

Abstract
X-ray crystallographic snapshots have shown that conformational changes of a tetraglycine loop in the active site of formyl-CoA: oxalate CoA transferase (FRC) play an important role in the catalytic cycle of the enzyme. Orthogonal space random walk (OSRW) simulations have been applied to obtain quantitative computational estimates of the relative free energy of the “open” and “closed” conformations of this loop together with the energetic barrier for interconversion of these states in wild type FRC. These OSRW calculations not only show that the two conformations have similar free energies but also predict a barrier that is consistent with the observed turnover number of the enzyme. In an effort to quantitate the importance of specific residues in the tetraglycine loop, OSRW simulations have also been performed on the G258A, G259A, G260A and G261A FRC variants both to examine the energetic effects of replacing each glycine residue and to correlate the computed energies with kinetic and structural observations. In enzymes with substantially reduced catalytic efficiency (kcat/KM), the OSRW simulations reveal the adoption of additional low energy loop conformations. In the case of the G260A FRC variant, the new conformation identified by simulation is similar to that observed in the X-ray crystal structure of the protein. These results provide further evidence for the power of the OSRW method in sampling conformational space, and hence in providing quantitative free energy estimates for the conformations adopted by functionally important active site loops. In addition, these simulations model the motions of side chains that are correlated with changes in loop conformation thereby permitting access to long time-scale motions through the use of nanosecond simulations.
The importance of understanding the conformational energetics and dynamical properties of active site loops has driven the development of a number of sophisticated experimental biophysical methods to monitor the conformational transitions of these regions and their role in catalysis and/or binding.1,2 Computational methods offer an alternate approach to evaluating these motions, but the determination of free energies using molecular dynamics is greatly complicated by the “lagging Hamiltonian” problem, which arises from the relatively long times that are needed for the protein environment to relax about a given loop conformation.3 As a result, substantial effort is required to ensure adequate sampling for calculations employing perturbation4 or thermodynamic integration5 methods, limiting their application to studies on large molecular systems. In an effort to overcome this problem, Yang and co-workers have introduced an “Orthogonal Space Random Walk” (OSRW) sampling algorithm,6,7 which allows synchronous acceleration of the sampling of order parameter moves and their coupled protein reorganizations. We now report the application of this method to compute the conformational energetics of the functionally important tetraglycine loop in the active site of Oxalobacter formigenes formyl-CoA:oxalate transferase (FRC).8 Thus, OSRW-derived free energy profiles were computed for the interconversion of “open” and “closed” loop structures in FRC and correlated with the steady-state kinetic properties of the enzyme. In addition, the energetic and structural effects of introducing an alanine residue at each position of the tetraglycine loop were calculated. Taken overall, this comparison study demonstrates the capability of the OSRW strategy in predicting the free energy surfaces associated with active site loop motions.
FRC catalyzes the formation of oxalyl-CoA and formate from formyl-CoA and oxalate, which is an essential step for ATP synthesis in the bacterium.9 As a member of a new family of CoA-transferases,10 FRC is active as a homodimer in which the two monomers adopt a complex, interlocked fold.11 Using a variety of conditions, our group recently reported an extensive set of crystal structures for FRC corresponding to various “snapshots” of the catalytic cycle of the enzyme.12 These studies have revealed conformational changes in a tetraglycine loop (Fig. 1), which not only participates in stabilizing intermediates but also probably controls substrate access and product release.13
Figure 1.

Cartoon representations of the FRC active site tetraglycine loop (Gly258Gly259Gly260Gly261) in its “open” (left) and “closed” (right) conformations. Coordinates are taken from the crystal structures of free FRC (1P5H) and the FRC/CoA complex (1P5R), respectively. Catalytic residues are also indicated although crystallographic waters are not shown for clarity.
OSRW simulations were performed on the wild type FRC dimer, and the G258A, G259A, G260A and G261A FRC variants. Each calculation employed an all-atom representation of the homodimer embedded in a truncated octahedral box of explicit TIP3P water molecules, and the systems were described by the CHARMM27 force field.14 The whole system was dynamically simulated and the enhanced-sampling treatment was applied to one of the two active site loops in the homodimer. The order parameter λ was defined as (RMSDo – ΔRMSD)/2RMSDo, where RMSDo is the after-superimposition RMS deviation between the loop structures in the “open” and “closed” conformations (Fig. 1); and ΔRMSD denotes the difference of the after-superimposition RMS deviations of the dynamically evolving loop from the ones in the reference (“open” and “closed”) structures. Each OSRW simulation was begun from an equilibrated homodimer in which the loop was in its “open” conformation, and continued until convergence of the free energy profile by sampling over the range λ = 0 → λ = 1 (Figs. S1 and S2, Supporting Information). In general, converged energy profiles (Fig. S3, Supporting Information) were generally achieved in about 2.0 ns.
All of the free energy profiles show two minima for the tetrapeptide loops, with the exception of G259A FRC variant, for which there are three stable conformations of similar energy. For WT FRC, the calculations showed a small free energy preference for the “open” loop conformation (consistent with the need to bind substrate). The barrier to interconversion was computed to be 13 kcal/mol (Table 1), which is lower than the experimental energy of 16.3 kcal/mol at 303.15 K based on the turnover number5 and assuming a value of 5 × 1012 s−1 for the pre-exponential factor.15 We are aware, however, that the use of a ΔRMSD-type order parameter may imperfectly describe the target events; therefore the use of this parameter may underestimate free energy barrier heights.16 OSRW simulations of the loop conformational interconversion for the G261A FRC mutant gave a similar free energy profile (Table 1). This similarity in the two free energy profiles is consistent with the experimental observation that the experimental kinetics of the G261A FRC variant are essentially unchanged from those of wild type enzyme. OSRW free energy profiles calculated for the tetrapeptide loop conformations in the G258A, G259A and G260A FRC mutants were different to those determined for wild type enzyme and G261A. Most strikingly, these simulations identified additional conformational minima that were intermediate in shape between the “open” and “closed” loop conformations and comparable in free energy (Fig. S3, Supporting Information). The barriers to conformational interconversion in the G259A and G260A FRC mutants, however, were slightly lower than FRC. Experimentally, both the G259A and G260A FRC mutants exhibit reduced catalytic activity relative to wild type FRC (Table 1),12 consistent with the idea that catalytically important loop conformations are no longer energetically preferred due to the alanine residue at either position. Perhaps because Gly-259 adopts backbone angles that are disallowed for alanine in both loop conformations seen in free FRC (Table S1, Supporting Information), the tetrapeptide loop segments in the G259A and G260A variants are computed to have a substantially lower preference for the “open” structure, lower barriers to conformational interconversion, and adopt an additional loop structure. These energetic changes may underlie the observed reductions in catalytic efficiency for these FRC mutants (kcat/Km) (Table 1) because the tetraglycine loop stabilizes anhydride intermediates formed during catalytic turnover,8,12. In an effort to calibrate the predictive power of our OSRW calculations on a firm structural basis, we superimposed the “open”, “closed” and “intermediate” loop conformations computed for the G260A FRC variant (Fig. 2) with the loop structure observed by X-ray crystallography (2VJN).12 We were gratified to find extremely good agreement between the “intermediate” conformation and that seen in the G260A structure (Table S2, Supporting Information) even though no information about this new loop conformation had been included in our OSRW simulations. In addition, the non-interpolating reorientation of the Trp-48 side chain that takes place as the loop changes conformation was also correctly reproduced in these simulations. Both these observations engender confidence in the computed thermodynamic properties. Efforts to correlate the OSRW free energy profile computed for G258A with experiment have been precluded by repeated failures to express this protein. The presence of (i) an intermediate state that is lower in energy than both the “open” and “closed” loop conformations and (ii) an increased barrier to interconversion predicts, however, that this mutant should exhibit little activity.
Table 1.
Calculated free energy values (kcal/mol) for conformational interconversion of the tetrapeptide loop and steady-state kinetic parameters for free FRC and four alaninecontaining FRC mutants.
| Enzyme | ΔΔGa | ΔΔG† | kcat (s−1)b | kcat/Km (M−1s−1)c |
|---|---|---|---|---|
| FRC | 4.8 | 13.0 | 5.3 ± 0.1 | 1.4 × 103 |
| G258A | 5.3 | 23.0 | -d | -d |
| G259A | 2.2 | 9.0 | 1.9 ± 0.1 | 160 |
| G260A | 2.5 | 9.3 | 0.23 ± 0.02 | 12 |
| G261A | 3.7 | 10.5 | 1.65 ± 0.01 | 3.5 × 103 |
All calculated free energies are in kcal/mol. ΔΔG = ΔG(“closed”) – ΔG(“open”), and ΔΔG† is the barrier relative to the “open” conformation.
Experimental steady-state kinetic parameters at 30 °C have been published previously,12 and are included here for ease of comparison.
These kcat/Km values are for oxalate.
Technical problems in expressing and purifying the G258A FRC mutant have precluded its characterization.
Figure 2.
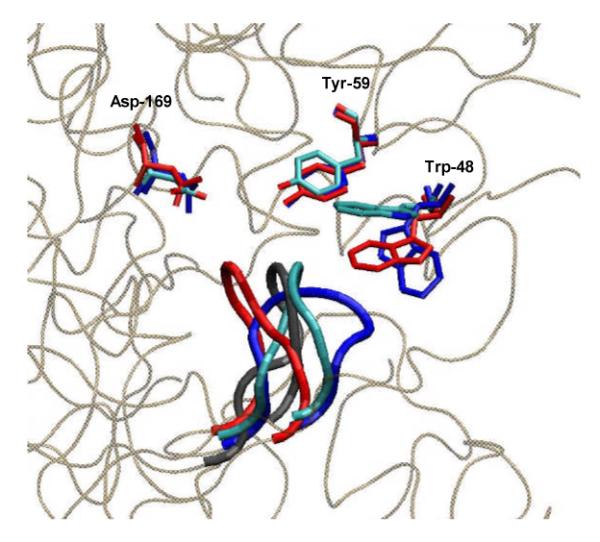
Cartoon showing superimposed active site tetrapeptide loops for the observed (2VJN) (cyan) and the “open” (blue), “intermediate” (black) and “closed” (red) loop conformations calculated for the G260A FRC mutant, together with the associated positions of the active site residues Trp-48, Tyr-59 and Asp-169.
Finally, relaxation of the protein environment about the loop conformation at a given λ value in only a small number of time steps, permits an assessment of side chain motions that are correlated with changes in loop structure. In the high-resolution X-ray crystal structure of wild type FRC, the side chain of Trp-48 undergoes a 90° rotation when the tetraglycine loop is in the “open” rather than the “closed” conformation. Detailed analysis of structures generated during the OSRW simulations showed the sampling algorithm to model side chain reorientation correctly, even though these motions were not defined explicitly in the reaction coordinate, or “driven” by prior knowledge of their positions in the FRC crystal structure (Fig. S3, Supporting Information). Because such changes represent motions that usually occur on a microsecond to millisecond time scale, the ability to obtain such information using nanosecond simulations is a significant advantage of the OSRW sampling strategy.
Supplementary Material
Acknowledgements
We thank the National Institutes of Health (DK61666) and the National Science Foundation (MCB0919983) for financial support.
Footnotes
Supporting Information Available. The full citation for reference 14, details of the theoretical and computational methods, tables S1 and S2, and additional figures relevant to the simulations are given as supporting information (14 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Loria JP, Berlow RB, Watt ED. Acc. Chem. Res. 2008;41:214–221. doi: 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]
- 2.Boehr DD, McElheny D, Dyson HJ, Wright PE. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 3.Kollman PA, Pearlman DA. J. Chem. Phys. 1989;91:7831–7839. [Google Scholar]
- 4.Zwanzig RW. J. Chem. Phys. 1954;22:1420–1426. [Google Scholar]
- 5.Straatsma TP, Berendsen HJC. J. Chem. Phys. 1988;89:5876–5886. [Google Scholar]
- 6.Zheng L, Chen M, Yang W. J. Chem. Phys. 2009;130:234105. doi: 10.1063/1.3153841. [DOI] [PubMed] [Google Scholar]
- 7.Zheng L, Chen M, Yang W. Proc. Natl. Acad. Sci., USA. 2008;105:20227–20232. doi: 10.1073/pnas.0810631106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jonsson S, Ricagno S, Lindqvist Y, Richards NGJ. J. Biol. Chem. 2004;279:36003–36012. doi: 10.1074/jbc.M404873200. [DOI] [PubMed] [Google Scholar]
- 9.Anatharam V, Allison MJ, Maloney PC. J. Biol. Chem. 1989;264:7244–7250. [PubMed] [Google Scholar]
- 10.Heider J. FEBS Lett. 2001;509:345–349. doi: 10.1016/s0014-5793(01)03178-7. [DOI] [PubMed] [Google Scholar]
- 11.Ricagno S, Jonsson S, Richards N, Lindqvist Y. EMBO J. 2003;22:3210–3219. doi: 10.1093/emboj/cdg333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berthold CL, Toyota CG, Richards NGJ, Lindqvist Y. J. Biol. Chem. 2008;283:6519–6529. doi: 10.1074/jbc.M709353200. [DOI] [PubMed] [Google Scholar]
- 13.Toyota CG, Berthold CL, Gruez A, Jonsson S, Lindqvist Y, Cambillau C, Richards NGJ. J. Bacteriol. 2008;190:2556–2564. doi: 10.1128/JB.01823-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacKerell A, et al. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 15.Himo F. Theor. Chem. Acc. 2006;116:232–240. [Google Scholar]
- 16.Rosta E, Woodcock HL, Brooks BR, Hummer G. J. Comput. Chem. 2009;30:1634–1641. doi: 10.1002/jcc.21312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.