

Abstract

A model for the spiroiminal moiety of marineosins A and B was prepared starting from methylvalerolactone. Addition of vinylmagnesium bromide, protection of the alcohol, and reaction of the vinyl ketone with a protected pyrrole-2-carbonitrile N-oxide gave an isoxazoline. Hydrogenolysis of the N-O bond with Raney nickel gave a keto imine that cyclized to a hemi-iminal. O-Methylation, acid-catalyzed cleavage of the TES group and spiroiminal formation, and deprotection completed a seven-step synthesis.
Fenical and co-workers recently reported the isolation of the cytotoxic spiroiminals marineosins A (1) and B (2) from a marine-derived Streptomyces-related actinomycete (see Scheme 1).1 The structures were assigned by analysis of the NMR spectra and the stereochemistry was assigned from the NOESY spectra. These compounds are clearly members of the prodigiosin family of bacterial pigments.2 It is noteworthy that marineosins A (1) and B (2) differ in stereochemistry at both C-7 and C-8. Analysis of models and molecular mechanics calculations suggest that the isomers of the marineosins at the spiroiminal center (C-8) are much less stable than marineosins A and B because of steric interactions between the methoxy group and the adjacent pyrrole that is part of the macrocycle in the two isomers that were not isolated. The major isomer, marineosin A (1) inhibits human colon carcinoma HCT-116 with an IC50 of 0.5 μM. Testing in the NCI 60 cell line panel showed considerable selectivity against melanoma and leukemia cell lines.
Scheme 1.
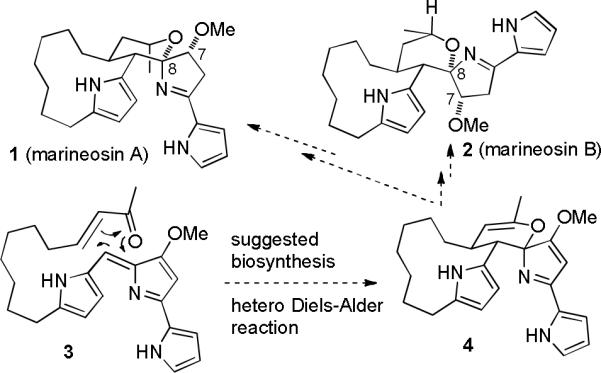
Structures of Marineosins A and B and Biosynthesis Suggested by Fenical
Fenical suggested that the marineosins might be biosynthesized by an intramolecular hetero Diels–Alder reaction of 3 to give 4, which would then be reduced to give marineosins A and B. However, this biosynthetic pathway seemed unlikely to us since it would require a six-electron oxidation of a likely precursor, undecylprodigiosin (5) (See Scheme 2) to form 3, followed by a four-electron reduction of 4 to give marineosins A and B. Lindsley recently reported that the conversion of 3 (prepared by an eight-step route) to 4 could not be achieved in the laboratory.3
Scheme 2.

Alternative Biosynthesis of the Marineosins
The gene cluster responsible for the biosynthesis of undecylprodigiosin (5) and butyl-meta-cycloheptylprodigiosin (6) in Streptomyces coelicolor has been sequenced (see Scheme 2).4 A single enzyme RedG, which is probably a non-heme iron-dependent dioxygenase, appears to convert 5 to 6 by oxidation of the CH2 group to a radical or cation that undergoes an intramolecular Friedel-Crafts reaction to the pyrrole to form 6. A related enzyme could easily oxidize 5 at the adjacent carbon to give radical 7, which could add to the exocyclic double bond to give macrocyclic radical 8. Radical 8 could undergo a facile 1,5-hydride shift to give macrocycle 9 with the radical at the side chain carbon adjacent to the methyl group. This radical is close to the enzyme active site that generated radical 7 and could therefore be easily oxidized to form macrocyclic alcohol 10. 2H-Pyrrole 10 could undergo a facile 1,5-sigmatropic hydride shift to form 1H-pyrrole 11, which could cyclize to form marineosins A and B. However, cyclization of 11 to 1 and 2 would require the loss of aromaticity of pyrrole 11. Alternatively, a 1,5-sigmatropic hydride shift from 10 could also give 3H-pyrrole 12, which should rapidly cyclize to form spiroaminal 13. Tautomerization of the enamine to an imine would give marineosins A (1) and B (2). This scheme is appealing because only a single enzyme would be needed to convert undecylprodigiosin (5) to the marineosins.
There are numerous examples of spiroaminals and many examples of iminals, but the spiroiminal moiety of the marineosins appears to be unprecedented. We therefore chose to start our synthetic planning for the marineosins by exploring routes to the spiroiminal moiety. We were particularly interested in routes in which the dihydropyrrole ring of the spiroiminal was formed before the tetrahydropyran because this approach is related to the biosynthesis proposed in Scheme 2. Our retrosynthesis is shown in Scheme 3. The marineosins should be available by spiroiminal formation from 14, which should be readily available from keto isoxazoline 15 by hydrogenolysis of the N-O bond over Raney nickel with spontaneous formation of the hemi-iminal5 and then O-methylation. Isoxazoline 15 will be prepared by nitrile N-oxide cycloaddition to the vinyl ketone prepared by addition of vinylmagnesium bromide to lactone 16 and protection of the secondary alcohol.
Scheme 3.
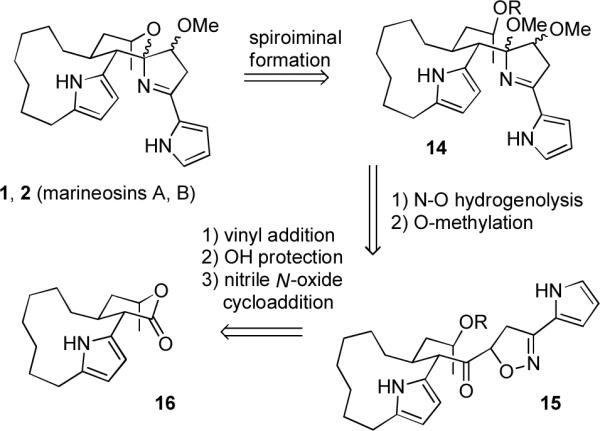
Retrosynthesis of the Marineosins
We chose to investigate this sequence with the readily available model lactone 17, which lacks the macrocyclic ring of 16 (see Scheme 4). Addition of vinylmagnesium bromide to lactone 17 afforded the known hydroxy ketone 18 in 85% yield,6 which was protected as its triethylsilyl ether to give enone 19 in 96% yield. The pyrrole ring is relatively unstable7 and susceptible to methylation so we initially investigated the reaction of 19 with benzonitrile N-oxide. Reaction of 19, benzaldehyde oxime, NCS, and Et3N in THF at −78 to 25 °C provided isoxazoline 20a in 78% yield. Hydrogenolysis (1 atm) over Raney nickel 2800 in MeOH for 45 min afforded hemi-iminal 21a as a mixture of isomers. Methylation with NaH and MeI in THF at 25 °C gave methyl ether iminal 22a in 58% yield from 20a.
Scheme 4.

Preparation of Iminal 22
Treatment of 22a with 2 M aqueous hydrochloric acid in 1:1 THF/CH3CN hydrolyzed the triethylsilyl ether and effected loss of methanol to give the desired spiroiminals 24a (41%), 25a (13%), and 27a (12%) and the undesired pyrrole 23a (8%) (see Scheme 5). Solutions of either pure 25a or 27a equilibrated to an identical 3:1 mixture of 25a and 27a in CDCl3 (containing adventitious HCl) over 2 to 3 weeks, establishing that these two compounds have the identical relative stereochemistry at C-4 and C-7 and differ only at the iminal center C-5. The major isomer 24a equilibrated in 2 weeks to give a 19:1 mixture of 24a and 26a, establishing that these two compounds have the identical relative stereochemistry at C-4 and C-7. We were unable to convert methoxypyrrole 23a to the desired spiroiminals 24a–27a under a variety of acidic conditions, which resulted in either no reaction or extensive decomposition. This suggests that methoxypyrrole 11 may not be an intermediate in the biosynthesis of the marineosins (see Scheme 2).
Scheme 5.
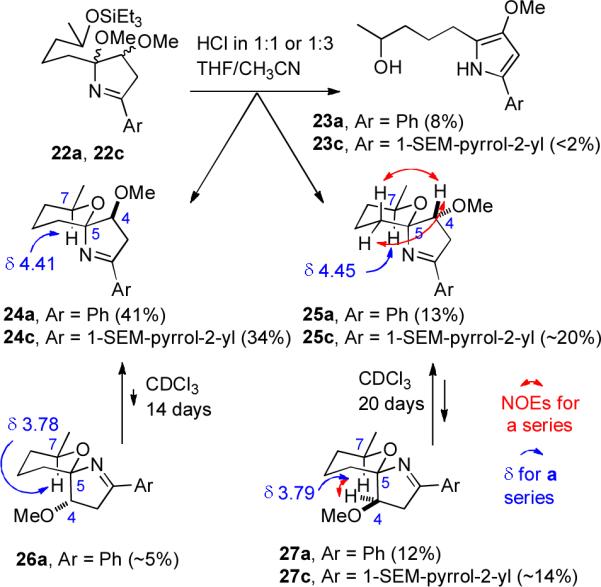
Preparation of Spiroiminals 24a,c, 25a,c and 27a,c
The presence of the imine double bond makes the formation of spiroiminals from 22 quite different from that of spiroketals and spiroaminals. Protonation of the methoxy group and loss of MeOH would give a cumulene (C=N+=C) in a five-membered ring. Spiroiminal formation may proceed by tautomerization to an enamine analogous to 13, which can easily lose MeOH. Other pathways are presented in the Supporting Information. Equilibration of the spiroiminals can also occur by formation of enamines or by protonation on the imine nitrogen and ring opening of the dihydropyrrole ring to give a six-membered oxocarbenium ion. Remarkably, very little pyrrole 23a was formed from 22a or during the equilibration of the spiroiminals.
The structure of 27a was established by an NOE between proton H-4 adjacent to the methoxy group and proton H-7 adjacent to the methyl group, as shown. These protons are too far apart in the other three isomers for an NOE to be observed. This NOE also allowed us to assign the structure of 25a, which differs from 27a only in the stereochemistry at the spiroiminal center. The structure of 25a was confirmed by NOEs between proton H-4 adjacent to the methoxy group and the adjacent CH2 group on the tetrahydropyran group as shown. The axial nitrogen deshields proton H-7 adjacent to the methyl group in 24a (δ 4.41) and 25a (δ 4.45), which absorbs much further downfield than H-7 in 26a (δ 3.78) and 27a (δ 3.79) with an axial carbon.8 As expected, the major isomer 24a has no NOEs between the protons on the tetrahydropyran ring and those on the dihydropyrrole ring.
Having developed a route to phenyl substituted spiroiminals, we turned our attention to the problem of a pyrrole substituent. Treatment of 19 with the oxime of pyrrole-2-carboxaldehyde in THF with NCS and Et3N at −78 °C afforded isoxazoline 20b in 62% yield. Hydrogenolysis of the N-O bond over Raney Ni proceeded uneventfully to give 21b, but methylation with NaH and MeI methylated the pyrrole NH in addition to the two hydroxy groups to give 22d in 41% yield from 20b. Therefore the pyrrole NH needed to be protected.
A SEM-protected pyrrole appeared to be compatible with the reagents in this sequence.9 N-SEM-Pyrrole-2-carboxaldehyde10 was converted to the oxime in 84% yield with NH2OH•HCl and NaOAc in aqueous MeOH. However, treatment of this oxime with NCS and enone 19 in THF at −78 °C gave 20c in <30% yield. Fortunately, reaction of this oxime in CH2Cl2 at 25 °C with 5% aqueous NaOCl11 generated the nitrile N-oxide, which added cleanly to enone 19 to give 20c in 73% yield. Both hydrogenolysis over Raney Ni and methylation now proceeded uneventfully to give 22c in 42% yield from 20c. Treatment of 22c with 2 M aqueous hydrochloric acid in 1:3 THF/CH3CN hydrolyzed the triethylsilyl ether and effected loss of methanol to give the protected spiroiminal 24c (34%), an inseparable equilibrium 3:2 mixture of spiroiminals 25c and 27c (34%), and the undesired pyrrole 23c (<2%). The stereochemistry of these spiroiminals was assigned by analogy to 24a to 27a, which have virtually identical 1H and 13C NMR spectra in the aliphatic region.
Deprotection of 24c with TBAF and molecular sieves in THF at 60 °C for 3 hours provided spiroiminal 24b in 54% yield (see Scheme 6). A similar sequence on the 3:2 mixture of 25c and 27c afforded a 7:3 mixture of 25b and 27b in 56% yield. The stereochemistry of 24b to 27b was again assigned by analogy to 24a to 27a.
Scheme 6.
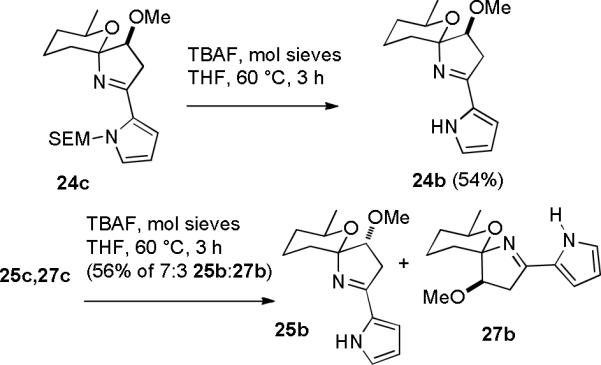
Preparation of Spiroiminals 24b, 25b and 27b
Comparison of the structures of model spiroiminals 24–27 with those of marineosins A (1) and B (2) is complicated by the trans-fused macrocyclic ring of the marineosins that forces the methyl group into an axial conformation, whereas it is equatorial in the models lacking the fused macrocyclic ring. Spiroiminal 25b has the same relative stereochemistry as marineosin B (2). Although 27b is only slightly less stable than 25b, examination of models and molecular mechanics calculations suggest that steric interactions between the methoxy group and macrocyclic ring will make the stereoisomer of marineosin that corresponds to 27b much less stable than marineosin B (2). Marineosin A (1) has the same relative stereochemistry as 26a, which was only observed as a minor isomer in equilibrium with 24a. However, steric interactions with the axial methyl group should strongly favor the spiroiminal isomer with an axial nitrogen (marineosin A) over that with the stereochemistry corresponding to 24a, which must exist in the conformation with a large axial carbon and also has steric interactions between the methoxy group and the macrocyclic ring. Therefore, treatment of 14 with acid should afford mainly marineosins A (1) and B (2) rather than the other two, presumably less stable, isomers.
In conclusion, we have developed a seven-step route to spiroiminals 24b, 25b, and 27b from lactone 17 that should be amenable to the conversion of lactone 16 to marineosins A (1) and B (2). A plausible biosynthesis from undecylprodigiosin (5) to the marineosins is proposed that only requires a single two-electron oxidation.
Supplementary Material
Acknowledgment
We are grateful to the National Institutes of Health (GM-50151) for support of this work.
Footnotes
Supporting Information Available: Complete experimental procedures, spectral assignment of 24a–27a, discussion of spiroiminal formation and equilibration, and copies of 1H and 13C NMR spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Boonlarppradab C, Kauffman CA, Jensen PR, Fenical W. Org. Lett. 2008;10:5505–5508. doi: 10.1021/ol8020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Fürstner A. Angew. Chem., Int. Ed. 2003;42:3582–3603. doi: 10.1002/anie.200300582. [DOI] [PubMed] [Google Scholar]; (b) Williamson NR, Fineran PC, Gristwood T, Chawrai SR, Leeper FJ, Salmond GPC. Future Microbiol. 2007;2:605–618. doi: 10.2217/17460913.2.6.605. [DOI] [PubMed] [Google Scholar]
- 3.Aldrich LN, Dawson EW, Lindsley CW. Org. Lett. 2010;12 doi: 10.1021/ol100034p. ASAP. [DOI] [PubMed] [Google Scholar]
- 4.(a) Cerdenõ AM, Bibb MJ, Challis GL. Chem. Biol. 2001;8:817–829. doi: 10.1016/s1074-5521(01)00054-0. [DOI] [PubMed] [Google Scholar]; (b) Williamson NR, Fineran PC, Leeper FJ, Salmond GPC. Nat. Rev. Microbiol. 2006;4:887–899. doi: 10.1038/nrmicro1531. [DOI] [PubMed] [Google Scholar]; (c) Mo S, Sydor PK, Corre C, Alhamadsheh MM, Stanley AE, Haynes SW, Song L, Reynolds KA, Challis GL. Chem. Biol. 2008;15:137–148. doi: 10.1016/j.chembiol.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 5.Hydrogenolysis of a keto isoxaoline afforded a compound whose spectral data are in agreement with those of a hemi-iminal. See compound 12 in: Andersen SH, Sharma KK, Torssell KBG. Tetrahedron. 1983;39:2241–2245.
- 6.Cohen N, Banner BL, Blount JF, Weber G, Tsai M, Saucy G. J. Org. Chem. 1974;39:1824–1833. [Google Scholar]
- 7.Cycloadditions with the nitrile N-oxide derived from benzaldehyde oxime can be carried out at room temperature, whereas those with the nitrile N-oxide derived from the oxime of pyrrole-2-carboxaldehyde must be carried out at −70 °C. See: Ghabrial SS, Thomsen I, Torssell KBG. Acta Chem. Scand. B. 1987;B41:426–434.
- 8.For a similar effect by an axial oxygen in spiroketals, see: Doubský J, Šaman D, Zedník J, Vašíčková S, Koutek B. Tetrahedron Lett. 2005;46:7923–7926.
- 9.Jolicoeur B, Chapman EE, Thompson A, Lubell WD. Tetrahedron. 2006;62:11531–11563. [Google Scholar]
- 10.Muchowski JM, Solas DR. J. Org. Chem. 1984;49:203–205. [Google Scholar]
- 11.Lee GA. Synthesis. 1982:508–509. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.