

Summary
Background
Subtelomeres, regions proximal to telomeres, exhibit characteristics unique to eukaryotic genomes. Genes residing in these loci are subject to epigenetic regulation and elevated rates of both meiotic and mitotic recombination. However, most genome sequences do not contain assembled subtelomeric sequences, and, as a result, subtelomeres are often overlooked in comparative genomics.
Results
We study the evolution and functional divergence of subtelomeric gene families in the yeast lineage. Our computational results show that subtelomeric families are evolving and expanding much faster than families that do not contain subtelomeric genes. Focusing on three related subtelomeric MAL gene families involved in disaccharide metabolism that show typical patterns of rapid expansion and evolution, we show experimentally how frequent duplication events followed by functional divergence yields novel alleles that allow metabolism of different carbohydrates.
Conclusions
Taken together, our computational and experimental analyses show that the extraordinary instability of eukaryotic subtelomeres supports rapid adaptation to novel niches by promoting gene recombination and duplication followed by functional divergence of the alleles.
Introduction
Subtelomeres are repeat-rich and gene-poor regions proximal to the telomeres [1]. A precise definition of a subtelomere is difficult because the length of the subtelomeric region varies from 20 kb in some yeasts to several hundred kb in higher eukaryotes [2,3]. Apart from the low gene density, subtelomeres are characterized by epigenetic silencing [4,5] and increased rates of recombination and mutation [3,6,7,8,9], with exception to flies [10,11]. These regions are often lacking from so-called “whole genome” sequences because their high repeat content and extensive sequence similarity [12] make it difficult to assemble these regions and to distinguish orthologs and paralogs [2,13,14]. As a result, subtelomeres remain relatively understudied. For example, several landmark studies that reconstruct the evolution of gene families could not comprehensively analyze subtelomeric gene families [14,15,16,17]. From the few examples we have, subtelomeres seem to contain specific gene families that reflect the organism′s lifestyle. In yeasts, genes involved in biofilm formation and carbohydrate utilization have been mapped to subtelomeres [18,19,20,21,22,23]. In parasitic eukaryotes such as Plasmodium spp., trypanosomes and pathogenic fungi, many virulence genes reside at subtelomeres. Variegated expression of these cell surface genes allows these pathogens to continuously change their outer surface and evade the host immune response [24,25]. In primates, multiple genes encoding olfactory receptors [26] and members of the WASP family [27] have been mapped to subtelomeres. Moreover, promiscuous rearrangements of these regions have been implicated in human genetic disorders [28,29]. These anecdotal examples support the hypothesis that subtelomeres are variable loci harboring specific and fast-evolving gene families. Indeed, other authors have noted the rapid turnover of genes at subtelomeres [30,31,32], but a comprehensive analysis is lacking. Here, we use the genome sequences of eight ascomycete fungi to study the evolution of subtelomeric genes. We use comparative genomics to show that subtelomeric gene families evolve faster than their non-subtelomeric counterparts and then focus on three related gene families to analyze how they have evolved and functionally diverged. Together, our results underpin the unique role of subtelomeres as hotbeds for genomic evolution and innovation.
Results
In silico analysis of subtelomeric gene families
Using the definition of subtelomeres as gene-depleted regions [33], we investigated the gene density across the chromosomes of various yeast species and found that the average gene density is significantly lower up to 33kb away from the telomeres (Figure S1). This 33kb region agrees with previous studies on the telomere position effect [5] and sequence similarity between nonhomologous chromosome ends [2,3]. For our analyses, we classified each gene as subtelomeric or non-subtelomeric based on its distance from the chromosome end. While we only show results for a telomere length of 33kb as defined above, all our results are robust for subtelomere lengths between 10 and 50kb (see below).
To investigate which genes are enriched and depleted at subtelomeres, we used the gene ontology (GO) classification [34] (Table S1). Subtelomeres show significant enrichment for genes involved in response to stress and toxins, metabolism of a broad spectrum of compounds, and transporters involved in metal, amino acid, and carbohydrate uptake. By contrast, genes responsible for typical “housekeeping” functions such as ribosomal function, RNA processing, cell cycle control, mitosis, DNA repair and DNA replication are depleted at subtelomeres.
In order to compare the evolution of subtelomeric and non-subtelomeric genes, we used the Markov Cluster Algorithm (MCL) algorithm [15,35,36] to divide all genes across different fungi into gene families based on their sequence similarity (see Methods). Gene families that contain at least one gene located in the subtelomeric region were considered subtelomeric families. On average, gene families that do not contain any subtelomeric gene show a small number of genes per species with only few genes located within 200k from the chromosome ends and very little difference in copy number between species. Subtelomeric gene families (i.e. families that contain at least one subtelomeric gene), on the other hand, often show several genes within 33 kb of the chromosome end, and even more genes within 200kb. Moreover, subtelomeric families also show drastic copy number variation between the different yeast species used in this analysis (Figure 1A).
Figure 1. Overview of subtelomeric families vs. non-subtelomeric families.
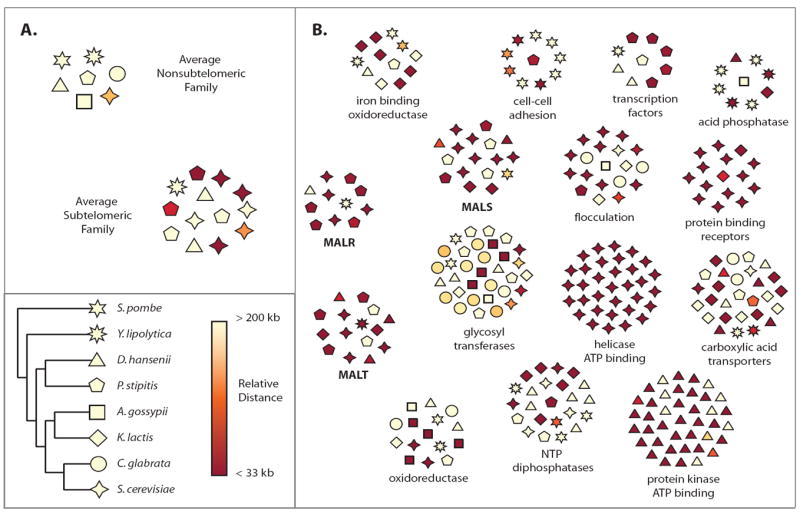
Subtelomeric families are more volatile than non-subtelomeric families. Every gene family (orthogroup) is shown as a number of polygons (individual genes per gene family) spatially arranged in a circular area. The polygons are colored according to their closest distance (denoted as relative distance in the figure) to a chromosome end going from pale yellow (> 200 kb away from the nearest chromosome end) to dark red (< 33 kb from the nearest chromosome end). The individual species are denoted by the eight differently shaped polygons (e.g. triangle for D. hansenii). A. The average composition of non-subtelomeric gene families and subtelomeric gene families is represented by two clusters (artificial clusters representing the mean copy number and mean distance to the telomere for subtelomeric and non-subtelomeric gene families). Non-subtelomeric gene families show small differences in copy number between species such that species contain around the same number of genes, and show few genes within 200 kb of chromosome end. Strikingly, subtelomeric gene families show high copy number variation between species, and have more genes within 200 kb (and especially within 33 kb) of the chromosome end. B. Some representative subtelomeric gene families are shown along with their functional annotation. Common characteristics among subtelomeric gene families can be seen; high copy number variation can be seen between species as well as multiple members less than 200 kb from the nearest chromosome end. The three MAL gene families, MALR, MALT, and MALS are shown in bold text. See Figure S1 and Table S1 for more information.
Statistical analysis of the gene families shows that within a species, there are far fewer subtelomeric gene families than would be expected if subtelomeric and non-subtelomeric genes were distributed randomly among the families. In other words, subtelomeric genes cluster together in a small number of families, and families that contain at least one subtelomeric member are more likely to contain multiple subtelomeric members (p-value < 10-10) (Figure 1B, Figure 2A and Table S2); this signal remains even after controlling for tandem or local duplications (p-value < 10-10) (Table S2). Even more striking, subtelomeric gene families are on average much larger than non-subtelomeric families; containing two to four times more genes than non-subtelomeric families (p < 10-10)(Figure 1B, Figure 2B and Table S2).
Figure 2. Computational Results.
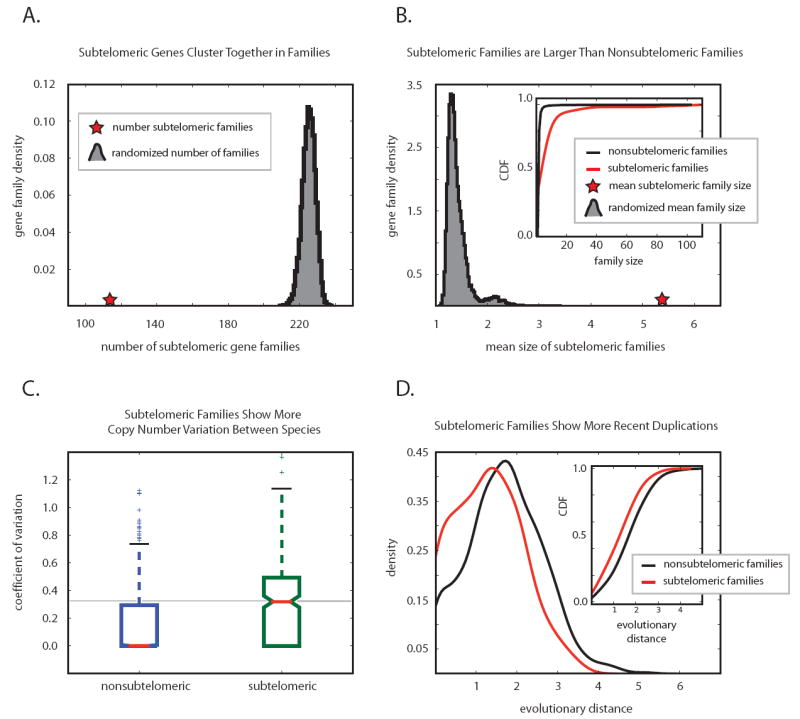
Subtelomeric gene families are larger and evolve faster. All S. cerevisiae genes were divided into gene families based on their homology (see text for details). Families containing at least one gene located within 33kb of a telomere were classified as subtelomeric gene families, all other families are non-subtelomeric families. Although we use the same 33 kb cutoff and the same MCL clustering parameters for our analyses, our results remain unchanged when we alter the definition of the subtelomeric region, and also when we alter the parameters for MCL gene clustering. A. The total number of subtelomeric gene families (114, red star) is smaller than what would be expected if all genes (subtelomeric and non-subtelomeric) were randomly distributed among gene families (grey curve, representing the number of gene families with subtelomeric members after 10000 randomizations). B. Subtelomeric gene families on average contain two to four times more genes than non-subtelomeric families. The red star represents the average family size of subtelomeric families in Saccharomyces cerevisiae at a 33 kb cutoff. The grey distribution shows the mean size of 10000 gene families that were chosen randomly amongst all gene families. A complementary analysis contrasting the cumulative distribution functions of subtelomeric gene family size (red) and non-subtelomeric gene family size (black) is consistent with the larger size of subtelomeric families. C. Subtelomeric gene families show increased copy number variation. The distribution of coefficients of variation (standard deviation normalized by mean, a metric of the extent variation) of the number of genes for all subtelomeric and non-subtelomeric gene families shows that subtelomeric gene families exhibit drastically higher copy number variation than non-subtelomeric gene families. D. The distribution of intraspecies protein distances (similarities) is compared for subtelomeric gene families (red) versus non-subtelomeric gene families (black). Subtelomeric gene families contain more closely related intraspecies proteins (recent duplications) than non-subtelomeric gene families, which is reflected in a shift of the distribution to the left. The inset compares the cumulative distribution functions of the intraspecies protein distances and shows a significant shift of the subtelomeric families towards newer duplications. See Figure S2 and Table S2 for more information.
Together, these analyses suggest that subtelomeric genes tend to spawn new subtelomeric genes, possibly as a result of the elevated recombination frequencies found at subtelomeres [3,6,7,37]. This hypothesis prompted us to ask whether subtelomeric families also show more copy number variation than their non-subtelomeric counterparts. When comparing gene family size across the fungal tree, subtelomeric families show significantly greater copy number variation between species (p < 10-10)(Figure 1B and Figure 2C). Moreover, subtelomeric families contain many genes that show greater similarity to other genes in the same species than to genes in all other species; a signature for recent duplication events that occurred after the different species diverged (p < 10-10)(Figure 2D). The extraordinarily rapid evolution of subtelomeric gene families was further investigated using the CAFE birth/death model [15]. This model uses a clever algorithm to quantify the rate at which new members of a gene family are being formed, or lost (see supplemental data for more information). The model confirms that subtelomeric families show remarkably aberrant birth/death rates (p < 10-10) (Table S2 and Figure S2), further demonstrating the rapid evolution of these families.
After demonstrating that subtelomeric gene families show both elevated copy number variation and gene family size, we revisited our initial GO enrichment analysis. We wondered whether the rapid gene turnover at subtelomeres is a property of the types of genes found at subtelomeres or rather a property of the subtelomeric region. We therefore analyzed copy number variation and family size of non-subtelomeric gene families to subtelomeric gene families belonging to the same functional GO category, and repeated this analysis for all GO categories that are enriched for subtelomeric genes. We found that for almost all families of a specific category (99% and 98% of non-subtelomeric and subtelomeric families tested, respectively), subtelomeric gene families show both higher copy number variation and average family size. In some cases we did not find statistically significant differences, but this seems to be due to the low numbers of genes in these GO categories. Taken together, these results indicate that the subtelomeric location rather than the functional enrichment is the causal driving force for the rapid gene turnover, with frequent duplication events (Table S1).
Duplication of subtelomeric genes involved in disaccharide utilization
Gene duplication is recognized as a crucial mechanism in evolution. The extra copy resulting from duplication events provides a dispensable copy of a gene that can acquire new function (neofunctionalization) without being restrained by purifying selective pressure on its original function [38]. An alternative view is that gene duplication allows asymmetric evolution of preexisting promiscuous function in a protein, such that these prior functions can be further optimized (subfunctionalization) [39]. Another putative advantage of subfunctionalization is that the expression of the two copies can be independently regulated, which further increases the evolutionary potential.
To begin investigating if members of subtelomeric gene families show signs of functional divergence, we studied their expression divergence (a measure for how differently the genes are regulated, see [40]) and responsiveness (a measure for how strongly a gene′s expression is influenced by the environment [40] (see Materials and Methods). The results show that subtelomeric genes show higher average expression divergence (0.250 vs. -0.007, p = 0.035) and higher average responsiveness (1961 vs. 1491; p < 10-10) when compared to non-subtelomeric genes, agreeing with the hypothesis that subtelomeric duplicates show rapid divergence.
To further investigate whether the frequent duplication of subtelomeric genes provides the raw material for functional divergence, we examined three related typical subtelomeric gene families involved in maltose metabolism [41] (see Figure 1). For each of these families, we investigated if and how the genes have been duplicated, and if these duplication events were followed by functional divergence. The first family, called MALT, contains transporters to import maltose into the cell, the second family, MALS, encodes maltases, enzymes that hydrolyze maltose into two glucose units, and the third family, MALR, encodes regulator proteins that induce the expression of MALR, MALT, and MALS genes when maltose is present [41].
We first manually mapped all MAL genes in completely assembled yeast genomes, as well as in available contigs of other (non-assembled) high-coverage genomes. We identified seven unannotated MAL genes (two from the MALR family and five from the MALS family) out of a total 14 MAL genes in the S. cerevisiae S288c genome that were present as unannotated ORFs. Second, consistent with our in silico analysis, we noted extraordinary fluctuations in the chromosomal location and number of MAL genes between different species and even strains (Figure 3, Figure 4, and Figure S4). These copy number variations are not a direct result of the whole genome duplication that occurred during the evolution of the hemiascomycetes [42]. Candida glabrata, Saccharomyces castelli, and Kluyveromyces polysporus underwent the whole genome duplication, but do not have any MAL loci. The protein phylogeny indicates that the common ancestor of these yeasts had only few MAL genes, which were completely lost in some lineages and expanded in other lineages (Figure S3).
Figure 3. MAL CNV in Fungal Lineage.
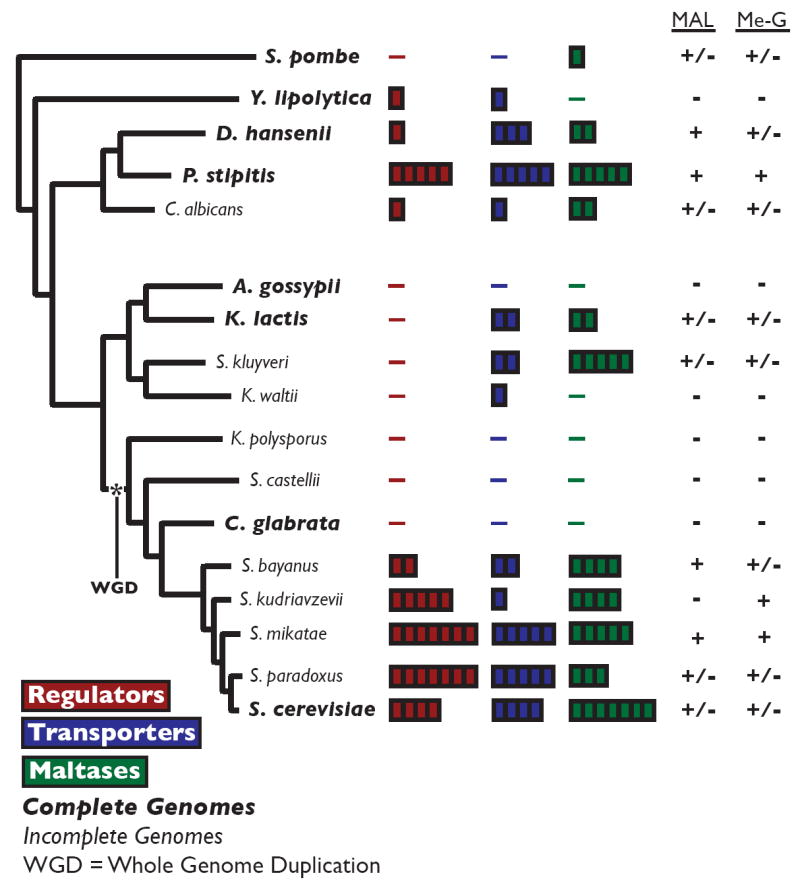
The MAL gene family shows extreme copy number variation in yeasts, and the presence of MAL genes correlates with the ability to grown on maltose and methyl-α-glucoside. The species names of genomes that have been completely sequenced and assembled are indicated in bold. The number of MAL regulator genes (red), MAL transporter genes (blue) and MAL maltase genes (green) is denoted by the number of blocks to the right of the species name. The panel on the right indicates whether (+) or not (-) individual strains grow on maltose (MAL) and methyl-α-glucoside (Me-G). Strain-dependent growth is denoted as +/-. See Figure S3 and Table S3 for more information.
Figure 4. Phylogeny of MAL genes in S. cerevisiae.
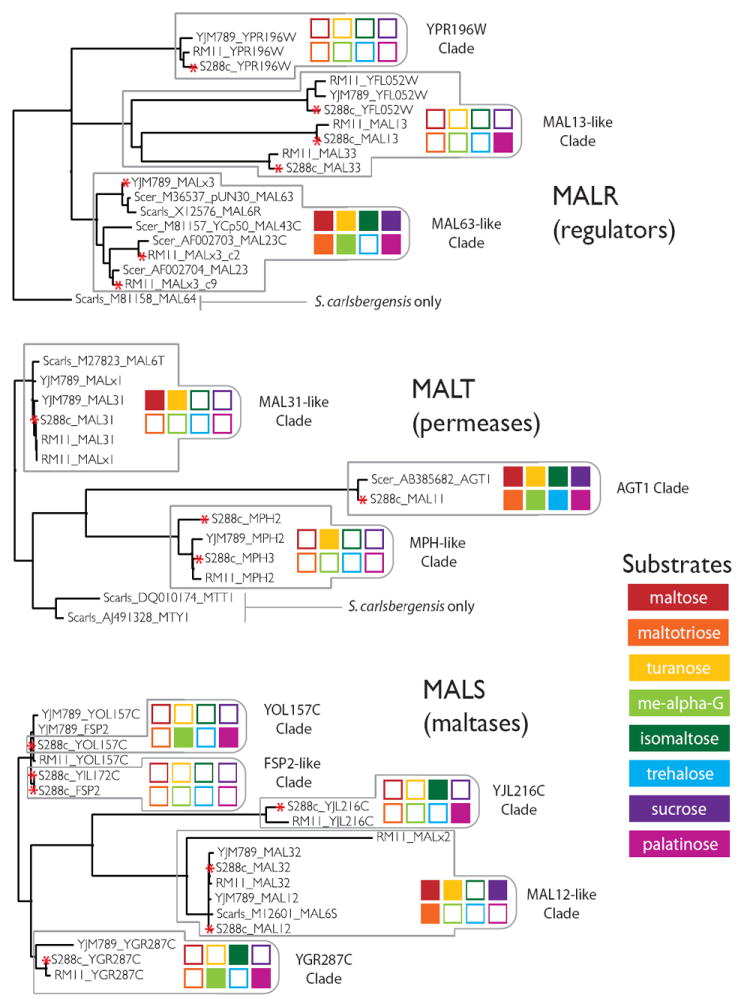
The MAL gene families sub-divide into tight subfamilies (clades) that correlate with specificity towards specific substrates. The phylogeny of the MAL regulators, transporters, and maltases, determined from multiple protein sequence alignments, is shown for alleles from Saccharomyces cerevisiae strains S288c, YJM789, and RM11, as well as alleles deposited in GenBank from Saccharomyces cerevisiae and Saccharomyces pastorianus. Individual subfamilies are outlined with grey boxes, in which specificity is denoted by a colored barcode. A red asterisk to the left of the allele name denotes that the function of this allele was experimentally investigated. The function of alleles that are not marked by an asterisk was not experimentally verified (in these cases, the function was only inferred from the sequence similarity with other alleles in the same clade). The specificity of the individual families was determined as follows. Combinatorial knockouts of MALR alleles in S288c (Figure 5C), knockins of MALR alleles from RM11 and YJM789 into S288c (Figure 5A & 5B), and combinatorial knockouts of MALR alleles in RM11 and YJM789 (Figure 5A), were used to determine MALR allele specificity. Overexpression of MALT alleles in S288c (Figure 6A & 6B) and knockouts of MALT alleles in S288c (Figure 5F) were used to determine MALT allele specificity. Combinatorial knockouts of MALS alleles in S288c (Figure 5D & 5E), overexpression of MALS alleles in S288c (Figure 6A & 6B), and purified enzyme assays of MalS proteins (Figure 6A & 6B), were used to determine MALS allele specificity. For more detailed information about assays, see Materials and Methods. Activity of a subfamily is summarized for maltose (red), maltotriose (orange), turanose (yellow), methyl-α-glucoside (lime green), isomaltose (green), trehalose (light blue), sucrose (purple), and palatinose (magenta). Activity towards a specific substrate is indicated by a solid colored square, while lack of activity for a specific substrate is depicted by white boxes with colored outlines. See Figure S4 for more information.
Further phylogenetic analysis revealed the existence of multiple subfamilies (“clades”) of the MALT, MALS, and MALR families that cluster tightly together based on their sequence similarity (Figure 4 and Figure S3). Genes within one subfamily do not only represent orthologs (i.e. copies that diverged independently after they were separated by speciation events; no gene duplication involved), but also recent paralogs (i.e. copies generated in duplication events within the species). Members of different subfamilies, on the other hand, show more sequence divergence and are usually ancient orthologs. Hence, the MAL genes show a remarkable instability in copy number and genomic location, even between evolutionary closely related S. cerevisiae strains. These characteristics of the different MAL genes agree very well with the results of our global in silico analysis of all subtelomeric genes (above). It is important to stress that we only based our analyses on the available fully sequenced yeast species. However, analysis of the MAL gene families in as many as 76 other (partly) sequenced S. cerevisiae and S. paradoxus strains confirm the trends observed in the fully assembled genomes (Table S3).
Functional divergence in the MAL gene families
Given the rapid expansion of the MAL gene families in S. cerevisiae, we asked whether the duplication events resulted in sub- or neofunctionalization. We screened three sequenced S. cerevisiae strains for their ability to grow on maltose and other related carbohydrates. Our systematic analysis extended previous work [41,43,44,45,46] and uncovered many novel functions for the different MAL genes. The laboratory strain, S288c, failed to grow on maltose while two feral isolates, RM11 (from a vineyard) and YJM789 (from an AIDS patient), both grew. Further analysis showed that this difference depends on the absence of one specific MALR subfamily (clade) from S288c (Figure 4). Expressing members of the MAL63-like subfamily (MAL63c9, MAL63c2 from RM11 and MALx3 from YJM789) in S288c restored growth on maltose. Conversely, deleting all members of this subfamily in strains RM11 and YJM789 abolished their capacity to ferment maltose (Figure 5A). Further growth assays show that these regulators are also required for growth on turanose, maltotriose, methyl-α-glucoside, isomaltose, palatinose and sucrose (Figure 5B and Figure S5).
Figure 5. Growth Data for MAL mutants.
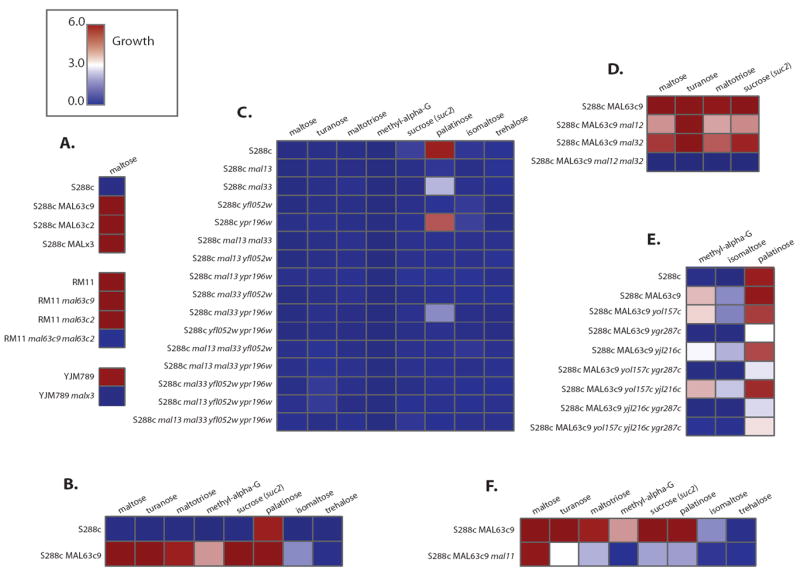
MAL deletion mutants confirm phenotypes and functional divergence. Growth of various MAL mutants is portrayed in a heatmap going from no growth (dark blue, 0.0) to strong growth (dark red, 6.0). A. S288c wild-type strain fails to grow on maltose while RM11 and YJM789 both grow on maltose. Transforming either a functional regulator from RM11 or YJM789 confers growth in S288c. Conversely, removing the functional regulator from YJM789, or both functional regulators from RM11 renders both strains unable to grow on maltose. B. The functional regulator from RM11, MAL63c9 (MAL63 found on supercontig 9 in RM11 (see Figure S5)), is not only required for growth on maltose, but is also required for growth on turanose, maltotriose, methy-α-glucoside, sucrose (suc2 mutant), palatinose, and isomaltose. C. All possible combinatorial knockouts of MAL regulators in S288c reveal that MAL13 and YFL052W are required for growth on palatinose, while the absence of MAL33 reduces growth on this carbon source. D. Two maltases, MALS genes, MAL12 and MAL32, are required for growth on maltose, turanose, maltotriose, and sucrose (the latter tested in a suc2 mutant), while the other MALS family members don’t affect the phenotype. E.YGR287C, a MALS gene, is the only MALS family member required for growth on palatinose, isomaltose, and methyl-α-glucoside. F. Removal of MAL11 permease renders strains unable to grow on most α-glucosides. See Figure S5 for more information.
A phylogeny of the MALR proteins shows three distinct subgroups of regulators. The previous results show that one MALR clade is vital for the consumption of many α-glucosides. Why then are the other regulator clades maintained? Further tests revealed that other regulators (YFL052W, MAL13, MAL33 in MAL13-like clade) evolved specificity for palatinose, a disaccharide naturally occurring in sugar cane and honey (Figure 4 and Figure S5C). Together, these results indicate that the function of the different MALR paralogs have diverged to regulate cellular metabolism in response to various distinct carbohydrates.
Next, we asked if the MALT and MALS families show similar sub-/neofunctionalization towards different carbohydrates. Phylogenetic analysis revealed three distinct MALT clades and five MALS clades (Figure 4). To investigate the specificity of individual transporters and maltases, we created a yeast strain without active MALR genes (so that all MALS and MALT genes are silent). Using this strain, we constructed yeast mutants that constitutively express different combinations of one MALT and one MALS gene each and tested their growth on a series of carbohydrates (Figure S6). Certain combinations of MALS/MALT pairs allowed growth on specific sugars. For example, expressing MAL11 (MALT family member) in combination with MAL12 or MAL32 (MALS family members) allows S288c to grow on maltotriose, while expressing MAL11 in combination with YOL157C or YGR287C (MALS family members) allows growth on methyl-α-glucoside (Figure 6A). Together, the tests indicate that the different MALT and MALS subgroups allow import and hydrolysis of specific α-glucosides. Some clades encode proteins with broad substrate specificity (e.g. MAL11 member of the MALT family), while others are more specific (e.g. YOL157C member of the MALS family)(Figure 4, Figure 5D-F, and Figure 6).
Figure 6. Growth & Enzyme Assays.
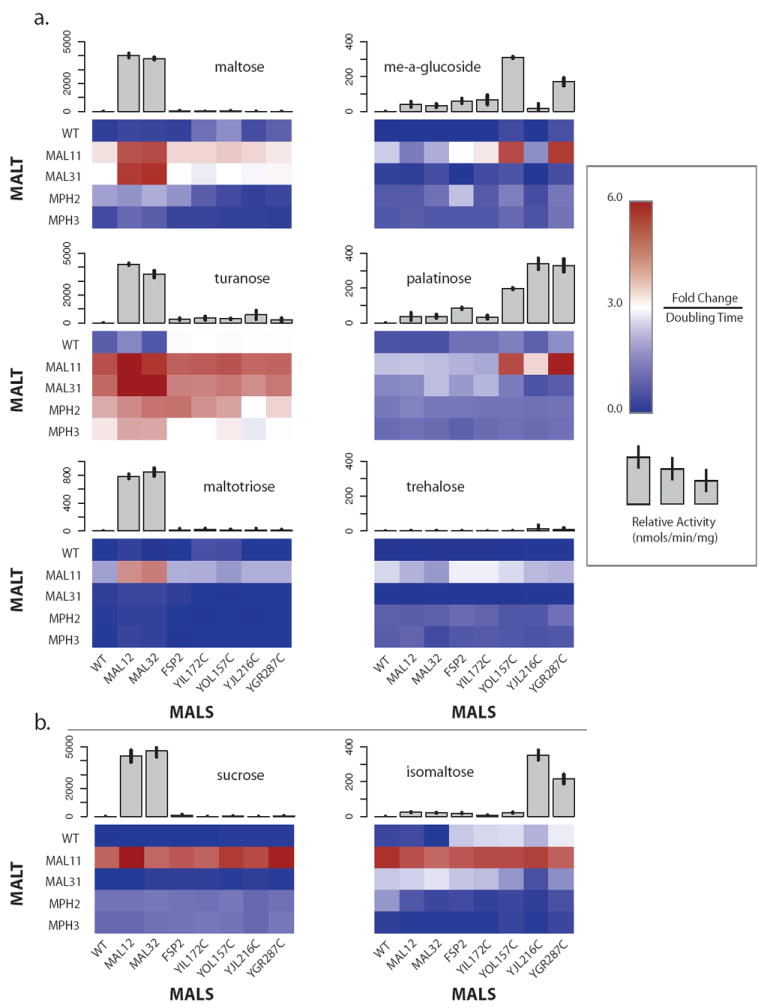
Growth and enzyme assays of MAL overexpression mutants show the functional divergence in the MAL families. Growth in various α-glucosides of S. cerevisiae S288c diploids resulting from a cross of a MALT (rows) overexpression strain and a MALS (columns) overexpression strain is shown as a heatmap. It is important to note that the upper left entry in the heatmap is a WT S288c diploid that is a control for growth. For all of the sugars except palatinose and sucrose, the genotype of the diploids is S288c, with exception of the MALS and MALT modifications. For palatinose, S288c mal13/mal13 diploids were used, while for sucrose S288c suc2/suc2 diploids were used. Relative activity in units of nmols/min/mg of purified MALS proteins is shown as grey bar graphs above the respective MALS column in the heatmap. Individual family members are denoted beside their row or column in the heatmap. A. Growth of the diploids in maltose, maltotriose, methyl-α-glucoside, palatinose, trehalose, and turanose correlates well with the relative activity of the purified enzymes, and implicates specific combinations of MALT and MALS alleles. B. Growth of the diploids in sucrose and isomaltose is dependent on the MAL11 family member of MALT and no specific MALS family members, while the enzyme assays indicate specific MALS proteins. This is most likely due to MALR alleles responding to the imported sugars and upregulating the pertinent MALS alleles once the native regulation of the MAL11 allele of MALT has been bypassed. Deletion of the putative MALS alleles indicated by the enzyme assays confirms their specificity for sucrose (Figure 5D) and isomaltose (Figure 5E). See Figure S6 for more information.
To further confirm the substrate specificity of the MALS family members, we purified all seven maltase proteins (Mal12p, Mal32p, Fsp2p, Yil172cp, Yol157cp, Yjl216cp, and Ygr287cp) from S. cerevisiae S288c, and measured their ability to hydrolyze different α-glucosides. The results confirm that MAL12-like clade genes (e.g. Mal12p and Mal32p) (Figure 4) have evolved specificity for maltose, maltotriose, turanose and sucrose (Figure 5D, and Figure 6A), while other clades (YOL157C, YJL216C, and YGR287C) (Figure 4) have evolved specificity for other carbohydrates, such as palatinose, isomaltose, and methyl-α-glucoside (Figure 5E and Figure 6). These results agree perfectly with the previous assays in which genes were deleted or overexpressed.
Discussion
Our results uncover the extraordinary dynamics of subtelomeric gene families. Genes residing near the telomeres undergo frequent recombination and duplication, which may allow evolutionary adaptation and innovation. Detailed analysis of three gene families that were historically linked with maltose metabolism confirms our genome-wide in silico analysis. In some yeasts, the MAL genes have completely disappeared, while in others, they show multiple recent duplication events. Moreover, the evolutionary rate at which these changes have taken place is exceptional, with wide differences in copy number within closely related species of the Saccharomyces sensu stricto group (Fig. 2) and even within one species (Fig. S4). Moreover, the various MAL loci reveal a surprisingly broad activity, with certain previously unidentified new family members showing no activity towards maltose, but instead degrading several other α-glucosides. Because of the remarkable evolutionary rate of these gene families, it is difficult to predict the specificity of the ancestral enzymes, and it remains a future direction to determine the extent that neofunctionalization and subfunctionalization have shaped their evolution.
It is interesting to hypothesize about the origins of variability of subtelomeric genes and gene families. In our analysis, we noted specific functional categories of genes that are enriched in subtelomeres. Non-subtelomeric genes that belong to the same functional categories do not show a similar variability, suggesting that the rapid turnover of subtelomeric genes is an inherent property of these regions, and not of the functional categories of genes. It is difficult to deduce whether certain rapidly evolving genes are adaptively relocated to subtelomeres, or whether genes are relocated purely randomly to the telomeres, which results in rapid evolution. While our results do not allow us to differentiate between these two scenarios, we hypothesize that both scenarios may be true. Genes may be relocated to the telomeres more or less randomly, but only those genes for which the local elevated dynamics are associated with a selective advantage will be retained in the subtelomeres.
Since most subtelomeric gene families are involved in niche-specific processes, including carbohydrate metabolism [18,19,21,41,47], stress response and cell surface properties [22,23,48], it is tempting to speculate that their “evolvability” allows rapid adaptation to novel niches and population structures [49,50]. In the case of the MAL genes, expansion of the gene families in Saccharomyces sensu stricto may have allowed metabolism of carbohydrates found in plants and fruits, while further selection by brewers has probably led to the other observed expansions [51]. Table S5 shows a significant amplification in S. paradoxus of specific MAL alleles involved in metabolism of sucrose, palatinose and other sugars found in tree sap and honey, from which S. paradoxus is often isolated [16,52,53]. In yeasts that colonize mammals, such as Candida spp., the MAL genes were completely lost, presumably because these yeasts encounter enough simple, preferred sugars present in blood and the digestive tract. Similarly, expansion of subtelomeric gene families may have supported an elegant immune evasion system in pathogens, while the contraction of olfactory receptors in humans may explain our inability to detect certain smells [9,26]. Interestingly, a recent study in which yeast cells were evolved under nutrient-limited continuous cultures of Saccharomyces cerevisiae identifies frequent duplications of SUL1, a subtelomeric gene encoding a sulfate permease located near a MAL locus on the right arm of chromosome II [54].
Recent studies have noted the importance of whole-genome duplication events for evolutionary innovation [17,42,55,56]. While these duplication events are rare, our results indicate that small-scale duplication events in the subtelomeric regions may also serve an important evolutionary role. Whereas the number of subtelomeric genes is much smaller than all genes involved in whole-genome duplication events, innovation at subtelomeres is a continuous process rather than a rare event. Furthermore, subtelomeric specific epigenetic effects, including chromatin-dependent silencing, may further add to the evolutionary potential of these interesting regions, for example, by allowing swift divergence of the transcriptional regulation of the duplicated copies, a crucial but often overlooked process in evolution [57].
Experimental Procedures
Microbial strains, growth conditions, molecular techniques
All yeast strains used and all oligonucleotides (Sigma-Genosys and IDT) used are listed in Supplemental Experimental Procedures. Yeast cultures were grown as described before (Sherman et al., 1991). All strains were grown in rich media consisting of 2% peptone (Difco), 1% yeast extract (Difco), and 2% sugar (Sigma-Aldrich). All strains were grown overnight for ~16 hours in 3 mL YP with 2% glucose at 30°C in a rotating wheel unless otherwise noted. Plated cultures were grown for three days at 30°C. The sugars used in this study were purchased to their highest available purity and were filter-sterilized before adding to rich media. Plasmid sets were obtained from EUROSCARF (http://web.uni-frankfurt.de/fb15/mikro/euroscarf/) for reusable markers (Deletion Marker Plasmids) and overexpression/epitope tagging (A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes). Plasmids were used as indicated in the references for use (PCR reaction mixture, primer design, etc.) found at EUROSCARF. Standard cloning and molecular biology procedures were used (Sambrook et. al. 1989).
Growth assays
Growth assays were preformed in a BioScreen C MBR system (Oy Growth Curves Ab Ltd.). Overnight cultures were then diluted 1:50 into YP without sugar. A final dilution of 1:10 into YP with 2% sugar was made into the BioScreen C plates. Cells were grown in duplicate for 48 hours at 30°C with continuous shaking, during which the optical density at 600nm (OD600) was collected every 15 minutes. After 48 hours, the machine was stopped and the doubling time and fold change (OD600final/OD600initial) were calculated for each strain using in house software. Growth was then expressed as fold change normalized by doubling time.
Protein purification
Yeast strains carrying pGPD-3xHA-MalS proteins (strains KV2325 – KV2331 in Supplemental Experimental Procedures) as well as a wildtype strain (KV447) were inoculated into one liter of YP with 2% glucose from overnight cultures to a starting OD600 of 0.1. When the cultures reached an OD600 of 0.8, the cells were pelleted and washed twice with cold water. The pellets were then frozen at -80°C for later processing. The frozen pellets were subsequently resuspended in cold water and split between 10 screw-top Eppendorf tubes. After spinning down and removing the supernatant, the pellets were mixed with 300 μL of lysis buffer with protease inhibitor (50 mM Tris-HCl, pH 7.6, 25 mM CaCl2, 5 mM MgCl2, 1 mM EDTA, 5% glycerol, 250 mM KCl, 0.5 mL protease inhibitor cocktail (Sigma)) and 150 μL of acid-washed glass beads. The mixture was bead-beat in a Fastprep-24 (MP Biomedicals) for 30 seconds, and then spun-down at 10,000 rpm for 15 minutes. After removing the crude extract for immunoprecipitation, an additional 300 μL of lysis buffer with protease inhibitors was added to the bead mixture, bead-beat, and pooled. The crude extract was mixed with 200 μL of EZ-view Red Anti-HA Affinity Gel (Sigma) that had been equilibrated with lysis buffer with protease inhibitor. The extract/affinity gel mixture was incubated and gently inverted at 4°C for 3 hours on a Roto-Shake Genie (Scientific Industries), after which the mixture was spun-down at 10,000 rpm. The supernatant was removed and the bound affinity gel was washed three times with 1 mL aliquots of lysis buffer without protease inhibitor. The bound 3xHA-MalS proteins were eluted twice with 500 μL aliquots of lysis buffer without protease inhibitors + 100 μg/mL HA peptide (Sigma), and the two aliquots were pooled. The elution was carried out at 37°C for 15 minutes with simple inversion. To check the quality of the immunoprecipitations in addition to quantifying the amount of purified protein, 10 μL of the elutions were added to 4 μL of 4x SDS sample buffer (Novagen) and 2 μL of water, and incubated at 95°C for 10 minutes. The samples were then loaded alongside a dilution series of a BSA standard (Fluka) and a 6-200 kDa protein ladder (AppliChem) on a NuPage Novex Bis-Tris Mini Gel (Invitrogen) and run 1h at 200V. The gel was washed three times with water, stained with SimplyBlue Safe Stain (Invitrogen), and destained for 2 hours.
Enzyme assays
The relative activities of the MalS proteins were determined by measuring glucose release using a GOD-PAP kit (Dialab). A standard curve was constructed for glucose using available standards (Sigma). The amount of glucose measured in the assay can be used to determine the amount of substrate hydrolyzed by normalizing for the number of glucose moieties in the substrate. Reaction mixtures consisted of 3 μL of purified protein in 27 μL of a 100 mM phosphate buffer at pH 6.8 with 0.5 mg/mL BSA and 500 mM of a given sugar. The reactions were incubated anywhere from 1 – 6 hours at 30°C, heat-killed at 98°C for 2 minutes, and then assayed at 500 nm on a plate reader. The mean and standard deviation of the relative activity (nmols of product hydrolyzed/min/mg protein) was calculated from three independent reactions. Initially, we attempted to measure Km and Vmax for the sugars, but found that for the sugars with weaker activity (methyl-α-glucoside, isomaltose, and palatinose) our measurements were very noisy, making it intractable to compute an accurate Km and Vmax with our experimental setup and available sugars.
Computational datasets
The proteomes of the eight completely sequenced ascomycetes (Saccharomyces cerevisiae S288c, Candida glabrata CBS138, Kluyveromyces lactis NRRL Y-1140, Ashbya gossypii ATCC 10895, Pichia stipitis CBS 6054, Debaryomyces hansenii CBS767, Yarrowia lipolytica CLIB122, and Schizosaccharomyces pombe 972h-) were downloaded from NCBI (www.ncbi.nlm.nih.gov/). The coordinates of the genes encoding each protein along with the respective chromosome length were used to determine relative coordinates (distance to nearest chromosome end) for each protein product (i.e. relative coordinate = min(coordinate, chromosome length – coordinate)). This relative coordinate was then used to determine whether a gene is subtelomeric or not based off of a cutoff (e.g. 30 kb). A subtelomeric cutoff based of off gene density was determined as follows (see Figure S1). A sliding window (varied from 50 – 70 kb to control for artifacts from the sliding window size) initially placed at the chromosome ends was moved towards the middle of the chromosomes. The density of genes (utilizing the relative coordinate from above) within that window was compared to a uniform density of equal sample size by calculating the Kolmogorov-Smirnov distance (KS-distance). This was repeated 500 times to obtain a mean and standard deviation of KS-distance. A baseline to control for finite sampling effects was constructed by comparing a uniform density to itself and then calculating the KS-distance (500 times with same number of points as before). The sliding window was moved incrementally (1 kb step size) from chromosome end to chromosome middle and the real KS-distance distribution was compared to the baseline KS-distance distribution using a two-sample Mann-Whitney test. The subtelomeric cutoff was determined as the first coordinate at which the p-value increased above 0.05. This process was repeated ~100 times and a subtelomeric cutoff of 33 kb was found. Although we use this cutoff for most of our analyses in the current manuscript, we altered this cutoff (see Table S2) and our results remain unaltered. After dividing genes into two groups (subtelomeric vs. nonsubtelomeric based on subtelomeric cutoff) we calculated GO term enrichment and depletion for individual GO terms by comparing the number of genes in each group using Fisher’s exact test. The p-values were corrected using the Bonferroni correction for multiple hypothesis testing. In order to control for individual GO categories in our enrichment calculation, we compared the copy number variation and size of non-subtelomeric vs. subtelomeric families for a given GO category that was significantly enriched in subtelomeres (p-value < 0.05). The statistical significance of the increased copy number variation and family size of subtelomeric gene families compared to non-subtelomeric gene families was determined with a Kolmogorov-Smirnov test with Bonferroni correction. Expression divergence and responsiveness of S. cerevisiae genes was taken from [40]. The distributions of expression divergence and responsiveness were compared, using a Kolmogorov-Smirnov test, between subtelomeric gene families and non-subtelomeric gene families.
Gene family statistics
Gene families were determined by using the Markov Cluster Algorithm (MCL) (www.micans.org/mcl/). Although we use an inflation parameter of 2 for most of our analyses, the MCL inflation parameter was varied (1.5 – 5) and our results remain unaltered (see Table S2). These gene families were then used for all of our analyses. To determine whether subtelomeric gene families (by definition contain at least one subtelomeric gene) contained more subtelomeric genes than expected, for a given species, we randomly placed subtelomeric and non-subtelomeric genes into families utilizing the empirical family size distribution. After randomly assigning genes to families we counted the number of families that contained at least one subtelomeric gene. After repeating this 10000 times, we compared the randomized distribution to the actual number of subtelomeric gene families with a z-test. To further control for tandem/local duplications that could influence this calculation, we removed all local duplicates that were within 10 kb of a gene, for each gene, and repeated the randomization above; this had a negligible effect on our original signal. To determine whether subtelomeric gene families are larger than non-subtelomeric gene families, we randomly selected gene families (same number as the observed number of subtelomeric gene families) from the empirical family size distribution within a species, and calculated the mean size of the family. This was repeated 10000 times and compared to the mean size of subtelomeric gene families within a species using a z-test. Similarly, the distribution of subtelomeric gene family size within a species was compared to the distribution of gene family sizes within a species using a Kolmogorov-Smirnov test. To determine whether subtelomeric gene families show more copy number variation between species than non-subtelomeric gene families, we calculated the coefficient of variation (standard deviation / mean) of gene family copy number in families with at least two species. The coefficient variation controls for the fact that subtelomeric gene families are larger than non-subtelomeric families by normalizing by the mean. If a species didn’t have a copy of a gene, then it wasn’t considered in the calculation of the coefficient of variation; this prevents the mean from dominating and makes it strictly greater than or equal to one. The distribution of coefficients of variation for subtelomeric and non-subtelomeric gene families were then compared using a Kolmogorov-Smirnov test. To compare the similarity of protein sequences in subtelomeric families versus nonsubtelomeric families, multiple sequence alignments of protein families were made using MUSCLE (Edgar 2004) with default parameters for families that contained at least two different species. The protdist package of PHYLIP by Felsenstein (http://evolution.genetics.washingtion.edu/phylip/). with JTT model was used to calculate the protein distances. Intraspecies protein distances were then pooled for subtelomeric gene families and non-subtelomeric gene families, respectively. The distributions were compared using the Kolmogorov-Smirnov test. The CAFE package (Hahn 2005) was used to compare the evolutionary rate of subtelomeric gene families to non-subtelomeric gene families. A high confidence (bootstrap values > 96%) species tree phylogeny was constructed by concatenating proteins from 75 different families, which were single copy in all species. Distances were calculated assuming a molecular clock using the kitsch package of PHYLIP by Felsenstein (http://evolution.genetics.washingtion.edu/phylip/), and the tree lengths in millions of years (Mya) were calibrated using a divergence of 1140 Mya between S. cerevisiae and S. pombe (Hedges 2002). For the single birth-death parameter model, the maximum likelihood routine was used to first determine a birth-death parameter (lambda) for each family, and p-values were then calculated using this lambda. For the two-parameter model, the same approach was taken except that a second parameter was assigned to the branch undergoing the largest gain or loss (i.e. largest difference from the mean observed copy number). The statistical significance of enrichment of subtelomeric genes in the top 10% of genes ranked by p-value was determined using Fisher’s exact test. The statistical significance of the difference between subtelomeric and non-subtelomeric lambda distributions was calculated using the Kolmogorov-Smirnov test. To investigate whether our single species samples represented larger population averages, we used the recently sequenced S. cerevisiae and S. paradoxus strains from the Sanger Institute [16]. We determined the total MALR, MALT, and MALS alleles as well as clade specific alleles for both species. Due to low sequence coverage, some alleles contained long stretches of undetermined bases, so they were not included in the analysis, although this represented only a small fraction of the total MALR, MALT, and MALS alleles (less than 5% per species per family).
Supplementary Material
Acknowledgments
The authors thank Verstrepen Lab members, G.R. Fink, E.K. O’Shea, C. Michels, M. Hahn, S. Maere, and B. Stern for their valuable advice. CAB acknowledges financial support from the NSF, Harvard University HILS, and a Harvard Sheldon Fellowship. Research in the lab of KJV is supported by NIH grant P50GM068763, Human Frontier Science Program HFSP RGY79/2007, ERC Young Investigator Grant 241426, VIB, K.U.Leuven, the FWO-Odysseus program and the AB InBev Baillet-Latour foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pryde FE, Gorham HC, Louis EJ. Chromosome ends: all the same under their caps. Curr Opin Genet Dev. 1997;7:822–828. doi: 10.1016/s0959-437x(97)80046-9. [DOI] [PubMed] [Google Scholar]
- 2.Cohn M, Liti G, Barton DBH. Telomeres in fungi. Top Curr Genet. 2006;15:101–130. [Google Scholar]
- 3.Mefford H, Trask B. The complex structure and dynamic evolution of human subtelomeres. Nat Rev Genet. 2002;3:91–102. doi: 10.1038/nrg727. [DOI] [PubMed] [Google Scholar]
- 4.Pryde F, Louis E. Limitations of silencing at native yeast telomeres. EMBO J. 1999;18:2538–2550. doi: 10.1093/emboj/18.9.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell. 1990;63:751–762. doi: 10.1016/0092-8674(90)90141-z. [DOI] [PubMed] [Google Scholar]
- 6.Friedman C, Parghi SS, Linardopoulou EV, Hsu L. Elevated rates of sister chromatid exchange at chromosome ends. PLoS Genet. 2007;3:319–323. doi: 10.1371/journal.pgen.0030032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linardopoulou E, Williams EM, Fan Y, Friedman C, Young J, et al. Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature. 2005;437:94–100. doi: 10.1038/nature04029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barton AB, Pekosz M, Kurvathi RS, Kaback DB. Meiotic recombination at chromosome ends in Saccharomyces cerevisiae. Genetics. 2008;179:1221–1235. doi: 10.1534/genetics.107.083493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mefford HC, Linardopoulou E, Coil D, van den Engh G, Trask BJ. Comparative sequencing of a multicopy subtelomeric region containing olfactory receptor genes reveals multiple interactions between non-homologous chromosomes. Hum Mol Genet. 2001;10:2363–2372. doi: 10.1093/hmg/10.21.2363. [DOI] [PubMed] [Google Scholar]
- 10.Anderson JA, Song YS, Langley CH. Molecular population genetics of Drosophila subtelomeric DNA. Genetics. 2008;178:477–487. doi: 10.1534/genetics.107.083196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kern AD, Begun DJ. Recurrent deletion and gene presence/absence polymorphism: telomere dynamics dominate evolution at the tip of 3L in Drosophila melanogaster and D. simulans. Genetics. 2008;179:1021–1027. doi: 10.1534/genetics.107.078345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Britten RJ. Precise sequence complementarity between yeast chromosome ends and two classes of just-subtelomeric sequences. Proc Natl Acad Sci. 1998;95:5906–5912. doi: 10.1073/pnas.95.11.5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eichler EE. Segmental duplications: what’s missing, misassigned, and misassembled--and should we care? Genome Res. 2001;11:653–656. doi: 10.1101/gr.188901. [DOI] [PubMed] [Google Scholar]
- 14.Kellis M, Patterson N, Endrizzi M, Birren B, Lander E. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- 15.Hahn M, De Bie T, Stajich JE, Nguyen C, Cristianini N. Estimating the tempo and mode of gene family evolution from comparative genomic data. Genome Res. 2005;15:1153–1160. doi: 10.1101/gr.3567505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liti G, Carter D, Moses A, Warringer J, Parts L, et al. Population genomics of domestic and wild yeasts. Nature. 2009;458:337–341. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wapinski I, Pfeffer A, Friedman N, Regev A. Natural history and evolutionary principles of gene duplication in fungi. Nature. 2007;449:54–61. doi: 10.1038/nature06107. [DOI] [PubMed] [Google Scholar]
- 18.Carlson M, Celenza J, Eng F. Evolution of the dispersed SUC gene family of Saccharomyces by rearrangements of chromosome telomeres. Mol Cell Biol. 1985;5:2894–2902. doi: 10.1128/mcb.5.11.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denayrolles M, de Villechenon E, Lonvaud-Funel A, Aigle M. Incidence of SUC-RTM telomeric repeated genes in brewing and wild wine strains of Saccharomyces. Curr Genet. 1997;31:457–461. doi: 10.1007/s002940050230. [DOI] [PubMed] [Google Scholar]
- 20.Michels C, Needleman R. The dispersed, repeated family of MAL loci in Saccharomyces spp. J Bacteriol. 1984;157:949–952. doi: 10.1128/jb.157.3.949-952.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naumov G, Turakainen H, Naumova E, Aho S, Korhola M. A new family of polymorphic genes in Saccharomyces cerevisiae: α-galactosidase genes MEL1-MEL7. Mol Genet Genomics. 1990;224:119–128. doi: 10.1007/BF00259458. [DOI] [PubMed] [Google Scholar]
- 22.Teunissen A, Steensma H. The dominant flocculation genes of Saccharomyces cerevisiae constitute a new subtelomeric gene family. Yeast. 1995;11:1001–1015. doi: 10.1002/yea.320111102. [DOI] [PubMed] [Google Scholar]
- 23.Verstrepen K, Klis F. Flocculation, adhesion and biofilm formation in yeasts. Mol Micro. 2006;60:5–15. doi: 10.1111/j.1365-2958.2006.05072.x. [DOI] [PubMed] [Google Scholar]
- 24.Verstrepen KJ, Fink GR. Genetic and epigenetic mechanisms underlying cell-surface variability in protozoa and fungi. Annu Rev Genet. 2009;43:1–24. doi: 10.1146/annurev-genet-102108-134156. [DOI] [PubMed] [Google Scholar]
- 25.Barry J, Ginger M, Burton P, McCulloch R. Why are parasite contingency genes often associated with telomeres? Int J Parasit. 2003;33:29–45. doi: 10.1016/s0020-7519(02)00247-3. [DOI] [PubMed] [Google Scholar]
- 26.Hasin Y, Olender T, Khen M, Gonzaga-Jauregui C, Kim P, et al. Highresolution copy-number variation map reflects human olfactory receptor diversity and evolution. PLoS Genet. 2008;4:e1000249. doi: 10.1371/journal.pgen.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linardopoulou EV, Parghi SS, Friedman C, Osborn GE, Parkhurst SM, et al. Human subtelomeric WASH genes encode a new subclass of the WASP family. PLoS Genet. 2007;3:2477–2485. doi: 10.1371/journal.pgen.0030237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knight SJL, Flint J. Perfect endings: a review of subtelomeric probes and their use in clinical diagnosis. J Med Genet. 2000;37:401–409. doi: 10.1136/jmg.37.6.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bailey J, Eichler E. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006;7:552–564. doi: 10.1038/nrg1895. [DOI] [PubMed] [Google Scholar]
- 30.Fairhead C, Dujon B. Structure of Kluyveromyces lactis subtelomeres: duplications and gene content. FEMS Yeast Res. 2006;6:428–441. doi: 10.1111/j.1567-1364.2006.00033.x. [DOI] [PubMed] [Google Scholar]
- 31.Gordon J, Byrne K, Wolfe K. Additions, losses, and rearrangements on the evolutionary route from a reconstructed ancestor to the modern Saccharomyces cerevisiae genome. PLoS Genetics. 2009;5:e1000485. doi: 10.1371/journal.pgen.1000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Louis E. The chromosome ends of Saccharomyces cerevisiae. Yeast. 1995;11:1553–1573. doi: 10.1002/yea.320111604. [DOI] [PubMed] [Google Scholar]
- 33.Winzeler EA, Castillo-Davis CI, Oshiro G, Liang D, Richards DR, et al. Genetic diversity in yeast assessed with whole-genome oligonucleotide arrays. Genetics. 2003;163:79–89. doi: 10.1093/genetics/163.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ashburner M, Ball C, Blake J, Botstein D, Butler H, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Enright A, Dongen SV, Ouzounis C. An efficient algorithm for large-scale detection of protein families. Nuc Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Dongen S. Graph clustering via a discrete uncoupling process. SIAM J Matrix Anal Appl. 2008;30:121–141. [Google Scholar]
- 37.Schober H, Kalck V, Vega-Palas M, Van Houwe G, Sage D, et al. Controlled exchange of chromosomal arms reveals principles driving telomere interactions in yeast. Genome Res. 2008;18:261–271. doi: 10.1101/gr.6687808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Force A, Lynch M, Pickett F, Amores A, Yan Y, et al. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohno S. Evolution by Gene Duplication. New York: Springer; 1970. [Google Scholar]
- 40.Tirosh I, Weinberger A, Carmi M, Barkai N. A genetic signature of interspecies variations in gene expression. Nat Genet. 2006;38:830–834. doi: 10.1038/ng1819. [DOI] [PubMed] [Google Scholar]
- 41.Charron MJ, Read E, Haut SR, Michels CA. Molecular evolution of the telomere-associated MAL loci of Saccharomyces. Genetics. 1989;122:307–316. doi: 10.1093/genetics/122.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kellis M, Birren BW, Lander ES. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 2004;428:617–624. doi: 10.1038/nature02424. [DOI] [PubMed] [Google Scholar]
- 43.Han EK, Cotty F, Sottas C, Jiang H, Michels CA. Characterization of AGT1 encoding a general α-glucoside transporter from Saccharomyces. Mol Microbiol. 1995;17:1093–1107. doi: 10.1111/j.1365-2958.1995.mmi_17061093.x. [DOI] [PubMed] [Google Scholar]
- 44.Cheng Q, Michels CA. MAL11 and MAL61 encode the inducible highaffinity maltose transporter of Saccharomyces cerevisiae. J Bacteriol. 1991;173:1817–1820. doi: 10.1128/jb.173.5.1817-1820.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jespersen L, Cesar LB, Meaden PG, Jakobsen M. Multiple alpha-glucoside transporter genes in brewer’s yeast. Appl Environ Micro. 1999;65:450–456. doi: 10.1128/aem.65.2.450-456.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Michels CA, Needleman RB. A genetic and physical analysis of the MAL1 and MAL3 standard strains of Saccharomyces cerevisiae. Mol Genet Genomics. 1983;191:225–230. [Google Scholar]
- 47.Ness F, Aigle M. RTM1: A member of a new family of telomeric repeated genes in yeast. Genetics. 1995;140:945–956. doi: 10.1093/genetics/140.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rachidi N, Martinez MJ, Barre P, Blondin B. Saccharomyces cerevisiae PAU genes are induced by anaerobiosis. Mol Microbiol. 2000;35:1421–1430. doi: 10.1046/j.1365-2958.2000.01807.x. [DOI] [PubMed] [Google Scholar]
- 49.Gore J, Youk H, Oudenaarden Av. Snowdrift game dynamics and facultative cheating in yeast. Nature. 2009;459:253–256. doi: 10.1038/nature07921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rando OJ, Verstrepen KJ. Timescales of genetic and epigenetic inheritance. Cell. 2007;128:655–668. doi: 10.1016/j.cell.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 51.Dunn B, Sherlock G. Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res. 2008;18:1610–1623. doi: 10.1101/gr.076075.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de la Fuente E, Sanz ML, Martinez I, Sanz J, Ruiz-Matute AI. Volatile and carbohydrate composition of rare unifloral honeys from Spain. Food Chem. 2007;105:84–93. [Google Scholar]
- 53.Heizmann U, Kreuzwieser J, Schnitzler J-P, Brueggemann N, Rennenberg H. Assimilate transport in the xylem sap of pedunculate oak (Quercus robur) saplings. Plant Biol. 2001;3:132–138. [Google Scholar]
- 54.Gresham D, Desai M, Tucker C, Jenq H, Pai D, et al. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet. 2008;4:e1000303. doi: 10.1371/journal.pgen.1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fawcett JA, Maere S, Van de Peer Y. Plants with double genomes might have had a better chance to survive the Cretaceous--Tertiary extinction event. Proc Natl Acad Sci. 2009;106:5737–5742. doi: 10.1073/pnas.0900906106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scannell DR, Wolfe KH. A burst of protein sequence evolution and a prolonged period of asymmetric evolution follow gene duplication in yeast. Genome Res. 2008;18:137–147. doi: 10.1101/gr.6341207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tirosh I, Barkai N, Verstrepen K. Promoter architecture and the evolvability of gene expression. J Biol. 2009;8:95.91–95.96. doi: 10.1186/jbiol204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.