 3-fold increase in total RNA (4, 21). The muscle-specific inactivation of the β-catenin gene completely prevented muscle growth in response to mechanical overload, clearly demonstrating the necessity of β-catenin and activation of its target genes i.e., c-Myc for muscle hypertrophy, though the latter point requires additional study.
Catabolic pathways
Skeletal muscle atrophy occurs when the rate of protein degradation exceeds the rate of protein synthesis. Such a shift in the equilibrium between protein synthesis and degradation can happen when there is an increase in the rate of protein degradation and/or a decrease in the rate of protein synthesis. In contrast to skeletal muscle hypertrophy, the loss of skeletal muscle mass is observed under many different conditions including disuse, various disease states (sepsis, cancer, AIDS, diabetes and renal failure) and aging. Accordingly, the proteolytic systems involved in muscle atrophy are responsive to a number of different triggers such as mechanical unloading (disuse), growth factors (myostatin), hormones (glucocorticoid), inflammatory cytokines (TNF-α and IL-6), oxidative stress (ROS and NO), metabolic stress (ATP levels) and nutrient availability (amino acids and glucose). The major catabolic pathways downstream of these triggers include the ubiquitinproteasome system, the lysosomal system, Ca2+- dependent calpains and caspases (for in depth review of each of these systems, readers are referred to (6, 22-24). Research conducted over the past few years has continued to further our understanding of these catabolic pathways by identifying new downstream targets, modulators and crosstalk between the pathways.
NF-κB signaling
A wealth of research has clearly shown the importance of NF-κB signaling in regulating skeletal muscle mass during periods of disease and inactivity (24). While the necessity of NF-kB signaling during muscle atrophy is unequivocal, current research has focused on defining the contribution of pathway components to muscle atrophy. Van Gammeren and coworkers extended earlier work in the mouse by showing that both IκB kinases, IKKα and IKKβ, are necessary and sufficient for muscle atrophy (25, 26). To arrive at this conclusion, these researchers over-expressed in the rat soleus muscle dominant negative or constitutively active forms of each IKK and found, respectively, disuse-induced muscle atrophy was reduced by 50% (individually) and 70% (in combination) or a ~40% reduction in fiber cross-sectional area (CSA) under weight-bearing conditions. An interesting question for future research will be whether or not the ability to blunt muscle atrophy with the dominant negative forms of IKK is observed under muscle wasting conditions associated with cachexia.
In a series of studies, the Judge laboratory has reported that heat shock protein 70 (Hsp70) and Hsp27 can prevent muscle atrophy associated with immobilization by modulating the signaling activities of NF-κB and FoxO pathways (27-29). A 5-fold over-expression of Hsp70 in the rat soleus muscle completely prevented the loss in fiber cross sectional area (CSA) in response to seven days of immobilization and attenuated the increase in MAFbx and MURF1 mRNA expression by ~70%. Further examination revealed that Hsp70 inhibited the transcriptional activities of both FoxO3a and NF-κB but that only FoxO3a inhibition blunted the increased expression of MAFbx and MURF1during immobilization (27). In a similar fashion, the over-expression of Hsp27 was able to ameliorate the loss in fiber CSA with immobilization and inhibit NF-κB activation, but not FoxO, by binding to IKKβ and presumably preventing the degradation of IκBα {Δ○δδ, 2009 #191}. Collectively, these studies indicate Hsp27 and Hsp70 can significantly modify the activity of catabolic pathways involved in muscle atrophy and highlight the need to identify the direct transcriptional targets of NF-κB during muscle atrophy.
FoxO signaling
The different mechanisms regulating MAFbx and MURF1 expression during muscle atrophy was further distinguished in studies examining their regulation by FoxO transcription factors. A central component of the ubiquitin/proteasome system in skeletal muscle is activation of the muscle-specific E3 ubiquitin ligase genes, MAFbx and MuRF1, by FoxO transcription factors. Employing a cell line deficient in FoxO1 DNA-binding, McLoughlin and colleagues showed activation of MAFbx gene expression required DNA-binding of FoxO1 but, in contrast, up-regulation of MURF1 expression occurred independently of DNA-binding (30). This finding was extended to FoxO3a by Senf (2009) and coworkers who reported that a FoxO3a DNA-binding mutant was capable of activating a MURF1reporter gene but failed to increase expression of a MAFbx reporter gene (29). In contrast to these results, chromatin immunoprecipitation analysis found that FoxO1 bound the endogenous MURF1 promoter and synergized with the glucocorticoid receptor to activate MURF1 gene expression (31). Beyond the importance of resolving this discrepancy, these findings clearly indicate MAFbx and MURF1 expression is regulated by distinct mechanisms, likely reflecting their unique roles during muscle atrophy.
In an effort to better understand the unique role of MAFbx and MURF1 in muscle atrophy, identifying the proteins they each target for degradation has been an ongoing challenge. Through the use of a mouse carrying a mutant form of MURF1 unable to ubiquitylate substrates, Cohen and others confirmed the myosin heavy chain protein is a substrate of MURF1(32, 33). The Leibovitch laboratory used a yeast two-hybrid screen to identify the eukaryotic initiation factor 3 subunit 5 (eIF3f) as a primary target of MAFbx in skeletal muscle atrophy (34). Extensive follow-up experiments revealed repression of eIF3f caused muscle atrophy whereas over-expression promoted a modest, but significant, 11% muscle hypertrophy. Based on these exciting findings, the authors have proposed a model in which eIF3f acts as a nodal point for upstream pathways regulating muscle hypertrophy and atrophy. The basis of this model is the proposal that eIF3f functions as a scaffold protein insuring mTOR activation of p70S6K (35). This idea is supported by studies which have shown that eIF3f physically interacts with mTOR and p70S6K (36, 37). The Leibovitch group has also reported that MAFbx targets MyoD for proteolysis and most recently, the inhibition of this process prevented muscle atrophy (38). These findings are intriguing as they suggest that MAFbx expression may contribute to regulation of muscle size through altering protein synthesis or the function of myogenic transcription factors. This model for the role of MAFbx in skeletal muscle atrophy is in contrast to the more common belief that MAFbx functions through mediating rates of protein degradation. Elucidation of the function of MAFbx on anabolic vs. catabolic pathways during muscle atrophy will be important for our basic understanding of the critical pathways regulating skeletal muscle size.
Myostatin signaling
In the past year a series of studies working to define the molecular mechanism of myostatin-induced muscle atrophy have revealed an interaction with Akt/mTOR signaling (39-44). Activation of TGF-β signaling by over-expression or treatment with myostatin was associated with decreased phosphorylation of Akt as well as reduced phosphorylation of other components of Akt/mTOR signaling such as ribosomal protein S6, p70S6K and 4E-BP1 (42, 44). Conversely, blocking myostatin activity through the use of an antibody increased protein synthesis and ribosomal protein S6 and p70S6K phosphorylation but curiously not Akt or 4E-BP1, suggesting the increase in protein synthesis was through an mTOR-independent pathway (39). In support of this latter finding, Sartori and colleagues reported inhibition of myostatin signaling by over-expression of a dominant negative ActRIIB construct resulted in a 29% hypertrophy that was modestly blocked by rapamycin (43). While these studies collectively indicate there is some degree of crosstalk between the Akt/mTOR and TGF-β pathways, they also present a scenario in which myostatin functions by inhibiting protein synthesis and not by promoting protein degradation via the ubiquitin-proteasome pathway, i.e., up-regulation of MAFbx and MURF1. Based on microarray analysis, Welle suggested future studies on myostatin function might benefit from investigating myostatin's impact on factors affecting translational capacity such as ribosomal biogenesis and turnover (40).
Conclusion
The main advances that have been made over the past few years have helped to refine our understanding of known anabolic and catabolic signaling pathways in adult skeletal muscle. While these advances have provided mechanistic details and demonstrated crosstalk between pathways, they have also shed light on the complexity of the processes involved in regulating skeletal muscle mass. Future studies will surely continue to enhance our understanding of the aforementioned pathways as well as begin to explore alternative pathways little considered in the field of muscle plasticity such as STARS (Striated activator of Rho signaling) and Hippo pathways (45, 46). In addition, work within our laboratory has begun to investigate the influence the role of microRNAs in the regulation of skeletal muscle mass. While there still remains much to learned, one perspective that is clear at this point is that the maintenance of skeletal muscle mass represents the orchestrated output of a collection of anabolic and catabolic signaling pathways.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Millward DJ, Garlick PJ, James WP, Nnanyelugo DO, Ryatt JS. Relationship between protein synthesis and RNA content in skeletal muscle. Nature. 1973 Jan 19;241(5386):204–5. doi: 10.1038/241204a0. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong DD, Wong VL, Esser KA. Expression of beta-catenin is necessary for physiological growth of adult skeletal muscle. Am J Physiol Cell Physiol. 2006 Jul;291(1):C185–8. doi: 10.1152/ajpcell.00644.2005. [DOI] [PubMed] [Google Scholar]
- 3.Nader GA, McLoughlin TJ, Esser KA. mTOR function in skeletal muscle hypertrophy: increased ribosomal RNA via cell cycle regulators. Am J Physiol Cell Physiol. 2005 Dec;289(6):C1457–65. doi: 10.1152/ajpcell.00165.2005. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong DD, Esser KA. Wnt/beta-catenin signaling activates growth-control genes during overload-induced skeletal muscle hypertrophy. Am J Physiol Cell Physiol. 2005 Oct;289(4):C853–9. doi: 10.1152/ajpcell.00093.2005. [DOI] [PubMed] [Google Scholar]
- 5.Miyazaki M, Esser KA. Cellular mechanisms regulating protein synthesis and skeletal muscle hypertrophy in animals. J Appl Physiol. 2009 Apr;106(4):1367–73. doi: 10.1152/japplphysiol.91355.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 2008 Jun;23:160–70. doi: 10.1152/physiol.00041.2007. [DOI] [PubMed] [Google Scholar]
- 7.Spangenburg EE, Le Roith D, Ward CW, Bodine SC. A functional insulin-like growth factor receptor is not necessary for load-induced skeletal muscle hypertrophy. J Physiol. 2008 Jan 1;586(1):283–91. doi: 10.1113/jphysiol.2007.141507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hornberger TA, Chu WK, Mak YW, Hsiung JW, Huang SA, Chien S. The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc Natl Acad Sci U S A. 2006 Mar 21;103(12):4741–6. doi: 10.1073/pnas.0600678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Neil TK, Duffy LR, Frey JW, Hornberger TA. The role of phosphoinositide 3-kinase and phosphatidic acid in the regulation of mammalian target of rapamycin following eccentric contractions. J Physiol. 2009 Jul 15;587(Pt 14):3691–701. doi: 10.1113/jphysiol.2009.173609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10**.Sun Y, Fang Y, Yoon MS, Zhang C, Roccio M, Zwartkruis FJ, et al. Phospholipase D1 is an effector of Rheb in the mTOR pathway. Proc Natl Acad Sci U S A. 2008 Jun 17;105(24):8286–91. doi: 10.1073/pnas.0712268105. The paper details the interaction between PLD1 and Rheb and the impact on mTOR signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci U S A. 2005 Oct 4;102(40):14238–43. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byfield MP, Murray JT, Backer JM. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem. 2005 Sep 23;280(38):33076–82. doi: 10.1074/jbc.M507201200. [DOI] [PubMed] [Google Scholar]
- 13*.MacKenzie MG, Hamilton DL, Murray JT, Taylor PM, Baar K. mVps34 is activated following high-resistance contractions. J Physiol. 2009 Jan 15;587(Pt 1):253–60. doi: 10.1113/jphysiol.2008.159830. First paper to describe the activation of Vps34 in skeletal muscle following high resistance exercise. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gulati P, Gaspers LD, Dann SG, Joaquin M, Nobukuni T, Natt F, et al. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008 May;7(5):456–65. doi: 10.1016/j.cmet.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008 Feb 15;410(1):1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 16.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008 Aug;10(8):935–45. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17**.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008 Jun 13;320(5882):1496–501. doi: 10.1126/science.1157535. This is the first paper to identify the Rag GTPase protein as the factor responsible for mediating amino acid stimulation of mTOR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunlop EA, Dodd KM, Seymour LA, Tee AR. Mammalian target of rapamycin complex 1-mediated phosphorylation of eukaryotic initiation factor 4E-binding protein 1 requires multiple protein-protein interactions for substrate recognition. Cell Signal. 2009 Jul;21(7):1073–84. doi: 10.1016/j.cellsig.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 19.Miyazaki M, Esser KA. REDD2 is enriched in skeletal muscle and inhibits mTOR signaling in response to leucine and stretch. Am J Physiol Cell Physiol. 2009 Mar;296(3):C583–92. doi: 10.1152/ajpcell.00464.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drummond MJ, Miyazaki M, Dreyer HC, Pennings B, Dhanani S, Volpi E, et al. Expression of growth-related genes in young and older human skeletal muscle following an acute stimulation of protein synthesis. J Appl Physiol. 2009 Apr;106(4):1403–11. doi: 10.1152/japplphysiol.90842.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCarthy JJ, Esser KA. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J Appl Physiol. 2007 Jan;102(1):306–13. doi: 10.1152/japplphysiol.00932.2006. [DOI] [PubMed] [Google Scholar]
- 22.Kandarian SC, Jackman RW. Intracellular signaling during skeletal muscle atrophy. Muscle Nerve. 2006 Feb;33(2):155–65. doi: 10.1002/mus.20442. [DOI] [PubMed] [Google Scholar]
- 23.Murton AJ, Constantin D, Greenhaff PL. The involvement of the ubiquitin proteasome system in human skeletal muscle remodelling and atrophy. Biochim Biophys Acta. 2008 Dec;1782(12):730–43. doi: 10.1016/j.bbadis.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Malhotra S, Kumar A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med. 2008 Oct;86(10):1113–26. doi: 10.1007/s00109-008-0373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25*.Van Gammeren D, Damrauer JS, Jackman RW, Kandarian SC. The IkappaB kinases IKKalpha and IKKbeta are necessary and sufficient for skeletal muscle atrophy. FASEB J. 2009 Feb;23(2):362–70. doi: 10.1096/fj.08-114249. Through use of dominant negative and constitutively active forms of IKK, the authors provide evidence that IKK is sufficient to alter muscle mass. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai D, Frantz JD, Tawa NE, Jr., Melendez PA, Oh BC, Lidov HG, et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004 Oct 15;119(2):285–98. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 27.Senf SM, Dodd SL, McClung JM, Judge AR. Hsp70 overexpression inhibits NF-kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J. 2008 Nov;22(11):3836–45. doi: 10.1096/fj.08-110163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dodd SL, Hain B, Senf SM, Judge AR. Hsp27 inhibits IKKbeta-induced NF-kappaB activity and skeletal muscle atrophy. FASEB J. 2009 Oct;23(10):3415–23. doi: 10.1096/fj.08-124602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Senf SM, Dodd SL, Judge AR. FOXO Signaling is Required for Disuse Muscle Atrophy and is Directly Regulated by Hsp70. Am J Physiol Cell Physiol. 2009 Oct 28; doi: 10.1152/ajpcell.00315.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLoughlin TJ, Smith SM, DeLong AD, Wang H, Unterman TG, Esser KA. FoxO1 induces apoptosis in skeletal myotubes in a DNA-binding-dependent manner. Am J Physiol Cell Physiol. 2009 Sep;297(3):C548–55. doi: 10.1152/ajpcell.00502.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waddell DS, Baehr LM, van den Brandt J, Johnsen SA, Reichardt HM, Furlow JD, et al. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am J Physiol Endocrinol Metab. 2008 Oct;295(4):E785–97. doi: 10.1152/ajpendo.00646.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, et al. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol. 2009 Jun 15;185(6):1083–95. doi: 10.1083/jcb.200901052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007 Nov;6(5):376–85. doi: 10.1016/j.cmet.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 34**.Lagirand-Cantaloube J, Offner N, Csibi A, Leibovitch MP, Batonnet-Pichon S, Tintignac LA, et al. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008 Apr 23;27(8):1266–76. doi: 10.1038/emboj.2008.52. This paper identifies the translation initiation factor, eIF3-f as a target for atrogin/MAFbx function in skeletal muscle atrophy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Csibi A, Tintignac LA, Leibovitch MP, Leibovitch SA. eIF3-f function in skeletal muscles: to stand at the crossroads of atrophy and hypertrophy. Cell Cycle. 2008 Jun 15;7(12):1698–701. doi: 10.4161/cc.7.12.6090. [DOI] [PubMed] [Google Scholar]
- 36.Harris TE, Chi A, Shabanowitz J, Hunt DF, Rhoads RE, Lawrence JC., Jr. mTOR-dependent stimulation of the association of eIF4G and eIF3 by insulin. EMBO J. 2006 Apr 19;25(8):1659–68. doi: 10.1038/sj.emboj.7601047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005 Nov 18;123(4):569–80. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 38.Lagirand-Cantaloube J, Cornille K, Csibi A, Batonnet-Pichon S, Leibovitch MP, Leibovitch SA. Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS One. 2009;4(3):e4973. doi: 10.1371/journal.pone.0004973. The result. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39*.Welle S, Burgess K, Mehta S. Stimulation of skeletal muscle myofibrillar protein synthesis, p70 S6 kinase phosphorylation, and ribosomal protein S6 phosphorylation by inhibition of myostatin in mature mice. Am J Physiol Endocrinol Metab. 2009 Mar;296(3):E567–72. doi: 10.1152/ajpendo.90862.2008. The results of this study provide evidence indicating myostatin functions by blocking protein synthesis and not breakdown. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welle SL. Myostatin and muscle fiber size. Focus on “Smad2 and 3 transcription factors control muscle mass in adulthood” and “Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size”. Am J Physiol Cell Physiol. 2009 Jun;296(6):C1245–7. doi: 10.1152/ajpcell.00154.2009. [DOI] [PubMed] [Google Scholar]
- 41.Welle S, Burgess K, Thornton CA, Tawil R. Relation between extent of myostatin depletion and muscle growth in mature mice. Am J Physiol Endocrinol Metab. 2009 Aug 4; doi: 10.1152/ajpendo.00179.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009 Jun;296(6):C1258–70. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- 43.Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, et al. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol. 2009 Jun;296(6):C1248–57. doi: 10.1152/ajpcell.00104.2009. [DOI] [PubMed] [Google Scholar]
- 44*.Amirouche A, Durieux AC, Banzet S, Koulmann N, Bonnefoy R, Mouret C, et al. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology. 2009 Jan;150(1):286–94. doi: 10.1210/en.2008-0959. First paper to demonstrate that myostatin functions to inhibit mTOR signaling in skeletal muscle. [DOI] [PubMed] [Google Scholar]
- 45.Lamon S, Wallace MA, Leger B, Russell AP. Regulation of STARS and its downstream targets suggest a novel pathway involved in human skeletal muscle hypertrophy and atrophy. J Physiol. 2009 Apr 15;587(Pt 8):1795–803. doi: 10.1113/jphysiol.2009.168674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Badouel C, Garg A, McNeill H. Herding Hippos: regulating growth in flies and man. Curr Opin Cell Biol. 2009 Oct 19; doi: 10.1016/j.ceb.2009.09.010. [DOI] [PubMed] [Google Scholar]