

Abstract
Database search algorithms are the primary workhorses for the identification of tandem mass spectra. However, these methods are limited to the identification of spectra for which peptides are present in the database, preventing the identification of peptides from mutated or alternatively spliced sequences. A variety of methods has been developed to search a spectrum against a sequence allowing for variations. Some tools determine the sequence of the homologous protein in the related species but do not report the peptide in the target organism. Other tools consider variations, including modifications and mutations, in reconstructing the target sequence. However, these tools will not work if the template (homologous peptide) is missing in the database, and they do not attempt to reconstruct the entire protein target sequence. De novo identification of peptide sequences is another possibility, because it does not require a protein database. However, the lack of database reduces the accuracy. We present a novel proteogenomic approach, GenoMS, that draws on the strengths of database and de novo peptide identification methods. Protein sequence templates (i.e. proteins or genomic sequences that are similar to the target protein) are identified using the database search tool InsPecT. The templates are then used to recruit, align, and de novo sequence regions of the target protein that have diverged from the database or are missing. We used GenoMS to reconstruct the full sequence of an antibody by using spectra acquired from multiple digests using different proteases. Antibodies are a prime example of proteins that confound standard database identification techniques. The mature antibody genes result from large-scale genome rearrangements with flexible fusion boundaries and somatic hypermutation. Using GenoMS we automatically reconstruct the complete sequences of two immunoglobulin chains with accuracy greater than 98% using a diverged protein database. Using the genome as the template, we achieve accuracy exceeding 97%.
Database search algorithms, such as Sequest (1), Mascot (2), and InsPecT (3), are the primary workhorses for the identification of tandem mass spectra. However, these methods are limited to the identification of spectra for which peptides are present in the database. It is well recognized that curated protein databases are, at best, an imperfect template for the extant peptides. For example, peptides arising from novel splice forms or fusion proteins would be difficult to identify using most protein databases.
Recent developments have extended the identifications to peptides that have diverged from the database entry. By allowing divergence, the methods enable the identification of small-scale mutations, and post-translational modifications, albeit with some loss of sensitivity (4–7). Among these tools, MS-Blast is able to determine a homologous protein in the related species but does not report the (diverged) protein in the target organism. The other tools consider variations, including modifications and mutations, in reconstructing the target sequence. However, these tools will not work if the template (homologous peptide) is missing in the database or comes from a novel splice form. In addition, these tools do not attempt to reconstruct the entire protein target sequence. De novo identification of peptide sequences (8, 9) is another possibility and does not require a protein database. However, these methods are prone to error.
The issue of discovering spliced peptides (more generally, eukaryotic gene structures) has been investigated using a combination of approaches, loosely termed proteogenomics. Often, these approaches start by creating specialized databases of splice forms, combining evidence from protein (e.g. NCBI nr (10)) and cDNA sequencing (11–13). To discover novel splicing events, the tools also search databases derived directly from the genome such as a six-frame translation or a compact encoding of multiple putative splicing events (14–17). For example, Castellana et al. (15) achieved this by constructing a database, represented as a graph (16), containing many putative exons and exon splice junctions.
However, this approach also has its shortcomings. The putative gene models are constructed based on prior assumptions about splice junctions and proximal exons. In addition, recent genomic discoveries point to extensive structural variation in the genome in the form of large-scale deletions, insertions, inversions, and translocations on the genome that might fuse different genic regions or create nonstandard splice forms (18, 19). Indeed, many cancers are characterized by such large-scale mutations of the genome (20). Other examples of variation that confound standard database identification techniques are immunoglobulins and antibodies. Here, recombination events fuse disparate regions of the genome, often inserting nontemplated sequence and creating many novel gene structures in every individual. The common theme in all of the scenarios described is that it is not possible to maintain all possible encodings in a database to allow for a standard proteogenomic search.
In this study, we sought to determine whether the imperfect template provided by the genome can be still used as a basis for peptide (and protein) identification. We are motivated in our approach by the work of Bandeira et al. (21), who were able to sequence monoclonal antibodies de novo, making no use of a database at all. In their method, an all-to-all comparison of spectra allowed the creation of spectral contigs, similar to sequence contigs in shotgun sequencing projects. The sequences of the spectral contigs were determined de novo. Using full antibody sequences as references, they were able to order the contigs and infer the missing sequence. Because the construction and sequencing of the contigs was performed completely de novo, Bandeira et al. (21) were able to sequence highly divergent proteins or proteins for which there is no database. However, the ordering of the sequenced contigs relies on a database of full antibody sequences for mapping. Sequences that cannot be mapped to an antibody in the database may be discarded. In contrast, the templates used in our method are not full proteins, but substrings of proteins, such as exons, which are combinatorially chained together to best explain the spectrometric evidence.
Liu et al. (22) have developed Champs, a method for sequencing a divergent protein using a homologous protein database. In their method, a single reference protein was chosen, and the de novo interpretations of spectra were mapped to the reference. They were able to sequence a protein with high accuracy using a reference protein with only 77% similarity to the target. Although Champs is able to map peptides that differ from the reference by one or two amino acids, it does not look for large insertions or deletions in the target sequence, as in a novel splice form. In our work, use of the database as an incomplete template lends additional confidence to the target sequencing without substantially limiting the ability to identify diverged sequences.
Here, we describe a novel method for template proteogenomics, implemented in the tool GenoMS. GenoMS takes as input a collection of spectra (acquired from multiple protease digests) and a collection of imperfect templates and constraints (defined under Experimental Procedures). It returns a target protein sequence. At the heart of the approach is a novel method of extending a target amino acid sequence by recruiting and aligning spectra that match it partially. By using spectral data sets with multiple protease digests, we are able to identify many overlapping peptides. We then align the overlapping spectra and produce an extended consensus spectrum. We are able to extend 89% of the target amino acid sequences. More than 40% of these extensions are three or more amino acids.
We test the performance of GenoMS in reconstructing monoclonal antibody sequences. Antibodies are an interesting test case because of their highly variable nature and because no complete antibody database exists. They are composed of four polypeptide chains: two identical heavy chains and two identical light chains (Fig. 1). An antibody's preference and efficiency in the detection and removal of encountered antigens is heavily dependent on its amino acid sequence. Consequently, antibodies are extremely diverse. A principal way in which antibody diversity is achieved is through genome rearrangement of the germline locus (Fig. 1). An antibody's heavy chain comprises four gene segments; a variable (V) segment, a diversity (D) segment, a joining (J) segment, and a constant (C) segment. Likewise, the light chain is composed of three gene segments: a V segment, a D segment, and a C segment. Each segment is chosen from potentially hundreds present in the genome, and many combinations of gene segments may be joined. Imprecise boundaries with the possible insertion of additional nucleotides allow the creation of many sequences from a single germline locus. Somatic hypermutation also plays a role in achieving antibody diversity. Although antibody sequence may be determined by sequencing the DNA of the source cell line, few direct protein-sequencing options exist when the source is unavailable or for ensuring antibody integrity. The antibody structure provides enough complexity to serve as a test case for template proteogenomics.
Fig. 1.

An overview of the production of a mature immunoglobulin. Bottom, the mature immunoglobulin protein structure contains two identical light chains and two identical heavy chains. The germline heavy-chain and light-chain loci (top) contain many different gene segments. During heavy-chain gene rearrangement, in B-cell differentiation, one V, one D, and one J gene segment are combined. For light-chain gene formation, a V and a J gene segment are combined. The combined VDJ or VJ segments are joined by splice junction to a constant region.
Using the technique of extending the peptide sequence without reference to a database, we are able to reconstruct the full protein sequence for the antibody raised against the B- and T-lymphocyte attenuator molecule (aBTLA1) (21). We also test our approach by using an available data set of spectra acquired using multiple protease digests for bovine serum album (BSA). The sequence of BSA is determined using the bovine genome as a template database. Both chains of aBTLA were sequenced using unrearranged gene segments as templates. An independent reconstruction of the aBTLA heavy chain was performed using the unrearranged heavy-chain genomic locus as a template.
EXPERIMENTAL PROCEDURES
Our goal is to reconstruct the target amino acid sequence, using a chain of templates. A template is defined as an amino acid sequence that may be present in the target protein, although possibly in a mutated or modified form. The target protein might contain multiple templates chained together. We provide additional abstraction to model constraints on the templates. First, the user can specify a partial order t1 → t2 to enforce that template t1 must precede t2 in the chain. Second, the user can provide mutual exclusion constraints on (t1, t2), a pair of templates, to enforce that only one of the two templates is in the chain. For example, in antibody sequences, all V, D, J, and C genes are templates. The constraints help specify the ordering of V, D, J, and C genes, and the exclusion of any pair of genes from the same class (e.g. V).
An anchor is defined as a substring of a template that is present in the target with no mutations. Each template may contain zero or more anchors. Fig. 2 describes an overview of our algorithm. GenoMS takes a collection of tandem MS spectra as input, along with a set of templates and their constraints, and requires at least one anchor sequence. It outputs a target protein sequence using a chain of templates as a guide. There are three stages: template-chain selection, anchor extension, and sequence construction, all described below.
Fig. 2.

The template proteogenomic method reconstructs a target protein sequence using tandem mass spectra and a template database in three steps: template-chain selection, anchor extension, and sequence construction. The template database specifies ordering and mutual exclusion constraints between templates. A set of templates is selected that obeys these constraints based on peptides identified on them. Anchors are peptides identified by searching spectra against the template database. Anchors are extended by aligning spectra that overlap the anchor. Finally, the sequence is reconstructed by merging the extended anchor sequences.
Template-Chain Selection
We create a custom database of all template sequences and use the database search tool InsPecT to search all spectra against the database (3) (see supplemental Methods). The best templates to use as guides are those that show a good match to the spectra. Coverage[t] is defined as the number of amino acids on t that were confirmed by the database search. Peptides that appear in multiple templates count toward the coverage of all of them. This reuse is eliminated in the next step. The goal of the template-chain selection phase is to select a chain of templates with maximum coverage while satisfying all constraints.
To find the chain of templates, we define a graph in which the nodes are templates. There are two sets of edges. Directed edges t1 → t2 model the ordering constraints, while a set of undirected edges, (t1,t2) ∈ Ef, models the exclusion. In addition to the constraints specified by the user, we also create forbidden edges between templates that share more than the minimum of two peptides or half of the peptides belonging to one of the templates. A chain T = {t1,t2,…,tk} is valid if (ti,tj ∉)Ef for all ti,tj in T, and t1 → t2 → … → tk. The objective is to compute a valid chain so that Σi=1k is maximized.
Solving this problem generally is hard. We use a heuristic method based on dynamic programming to find a valid chain (see supplemental Methods). Let Vj denote the maximum score of a valid chain ending at tj, and Tj denote the corresponding chain. Then,
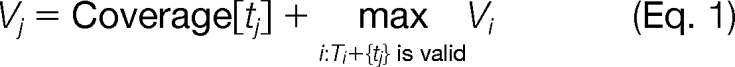 |
and Tj is constructed by chaining tj to the optimal Ti. The template-chain determined by this heuristic is considered for subsequent stages of GenoMS. For an antibody, the template chain will often link V(D)JC together in that order. However, all templates are not required. Missing templates will be filled in by anchor extension. Second, we are not limited to a single chain. A variant of this heuristic can output multiple chains when needed (e.g. alternative splicing).
Anchor Identification and Extension
Recall that the template chain was created by connecting templates that were well covered by target peptides. For each selected template in the chain, anchors are created by merging overlapping peptides. Anchors are ordered by their position on the chain. Spectra not annotated using the database search are reconsidered in the subsequent phases of the algorithm.
In the second step, we extend the sequence of each anchor. Before extension, all spectra are first clustered to reduce the overall number of spectra and improve spectrum quality (23). The clustered spectra are converted to prefix residue mass (PRM) spectra (8). A PRM spectrum is represented by a list of mass values, and a PRM-score function ϕ that computes the likelihood that a mass value is a PRM. The procedure for extending the sequence of an anchor is shown in ExtendAnchor below.
procedure EXTENDANCHOR
-
Recruit PRM spectra that overlap the N/C-terminal of the anchor
repeat
1.1 Align the recruited spectra
1.2 Construct a consensus spectrum from the aligned spectra
1.3 Recruit spectra that ovelap the N/C-terminal of the consensus spectrum
while
Sequence the consensus spectrum
Recruiting PRM Spectra
All spectra that do not contribute to an anchor and have not already been recruited are examined for overlap with each anchor. Any spectra that have been recruited in previous rounds to the same terminus of the anchor are eligible for recruitment in subsequent rounds of recruitment for the terminus as well. We determine overlap by using a modified spectral alignment method (24). When aligning a spectrum to an anchor, we allow the spectrum to only partially overlap the anchor (Fig. 3). Because the extended target sequence is determined by aligning the recruited spectra, it is critical to reduce false-positive recruitment and maintain enough coverage to reliably extend the sequence.
Fig. 3.

The partial alignment of a spectrum to an anchor sequence and consequent extension of the anchor sequence. Top, a PRM spectrum is shown with a partial alignment to the theoretical PRM spectrum of an anchor. The C terminus of the spectrum is not aligned. Bottom, the overhanging peaks enable the extension of the anchor sequence by two amino acids (QT).
We consider three parameters: the minimum additive score of the spectral alignment, Q (24); the minimum number of overlapping peaks, β, for a spectral alignment to be considered, and the exact number of spectra recruited, NS. Q could be learned by the algorithm independently for each experiment by looking at the alignment score of spectra identified by InsPecT (supplemental Methods). We tested for the dependence on β and NS using a training set of 206 uniformly selected anchor ends from the aBTLA heavy-chain sequence (supplemental Figs. 1 and 2). Values β = 4 and NS = 5 were chosen to balance the accuracy (fraction of recruited spectra that are correct) and sensitivity (fraction of true spectra recruited).
The recruited spectra and the anchor sequence must then be aligned. The sequence helps to anchor the spectral alignment, and the spectral alignment is then used to produce a consensus extension of the sequence. We do this using hidden Markov models (HMMs).
Multiple Spectrum Alignment
Profile HMMs are a popular tool for performing multiple sequence alignment (25). We alter the scheme slightly to perform multiple spectrum alignment. The use of HMMs for scoring peptide-spectrum alignments has previously been proposed (26). A novel part of our approach is that the HMM is not static, but is updated by model surgery, as we extend the anchor sequence.
Recall that the anchor sequence can also be interpreted as a list of PRMs [m1,m2,m3,…]. For example, the anchor VCAK corresponds to the PRM list [0,99.07,259.21,330.28,458.32]. Intuitively, the HMM is an automaton that generates these PRMs (Fig. 4A). In the absence of noise, we have a set of Match states (M1,M2,…). The automaton starts in Match state M1. In each Match state Mi, the PRM mi is emitted, followed by a transition to the next Match state. An HMM is formally described by a 5-tuple M = (Ω,A,B,π,Σ), where Ω is the set of states. The HMM is initially in state ωi ∈ Ω according to the distribution π. In state ωi, M emits a symbol o ∈ Σ according to the distribution Bi,o, and transitions to state ωj, according to the transition probability Ai,j. To model measurement errors, the Match state Mj outputs a mass m according to BMj,m ∼ N(mj,σ), where σ (i.e. S.D.) is obtained by empirically measured instrument accuracy. Noise peaks are modeled by Insert states in between each adjacent pair of Match states, with the emission probabilities defined by
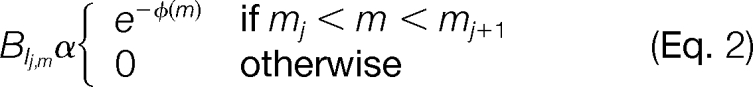 |
Missing peaks in spectra are modeled by moving from a Match state to a Delete state, where no symbol is emitted. The transition probabilities Ai,j are initialized to favor match transitions, and penalize delete transitions (supplemental Methods). All parameters are updated at each iteration using a Bayesian approach described in the next section.
Fig. 4.

The profile HMM used to align spectra and produce a consensus spectrum. A, the spectrum profile HMM derived from the anchor “VCAK” after aligning four spectra, all of which are aligned to the state I5. The peaks aligned to that state are shown and suggest a candidate Match state at mass 515.33 Da. B, the same HMM after we performed model surgery to add the new Match state.
In this generative model, each spectrum is produced by traversing a (hidden) path through the states of the HMM. Reconstructing the most likely path is equivalent to aligning the spectrum to the HMM and can be determined using the Viterbi algorithm (27). An Insert state is created after the final Match state for C-terminal extension or before the initial Match state for N-terminal extension. Model surgery is performed to generate additional Match states from these terminal Insert states, which are used to reconstruct the template extension. The procedure for learning the HMM by aligning recruited spectra is shown in AlignSpectrum below.
procedure ALIGNSPECTRUM
Create an initial HMM using the anchor
-
For each recruited spectrum, S
2.1 Align S to the model using the Viterbi algorithm
2.2 Update model parameters
2.3 Perform model surgery
Updating Model Parameters
Transitions Ai,j are updated according to
 |
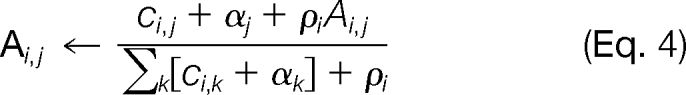 |
where ci,j is the number of aligned spectra with transition from ωi to ωj and ρi is the “learning rate.” Low values of ρ favor the observed transitions, whereas high values of ρ favor the current transition probability. αj is the pseudocount for ωj, described empirically by
 |
To update BMj,m, the mean is recomputed in each step by using spectral PRMs that were emitted in state Mj. The variance remains unchanged.
Model Surgery
The initial HMM is constructed using the anchor PRMs. The aligned spectra overlap only partially. The PRMs preceding the N-terminal Match state (or succeeding the C-terminal Match state in the case of right extension) are emitted by Insert states. The observed masses emitted by an Insert state cluster around certain PRM values, specifically at the preceding (or succeeding) PRMs of the target sequence. Model surgery is used create a Match state that can emit the cluster of PRMs (see Fig. 4B). In this way, the HMM is extended to better represent the target sequence.
Let WI denote the set of mass values emitted by Insert state I. Consider a subset W′ ⊆ WI. Let μW′ and σW′ denote the mean of the values in W′ and the S.D., respectively. Define
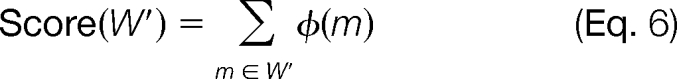 |
We compute
 |
Note that the computation can be done efficiently by sorting the mass values, and looking at intervals.
If Score(W*) exceeds the minimum PRM score ϕ(m) for any spectrum, we add a new Match state with mean μW*, along with the corresponding Delete and Insert states (Fig. 4B). All spectra are realigned to the new HMM.
Building a Consensus Spectrum and Extending the Anchor Sequence
The HMM, once learned from the recruited spectra, is used to produce a consensus spectrum. The consensus PRM spectrum is produced by finding the maximum likelihood path constrained to those paths that begin at the initial Match or Delete state and end at the final Match or Delete state. The peak emissions of this path, omitting noise peaks emitted from Insert states, produce the consensus spectrum. Each peak in the consensus spectrum is associated with a peak score. The PRM score for the mass emitted from state Mi is
where WMi is the set of peaks aligned to state Mi and WDi is the set of spectra aligned to state Di. λ is a constant. The peak scores are likelihoods, and λ is the likelihood that a true peak will not be observed. We chose λ to be the average score of a PRM in the data set. The consensus spectrum is then used as the anchor for subsequent rounds of extension. The sequence of the final consensus spectrum, once no more spectra can be recruited, is determined de novo by constructing a spectrum graph allowing edges for single- and double-amino acid masses (28). The sequence is then recovered from the highest scoring path in the spectrum graph.
Protein Sequence Reconstruction
Once anchors have been extended until no spectra can be recruited, the extended anchor sequences are merged into a single protein sequence. If confident overlap between extended anchor sequences exists, then the sequences are merged (supplemental Methods). Each extended anchor sequence is considered for merging with all eligible anchors; that is, the C-terminal extension of an anchor can only be merged with the N-terminal of another anchor. Likewise, if template ordering is provided, anchors can only be merged in accordance with the template order constraints.
Confidence Estimates
The output of GenoMS is a sequence of mass intervals, some of which are represented by amino acids in the final sequence, whereas others, which can be explained by two AAs, are presented as masses. Each interval is inferred from a pair of adjacent PRMs. All spectra that were used to create an anchor or extend an anchor can be mapped to the sequence of intervals. We compute the confidence of a mass-interval as the fraction of overlapping spectra with PRMs mapping to the two adjacent PRMs. Fig. 5 illustrates the computation of the site-wise confidence from the set of mapped spectra.
Fig. 5.

A set of spectra is shown overlapping a region of the predicted sequence. A spectrum supports a mass interval in the predicted sequence if both adjacent PRMs to the interval are matched in the spectrum. The confidence of each mass interval is the fraction of overlapping spectra that support the interval (with pseudocounts). The PRMs of the overlapping spectra that are necessary to support the mass interval corresponding to “C” are circled.
For spectra that are identified using InsPecT and are used for anchor creation, all mass intervals that are overlapped are also considered supported. In some cases, the confidence estimate is conservative, because many spectra may support a larger mass interval that contains the correct one. For example, the first spectrum in Fig. 5 supports the large interval SR, but is not counted toward either mass interval because it is missing the PRM between S and R. We use pseudo-counts 1 and 1.5 for the number of supporting and overlapping spectra, respectively.
Genomic Templates
Immunoglobulins are an excellent candidate for template proteogenomics, with templates selected from translated germline segments. However, for other applications of protein sequencing, such as gene annotation, a protein template database may be difficult to produce. To handle these situations, GenoMS also accepts genomic sequence as input. It automatically generates templates and constraints as follows: the template database is a six-frame translation of a gene locus, with each open reading frame (ORF) describing a template. Templates that overlap or are on different strands are mutually exclusive. Templates are ordered according to their genomic coordinates. Once the template database and constraint are produced, the same algorithm is used to reconstruct the target. For flexibility, we do not consider splice-junction signals in selecting template boundaries. However, users have the option to input customized template and constraint files.
The output of the GenoMS from genomic templates is the sequence of the target protein, as well as the genomic coordinates of the exons or gene segments selected as templates. In this way, the precise exon and splice boundaries for a gene may be discovered. Because template proteogenomics does not require the template genome to be an exact match for the target protein, it is possible to sequence the target protein of one species using the genomic template database derived from the genome of a related and more comprehensively studied species.
Constructing a Divergent Sequence Database
To assess the ability of GenoMS to sequence more divergent proteins, we alter the template database to contain sequences with less similarity to the target. We construct the divergence sequence database from the known target protein sequence appended with the Mouse IPI database (version 3.54), which contains 56,551 proteins once immunoglobulin sequences are removed. We simulated degrees of mutation by replacing regions of the target sequence with nonsense amino acids, “XXXX.” The regions were selected at random positions on the heavy chain, with a normal length distribution, with mean 7, S.D. 2, and a minimum length of four amino acids, similar to the hypervariable complementarity defining regions (29).
Mass Spectrometry Analysis
Spectral data sets derived from aBTLA described in Bandeira et al. (21) were used for evaluating anchor extension and for full antibody sequence reconstruction. The data set consisted of 44,985 tandem MS from the heavy chain and 39,135 tandem MS from the light chain acquired on either an LTQ-Orbitrap or LTQ-FTMS instrument. Heavy-chain samples were prepared using four different protease digestions (trypsin, chymotrypsin, pepsin, and AspN), whereas light-chain samples were prepared with three different proteases (trypsin, chymotrypsin, and AspN). To determine the gene structure of BSA 5,154 tandem MS spectra acquired on an Orbitrap instrument from three digestion conditions using GluC, LysC, and trypsin (22) were used. All spectra were first clustered to reduce the overall number of spectra and improve spectrum quality (23) and converted to PRM spectra (8) (supplemental Methods).
RESULTS
Anchor Extension
Anchor extension is the mainstay of our algorithm. We measured the accuracy and length of extension of arbitrary anchors by GenoMS. Certain discrepancies between the target sequence and the predicted sequence were not considered errors, such as substitutions of amino acids with similar mass or mass shifts caused by common post-translational modifications (supplemental Methods).
From the known sequence of the aBTLA heavy chain, we selected every possible 10 amino acid sequence as an anchor. Q was fixed at 70, which is approximately the score cutoff chosen for the heavy chain. We then performed one round of recruitment, alignment, and reconstruction, as described above. Of the 413 anchors, 89% were extended. The average extension length was 2.56 amino acids. Of the extendable anchors, 41% were extended by three or more amino acids, whereas 12% were extended by five or more amino acids. Across all extensions, 95% of the amino acids were correctly predicted. Errors generally occurred in regions with one or more prolines, which hinders peptide fragmentation. We found that the quality of the extension depends greatly on the quality of the spectra. Beyond the two spectra per extension required by the algorithm, we found no real improvement with increased spectral counts.
The accuracy depends greatly on the position in the extension. Fig. 6 shows the accuracy as a function of the position from the tail of the extension. To explain, consider an anchor that is extended by two amino acids, g1g2, and another that is extended by three amino acids, h1h2h3. The accuracy at position −1, the last position of the extension, considers the accuracy (fraction of residues predicted accurately) of g2 and h3. The accuracy at position −2 is determined by the accuracy of g1 and h2, whereas the accuracy at position −3 is only determined by the accuracy of h1. As expected, the number of predictions decreases from −1 onward, whereas the accuracy increases. The length of extension depends upon the availability of overlapping peptide spectra, which in turn depends upon the protease mixtures. Our results indicate that with a large number of overlapping peptides, the extensions are accurate.
Fig. 6.

The average accuracy of each position in the extension. The accuracy of the extension degrades for positions close to the end of the extension, whereas the number of predictions increases. Each data point is annotated with the total number of anchors extended to that position or further.
Complete Protein Sequence Reconstruction
The International Immunogenetics Information System (IMGT) GENE-DB (30) contains immunoglobulin genes observed in human, mouse, rat, and rabbit. We used the mouse genes in GENE-DB as templates for full protein sequencing. These templates contain sequences that are highly similar but not identical to the specific aBTLA antibody used to acquire spectra. The heavy chain sequence and light chain sequence of the target, determined previously by Edman sequencing, are 443 and 221 AAs in length, respectively. We tested whether GenoMS could reconstruct the aBTLA targets using the tandem MS spectra and the GENE-DB templates.
We constructed a database, IgH-DB, containing all mouse immunoglobulin heavy chain genes in GENE-DB (version 20090331), and a database, IgLK-DB, containing all mouse immunoglobulin light chain genes in GENE-DB (version 20090320). IgLK-DB contained both λ and κ light chain genes. Each V, D, J, and C segment was a template, and constraints were created according to two rules. Templates of the same type (e.g. V segments) were mutually exclusive, and the templates were ordered so that all V segments preceded D segments, D segments preceded J segments, and J segments preceded C segments. IgH-DB contained 479 templates, and IgLK-DB contained 177 templates. Templates with peptide identifications for all protein reconstructions can be found in the supplemental material.
Fig. 7A contains the results of full protein sequencing for the heavy and the light chains. The gray boxes correspond to anchors, annotated by the Genbank GI number and position in the sequence. Arrows extending and linking anchors in Fig. 7 are annotated with the sequence that was determined by anchor-extension and sequence-reconstruction. A red sequence indicates error in extension. If the arrow is continuous from one anchor to the next, then there was sufficient overlap in the extensions to allow merging of anchor sequences. Mass gaps in the consensus spectrum that could not be resolved to a single amino acid are indicated with brackets ([XX]). If the mass gap correctly identifies a pair of amino acids from the sequence, the mass in brackets is replaced by the amino acid symbols in brackets.
Fig. 7.

The accuracy of extension as a function of the position from the end of the extended sequence. A, the aBTLA heavy chain and light chains reconstructed from protein template databases. The gray rectangles are anchors; the arrows, annotated with sequence, are the extended and merged sequences. Text above the anchors indicates the GI number of the template used, and coordinates within or below the anchors indicate their position within the template. Red amino acids were incorrectly predicted. B, the aBTLA heavy chain identified using a genomic template database. The anchors were identified using templates from the locus reverse strand. Anchor ordering and genomic position is annotated with reference to the forward strand. The coordinates of each anchor on the chromosome are shown. Red portions of the anchors are incorrectly incorporated anchor sequence. C, the heavy chain sequence produced by using increasingly divergent templates. The reconstructions at 85, 75, and 65% similarity to the aBTLA heavy chain sequence are shown.
Nearly all (99%) of the heavy chain sequence was recovered with 99% accuracy. One pair of anchors had sufficient overlap to be merged. The C-terminal extension of the third anchor and the N-terminal extension of the fourth anchor also overlap and could be merged by eye but not by our conservative merging criteria. The chain consisted of a template from each V (gene IGHV2–3, GenBank accession number AC090887), J (IGHJ4*01, GenBank accession number V00770), and C (IGHCG1*02, GenBank accession number L35252) segment. No database sequence matched the D gene, but the automated extension could reconstruct much of it. The only error in the D gene sequence, which appears in the C-terminal extension of the first anchor, has one incorrect PRM. “[174]E” has the same mass as the correct sequence “RF.” The incorrect intervals receive a lower site-level confidence (0.62 and 0.73, respectively) than the rest of the sequence. The full sequences with site-level confidence from all reconstructions can be found in the supplemental material. The missing sequence occurred at the N terminus (three AAs) because of modification of the leading glutamine to glutamic acid.
Light chain templates were chosen in the same manner as for the heavy chain. The V, J, and C templates correspond to genes IgKV8–21*01 (GenBank accession number Y15982), IgKJ4*02 (GenBank accession number V00777), and IgKC*01 (GenBank accession number V00807) (Fig. 7A). Sequencing construction determined 96% of the sequence with 99% accuracy.
There was little gap between any of the anchors. A gap of two amino acids between the end of the first anchor and the start of the second anchor could be filled in correctly by inferring the sequence from the template. The N-terminal extension of the first anchor “NN” has the same mass as the correct sequence “DI,” but the internal PRM is off by 1 Da. These two incorrect amino acids get very low site-level confidence (0.08 and 0.16, respectively). Five amino acids are missing between the end of the extension of the second anchor and the start of the third anchor. This gap corresponds to the joining boundary of the V and J gene segments. N-terminal extension of the third anchor is prevented by the incorrect incorporation of “L” during anchor construction as the first AA in the anchor. The third and fourth anchors are directly abutting, with no missed amino acids.
aBTLA Heavy Chain: Genomic Templates
We tested whether the target could be reconstructed in the absence of protein templates. The mouse heavy-chain genomic locus, GenBank accession number NG005838, was used to construct a template database, as described under Genomic Templates. The database contained 87,265 templates. Spectrum identifications were filtered to a 1% false discovery rate. Fig. 7B shows the reconstruction of the heavy chain. We identified seven anchors comprising 95% of the target sequence. Each anchor was identified from a different ORF template on the reverse strand except for the fifth and sixth anchors, which were from the same ORF. Each ORF revealed an exon in the final rearranged immunoglobulin heavy-chain gene. Gaps between anchors, which are sequenced via anchor extension, determine the exact splice boundaries between exons.
The predicted target antibody sequence contained 443 AA, with 98% accuracy. Two pairs of anchors could be merged. The first anchor of the sequence was identical to the first anchor identified against the IMGT database; consequentlym the error at the C-terminal extension of the anchor is also the same. The boundaries of two anchors were misidentified by InsPecT. Four and three amino acids were incorporated into the third and fifth anchors, respectively, and are denoted by the red shaded portions of those anchors. The summed masses of the incorrect amino acids were no more than 1 Da different from the true sequence, but they prevented the N-terminal extension of both anchors. The second anchor's C-terminal extension overlaps the correct portion of the third anchor, but the incorrect anchor boundary prevents merging.
Protein Sequence Reconstruction with Template Divergence
The template databases used contained sequences that were highly similar to the target sequence, presenting us with an easier test case. We tested accuracy by comparing against a diverged template database. Various levels of divergence (based on similarity to the original template) were created by introducing nonsense mutations in the template database (see Experimental Procedures). At each level of similarity, 20 independent divergent database results were averaged in Table I. Q varied between experiments, with a mean of 68.9.
Table I. Target sequence reconstruction length and accuracy at various levels of target divergence.
“Sequence similarity” is the identity of the target sequence to the closest mutated sequence in the database. “No. of peptides” and “No. of anchors” refer to the number of unique peptides and anchors, respectively, identified on the mutated templates. “Target Length” refers to the length of the reconstructed sequence, whereas “Anchor sequence” refers to the fraction recovered from the anchors. “Target accuracy” is the percentage of amino acids predicted correctly. Although the anchor sequence drops rapidly, a significant fraction of the target is reconstructed accurately.
| Sequence similarity | No. of peptides | No. of anchors | Anchor sequence | Target length | Target accuracy |
|---|---|---|---|---|---|
| % | % | ||||
| 95% | 527 | 4 | 90.7 | 429 | 99.1 |
| 90% | 443 | 6 | 83.6 | 406 | 98.3 |
| 85% | 364 | 8 | 77.1 | 402 | 97.6 |
| 80% | 286 | 9 | 68.2 | 375 | 96.9 |
| 75% | 245 | 10 | 62.8 | 368 | 95.1 |
| 70% | 201 | 10 | 56.1 | 337 | 96.1 |
| 65% | 181 | 10 | 52.2 | 324 | 95.3 |
As the similarity of the database to the target decreases, more of the sequence was determined by automated de novo extension (Table I). Three reconstructions with 85, 75, and 65% database similarity to the target sequence are shown in Fig. 7C. Table I demonstrates how the number of peptides decreases with greater target sequence divergence.
The increasing number of anchors indicates the disjointedness of the peptides. Although the accuracy is diminished as the target sequence becomes more divergent, it is never below 95%. As the amount of sequence recovered in anchors decreases (“Anchor Sequence” in Table I), the portion of the sequence recovered by extension increases. In Fig. 7C, the longest extension in the case of 85% similarity is 14 AA, and most of the anchor extensions could be merged. Once the similarity drops to 65%, the longest extension is 22 AA, and fewer extensions could be merged.
Gene Annotation
Liu et al. (22) published an algorithm for sequencing a diverged protein using a homologous protein. The target sequence used was BSA, and purified spectra were derived from three protease digestions. The complete BSA protein consists of 608 AAs; however, the first 25 residues are cleaved as a signal peptide. Therefore, we consider only the 583 AAs following the cleavage site. Liu et al. (22) were able to sequence the target protein with 100% accuracy and over 99% coverage by using a close homolog in sheep (>90% similarity). In contrast, our method does not require a protein reference sequence but can build from the genome directly.
In addition to sequencing the target protein, identifying templates from a genomic template database gives the positions of exons in the gene whose product is the target protein. We sequenced BSA using a database constructed from the bovine genome. We created a genomic template database from the six-frame translation of the BSA locus (GenBank accession number NC_007304.3:91,461,065–91,479,638) containing 559 templates. From the genomic template database, we identified 91% of the sequence with 98% accuracy. We recovered ORFs for 12 exons (Fig. 8). The lack of overlapping spectra prevented the merging of all but one pair of anchors; however, some splice junctions could still be determined. For example, the N-terminal extensions “PCTEDY” and “[LGE]YGFQNA” span the splice junctions between the tenth and eleventh exons and the ninth and tenth exons, respectively. This allows us to determine boundaries of the splice junctions and infer the missing exon sequence from the template.
Fig. 8.

The annotation of the BSA gene using a genomic template database. Twelve exons for the gene are shown, with corresponding extensions. Each anchor is annotated with its genomic coordinates.
DISCUSSION
Since the first sequencing of the human genome in 2001 (31), we have witnessed an explosion in the number of species with partial and fully sequenced genomes. Gene and proteome annotation, however, have not been able to keep pace. Mass spectrometry and advancing computational tools, as a complement to cDNA sequencing, have been shown to greatly improve the accuracy and efficiency of the annotation process (14, 15, 32). At their core, these methods rely on the assumption that the genome is an adequate database for the identification of peptides. It is nearly impossible to create a database that encodes all possible gene splice variants as well as small and large scale genome rearrangements. One alternative is to use de novo methods (8, 33) for peptide and protein sequencing. These algorithms make no such assumptions, but are plagued with low accuracy.
We have presented a novel method for protein sequencing that draws from the strengths of both the database and de novo approaches. Template proteogenomics improves upon prior proteogenomic efforts by eliminating the need for custom databases that anticipate splice junctions and mutations (16). Our method makes use of the genome as an imperfect template and employs de novo techniques to sequence the divergent portions of the protein. By using available sequence information, we are able to increase confidence in the final sequence while not relying on the existence of a complete and accurate database.
Antibodies are highly diverse proteins that have confounded past attempts to construct a complete sequence database. We are able to use known antibody gene segments as templates to sequence proteins with up to 35% sequence divergence from the templates. The utility of the template proteogenomic method for gene annotation has also been demonstrated. From the final protein sequence, we were able to determine many exon boundaries and splice-junctions by constructing a template database from the six-frame translation of the aBTLA heavy-chain locus.
The alignment of overlapping spectra derived from a mixture of proteases lends additional confidence to full protein sequence. However, the errant portions of the alignment provide useful information as well yet are often ignored. Post-translational modifications may be identified by observing both modified and unmodified spectra aligned. Complex protein mixtures containing both modified and unmodified spectra, or spectra from alternatively spliced peptides, may be lost when an alignment is reduced to a consensus spectrum. In these scenarios, the correct output of the template proteogenomic method would be multiple sequences, not a single protein. In future work, template proteogenomics will be extended to capture post-translational modifications and sequence higher complexity samples.
* This research was supported, in whole or in part, by National Institutes of Health Grant P41-RR024851.
 This article contains supplemental Methods and supplemental Figs 1 and 2.
This article contains supplemental Methods and supplemental Figs 1 and 2.
§ Supported by National Science Foundation IGERT Plant Systems Biology training Grant DGE-0504645.
1 The abbreviations used are:
- aBTLA
- antibody raised against the B- and T-lymphocyte attenuator molecule
- PRM
- prefix residue mass
- HMM
- hidden Markov model
- AA
- amino acid.
REFERENCES
- 1.Eng J., McCormack A., J. Y. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein data base. J. Am. Soc. Mass Spectrom 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 2.Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence data bases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 3.Tanner S., Shu H., Frank A., Wang L. C., Zandi E., Mumby M., Pevzner P. A., Bafna V. (2005) InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem 77, 4626–4639 [DOI] [PubMed] [Google Scholar]
- 4.Shevchenko A., Sunyaev S., Loboda A., Shevchenko A., Bork P., Ens W., Standing K. G. (2001) Charting the proteomes of organisms with unsequenced genomes by MALDI-quadrupole time-of-flight mass spectrometry and BLAST homology searching. Anal. Chem 73, 1917–1926 [DOI] [PubMed] [Google Scholar]
- 5.Tsur D., Tanner S., Zandi E., Bafna V., Pevzner P. A. (2005) Identification of post-translational modifications by blind search of mass spectra. Nat. Biotechnol 23, 1562–1567 [DOI] [PubMed] [Google Scholar]
- 6.Han Y., Ma B., Zhang K. (2005) SPIDER: software for protein identification from sequence tags with de novo sequencing error. J. Bioinform. Comput. Biol 3, 697–716 [DOI] [PubMed] [Google Scholar]
- 7.Searle B. C., Dasari S., Wilmarth P. A., Turner M., Reddy A. P., David L. L., Nagalla S. R. (2005) Identification of protein modifications using MS/MS de novo sequencing and the OpenSea alignment algorithm. J. Proteome Res 4, 546–554 [DOI] [PubMed] [Google Scholar]
- 8.Frank A., Pevzner P. (2005) PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem 77, 964–973 [DOI] [PubMed] [Google Scholar]
- 9.Ma B., Zhang K., Hendrie C., Liang C., Li M., Doherty-Kirby A., Lajoie G. (2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 10.Benson D. A., Karsch-Mizrachi I., Lipman D. J., Ostell J., Wheeler D. L. (2008) GenBank. Nucleic Acids Res 36, D25–D30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boguski M. S., Lowe T. M., Tolstoshev C. M. (1993) dbEST–data base for “expressed sequence tags”. Nat. Genet 4, 332–333 [DOI] [PubMed] [Google Scholar]
- 12.Fermin D., Allen B. B., Blackwell T. W., Menon R., Adamski M., Xu Y., Ulintz P., Omenn G. S., States D. J. (2006) Novel gene and gene model detection using a whole genome open reading frame analysis in proteomics. Genome Biol 7, R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menon R., Zhang Q., Zhang Y., Fermin D., Bardeesy N., DePinho R. A., Lu C., Hanash S. M., Omenn G. S., States D. J. (2009) Identification of novel alternative splice isoforms of circulating proteins in a mouse model of human pancreatic cancer. Cancer Res 69, 300–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baerenfaller K., Grossmann J., Grobei M. A., Hull R., Hirsch-Hoffmann M., Yalovsky S., Zimmermann P., Grossniklaus U., Gruissem W., Baginsky S. (2008) Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 320, 938–941 [DOI] [PubMed] [Google Scholar]
- 15.Castellana N. E., Payne S. H., Shen Z., Stanke M., Bafna V., Briggs S. P. (2008) Discovery and revision of Arabidopsis genes by proteogenomics. Proc. Natl. Acad. Sci. U.S.A 105, 21034–21038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanner S., Shen Z., Ng J., Florea L., Guigó R., Briggs S. P., Bafna V. (2007) Improving gene annotation using peptide mass spectrometry. Genome Res 17, 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edwards N. J. (2007) Novel peptide identification from tandem mass spectra using ESTs and sequence data base compression. Mol. Syst. Biol 3, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iafrate A. J., Feuk L., Rivera M. N., Listewnik M. L., Donahoe P. K., Qi Y., Scherer S. W., Lee C. (2004) Detection of large-scale variation in the human genome. Nat. Genet 36, 949–951 [DOI] [PubMed] [Google Scholar]
- 19.Sebat J., et al. (2004) Large-scale copy number polymorphism in the human genome. Science 305, 525–528 [DOI] [PubMed] [Google Scholar]
- 20.Campbell P. J., et al. (2008) Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet 40, 722–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bandeira N., Pham V., Pevzner P., Arnott D., Lill J. R. (2008) Automated de novo protein sequencing of monoclonal antibodies. Nat. Biotechnol 26, 1336–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X., Han Y., Yuen D., Ma B. (2009) Automated protein (re)sequencing with MS/MS and a homologous data base yields almost full coverage and accuracy. Bioinformatics 25, 2174–2180 [DOI] [PubMed] [Google Scholar]
- 23.Frank A. M., Bandeira N., Shen Z., Tanner S., Briggs S. P., Smith R. D., Pevzner P. A. (2008) Clustering millions of tandem mass spectra. J. Proteome Res 7, 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pevzner P. A., Dancík V., Tang C. L. (2000) Mutation-tolerant protein identification by mass spectrometry. J. Comput. Biol 7, 777–787 [DOI] [PubMed] [Google Scholar]
- 25.Durbin R., Eddy S., Krogh A., Mitchison G. (1998) Biological Sequence Analysis Cambridge University Press, Cambridge, UK [Google Scholar]
- 26.Wan Y., Yang A., Chen T. (2006) PepHMM: a hidden Markov model based scoring function for mass spectrometry data base search. Anal. Chem 78, 432–437 [DOI] [PubMed] [Google Scholar]
- 27.Viterbi A. (1967) Error bounds for convolutional codes and an asymptotically optimum decoding algorithm. IEEE Trans. Inf. Theory 13, 260–269 [Google Scholar]
- 28.Dancík V., Addona T. A., Clauser K. R., Vath J. E., Pevzner P. A. (1999) De novo peptide sequencing via tandem mass spectrometry. J. Comput. Biol 6, 327–342 [DOI] [PubMed] [Google Scholar]
- 29.MacCallum R. M., Martin A. C., Thornton J. M. (1996) Antibody-antigen interactions: contact analysis and binding site topography. J. Mol. Biol 262, 732–745 [DOI] [PubMed] [Google Scholar]
- 30.Lefranc M. P., Giudicelli V., Ginestoux C., Bodmer J., Müller W., Bontrop R., Lemaitre M., Malik A., Barbié V., Chaume D. (1999) IMGT, the international ImMunoGeneTics data base. Nucleic Acids Res 27, 209–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Venter J. C., et al. (2001) The sequence of the human genome. Science 291, 1304–1351 [DOI] [PubMed] [Google Scholar]
- 32.Gupta N., et al. (2008) Comparative proteogenomics: combining mass spectrometry and comparative genomics to analyze multiple genomes. Genome Res 18, 1133–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bandeira N., Clauser K. R., Pevzner P. A. (2007) Shotgun protein sequencing: assembly of peptide tandem mass spectra from mixtures of modified proteins. Mol. Cell Proteomics 6, 1123–1134 [DOI] [PubMed] [Google Scholar]