

Summary
Objective
Normal cartilage is resistant to vascular invasion and anti-angiogenic protease inhibitors may contribute to its avascular status. We hypothesized that dysregulated expression of four key anti-angiogenic protease inhibitors may contribute to increased osteochondral vascularity in osteoarthritis (OA).
Design
Medial tibial plateaux from OA patients (n = 40) were compared with those from non-arthritic controls collected post-mortem (PM, n = 10). Immunohistochemistry was performed for protease inhibitors TIMP-1, TIMP-3, PAI-1 and SLPI and the pro-angiogenic factor vascular endothelial growth factor (VEGF). Immunoreactivity in articular chondrocytes was scored. Chondropathy was measured as a modified Mankin score, and osteochondral vascular density as number of channels crossing each mm of tidemark. Non-parametric analyses were used for all data.
Results
All protease inhibitors and VEGF were localised to chondrocytes near the articular surface, less often in the middle zone, and rarely to deep chondrocytes. Scores for VEGF, TIMP-1, TIMP-3, SLPI and PAI-1 were all increased in OA compared with PM, and higher scores were associated with greater chondropathy. Chondrocyte expression of VEGF was associated with higher osteochondral vascular density (r = 0.32, P < 0.05), whereas protease inhibitors were not.
Conclusions
The resistance of normal articular cartilage to vascular invasion may be more due to its matrix environment than ongoing protease inhibitor expression. Upregulation of protease inhibitors by superficial chondrocytes in OA may moderate the angiogenic effects of growth factors such as VEGF. However, failure of deep chondrocytes to express anti-angiogenic protease inhibitors may permit vascular invasion into the articular cartilage.
Keywords: TIMP, Protease, Angiogenesis, Cartilage, Osteoarthritis
Abbreviations: VEGF, vascular endothelial growth factor; EDTA, ethylenediaminetetraacetic acid; ABC, avidin–biotin complex; DAPI, 4′-6′-diamidino-2-phenylindole; TIMP, tissue inhibitor of metalloproteinase; PAI-1, plasminogen activator inhibitor-1; SLPI, secretory leukocyte proteinase inhibitor; PM, post-mortem
Introduction
Osteoarthritis (OA) is one of the leading causes of pain and disability in aging populations. Current treatments improve symptoms, but joint replacement surgery is still often required in late-stage disease. Angiogenesis, the growth of new blood vessels from pre-existing ones1, is characteristic of OA. New blood vessels from the subchondral bone, breach the tidemark into the usually avascular cartilage2. This osteochondral angiogenesis may potentiate joint damage by stimulating ossification in the articular cartilage3–7. Angiogenesis also results in innervation of articular cartilage3, providing a possible source of pain in OA. We, and others, have previous reported the range of osteochondral angiogenesis3,4,7 but the cause of this heterogeneity is not known.
The extent of angiogenesis results from a balance between actions of angiogenic and anti-angiogenic factors. Angiogenic factors such as vascular endothelial growth factor (VEGF) are upregulated by chondrocytes8, and in subchondral bone in OA9. Normal articular cartilage resists vascular invasion due to its matrix composition, and also by producing diffusible angiogenesis inhibitors10. Matrix degradation is required to permit blood vessels to penetrate cartilage, and protease producing cells line vascular channels at the osteochondral junction11. Protease inhibitors reduce angiogenesis by inhibiting this extracellular matrix degradation, and the production by articular cartilage of tissue inhibitors of metalloproteinases (TIMPs) is believed to contribute to its resistance to vascular invasion12. Inhibitors of serine protease inhibitors may also inhibit angiogenesis13, and may be expressed by chondrocytes14–17. TIMP-1, TIMP-3, and the serine protease inhibitors secretory leukocyte proteinase inhibitor (SLPI) and plasminogen activator inhibitor (PAI)-1 are of particular interest due to their specificities for proteases responsible for cartilage matrix degradation, their putative localization in cartilage, and potential anti-angiogenic activities13–17.
TIMP-1 is the dominant cartilage TIMP16, and can inhibit all collagenases. TIMP-3 may have a particularly important role in inhibition of cartilage degradation as it is the only TIMP capable of inhibiting the A-Disintegrin And Metalloproteinase with Thrombospondin-like Repeats (ADAMTS)-4 and -5 enzymes18 which are the major proteases responsible for degradation of aggrecan, the main proteoglycan in cartilage. PAI-1 regulates activation of the serine protease plasmin, which, in turn, contributes to cartilage turnover by activating the pro-MMPs19. SLPI inhibits many cartilage degrading proteases20, and may, in addition, reduce angiogenesis by inhibiting endothelial cell migration13.
It has been suggested that osteoarthritic cartilage may lose the ability to produce protease inhibitors21,22 and that the subsequent imbalance between proteases and their inhibitors may contribute to osteochondral vascularisation23. Other data, however, have indicated that protease inhibitors may be upregulated in OA16,17. Previous work has often focused on individual protease inhibitors, often in animal models that may not adequately reflect human OA, and without reference to osteochondral vascularity. We aimed to address the hypothesis that altered protease inhibitor expression in articular cartilage is associated with increased osteochondral vascularity in human OA.
Methods
Patient recruitment and sample selection
Samples were collected from patients who fulfilled American Rheumatism Association diagnostic criteria for OA24 and were undergoing total knee replacement (TKR) surgery at King's Mill Hospital, Sutton-in-Ashfield, UK. All patients had no additional diagnosis of another rheumatic disease, did not display rheumatoid nodules and had had no previous surgery or fracture to the joint.
Post-mortem (PM) knee samples were collected upon consent from next-of-kin 25. PM samples were from patients who had no history of arthritis or knee pain according to both next-of-kin and medical records, were not diagnosed with OA and had not had surgery or fracture to that joint. PM cases had no Heberden's nodes, rheumatoid nodules or visible osteophytes.
All procedures were approved by Nottingham Research Ethics Committee 1 (05/Q2403/24 and 05/Q2403/61).
Sample processing
All samples were processed on site at King's Mill Hospital by an experienced laboratory technician following standardised procedures. Surgical samples were processed within 2 h of removal or after storage at 4°C overnight, and PM samples were immersed in formalin immediately after removal. PM cases were stored at 4°C prior to dissection, and median time from death to tissue fixation was 51 h (range 47–99 h). The middle one third (weightbearing region) of mid-coronal sections from medial tibial plateaux were fixed in neutral buffered formalin overnight and then decalcified in 10% ethylenediaminetetraacetic acid (EDTA) in 10 mM Tris buffer (pH 6.95) at room temperature prior to wax embedding.
Sample selection
A cohort of 40 patients with OA and a range of osteochondral vascular density suitable for correlational analysis was selected by screening haematoxylin and eosin stained index sections from medial tibial plateaux. Forty cases were selected with cartilage present along at least 70% of the length of the tidemark, 20 without tidemark breaching and 20 with high vascular densities at the osteochondral junction.
In addition, 10 PM cases without tidemark breaching on index sections were selected as a non-arthritic control group.
Histology
Polyclonal anti-TIMP-1 (1:200 dilution) was used with antigen retrieval in 10 mM citrate buffer (pH 6) heated to 90°C for 20 min. Monoclonal anti-TIMP-3 (1:100 dilution) was used with antigen retrieval in sodium phosphate/citrate buffer at 90°C for 10 min. Monoclonal anti-PAI-1 (diluted to 1:50) was used with antigen retrieval in 0.1% trypsin in phosphate buffered saline (PBS) at 37°C for 30 min. Polyclonal anti-SLPI (1:200 dilution) and polyclonal anti-VEGF (1:100 dilution) were used without antigen retrieval26. After application of biotinylated secondary antibody visualization was by avidin–biotin–peroxidase complex, developed with diaminobenzidine (DAB). All antibodies were diluted in 0.03% serum from the host species of the secondary antibody (horse or goat) in PBS, plus (except for SLPI immunohistochemistry) 0.05% bovine serum albumin. Negative controls were incubated in diluent without primary antibody. Immunohistochemistry for each antigen was undertaken on sections from all cases within a single staining run.
One section from each case was stained with haematoxylin and eosin. Safranin-O staining with Fast-green FCF was carried out to enable scoring of chondropathy as previously described3,4,27.
Analysis
Postero-anterior knee radiographs were allocated scores for joint space narrowing and osteophytosis using a standardised line atlas28. All histological scoring and visual analyses were undertaken blinded to disease group, using a Zeiss Axioscop-50 microscope, under ×10, ×20 or ×40 objective lens (Carl Zeiss Ltd, Welwyn Garden City, UK). Where loss of section adhesion precluded scoring for a case, actual numbers analysed are presented with the results.
Osteochondral vascular densities were determined as previously described3,4 as number of vascular channels terminating in the non-calcified cartilage divided by section length (measured by digital caliper, Mitutoyo, Japan). Use of duplicate tissue sections enabled analysis of vascular density as a continuous variable, as all but four OA cases displayed tidemark breaching on one or more sections. Safranin-O stained sections were scored for chondropathy according to the modification of Mankin's method excluding tidemark breaching4,27. This we refer to as chondropathy score.
Scoring systems for abundance of immunoreactivity were devised based on initial qualitative analysis (Table I). Samples were scored for abundance of immunoreactivity in superficial chondrocytes (TIMP-1, TIMP-3 and SLPI), or for the depth to which immunoreactive chondrocytes were localised in articular cartilage (PAI-1 and VEGF). All samples, except for PAI-1, were also categorised for presence or absence of immunoreactivity in deep chondrocytes. For PAI-1, the deep chondrocytes contributed to the overall cartilage scoring, and were not categorised separately.
Table I.
Criteria for scoring immunoreactivity in articular cartilage
| Score | Criteria |
|||
|---|---|---|---|---|
| TIMP-1 and TIMP-3 | SLPI | PAI-1 | VEGF | |
| 0 | No positive superficial chondrocytes | No positive superficial chondrocytes | No positive chondrocytes | No positive superficial chondrocytes |
| 1 | Some, but fewer than 10% of superficial chondrocytes positive | Fewer than 50% of superficial chondrocytes positive | Positive chondrocytes in the superficial zone | Positive chondrocytes in the superficial zone |
| 2 | Thin layer (up to three cells deep) of positive superficial chondrocytes | 50% or more of the superficial chondrocytes positive | Positive chondrocytes in the superficial and intermediate zones | Positive chondrocytes in the superficial and intermediate zones |
| 3 | Thick layer (>3 cells deep) of positive superficial chondrocytes | Positive chondrocytes in the superficial, intermediate and deep zones | ||
Associations between protease inhibitor expression and disease were explored by (1) comparing the extent of immunostaining between OA and PM cases, and (2) by assessing the strength of any associations between immunostaining and chondropathy scores. As it was not possible to definitively exclude early OA from PM cases (see above), initial correlation analyses combined PM and OA groups, thereby giving a wide range of chondropathy scores, and increased power to demonstrate statistical significance. In order to exclude the possibility that observed associations were mediated by disease group rather than chondropathy, we also undertook secondary analyses excluding PM cases.
Statistics
Data were analysed using the Statistical Package for Social Scientists (SPSS) version 14.0.1 (SPSS Inc, Chicago, IL, USA). Chi-square and Mann–Whitney U tests were used to compare binomial outcomes and scores between OA and PM. Associations between variables are expressed as Spearman's correlation coefficients. P < 0.05 was taken to indicate statistical significance. Data are presented as median (Interquartile Range (IQR)).
Materials
Polyclonal anti-TIMP-1 (Ab-6) was from Thermo Fisher Scientific, Warrington, UK. Monoclonal anti-TIMP-3 (clone 136-13H4) was from Merck, Nottingham, UK. Monoclonal anti-PAI-1 (3785) was from Axis-Shield UK, Kimbolton, UK. Polyclonal anti-SLPI (AF1274) was from R&D Systems, Abingdon, UK. Polyclonal anti-VEGF (A20) was from Insight Biotechnology, London, UK. Secondary biotinylated antibodies (BA-1000, BA2001 and BA9500) and the Elite avidin–biotin–peroxidase complex were from Vector Laboratories, Peterborough, UK. Haemotoxylin and eosin were from Raymond A Lamb, Eastbourne, UK. PBS was from VWR, Lutterworth, UK. All other chemicals were from Sigma Aldrich, Poole, UK.
Results
Patient and sample details
OA cases (n = 40, median age 72 (IQR; 67–75) years, 21 females) each reported knee pain for >5 years. Median joint space narrowing score was 5 (IQR; 5 to 5) and osteophyte score was 8 (IQR; 8–10). PM cases (n = 10, five females, median age 67 (IQR; 61–70) years) were younger than OA cases (t = −2.4, P = 0.03), but age was not significantly associated with any of the scores or measurements in this study. Sex distribution was similar in PM and OA (χ2 = 0.30, P = 0.86). Nine patients reported using regular oral non-steroidal anti-inflammatory agents, and two used glucosamine sulphate (all OA). Three cases (two PM, one OA) had been taking systemic corticosteroids for conditions unrelated to joint disease prior to tissue sampling.
OA samples displayed greater chondropathy (modified Mankin score median 8 (IQR; 6–10)) than PM samples (median 4.5 (IQR; 2.5–6), Z = −3.48, P < 0.001) and displayed greater vascular density at the osteochondral junction (OA: median 0.27 (IQR; 0.20–0.64) mm−1; PM: median 0.12 (IQR; 0.07–0.23) mm−1; Z = −2.6, P = 0.01) (Fig. 1).
Fig. 1.

Osteochondral vascularity and superficial cartilage appearances in medial tibial plateaux. Vascular channels (asterisks) breach the deepest tidemark (arrowheads) in a sample from a patient with OA (A), but not in one from a different patient with OA (C), nor from a PM case (E). Cartilage above the regions shown in A and C displays surface irregularity (B and D, respectively), chondrocyte depletion (B), or chondrocyte cloning (D, arrows). Cartilage above the region shown in E displays normal morphology (F). Haematoxylin and eosin stain. Bar = 250 microns.
TIMP-1
TIMP-1 positive chondrocytes were localised to the superficial zone of articular cartilage in all OA samples, but only occasionally in PM controls [Fig. 2(A, B)]. TIMP-1 positive superficial chondrocytes were more abundant in OA than PM, (Z = −3.1, P = 0.002) [Fig. 3(A)]. TIMP-1 expression in superficial chondrocytes was associated with greater chondropathy [Table II, Fig. 3(B)], but was not associated with vascular density. Occasional chondrocytes in the middle and deep zones displayed TIMP-1-immunoreactivity [Fig. 4(A)]. The presence of TIMP-1 positive chondrocytes in the deep zone did not differ between OA (22/39 cases) and PM (4/9 cases, χ2 = 0.42, P = 0.52). Vascular density did not differ between samples with positive deep chondrocytes (median vascular density 0.24 (IQR; 0.09–0.57) mm−1) and those without (median 0.28 (IQR; 0.19–0.57) mm−1; Z = −0.45, P = 0.65).
Fig. 2.

Immunoreactivities for protease inhibitors and VEGF in superficial chondrocytes in OA and PM. Photomicrographs represent OA (A, C, E, G, I) or PM (B, D, F, H, J) cases with median scores for TIMP-1 (A, B), TIMP-3 (C, D), SLPI (E, F), PAI-1 (G, H) or VEGF (I, J). Each immunoreactivity is more abundant in OA than PM. Scale bar = 100 microns.
Fig. 3.

TIMP-1 (A, B), TIMP-3 (C, D), SLPI (E, F) and PAI-1 (G, H) scores were each higher in OA than in PM (A, C, E, G) and were each associated with greater chondropathy (B, D, F, H). A, C, E and G: box and whisker plots. *P < 0.05, **P < 0.01 for differences between PM and OA. B, D, F and H: scatter plots of data from patients with OA (○) and PM controls (▴).
Table II.
Associations of immunoreactivities for protease inhibitors and VEGF with osteochondral vascular density and chondropathy
| Group(s) | TIMP-1 | TIMP-3 | SLPI | PAI-1 | VEGF | |
|---|---|---|---|---|---|---|
| Vascular density | OA + PM | 0.11 | 0.10 | 0.10 | 0.28 | 0.32* |
| OA only | −0.02 | 0.01 | 0.00 | 0.22 | 0.13 | |
| Chondropathy | OA + PM | 0.67** | 0.79** | 0.55** | 0.42** | 0.66** |
| OA only | 0.58** | 0.79** | 0.45** | 0.24 | 0.61** | |
| VEGF | OA + PM | 0.62** | 0.67** | 0.57** | 0.39** | – |
| OA only | 0.55** | 0.67** | 0.45** | 0.25 | – | |
Increased scores for each protease inhibitor and VEGF were associated with higher chondropathy scores. Increased VEGF, but not protease inhibitors, was associated with increased vascular density in the non-calcified cartilage. Increased protease inhibitor expression was associated with greater VEGF expression. Scoring criteria for protease inhibitors and VEGF are given in Table I. Values are Spearman's rank correlation coefficients for all cases (OA + PM), or OA subgroup only (OA). *P < 0.05 **P < 0.01 – significant values are in bold.
Fig. 4.
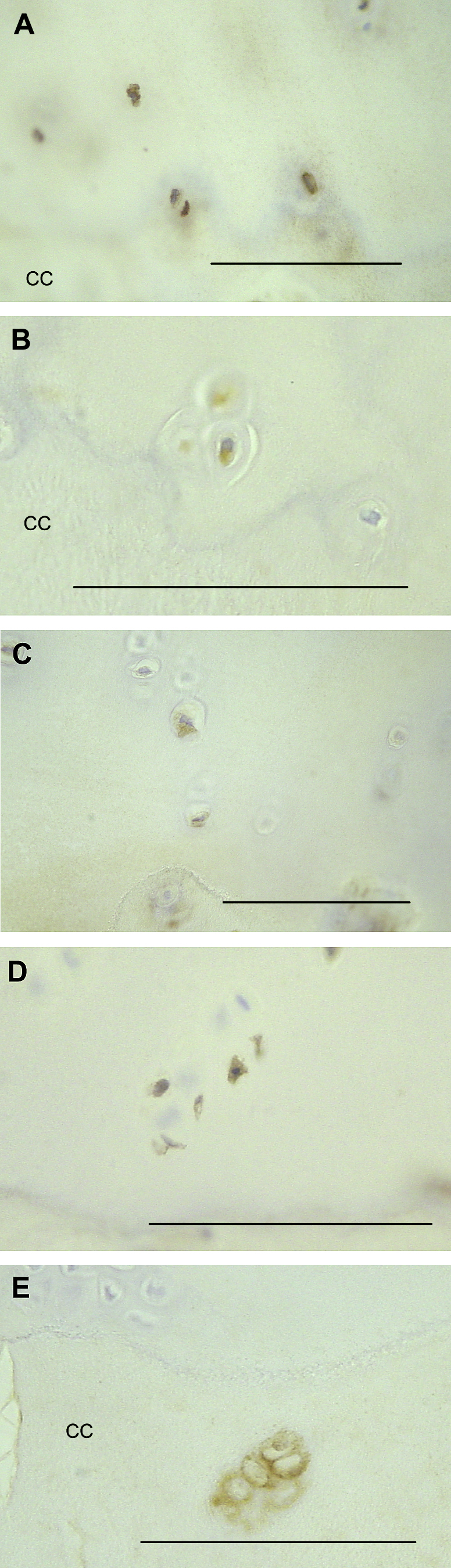
Chondrocytes in the deep and calcified zones of cartilage may display immunoreactivities for protease inhibitors and VEGF. (A) TIMP-1 positive chondrocytes in the non-calcified cartilage close to the tidemark in a patient with OA. (B) TIMP-3 positive hypertrophic chondrocytes in the non-calcified cartilage, just above the tidemark in a patient with OA. (C) SLPI-positive chondrocytes in the deep zone of the non-calcified cartilage of a patient with OA. (D) PAI-1 positive chondrocytes in the deep zone of the non-calcified cartilage of a patient with OA. (E) VEGF-positive chondrocytes in the calcified cartilage of a patient with OA. CC = calcified cartilage. Scale bars = 100 microns.
TIMP-3
TIMP-3-immunoreactivity was occasionally detected in samples from either disease group. TIMP-3-immunoreactivity was localised within and around chondrocytes in the superficial articular cartilage in OA and, less commonly in PM [Fig. 2(C, D)]. TIMP-3 positive superficial chondrocytes were more abundant in OA than PM (Z = −2.6, P = 0.02) [Fig. 3(C)], and were associated with more severe chondropathy [Table II, Fig. 3(D)] but not with vascular density. TIMP-3-immunoreactivity in deep chondrocytes was only occasionally observed [Fig. 4(B)], localised to regions with only a thin layer of overlying cartilage, or where fissures extended into the deep cartilage. Presence of TIMP-3 in the deep chondrocytes did not differ between OA (4/39 cases) and PM (1/9 cases, χ2 = 0.006, P = 0.94). Vascular density did not differ between samples that displayed TIMP-3 positive deep chondrocytes (median 0.22 (IQR; 0.10–1.39) mm−1) and those that did not (median 0.26 (IQR; 0.14–0.56) mm−1; Z = −0.22, P = 0.83).
SLPI
SLPI was most commonly found in superficial chondrocytes [Fig. 2(E, F)], where there was significant upregulation in OA samples compared to PM (Z = −2.8, P = 0.007) [Fig. 3(E)]. SLPI-positive superficial chondrocytes were associated with greater chondropathy [Table II, Fig. 3(F)] but not with vascular density. Expression in the deeper zones of articular cartilage was less extensive, and presence of positive cells in the deep zone [Fig. 4(C)] did not differ between OA (27/39 cases) and PM (7/9 cases, χ2 = 0.55, P = 0.76). Vascular density did not differ between samples that displayed SLPI-positive deep chondrocytes (median 0.27 (IQR; 0.10–0.64) mm−1) and those that did not (median 0.25 (IQR; 0.16–0.45) mm−1; Z = −0.31, P = 0.75).
PAI-1
Most samples displayed PAI-1 immunoreactive chondrocytes near the surface of the articular cartilage [Fig. 2(G, H)], with expression decreasing towards the tidemark, but sometimes extending to include deep chondrocytes [Fig. 4(D)]. The depth of articular cartilage in which PAI-1 positive cells were identified ranged from 1 or 2 cells thick to all chondrocytes being positive down to the tidemark. PAI-1 positive chondrocytes were observed in deeper regions of the articular cartilage in OA than PM (Z = −2.8, P = 0.007) [Fig. 3(G)]. Chondrocyte expression of PAI-1 was associated with greater chondropathy [Table II, Fig. 3(H)], but association with vascular density did not reach statistical significance (r = 0.28, P = 0.054).
VEGF
Most samples displayed VEGF-positive chondrocytes near the surface of the articular cartilage [Fig. 2(I, J)]. The layer of VEGF-positive chondrocytes sometimes extended into the intermediate zone of the cartilage, and isolated VEGF-positive chondrocytes or chondrocyte clusters were occasionally found in the deep zone and the calcified cartilage [Fig. 4(E)]. VEGF-positive chondrocytes were observed in deeper regions of the articular cartilage in OA (median score 1 (IQR; 1–2)) than PM (median 0 (IQR; 0–1)) (Z = −3.7, P < 0.001). Chondrocyte expression of VEGF was associated with greater chondropathy and with higher vascular density in the non-calcified cartilage (Table II, Fig. 5). The depth to which VEGF-positive chondrocytes were localised was associated with greater expression of each protease inhibitor (Table II). Vascular density did not differ between samples that displayed VEGF-positive deep chondrocytes (median 0.25 (IQR; 0.17–0.88) mm−1) and those that did not (median 0.24 (IQR 0.11–0.52) mm−1; Z = −0.69, P = 0.49).
Fig. 5.

Associations between VEGF, vascular invasion and chondropathy. Scatter plots showing that scores of VEGF in superficial chondrocytes increased with increasing (A) chondropathy and (B) density of vascular channels breaching the tidemark. OA (○) and PM controls (▴).
Discussion
We have shown that osteoarthritic articular cartilage contains chondrocytes that display increased expression of the angiogenic factor VEGF, and also of a variety of protease inhibitors that are known to have anti-angiogenic activity. Chondrocyte expression both of angiogenic and anti-angiogenic factors is greater in cases with more severe osteoarthritic structural change. Although superficial chondrocytes display a clear ability to express anti-angiogenic protease inhibitors in OA, those in the deep zone, the site of osteochondral angiogenesis, did not demonstrate protease inhibitor upregulation. When angiogenic stimuli are increased in OA, this apparent deficiency of deep chondrocytes may permit vascular invasion into the articular cartilage.
Our findings provide immunolocalisation data that extend previous studies on TIMP-1 and PAI-1 mRNA upregulation by superficial chondrocytes in human OA29. We show similar upregulation of two further anti-angiogenic protease inhibitors in OA, TIMP-3 and SLPI, with comparable expression patterns to TIMP-1. Our data do not support a role for these protease inhibitors in maintaining the avascular status of normal cartilage, as they were largely absent from PM cases. Constitutive factors within the normal cartilage matrix may be more important in maintaining its avascularity than is expression of anti-angiogenic protease inhibitors by chondrocytes. Protease inhibitor upregulation alongside increased angiogenic factor expression however may moderate osteochondral angiogenesis, for example by inhibiting receptor shedding or growth factor activation.
The tidemark in normal cartilage provides a barrier to macromolecule diffusion and mass transport. Chondropathy and tidemark breaching by vascular channels in OA may, however, permit growth factors such as VEGF from superficial cartilage to access the osteochondral junction. Stimulation of osteochondral angiogenesis by VEGF produced by superficial chondrocytes may explain our observed association between VEGF expression and osteochondral vascular density. Such an angiogenic drive from superficial chondrocytes would be additional to that from growth factors (including VEGF) produced within the subchondral bone marrow spaces9,30,31.
By contrast, osteochondral vascular densities in OA were not associated with protease inhibitor expression, indicating that protease inhibitors generated by superficial chondrocytes may not gain access to the deeper articular cartilage in concentrations that can prevent vascular invasion, particularly in the context of the increased angiogenic drive of OA.
Previous studies of VEGF expression by articular chondrocytes have not demonstrated associations with osteochondral angiogenesis8, and the relatively weak association in the current study (R = 0.32) was only observed when OA and PM groups were combined. Chondrocyte-derived VEGF may play roles other than supporting angiogenesis, and has been reported to increase MMP expression in immortalized chondrocytes32. We observed a coordinated upregulation of protease inhibitors and VEGF. VEGF may therefore contribute to OA severity both as an angiogenic factor and as a regulator of matrix turnover.
Our study was limited by a number of factors. Definition of a normal control group is difficult for a condition such as OA, where onset may be insidious, and pathological change may occur in the absence of symptoms. PM cases provide a source of non-arthritic tissues, but may display macroscopic or microscopic changes consistent with OA even in the absence of symptoms prior to death33. We attempted to avoid inclusion of cases with OA in the PM group, based on clinical history, macroscopic and microscopic appearances, but cannot exclude the possibility that some patients had early or mild OA. We also cannot exclude the possibility that protease antigenicity declined in PM samples prior to tissue harvesting. However, immunoreactivities in deep chondrocytes were similar in both PM and OA cases, increased protease immunoreactivities were associated with greater chondropathy within OA samples (Table II), and extent of immunoreactivity was not significantly associated with time from death to tissue processing (data not shown).
We used a targeted approach to identify protease inhibitors that may be downregulated in OA, focusing on those for which there was prior evidence of expression by chondrocytes or ability to inhibit angiogenesis. Future work should address changes in cartilage matrix and expression of other important anti-angiogenic factors. Osteochondral angiogenesis is accompanied by sensory nerve growth3. Inhibition of osteochondral angiogenesis may therefore improve symptoms by structural disease modification. It may be unnecessary for a disease modifying drug to prevent all changes of chondropathy, if preventing the structural changes leading to pain can be achieved alone. We speculate that facilitating the anti-angiogenic effects of protease inhibitors may achieve such a goal.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We are grateful to all the patients, the orthopaedic surgeons and the Bereavment Centre at the Sherwood Forest Hospitals NHS Trust for providing clinical material. We also thank the histopathology personnel at the King's Mill Hospital for their help with processing of tissue samples. We are grateful to AstraZeneca for financial support to assist the creation of the tissue repository used in this study, DM was supported by arc grant 14851.
References
- 1.Griffioen A.W., Molema G. Angiogenesis: potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacol Rev. 2000;52:237–268. [PubMed] [Google Scholar]
- 2.Walsh D.A., Haywood L. Angiogenesis: a therapeutic target in arthritis. Curr Opin Investig Drugs. 2001;2:1054–1063. [PubMed] [Google Scholar]
- 3.Suri S., Gill S.E., Massena de Camin S., Wilson D., McWilliams D.F., Walsh D.A. Neurovascular invasion at the osteochondral junction and in osteophytes in osteoarthritis. Ann Rheum Dis. 2007;66:1423–1428. doi: 10.1136/ard.2006.063354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walsh D.A., Bonnet C.S., Turner E.L., Wilson D., Situ M., McWilliams D.F. Angiogenesis in the synovium and at the osteochondral junction in osteoarthritis. Osteoarthritis Cartilage. 2007;15:743–751. doi: 10.1016/j.joca.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 5.Walsh D.A. Angiogenesis and arthritis. Rheumatology (Oxford) 1999;38:103–112. doi: 10.1093/rheumatology/38.2.103. [DOI] [PubMed] [Google Scholar]
- 6.Lane L.B., Bullough P.G. Age-related changes in the thickness of the calcified zone and the number of tidemarks in adult human articular cartilage. J Bone Joint Surg Br. 1980;62:372–375. doi: 10.1302/0301-620X.62B3.7410471. [DOI] [PubMed] [Google Scholar]
- 7.Lane L.B., Villacin A., Bullough P.G. The vascularity and remodelling of subchondrial bone and calcified cartilage in adult human femoral and humeral heads. An age- and stress-related phenomenon. J Bone Joint Surg Br. 1977;59:272–278. doi: 10.1302/0301-620X.59B3.893504. [DOI] [PubMed] [Google Scholar]
- 8.Pufe T., Petersen W., Tillmann B., Mentlein R. The splice variants VEGF121 and VEGF189 of the angiogenic peptide vascular endothelial growth factor are expressed in osteoarthritic cartilage. Arthritis Rheum. 2001;44:1082–1088. doi: 10.1002/1529-0131(200105)44:5<1082::AID-ANR188>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 9.Hashimoto S., Creighton-Achermann L., Takahashi K., Amiel D., Coutts R.D., Lotz M. Development and regulation of osteophyte formation during experimental osteoarthritis. Osteoarthritis Cartilage. 2002;10:180–187. doi: 10.1053/joca.2001.0505. [DOI] [PubMed] [Google Scholar]
- 10.Eisenstein R., Sorgente N., Soble L.W., Miller A., Kuettner K.E. The resistance of certain tissues to invasion: penetrability of explanted tissues by vascularized mesenchyme. Am J Pathol. 1973;73:765–774. [PMC free article] [PubMed] [Google Scholar]
- 11.Shibakawa A., Yudoh K., Masuko-Hongo K., Kato T., Nishioka K., Nakamura H. The role of subchondral bone resorption pits in osteoarthritis: MMP production by cells derived from bone marrow. Osteoarthritis Cartilage. 2005;13:679–687. doi: 10.1016/j.joca.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 12.Moses M.A., Sudhalter J., Langer R. Identification of an inhibitor of neovascularization from cartilage. Science. 1990;248:1408–1410. doi: 10.1126/science.1694043. [DOI] [PubMed] [Google Scholar]
- 13.Sugino T., Yamaguchi T., Ogura G., Kusakabe T., Goodison S., Homma Y. The secretory leukocyte protease inhibitor (SLPI) suppresses cancer cell invasion but promotes blood-borne metastasis via an invasion-independent pathway. J Pathol. 2007;212:152–160. doi: 10.1002/path.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davidson R.K., Waters J.G., Kevorkian L., Darrah C., Cooper A., Donell S.T. Expression profiling of metalloproteinases and their inhibitors in synovium and cartilage. Arthritis Res Ther. 2006;8:R124. doi: 10.1186/ar2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacoby A.S., Melrose J., Robinson B.G., Hyland V.J., Ghosh P. Secretory leucocyte proteinase inhibitor is produced by human articular cartilage chondrocytes and intervertebral disc fibrochondrocytes. Eur J Biochem. 1993;218:951–957. doi: 10.1111/j.1432-1033.1993.tb18452.x. [DOI] [PubMed] [Google Scholar]
- 16.Kevorkian L., Young D.A., Darrah C., Donell S.T., Shepstone L., Porter S. Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum. 2004;50:131–141. doi: 10.1002/art.11433. [DOI] [PubMed] [Google Scholar]
- 17.Treadwell B.V., Pavia M., Towle C.A., Cooley V.J., Mankin H.J. Cartilage synthesizes the serine protease inhibitor PAI-1: support for the involvement of serine proteases in cartilage remodeling. J Orthop Res. 1991;9:309–316. doi: 10.1002/jor.1100090302. [DOI] [PubMed] [Google Scholar]
- 18.Kashiwagi M., Tortorella M., Nagase H., Brew K. TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5) J Biol Chem. 2001;276:12501–12504. doi: 10.1074/jbc.C000848200. [DOI] [PubMed] [Google Scholar]
- 19.Milner J.M., Elliott S.F., Cawston T.E. Activation of procollagenases is a key control point in cartilage collagen degradation: interaction of serine and metalloproteinase pathways. Arthritis Rheum. 2001;44:2084–2096. doi: 10.1002/1529-0131(200109)44:9<2084::AID-ART359>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 20.Williams S.E., Brown T.I., Roghanian A., Sallenave J.M. SLPI and elafin: one glove, many fingers. Clin Sci (Lond) 2006;110:21–35. doi: 10.1042/CS20050115. [DOI] [PubMed] [Google Scholar]
- 21.Brown R.A., Weiss J.B. Neovascularisation and its role in the osteoarthritic process. Ann Rheum Dis. 1988;47:881–885. doi: 10.1136/ard.47.11.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stephens R.W., Ghosh P., Taylor T.K. The pathogenesis of osteoarthrosis. Med Hypotheses. 1979;5:809–816. doi: 10.1016/0306-9877(79)90041-0. [DOI] [PubMed] [Google Scholar]
- 23.Joronen K., Kahari V.M., Vuorio E. Temporospatial expression of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in mouse antigen-induced arthritis. Histochem Cell Biol. 2005;124:535–545. doi: 10.1007/s00418-005-0011-2. [DOI] [PubMed] [Google Scholar]
- 24.Altman R., Asch E., Bloch D., Bole G., Borenstein D., Brandt K. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 1986;29:1039–1049. doi: 10.1002/art.1780290816. [DOI] [PubMed] [Google Scholar]
- 25.Walsh D.A., Wilson D. Post-mortem collection of human joint tissues for research. Rheumatology (Oxford) 2003;42:1556–1558. doi: 10.1093/rheumatology/keg406. [DOI] [PubMed] [Google Scholar]
- 26.Haywood L., McWilliams D.F., Pearson C.I., Gill S.E., Ganesan A., Wilson D. Inflammation and angiogenesis in osteoarthritis. Arthritis Rheum. 2003;48:2173–2177. doi: 10.1002/art.11094. [DOI] [PubMed] [Google Scholar]
- 27.Mankin H.J., Dorfman H., Lippiello L., Zarins A. Biochemical and metabolic abnormalities in articular cartilage from osteo-arthritic human hips. II. Correlation of morphology with biochemical and metabolic data. J Bone Joint Surg Am. 1971;53:523–537. [PubMed] [Google Scholar]
- 28.Nagaosa Y., Mateus M., Hassan B., Lanyon P., Doherty M. Development of a logically devised line drawing atlas for grading of knee osteoarthritis. Ann Rheum Dis. 2000;59:587–595. doi: 10.1136/ard.59.8.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walter H., Kawashima A., Nebelung W., Neumann W., Roessner A. Immunohistochemical analysis of several proteolytic enzymes as parameters of cartilage degradation. Pathol Res Pract. 1998;194:73–81. doi: 10.1016/S0344-0338(98)80073-3. [DOI] [PubMed] [Google Scholar]
- 30.Hulejova H., Baresova V., Klezl Z., Polanska M., Adam M., Senolt L. Increased level of cytokines and matrix metalloproteinases in osteoarthritic subchondral bone. Cytokine. 2007;38:151–156. doi: 10.1016/j.cyto.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 31.Lisignoli G., Toneguzzi S., Pozzi C., Piacentini A., Grassi F., Ferruzzi A. Chemokine expression by subchondral bone marrow stromal cells isolated from osteoarthritis (OA) and rheumatoid arthritis (RA) patients. Clin Exp Immunol. 1999;116:371–378. doi: 10.1046/j.1365-2249.1999.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pufe T., Harde V., Petersen W., Goldring M.B., Tillmann B., Mentlein R. Vascular endothelial growth factor (VEGF) induces matrix metalloproteinase expression in immortalized chondrocytes. J Pathol. 2004;202:367–374. doi: 10.1002/path.1527. [DOI] [PubMed] [Google Scholar]
- 33.Walsh D.A., Yousef A., McWilliams D.F., Hill R., Hargin E., Wilson D. Evaluation of a Photographic Chondropathy Score (PCS) for pathological samples in a study of inflammation in tibiofemoral osteoarthritis. Osteoarthritis Cartilage. 2009;17:304–312. doi: 10.1016/j.joca.2008.07.016. [DOI] [PubMed] [Google Scholar]